Abstract
Vindoline and catharanthine are the major alkaloids of Catharanthus roseus and are extracted in large quantities to prepare the pharmaceutically important Vinca type alkaloids vincaleukoblastine, vincristine and navelbine. The higher yield of vindoline relative to catharanthine makes it an attractive substrate for developing new chemistry and adding value to the plant. In this context, we have reacted vindoline with a selection of electrophiles among which benzoquinone. Conditions were developed to optimize the synthesis of a mono-adduct, of five bis-adducts, and of tri-adducts and tetra-adducts, several of these adducts being mixtures of conformational isomers. Copper(II) was added to the reactions to promote reoxidation of the intermediate hydroquinones and simplify the reaction products. The structures were solved by spectroscopic means and by symmetry considerations. Among the bis-isomers, the 2,3-diadduct consists of three unseparable species, two major ones with an axis of symmetry, thus giving a single set of signals and existing as two different species with indistinguishable NMR spectra. The third and minor isomer has no symmetry and therefore exhibits nonequivalence in the signals of the two vindoline moieties. These isomers are designated as syn (minor) and anti (major) and there exists a high energy barrier between them making their interconversion difficult. DFT calculations on simplified model compounds demonstrate that the syn-anti interconversion is not possible at room temperature on the NMR chemical shift time scale. These molecules are not rigid and calculations showed a back-and-forth conrotatory motion of the two vindolines. This “windshield wiper” effect is responsible for the observation of exchange correlations in the NOESY spectra. The same phenomenon is observed with the higher molecular weight adducts, which are also mixtures of rotational isomers. The same lack of rotations between syn and anti isomers is responsible for the formation of four tri-adducts and of seven tetra-adducts. On a biological standpoint, the mono adduct displayed anti-inflammatory properties at the 5 μM level while the di-adducts and tri-adducts showed moderate cytotoxicity against Au565, and HeLa cancer cell lines.
1. Introduction
Vindoline 1 is by far the most abundant alkaloid of Catharanthus roseus [1]. The molecule per se does not show any significant biological activity, but some combinations of vindoline with pharmacophores do present a suitable level of activity [2,3]. Vindoline is part of the so-called antileukemic Vinca alkaloids and, as such, has received considerable attention in coupling reactions with cleavamine type alkaloids. This approach was first proposed by Professor Atta-ur-Rahman, who suggested the use of catharanthine as the partner of vindoline in 1967 [4,5]. A breakthrough was made with Potier’s discovery that the Polonovski reaction was an efficient means of coupling vindoline and catharanthine to afford anhydrovinblastine en route to the other Vinca alkaloids [6]. Vindoline and catharanthine are extracted on an industrial scale in a process aiming at preparing the pharmacologically important navelbine. VLB and VCR are simultaneously obtained. Since the amount of vindoline exceeds the amount of catharanthine, it is of interest to find new uses for vindoline.
Vindoline is present in several bisindoles and in a few other natural products such as vindolicine [7], vindogentianine [8] and bannucine [9], for example. The biosynthesis of these molecules is based on the high nucleophilic character of C-10, which we found reactive towards aldehydes, ketals, orthoesters and quinones inter alia. In this article, we wish to describe the reaction with benzoquinone, which, depending on stoichiometry, gives a variety of adducts.
2. Results
The addition of nucleophiles to 1,4-benzoquinone is not a simple reaction. The initially formed products are dihydroquinones, which can be reoxidized by excess quinone to give substituted benzoquinones. Depending on time, reaction conditions and the stoichiometry of the reagents, the reaction may proceed further and yield a di-adduct, a tris-adduct and a tetrasubstituted benzoquinone (Scheme 1). The addition reaction of amines and proteins with benzoquinone was studied long ago, with emphasis on the oxido-reduction properties of the adducts in biological media [10]. An excellent account of the possibilities offered by the addition of thio-nucleophiles to benzoquinones has been published by Katritzky et al. [11]. The reaction usually works under acid catalyst and recently triflic acid was proposed as a reagent of choice selectively giving mono adducts [12]. Depending on reaction time and stoichiometry, mono- or bis-adducts were obtained with Indium triflate in water [13].
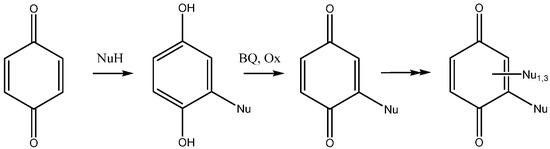
Scheme 1.
The addition of nucleophiles to benzoquinone (BQ = benzoquinone, Ox is an oxidant).
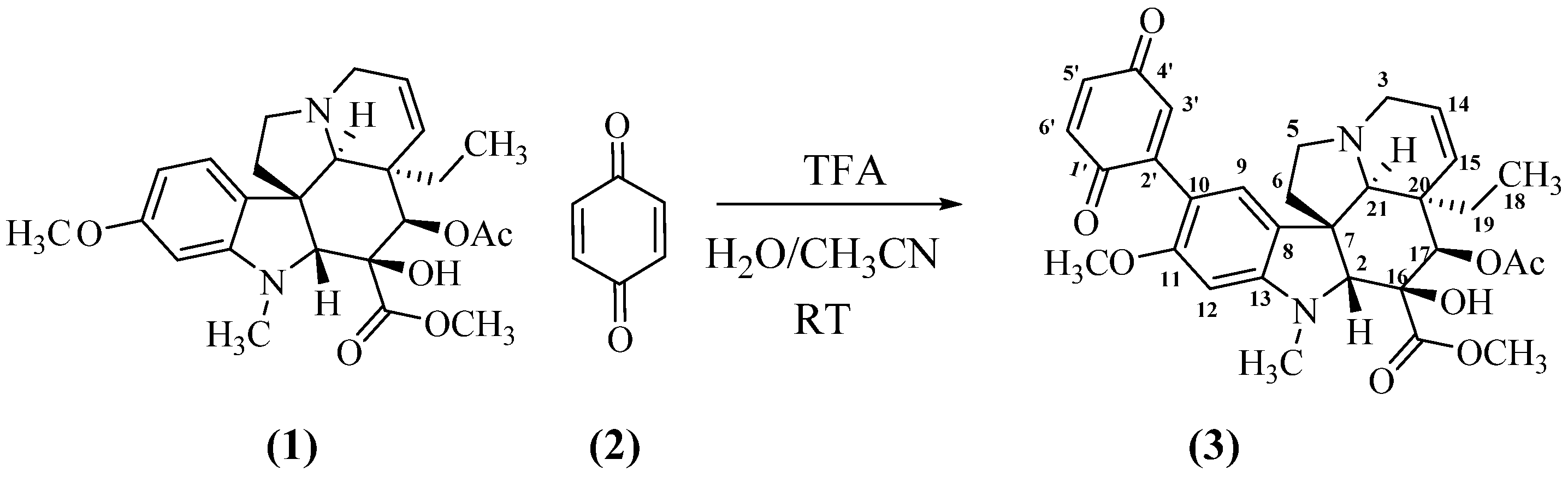
2.1. Preparation of the Mono-Adduct 3 of Vindoline and Benzoquinone
In a first round of experiments, vindoline 1 was mixed with benzoquinone 2 in aqueous acetonitrile and nothing occurred (Table 1, entry 1). Addition of a base (NaOH) had no effect (entry 2). The reaction started to proceed with an acid catalyst, HCl (entry 3) or TFA (entry 4). After optimization of reaction conditions, mono adduct 3 was obtained in a 95% yield, when a 4 to 1 ratio of benzoquinone to vindoline and a large excess of TFA were used (entry 8, Scheme 2). Compound 3 (see Appendix A) was a high melting point blueish solid, the MS of which corresponded to a 1:1 adduct ([M + H]+ at m/z 563.2393, calc. for C31H35N2O8 m/z = 563.2388). The UV showed the nearly unaffected three band pattern of vindoline (221, 250 and 309 nm) in overlap with the π-π* of benzoquinone, the n-π* band being shifted to 542 nm. The 1H NMR spectrum of 3 (see Supplementary Materials) displayed all the signals of the vindoline moiety except those of the aromatic part where one of the aromatic protons was missing. The observation of two aromatic singlets at δ 7.04 and 6.34 ppm confirmed that the reaction expectedly took place at C-10 of vindoline. Three protons of the quinone formed the expected three spin system around 6.80 ppm, while the 13C NMR spectrum showed signals for the two carbonyls of the quinone at δ 186.5 and 188.0 ppm. All the other signals were assigned through the regular 2D NMR experiments (see Section 5).

Table 1.
Products obtained with vindoline 1 addition to benzoquinone 2. Reaction conditions: all reactions were performed on 200 mg 1 (0.44 mmol) except entries 1, 2 (20 mg) and 9–11, 13, 14 (150 mg); catalyst (CuCl2): entries 1–8: none, entries 9, 10: 0.6 mmol, entries 11, 12: 6.6 mmol, entries 13, 14: 3.3 mmol. Reaction time was 24 h, except 10, 12 (8 h) and 13, 14 (48 h). TFA (1 mL) was added in reactions 6–14 and 0.3 mL in entries 4, 5. Solvent was MeOH (entries 1, 2), CH3CN:H2O 7:1 (entries 3, 9) or CH3CN:H2O 1:2 (entries 10–14).
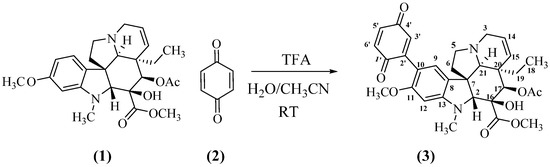
Scheme 2.
Formation of the mono-adduct of vindoline and benzoquinone.
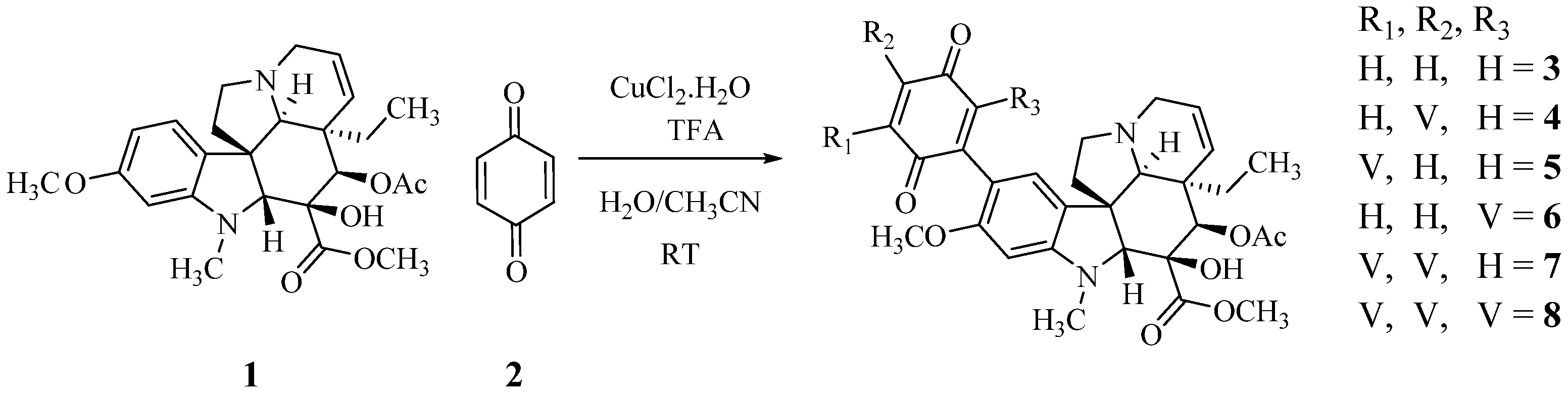
2.2. Preparation of Higher Stoichiometry Adducts
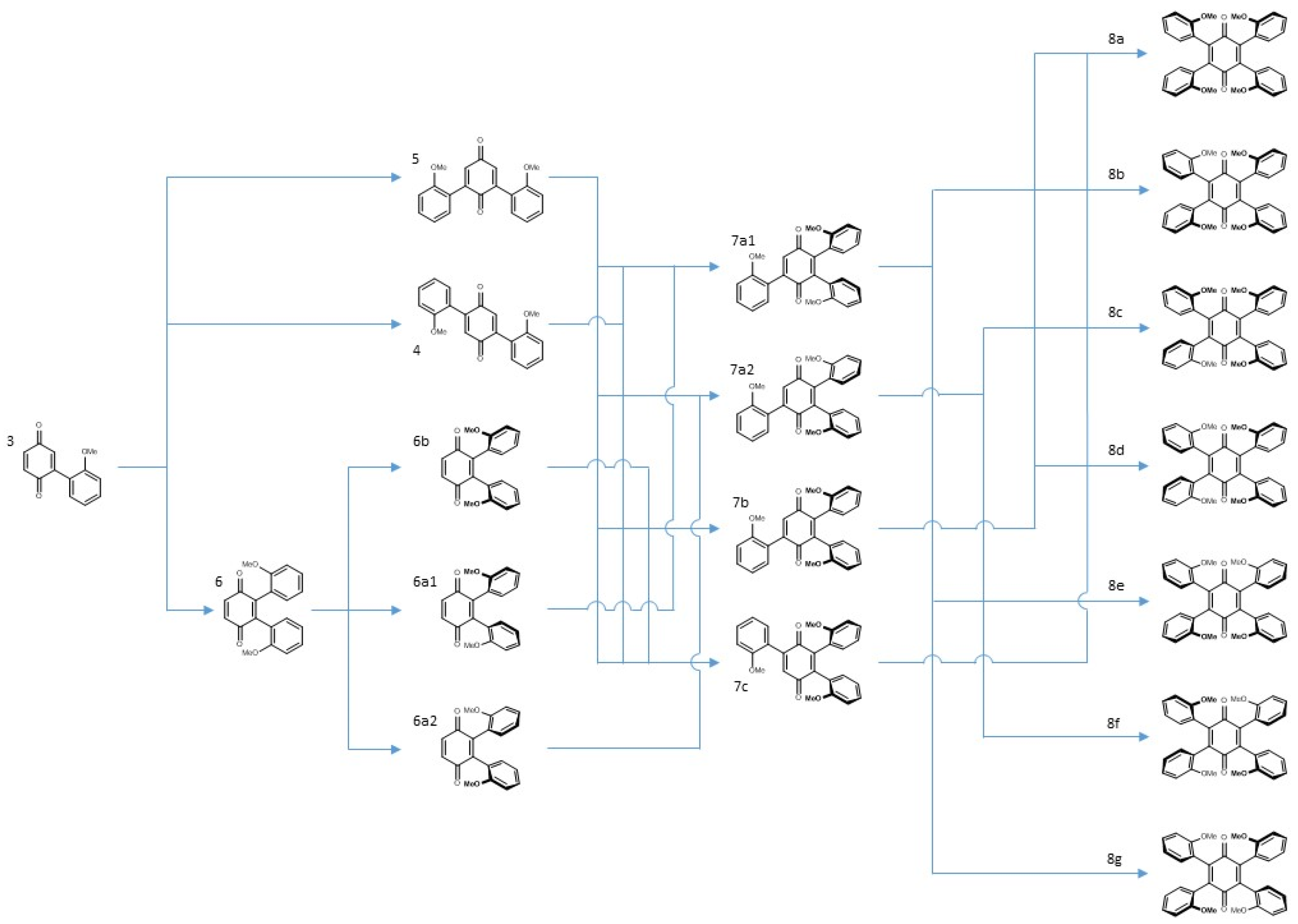
A large part of the excess of benzoquinone required in the reaction is consumed in the reoxidation of the initially formed dihydrobenzoquinone, a reaction that must be faster than the initial addition since the intermediate does not accumulate. To facilitate this reoxidation and to control the consumption of benzoquinone, Cu(II) was added to the reaction mixture, as proposed by Bäckvall for the reoxidation of benzoquinone in Pd catalyzed reactions [14]. Table 1 and Scheme 3 summarize these results. The use of a large excess of vindoline provided the high molecular weight tri-adduct 7 and tetra-adduct 8, while the optimum ratio for the production of the di-adducts (4–6) was slightly over 2. The different adducts were easily distinguished by TLC and the separations were achieved by column chromatography. Structures were determined by spectroscopic means and symmetry considerations.
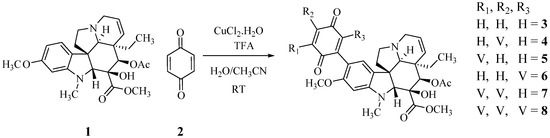
Scheme 3.
The preparation of poly-adducts of vindoline and benzoquinone.
2.2.1. The Bis Adducts. The Simple Adducts and General Considerations
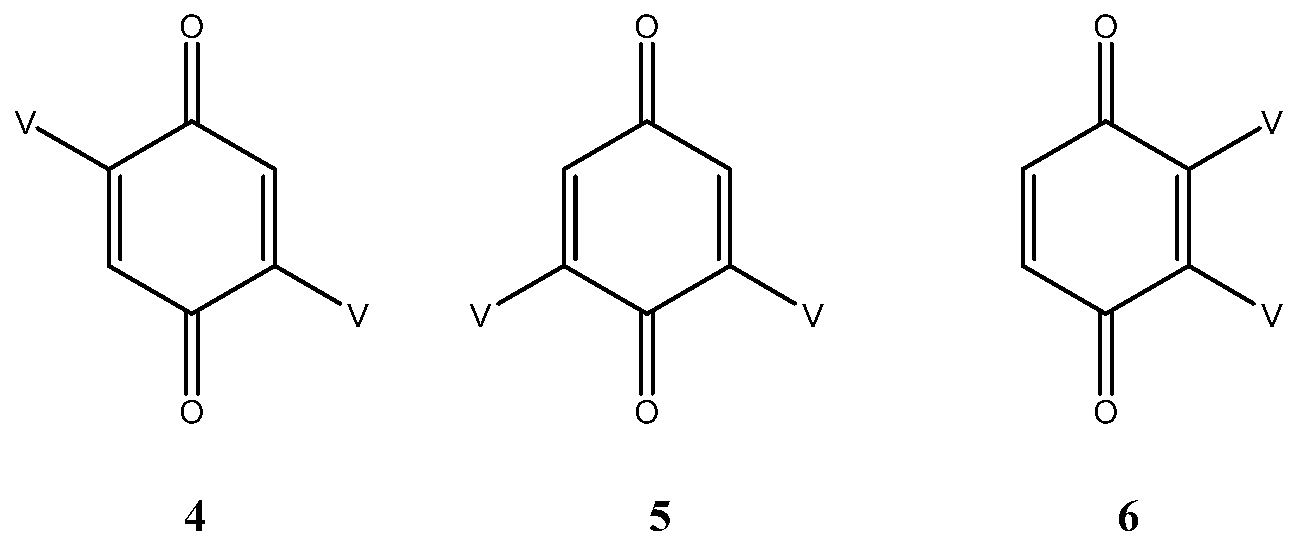
As expected, three bis adducts, 4, 5 and 6 were obtained with 5 being by far the most abundant. For the sake of clarity, they are represented below under the simplified drawings in Figure 1, where V stands for vindoline. They all showed the expected molecular ion at m/z 1017 [M + H]+ corresponding to a C56H65N4O14 formula, that is to say to the substitution of two hydrogen atoms of benzoquinone by vindolines.
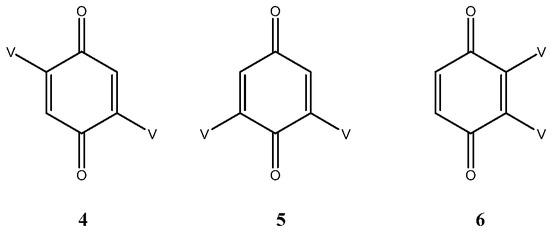
Figure 1.
The three bis adducts of vindoline (V) and benzoquinone.
Compound 4 is a centrosymmetric bis-adduct and as expected, it showed a single set of signals in the 1H and 13C NMR spectra. It is a pink solid with an extended UV chromophore (λmax at 526 nm). The low field part of the 1H NMR spectrum showed four two proton singlets for H-3′, H-9, H-12 and H-17 at δ 6.76, 6.35, 7.04 and 5.38. The quinone proton was identified by observing long range coupling with the ketone carbonyl at δ 188.3; it also coupled to two quaternary carbons at 114.6 and 146.7 respectively assigned to C-10 (vindoline) and C-2′ (quinone). All the vindoline signals were identified by comparison with the spectra of 1 and by suitable 2D NMR experiments.
In compound 5 (see Appendix B), the two vindoline moieties are exchangeable through an axis of symmetry and therefore give a single set of signals. This is not the case for the quinone carbonyls, which showed two signals at δ 189.6 and 187.8. In the 1H NMR spectrum, the four singlets were at δ 7.02 (H-12), 6.75 (H-3′), 6.34 (H-9) and 5.37 (H-17). Due to the symmetry H-3′ showed couplings in the HMBC experiment with C-5′ (equivalent with C-3′), C-10 (115.5) and C-2′ (148.7). In the UV spectrum the n-π* band was shifted to 550 nm indicating extension of the chromophore compared to compound 4.
The 13C NMR spectra of compounds 4 and 5 show a high degree of similarity but can be easily distinguished by their quinone carbonyl resonances. The 1H NMR spectra are not very different either, with noticeable and unexplained differences being observed for CH3-18 (0.05 ppm), H-14 (0.03 ppm) and H-21 (0.06 ppm).
2.2.2. The Vicinal Bis Adduct 6
Since the structures of compounds 4 and 5 are firmly established, there only remained the possibility of vicinal substitution for compound 6. Despite our efforts, and even though UPLC showed a highly predominant peak, compound 6 could not be completely purified, as judged by NMR. The mass spectrum showed the expected molecular ion and the UV was shifted to 534 nm. The 1H NMR spectra showed a major compound with the four typical singlets of the bis-adducts at δ 6.89, 6.30, 6.28 and 5.35 for H-5′ (H-6′), H-9, H-12 and H-17 respectively. These resonances were assigned as above by means of HSQC and HMBC experiments. In addition, the spectrum showed several other sets of singlets of smaller intensities, which could be assigned to other isomers or conformers.
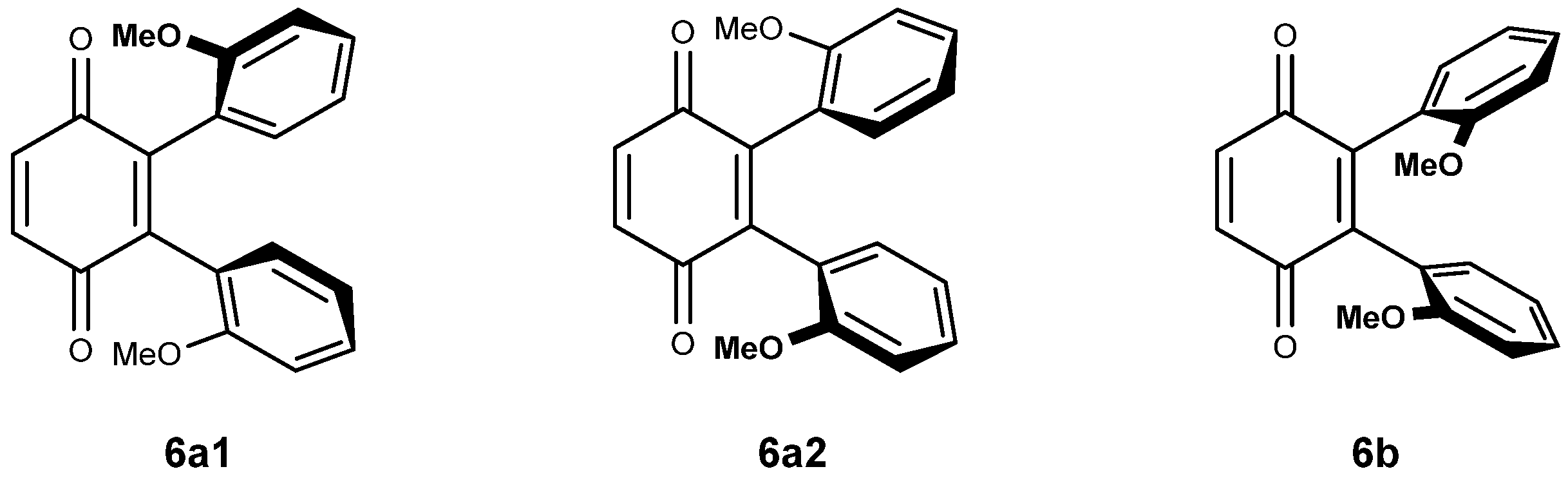
2.2.3. Vicinal Bis Adducts and Symmetry Considerations
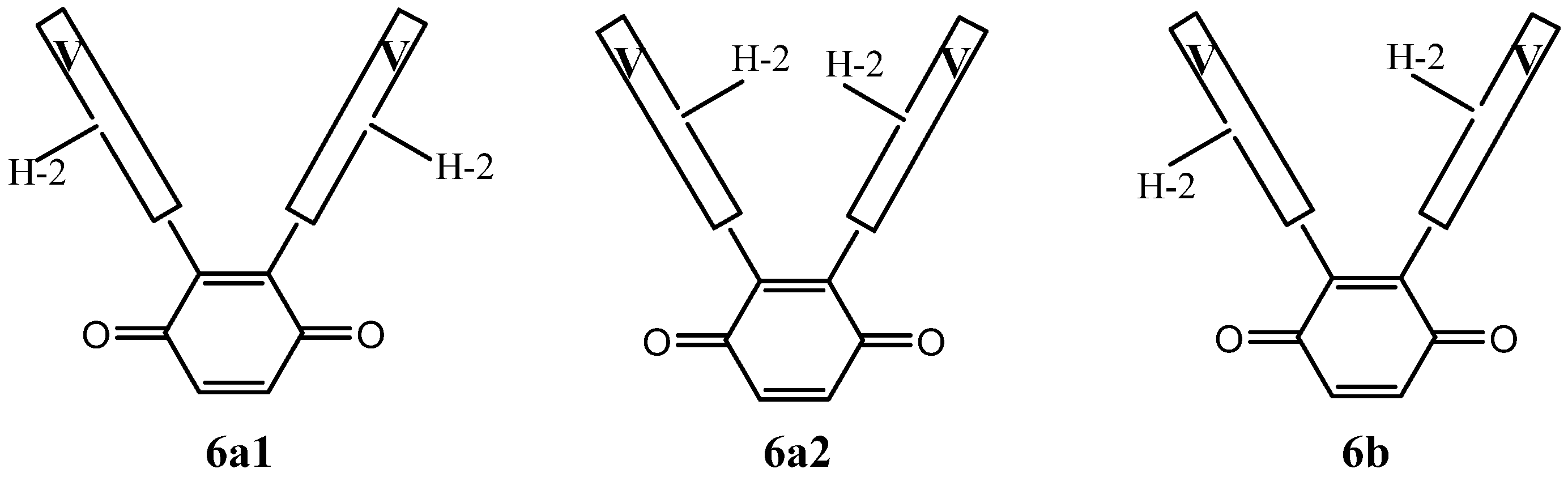
The di-substitution and the bulky nature of vindoline makes rotation around the C-10- C-2′ bond difficult and it might be that the vicinal di-adducts present a character of atropoisomerism. In this case, four vicinal di-adducts may be expected to form: two syn and two anti isomers. The anti adducts exist as two isomers while the syn adducts are one and the same compound (homomers). They will be denominated 6a1, 6a2 and 6b. Figure 2 is a simplified representation of these molecules in which, for the sake of clarity, an anisole ring is drawn instead of the vindoline. Compounds 6a1 and 6a2 possess an axis of symmetry passing through the middle of the C-2′-C-3′ and C-5′-C-6′ of the benzoquinone and exchanging the vindolines, which, therefore must present a unique set of signals. Due to the natural chirality of vindoline, compounds 6a1 and 6a2 are different. There is no element of symmetry in compound 6b and the two vindolines must give different sets of signals of respective equal intensity.
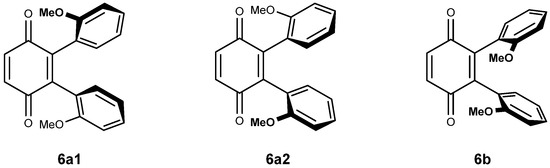
Figure 2.
Structures and molecular models for compounds 6a and 6b.
2.2.4. The Vicinal Adducts and NMR Assignments
Figure 3 is another simplified representation of the three vicinal dimers in which the vindolines are represented by the boxes marked V, where H-2 serves as a marker of the asymmetry of vindolines. This figure shows that the three structures are different, which is not obvious for 6a1 and 6a2, where the H-2 atoms are either inside or outside the branches of the U shaped molecules.
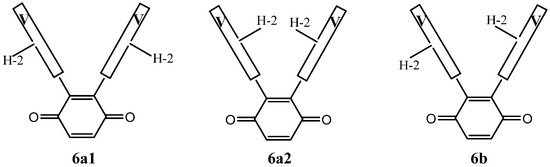
Figure 3.
A simplified representation of compounds 6a1, 6a2 and 6b.
As far as NMR is concerned, compounds 6a1 and 6a2 are expected to display a single set of resonances for the vindolines, with no reason for them to be identical, while 6b should show signals for two sets. As an example, H-2 may show up to four distinct resonances and the same is true for other observable signals such as those of CH3-18 and H-15.
2.2.5. Higher Order Adducts
It took forcing conditions to obtain the higher order adducts 7 and 8, i.e., a large excess of vindoline and a longer reaction time. For each of these compounds, there is only one possible regioisomer.
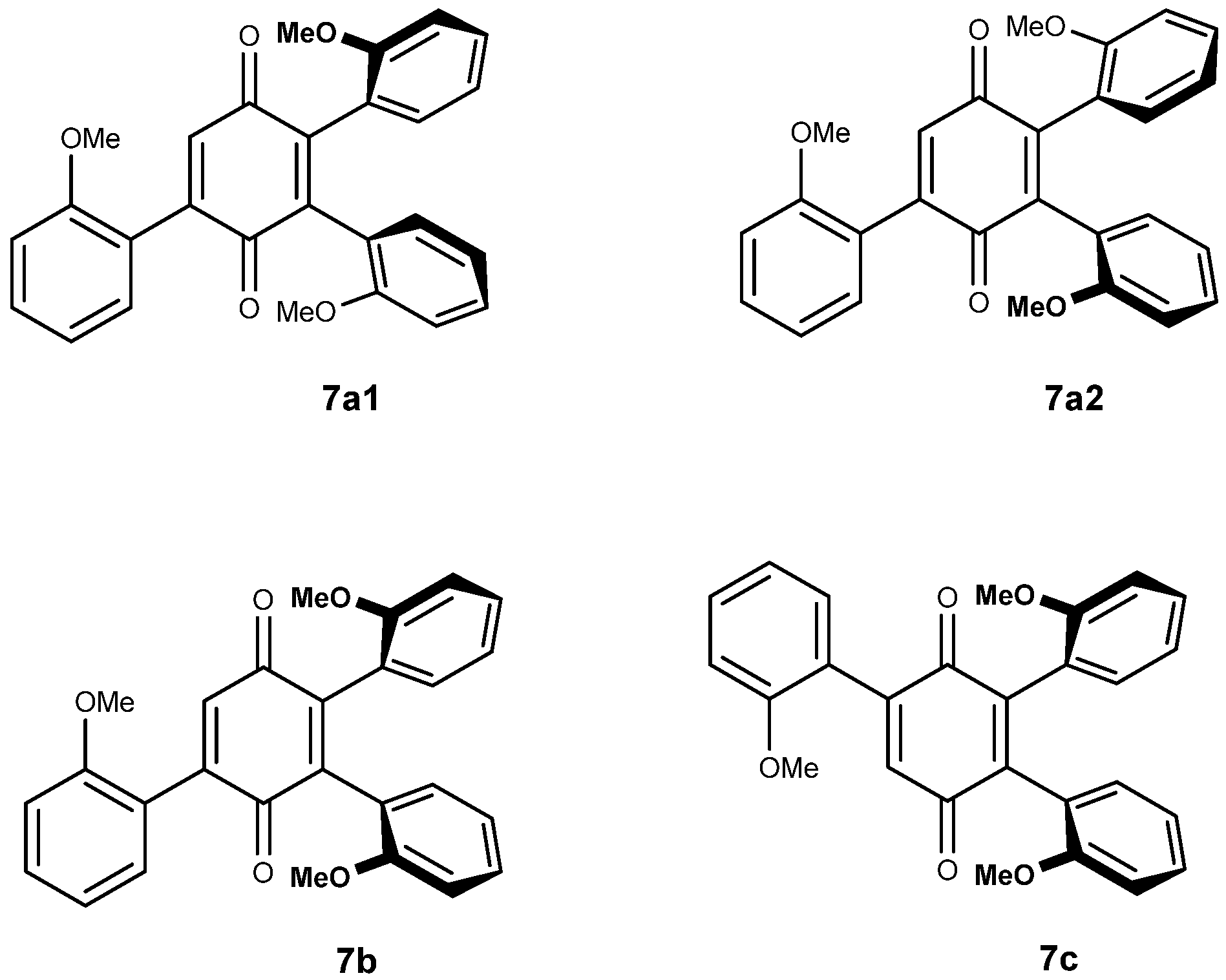
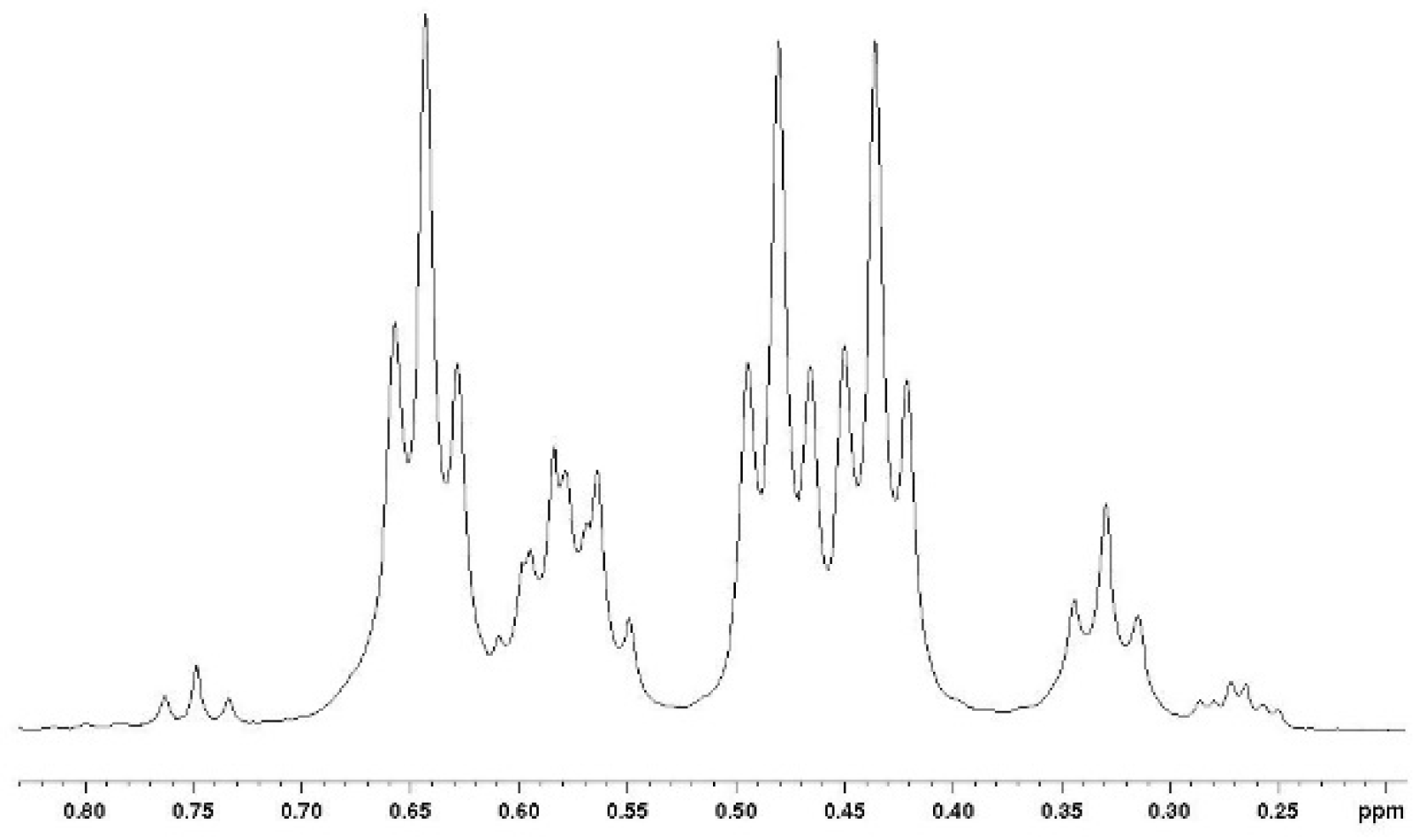
The tri-adducts may be seen as arising from the addition of a vindoline unit to vicinal bis-adducts 6a1, 6a2 and 6b, even though the intermediacy of di-adduct 5 is the most probable event. Compounds 6a1, 6a2 are different and they are expected to only give two tri-adducts: 7a1 and 7a2, since the positions on the quinone are equivalent. In the syn adduct however, the two quinone positions are different and therefore, one may expect two compounds 7b and 7c. This is represented in Figure 4 with the same simplified drawings as in Figure 2. As regards symmetries, none of the four adducts possess any element of symmetry and therefore each of the three vindolines is expected to give different signals. It is not easy to disentangle the resonances in the 1H NMR spectra, but at high field and for the major compound a set of three triplets of equal intensities is observable (Figure 5).
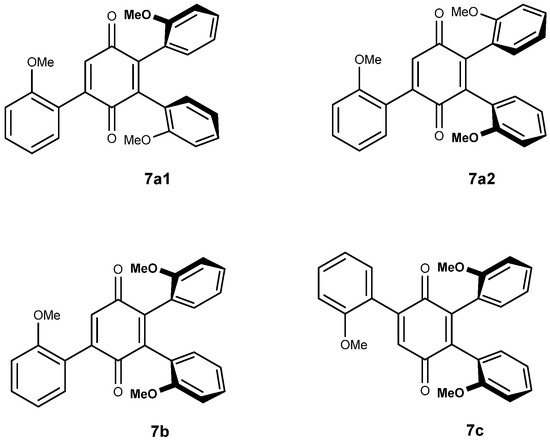
Figure 4.
The four possible tri-adducts 7a1, 7a2, 7b and 7c.
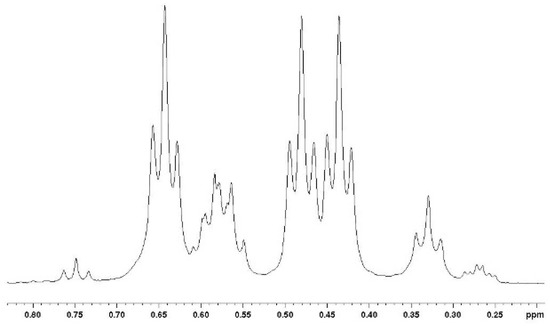
Figure 5.
High field part of the 1H NMR spectrum of the mixture of tri-adducts showing similar intensities for three CH3-18 in the major isomer.
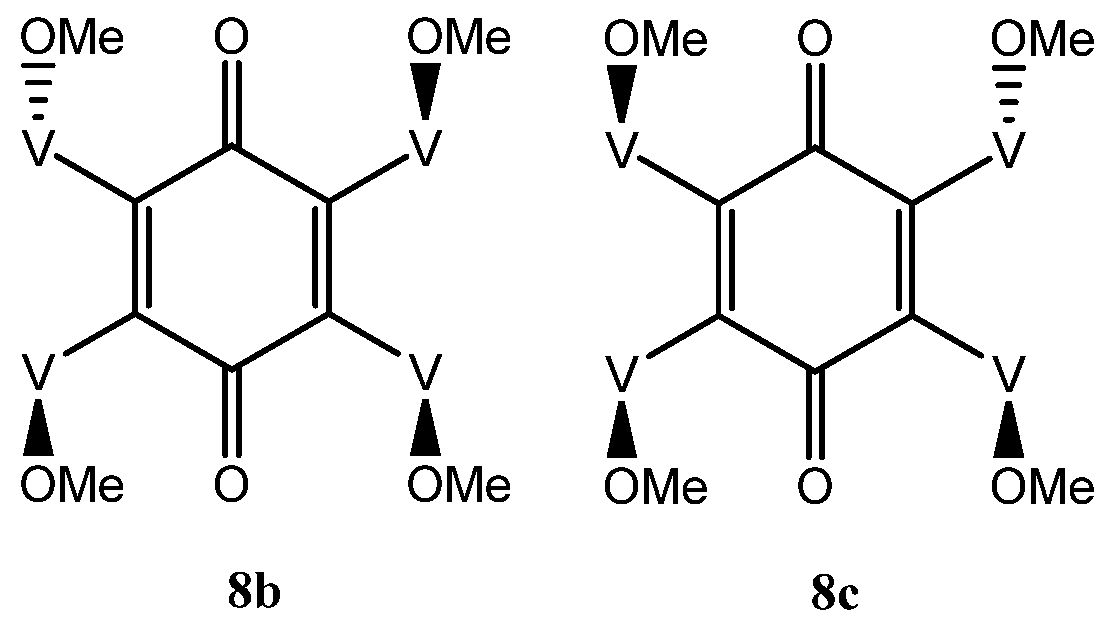
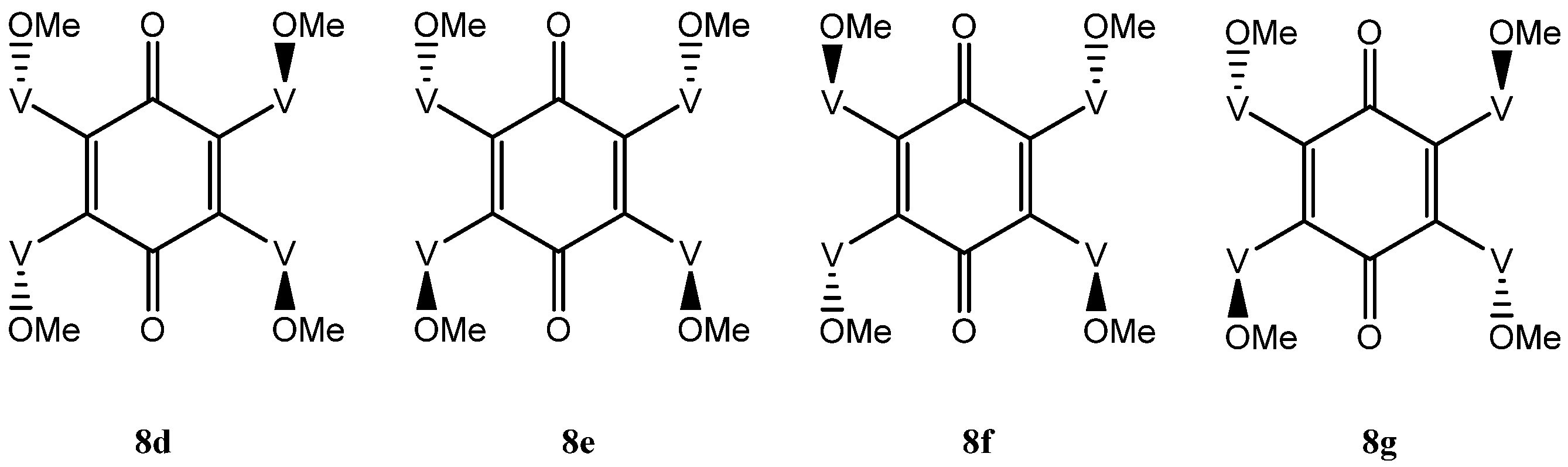
The situation with the tetra-adduct is more complicated with seven possible isomers: a single compound 8a with all vindolines oriented in the same manner, two compounds 8b and 8c with one vindoline oriented towards the other face with regards to the three others and four isomers with two vindolines oriented on the same side (2,3, 2,6, 2,5 and 3,6 with respect to the quinone) 8d–g. Isomers with the OMe top-oriented 2,5 and 3,6, 8f and 8g are not superimposable and therefore are different compounds) (Figure 6 and Figure 7).
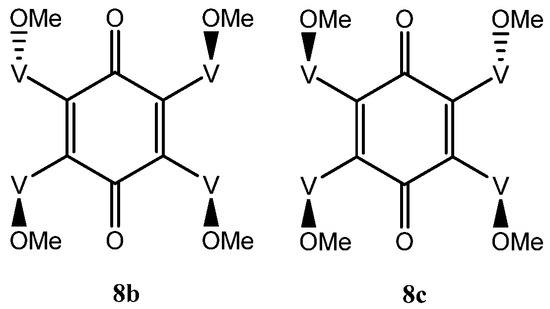
Figure 6.
Tetra-adducts with three vindolines with same orientation.
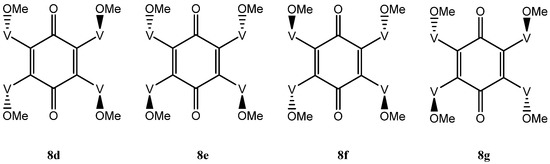
Figure 7.
Tetra-adducts with two vindolines with same orientation.
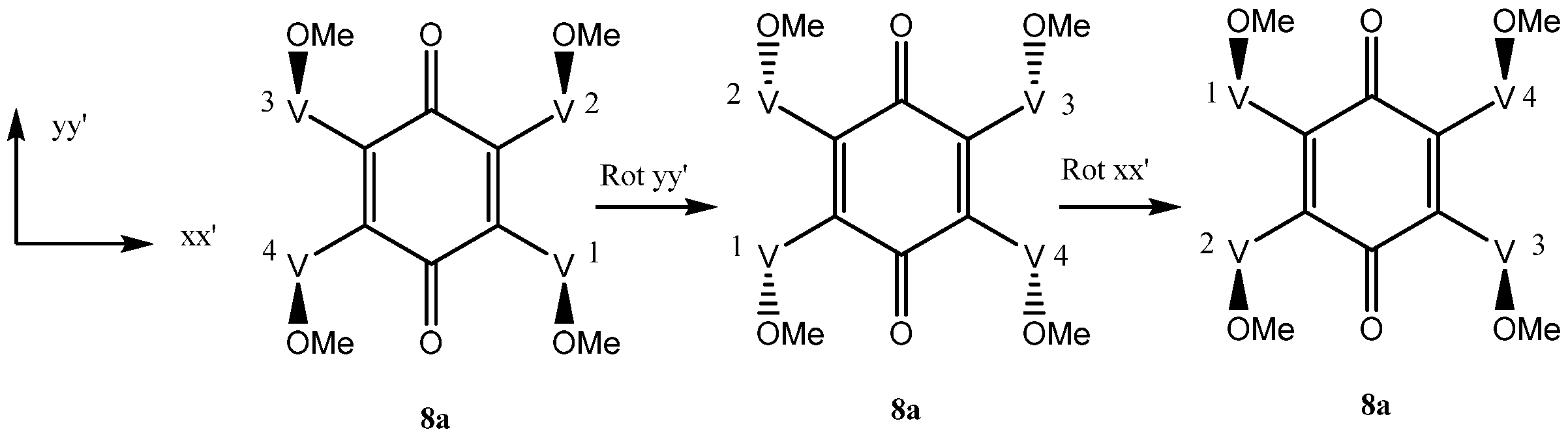
In isomer 8a since vindoline is chiral, none of the four vindolines is directly exchangeable through a plane of symmetry. However, if the molecule is rotated by 180° along the yy’ axis and then by 180° along the xx’ axis, then 8a is recovered unchanged (Scheme 5). V1 and V3, on one hand, V2 and V4, on the other, are thus exchanged and are equivalent (Scheme 4). The NMR spectra of compound 8a should show two sets of signals for vindoline but a single carbonyl for the benzoquinone.
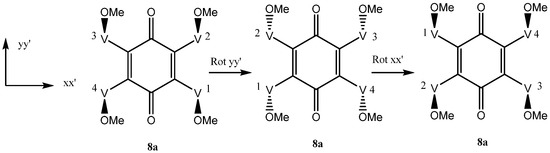
Scheme 4.
Vindoline exchanges by rotations around axes xx’ and yy’ in 8a.
There are two tetra-adducts 8b and 8c with only one vindoline moiety pointing downwards with the three other ones being upwards (Figure 6). Observation of 8b and 8c from the ketone side with the two vindolines with their OMe upward, brings the vindoline with Ome pointing downward either on the right or on the left side. They are not exchangeable since vindoline is chiral. Compounds 8b and 8c are therefore different and within each of them, the four vindolines are different and thus these isomers may be expected to show four sets of vindoline signals of equal intensity. A consequence of the absence of symmetry is that two signals should be detected for the benzoquinone carbonyls.
In isomer 8d, a rotation along the yy’ axis exchanges V1 and V4, V2 and V3, which are therefore equivalent pairs. There is no mechanism of exchange for the pairs V1, V2 and V3, V4. This isomer would be expected to give two sets of signals and a single benzoquinone carbonyl. In isomer 8e, the vindolines on the same side of the ketones are exchanged by a 180° rotation around axis xx’ and are therefore equivalent. There is no symmetry to exchange the other pairs and this molecule is expected to show two sets of signals for the four vindoline moieties. The quinone carbonyls are also exchanged in this rotation and should give a single signal. Finally in the last isomers 8f and 8g rotations around the two axis xx’ and yy’ exchange the four vindoline units and symmetry considerations suggest that these isomers will show a single set of signals and a single benzoquinone carbonyl (Figure 7).
2.2.6. The NOESY Experiment: Further Complexity and DFT Calculations
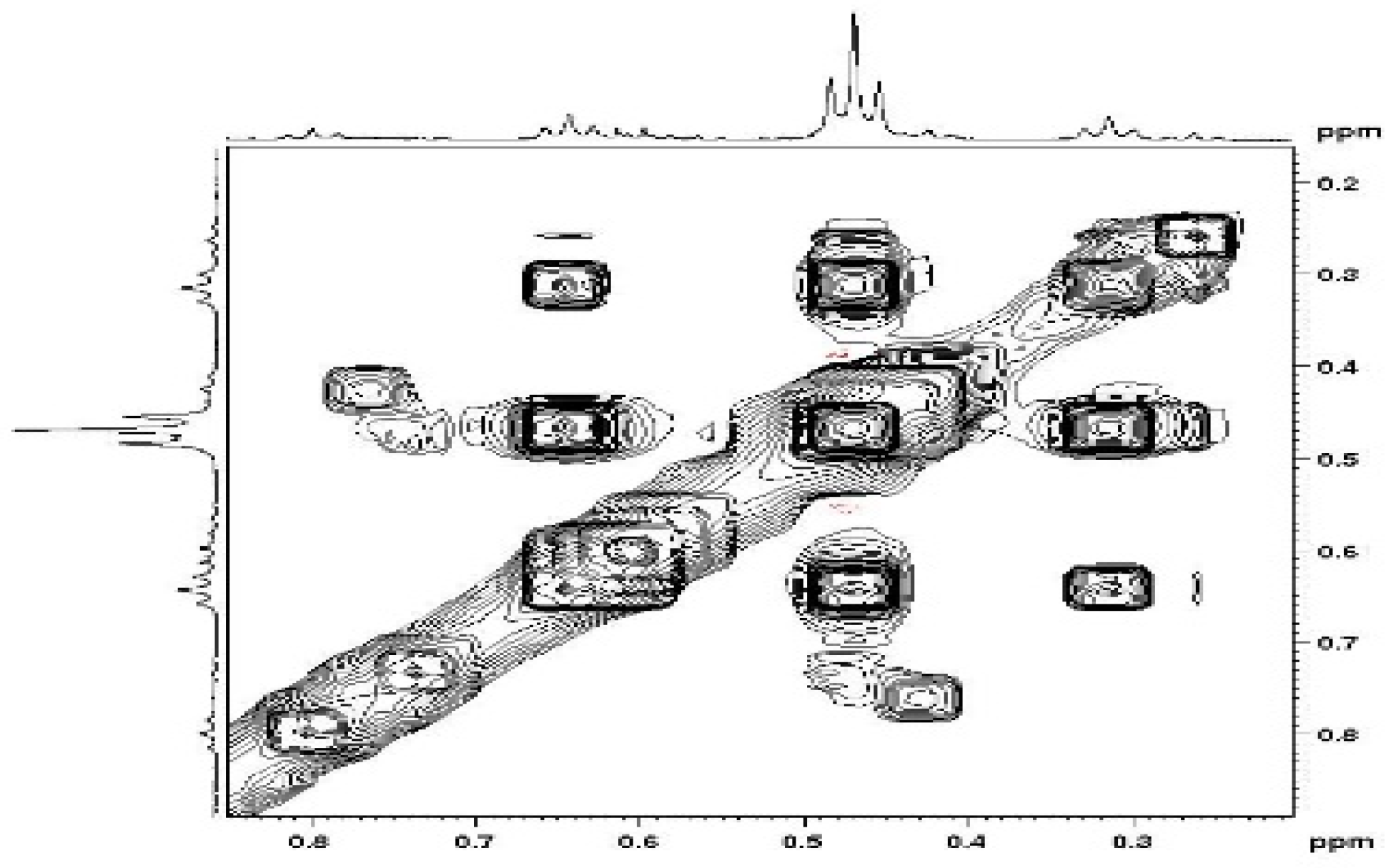
The NOESY maps obtained for the simple compounds 3, 4 and 5 showed the expected correlations of positive intensity for the vindoline protons interactions. The mixture of di-adducts 6, however gave positive and negative NOEs. This was rather unexpected since these molecules of the same molecular weight should have close correlation times (τC). The phenomenon was particularly intriguing in the high field area, which showed correlations between methyl groups, negative in sign (same as the diagonal) (Figure 8). In other parts of the spectra, regular (positive) NOEs were observed leading to conclude that these negative effects are due to a slow exchange process between conformers. The same situation happened with the trimer, but due to the relatively high molecular weight (1470), all correlations were negative. The existence of conformers stable enough to give rise to exchange at the NMR timescale, makes definitive assignments of signals to a particular species extremely difficult even for compound 6, which exists as three forms with two conformations each, amounting to eight different sets of signals for vindolines. Of course, the situation is even worse for the tri- and tetra-adducts.
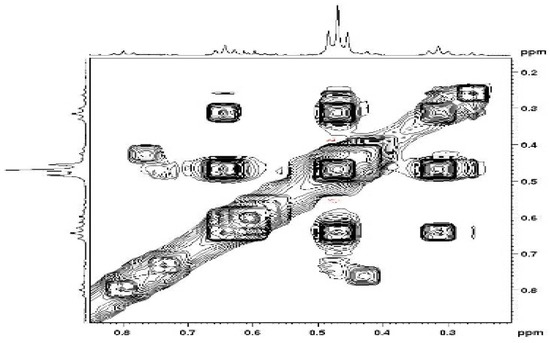
Figure 8.
NOESY correlations between methyl groups in compounds 6.
Density Functional Theory (DFT) M06-2X/6-311G** calculations were performed in methanol (implicit solvent) on simplified models corresponding to structures 6a1, 6a2 and 6b of Figure 2. In a first round of calculations, it was demonstrated that rotating one anisole ring by 180° around the single quinone-anisole bond involves a substantial energy barrier (ΔG°‡(298 K) = 21.6 kcal·mol−1). Actually, the ortho methoxy group knocks into the quinone carbonyl (steric repulsion), making the rotation difficult without a deformation of the quinone ring. Thus, the interconversion between 6a and 6b is certainly not possible under our observation conditions. Since an eventual interconversion between 6a1 and 6a2 must occur through the intermediacy of 6b, 6a1 and 6a2 are definitively different molecules. At that stage the NOESY exchange correlations could not be explained and it was decided to perform a potential energy surface (PES) scan of the conrotatory motion in which both anisole groups turn simultaneously in the same direction. The results showed that there exist for each isomer (6a1, 6a2, 6b) two distinct minima on the PES, the oscillation between them resembling a windshield wipers motion. In the case of the syn isomer, these two conformations are chemically equivalent while they are not for the anti since in that case either the methoxy or the phenyl comes close to each of the quinone carbonyls. The transition between these conformations does not require much energy (ΔG°‡(298 K) in the range 1.3–2.5 kcal·mol−1) and thus is observable at the NMR time scale.
3. Discussion
The addition of vindoline to benzoquinone is not different from that reported for amino and sulfur nucleophiles [10,11]. A mono adduct is formed first, followed by a di-adduct, either -2,4 or -2,5 in the case of sulfur and -2,5 in the case of nitrogen. If nothing is anticipated regarding the reoxidation of the initially formed dihydro-benzoquinones, the reaction mixtures are far more complicated and minor adducts may not be detected. It is worth noting that the 2,4 or -2,5 adducts are not easy to distinguish and 13C NMR, used here as a diagnostic tool, is not always reliable since, for example 2,5- and 2,6-bis(cyclohexylsulfanyl)(1,4)benzoquinones both showed a single carbonyl signal [11].
At variance with heteroatom nucleophiles, simple heterocycles seemed to give predominantly the 2,3-adducts and in a remarkable article by Escolastico et al. the presence of separable atropoisomers is described [15]. The difference in behavior with vindoline may be explained by the steric bulk in the mono-adduct, which prevents the approach of the second molecule of vindoline from the same side, even though the trajectory must be perpendicular to the plane of the benzoquinone. There were also differences in operating conditions: room temperature, acid and Cu(II) catalyst in our case, compared to no catalyst and reflux in dioxan, in the reference [15].
Vindoline does not follow the tendency of other nucleophiles and the 2,5-adduct is predominantly obtained as the second intermediate. Then, following what was observed for the bis adducts, the third addition occurs from the opposite face of the already present vindolines to give the anti compound 7a. This compound may be considered the major starting material for the synthesis of the tetra-adduct and should lead to the predominant formation of isomers 8d and 8e in equal proportions.
In summary, the reaction of vindoline with benzoquinone could lead to the formation of seventeen compounds. Three of them (3, 4, 5) were separated and characterized in a pure state and two pairs (6a, 6b and 7a, 7b) were separately characterized in the same NMR tube. The seven isomeric tetra-adducts could not be distinguished due to numerous superimpositions. Scheme 5 gives an overview of all the pathways leading to the tri-adducts and tetra-adducts. It shows that the “unoriented” and major di-adducts 4 and 5 are converted into the four tri-adducts, presumably with a preference for the anti compounds 7a1 and 7a2. The syn di-adduct 6b will lead to the two syn tri-adducts 7b and 7c while each of the anti compounds 6a1 and 6a2 will exclusively give a single anti tri-adduct, respectively 7a1 and 7a2. The conversion of the tri-adducts into tetra-adducts offers more possibilities but all syn compound 8a, may only be accessed from 7b and 7c.
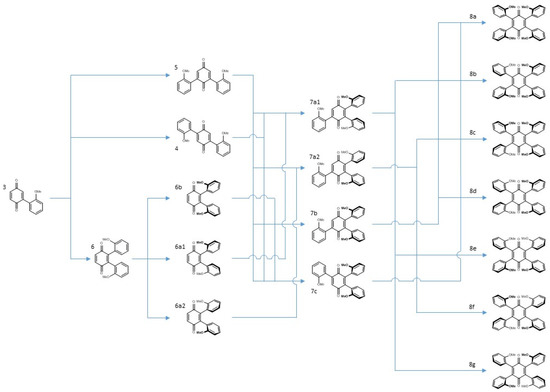
Scheme 5.
Sequence of events leading to tetra-adducts.
The ratio between di-adducts 4 and 5 depends on the polarization of the unsubstituted double bond and this clearly favors compound 5. Formation of the di-adducts 6 is doubly handicapped since both the steric hindrance and the polarization of the double bond disfavor the vicinal attack. In principle, since there are two reactions leading to the syn adduct, the yield should be twice the yield of the anti adducts. The same arguments could in principle be applied to the formation of the higher order adducts assuming that the tri-adduct is formed from the di-adducts and the tetra-adduct from the tri-adducts. While this last proposal is certainly true, it cannot be ruled out that reversibility plays a role in these sequences. This is currently under investigation.
4. Biological Evaluation of the Compounds
The mono-adduct 3, the di-adducts 4 and 5 and the mixture of tri-adducts were assayed for antifungal, anti-inflammatory, and anti-bacterial activities as well as cytotoxicity. Only compound 3 was found to have an anti-inflammatory activity (ROS) at the 5 μM level. A cytotoxicity assay was conducted against HeLa (cervical cancer), Au 565 (breast cancer) with 3T3 (mouse fibroblast) as reference. Activity was observed in the 10 μM range for the di-adducts and the tri-adduct, unfortunately with no selectivity (Table 2).
Table 2.
Cytotoxicity of compounds 3, 4, 5 and 7 against HeLa, Au565 cancer, and 3T3 normal cell lines.
5. Materials and Methods
5.1. General
Reactions were carried out in an open atmosphere, using distilled solvents and oven dried glassware. Acetonitrile was HPLC grade. Silica gel TLC plates (Merck silica gel 60 F254 plates) were used to monitor the reactions. Column chromatography CC) was carried out on silica gel 60 (Merck, mesh size 70–230, Billerica, MA, USA). UV data were measured on a Thermo-scientific model-300 spectrometer. IR data were recorded on a Bruker Vector-22 spectrophotometer on KBr disk. Optical rotations were recorded on JASCO P-2000 spectrometer. CD spectra were recorded on JASCO J-810 spectropolarimeter. 1H-NMR spectra were recorded on AV-500 MHz spectrometers in deuterated methanol. J-values (coupling constants) were expressed in Hertz (Hz). 13C-NMR spectra were recorded on Bruker AVIII-300, and 600 MHz spectrometers in deuterated chloroform or methanol. ESI-MS spectra were recorded on Bruker mass spectrometer Amazon ESI ion trap (Bruker, Billerica, MA, USA). The high-resolution electrospray ionization spectra (HR-ESI) were recorded on a mass spectrometer Bruker Daltonic Maxis II/ESI Q-TOF system with electrospray ionization (ESI) ion source.
5.2. Preparation of 2-(10-Vindolinyl)-benzoquinone 3
In a 50 mL round bottom flask containing 14 mL of a (7/1) mixture of acetonitrile and water, 200.5 mg (0.44 mmole) of vindoline and 1 mL TFA were added. The mixture was stirred for five minutes at room temperature, and 188.9 mg (1.74 mmole) p-benzoquinone was added in the reaction mixture, giving a reddish-brown coloration. The mixture was stirred for 24 h at room temperature, and the color became more pronounced. Acetonitrile was evaporated under reduced pressure and a solution of NaHCO3 was added until pH became basic. Then 20 mL CH2Cl2 was added, and the reaction mixture was transferred to a separating funnel. After shaking, the organic phase was separated. The process was repeated three times. The organic phases were combined, dried over Na2SO4, filtered and evaporated to give a blue residue, which was purified by flash chromatography with CH2Cl2 containing 1% MeOH as eluent. Compound 3 (235 mg, 95% yield) was obtained as a blue amorphous solid. G)
2-(10-Vindolinyl)-benzoquinone3, blue amorphous solid, melting point = 250 °C; Rf = 0.53 (1%/99% MeOH/CH2Cl2) 0.683; = +12 (c 0.0008, MeOH); UV (λmax MeOH): 221, 250, 309, 542 nm; IR (KBr): 3451, 2928, 2877, 1742, 1662, 1610, 1502, 1442, 1377, 1246, 1092, 1047 cm−1; 1H NMR (500 MHz, CD3OD): δ = 7.04 (1H, s, H-9), 6.86 (1H, d, J = 8.0 Hz, H-6′), 6.82 (1H, dd, J = 8.0, 2.0 Hz, H-5′), 6.75 (1H, d, J = 2.0 Hz, H-3′), 6.34 (1H, s, H-12), 5.91 (1H, ddd, J = 10.2, 4.9, 1.4 Hz, H-14), 5.38 (1H, s, H-17), 5.24 (1H, br d, J = 10.2 Hz, H-15), 3.81 (6H, s, C-11 OCH3 and COOCH3), 3.76 (1H, s, H-2), 3.51 (1H, dd, J = 16.2, 4.9 Hz, H-3β), 3.42 (1H, td, J = 9.5, 4.6 Hz, H-5β), 2.93 (1H, br d, J = 16.2 Hz, H-3α), 2.86 (1H, s, H-21), 2.79 (3H, s, N-CH3), 2.66 (1H, dt, J = 9.5, 7.6 Hz, H-5α), 2.33 (2H, m, 2H-6), 2.04 (3H, s, C-17 OCOCH3), 1.64 (1H, dq, J = 14.5, 7.2 Hz, H-19a), 1.22 (1H, dq, J = 14.5, 7.2 Hz, H-19b), 0.63 (3H, t, J = 7.2 Hz, H-18); 13C NMR (150 MHz, CDCl3): δ = 188.0 (C-1′), 186.5 (C-4′), 171.8 (COOCH3), 170.7 (OCOCH3), 159.5 (C-11), 155.1 (C-13), 145.1 (C-2′) 137.0 (C-6′) 136.1 (C-5′) 132.8 (C-3′), 130.3 (C-15), 125.0 (C-9), 124.4 (C-8), 124.2 (C-14), 112.6 (C-10), 92.9 (C-12), 83.3 (C-2), 79.4 (C-16), 76.2 (C-17), 66.8 (C-21), 55.8 (C-11 OCH3), 52.7 (C-7), 52.3 (COOCH3), 51.6 (C-5), 50.9 (C-3), 43.9 (C-6), 42.8 (C-20), 37.6 (N-CH3), 30.9 (C 19), 21.6 (OCOCH3), 7.6 (C-18); HRESI-MS calc. for: C31H35N2O8 [M + H]+ m/z = 563.2388, measured 563.2393.
5.3. Preparation of 2,5-di-(10-Vindolinyl)-benzoquinone 4, of 2,6-di-(10-Vindolinyl)-benzoquinone 5 and of 2,3-di-(10-Vindolinyl)-benzoquinone 6
In a 50 mL round bottom flask containing 4 mL distilled water, 1115 mg (6.56 mmoles) of CuCl2·2H2O was dissolved, followed by 18.9 mg (0.166 mmole) p-benzoquinone, and 200 mg (0.43 mmole) vindoline. After 5 min, 1 mL TFA and 2 mL acetonitrile was added and the mixture was stirred for 8 h at room temperature. The acetonitrile was evaporated under reduced pressure and the pH of the solution made basic with NaHCO3. The same operating procedure as above was followed and the mixture was separated by flash chromatography (2% MeOH in CH2Cl2). Three compounds were thus separated: 2, 5-di-(10-vindolinyl)-1,4 p-benzoquinone 4 (16 mg, 9%) 2,6-di-(10-vindolinyl)-1,4 p-benzoquinone 5 (134 mg, 75%) and 2,3-di-(10-vindolinyl)-1,4 p-benzoquinone 6 (14 mg, 7%).
2,5-di-(10-vindolinyl)-benzoquinone4, pink amorphous solid; melting point 266 °C; Rf = 0.47 (2%/98% MeOH/CH2Cl2); = +7.3 (c 0.00045, MeOH); UV (λmax MeOH): 223, 255, 312, 524; IR (KBr): 3460, 2926, 2878, 2852, 1743, 1651, 1614, 1505, 1461, 1433, 1374, 1335, 1244, 1158, 1092, 1040, 1047, 948, 892, 816, 747 cm−1; 1H-NMR (500 MHz, CD3OD): δ 7.04 (2H, s, H-9), 6.76 (2H, s, H-3′/H-6′), 6.35 (2H, s, H-12), 5.90 (2H, ddd, J = 10.2, 6.7 Hz, 4.6 Hz, H-14), 5.38 (2H, s, H-17), 5.24 (2H, br d, J = 10.2 Hz, H-15), 3.83 (6H, s, C-11 OCH3), 3.81 (6H, s, COOCH3), 3.75 (2H, s, H-2), 3.48 (2H, dd, J = 16.4 Hz, 4.6 Hz, H-3β), 3.42 (2H, dt, J = 8.9, 4.3 Hz, H-5β), 2.94 (2H, br. d, J = 16.4 Hz, H-3α), 2.89 (2H, s, H-21), 2.79 (6H, s, NCH3), 2.66 (2H, m, H-5α), 2.30 (4H, m, H-6), 2.04 (6H, s, OCOCH3), 1.63 (2H, dq, J = 14.5, 7.2 Hz, H-19a), 1.22 (2H, dq, J = 14.5, 7.2 Hz, H-19b), 0.64 (6H, t, J = 7.2 Hz, CH3-18); 13C-NMR (75 MHz, CD3OD): δ 188.3 (C-1′/C-4′), 173.4 (COOCH3), 172.5 (OCOCH3), 161.1 (C-11), 156.5 (C-13), 146.7 (C-2′/C-5′), 134.3 (C-3′/C-6′) 131.3 (C-15), 126.6 (C-9), 125.9 (C-8), 125.8(C-14), 114.6 (C-10), 94.4 (C-12), 84.5 (C-2), 80.9 (C-16), 77.6 (C-17), 67.6 (C-21), 56.4 (C-11 OCH3), 54.1 (C-7), 52.9 (COOCH3), 52.9 (C-5), 52.4 (C-3), 44.6 (C-6), 44.32 (C-20), 38.3 (N-CH3), 32.1 (C-19), 20.8 (OCOCH3), 8.1 (C-18); HRESI-MS calc. for: C56H65N4O14 [M + H]+ m/z 1017.4492, measured 1017.4496.
2,6-di-(10-vindolinyl)-benzoquinone5, blue amorphous solid; melting point 266 °C; Rf = 0.43 (2% MeOH/CH2Cl2); = +37 (c 0.0006, MeOH); UV (λmax MeOH): 222, 275, 310, 550; IR (KBr): 3458.4, 2925, 2877, 2852, 1743, 1641, 1613, 1504, 1462, 1433, 1448, 1430, 1374, 1246, 1166, 1114, 1077, 1040, 950, 892, 816, 785, 741, 644, 615, 584, 545, 481, 422 cm−1; 1H-NMR (500 MHz, CD3OD) δ 7.02 (2H, s, H-9), 6.75 (2H, H-3′/H-5′), 6.34 (2H, s, H-12), 5.89 (2H, ddd, J = 10.2, 5.0, 3.5 Hz, H-14), 5.37 (2H, s, H-17), 5.21 (2H, br d, J = 10.2 Hz, H-15), 3.86 (6H, s, C-11 OCH3), 3.81 (6H, s, COOCH3), 3.76 (2H, s, H-2), 3.50 (2H, dd, J = 16.3, 5.0 Hz, Hβ-3), 3.42 (2H, td, J = 9.0, 4.3 Hz, Hβ-5), 2.90 (2H, br d, J = 16.3 Hz, Hα-3), 2.83 (2H, s, H-21), 2.79 (6H, s, N-CH3), 2.63 (2H, m, Hα-5), 2.34 (4H, m, H-6), 2.04 (6H, s, OCOCH3), 1.64 (2H, dq, J = 14.5, 7.2 Hz, Ha-19),1.20 (2H, dq, J = 14.5, 7.2 Hz, Hb-19), 0.59 (6H, t, J = 7.2 Hz, H-18); 13C-NMR (600 MHz, CD3OD): δ 189.6 (C-1′), 187.8 (C-4′), 173.4 (COOCH3), 172.5 (OCOCH3), 161.2 (C-11), 156.7 (C-13), 148.7 (C-2′/C-6′), 132.4 (C-3′/C-5′), 131.2 (C-15), 126.5 (C-9), 125.9 (C-8), 125.8 (C-14), 115.5 (C-10), 94.4 (C-12), 84.4 (C-2), 80.8 (C-16), 77.5 (C-17), 67.9 (C-21), 56.6 (C-11 OCH3), 54.0 (C-7), 52.9 (COOCH3), 52.6 (C-5), 52.0 (C-3), 44.6 (C-6), 44.3 (C-20), 38.3 (N-CH3), 32.1 (C 19), 20.8 (OCOCH3), 8.1 (C-18); HRESI-MS calc. for: C56H65N4O14 [M + H]+ m/z 1017.4492, measured 1017.4495.
2,3-di-(10-vindolinyl)-benzoquinone6, brown amorphous solid; melting point 267 °C Rf = 0.36 (2% MeOH/CH2Cl2); = −10.8 (c 0.001, MeOH). UV (λmax MeOH): 300, 534 nm; IR (KBr): cm−1. Anti isomer 6a: 1H NMR (500 MHz, CD3OD) δ 6.89 (2H, s, H-5′/H-6′), 6.31 (2H, s, H-9), 6.28 (2H, s, H-12), 5.85 (2H, ddd, J = 10.2, 5.0, 3.5 Hz, H-14), 5.35 (2H, s, H-17), 5.18 (2H, br d, J = 10.2 Hz, H-15), 3.80 (6H, s, C-11 OCH3), 3.79 (6H, s, COOCH3), 3.58 (2H, s, H-2), 3.45 (2H, m, H-3β), 2.82 (2H, br d, J = 16.3 Hz, H-3α), 2.67 (6H, s, N-CH3), 2.54 (2H, s, H-21), 2.02 (6H, s, OCOCH3), 1.62 (2H, m, H-19a),1.15 (2H, m, H-19b), 0.47 (6H, t, J = 7.2 Hz, H-18); 13C NMR (150 MHz, CD3OD): δ 187.5 (C-1′, C-4′), 173.4 (CO2CH3), 172.4 (OCOCH3), 160.6 (C-11), 155.5 (C-13), 145.2 (C-2′/C-3′), 137.4 (C-5′/C-6′), 131.1 (C-15), 125.8 (C-9), 125.7 (C-8), 125.6 (C-14), 116.0 (C-10), 94.1 (C-12), 84.6 (C-2), 80.8 (C-16), 77.5 (C-17), 68.0 (C-21), 56.6 (C-11 OCH3), 53.6 (C-7), 52.9 (COOCH3), 52.7 (C-5), 51.9 (C-3), 44.4 (C-6), 44.2 (C-20), 38.7 (N-CH3), 32.1 (C 19), 20.8 (OCOCH3), 8.2 (C-18). Syn isomer 6b: 1H NMR (500 MHz, CD3OD), most characteristic peaks δ 6.88 (1H, s, H-5′ or H-6′), 6.87 (1H, s, H-3′ or H-5′), 6.84 (1H, s, H-9), 6.67 (1H, s, H-9), 6.24 (1H, s, H-12), 6.13 (1H, s, H-12), 5.43 (1H, s, H-17), 5.38 (1H, s, H-17), 3.67 (3H, s, C-11 OCH3), 3.79 (6H, s, COOCH3), 3.55 (3H, s, C-11 OCH3), 3.64 (1H, s, H-2), 3.52 (1H, s, H-2), 2.69 (3H, s, N-CH3), 2.65 (3H, s, N-CH3), 2.77 (1H, s, H-21), 2.55 (1H, s, H-21), 2.03 (3H, s, OCOCH3), 2.02 (3H, s, OCOCH3), 0.64 (3H, t, J = 7.2 Hz, CH3-18), 0.31 (3H, t, J = 7.2 Hz, CH3-18); 13C NMR (150 MHz, CD3OD) most characteristic peaks distinct from those of 6a: 126.4 (C-9), 126.1 (C-9), 125.7 (C-8), 125.6 (C-14), 116.0 (C-10), 94.1 (C-12), 84.6 (C-2), 80.8 (C-16), 77.5 (C-17), 68.0 (C-21), 56.6 (C-11 OCH3), 53.6 (C-7), 52.9 (COOCH3), 52.7 (C-5), 51.9 (C-3), 44.4 (C-6), 44.2 (C-20), 38.7 (N-CH3), 32.1 (C 19), 20.8 (OCOCH3), 8.2 (C-18).
5.4. Preparation of 2,3,5-tris-(10-Vindolinyl)-benzoquinone 7 and of 2,3,5,6-tetra-(10-Vindolinyl)-benzoquinone 8
In a 50 mL round bottom flask containing 4 mL distilled water, dissolve 560 mg (3.3 mmoles) of CuCl2·2H2O, followed by 9.5 mg (0.087 mmole) p-benzoquinone, and 150.8 mg (0.33 mmole) vindoline. After 5 min, 1 mL TFA and 2 mL acetonitrile were added and the mixture was stirred for 48 h at room temperature. The acetonitrile was evaporated under reduced pressure and the pH of the solution was made basic with NaHCO3. The same operating procedure as above was followed and the mixture was separated by flash chromatography (3% MeOH in CH2Cl2). Three compounds were thus separated: 2, 6-di-(10-vindolinyl)-1,4 p-benzoquinone 5 (6 mg, 5%), 2,3,5-tri-(10-vindolinyl)-1,4 p-benzoquinone 7, (60 mg, 37%), 2,3,5, 6-tetra-(10-vindolinyl)-1,4 p-benzoquinone 8 (26 mg, 15%)
2,3,5-tris-(10-vindolinyl)-benzoquinone 7, pink amorphous solid; melting point 275 °C; Rf = 0.54 (3%/97% MeOH/CH2Cl2); = +241 (c 0.0023, MeOH); UV (λmax MeOH): 229, 261, 310, 519, 529; IR (KBr): 3431, 2946, 1739, 1613, 1500, 1448, 1374, 1242, 1107, 1035, 889, 820, 743, 628, 481, 584, cm−1; 1H-NMR (500 MHz, CD3OD) major isomer δ = 7.07(3H, s, H-9), 6.81 (3H, s, H-6′), 6.36 (3H, s, H-12), 6.35 (3H, s, H-9), 6.32 (3H, s, H-9), 6.30 (3H, s, H-12), 6.29 (3H, s, H-12), 6.23, 6.18 (3H, s, H-12), 5.90–5.82 (3H, m, H-14), 5.38, 5.36, 5.35 (3H, 3s, H-17), 5.25–5.15 (3H, m H-15), 3.85, 3.82, 3.81 (9H, 3s, C(11)OCH3), 3.80, 3.79 (9H, s, C(16)COOCH3), 3.77, 3.59, 3.58 (3H, 3s, H-2), 2.88, 2.57, 2.50 (3H, 3s, H-21), 2.79, 2.68, 2.67 (9H, s, NCH3), 2.04, 2.02, 2.01 (9H, 3s, C(17)OCOCH3), 0.64, 0.48, 0.44, (9H, 3t, J = 7.2 Hz, CH3-18); 13C-NMR (150 MHz, CD3OD) major isomer δ = 187.9 (C-1′), 187.0 (C-4′), 173.4, (3 C-16-COOCH3), 172.43, 172.41 (3 C-17-OCOCH3), 161.3, 160.7, 160.5 (3 C-11), 156.6, 155.4, 155.3 (3 C-13), 147.9 (C-2′ or C-5′), 146.2 (C-3′) 144.3 (C-2′ or C-5′), 133.2 (C-6′), 131.2, 131.1, 131.0 (3 C-15), 126.5, 126.0, 125.9 (3 C-9), 125.8, 125.6 (3 C-14),125.9, 125.8 (3 C-8), 117.0, 116.2, 115.4 (3 C-10), 94.5, 94.2, 94.1, (3 C-12), 84.61, 84.60, 84.5, (3 C-2), 80.86, 80.85, 80.78 (C-16), 77.56, 77.53, 77.51 (3 C-17), 68.1, 67.9, 67.7 (3 C-21), 56.8, 56.65, 56.64, (3 C-11OCH3), 54.1, 53.65, 53.55 (3 C-7), 52.9 (3 C-16-COOCH3), 52.8, 52.6, 52.5, (3 C-5), 52.0, 51.9, 51.8 (3 C-3), 45.5, 45.4, 44.7 (3 C-6), 44.7, 44.55, 4.45 (3 C-20), 38.8, 38.4 (3 C-1), 32.2, 32.1 (3 C 19), 20.8, 20.7 (3 C-17-OCOCH3), 8.23, 8.1 (3 C-18); HRESI-MS calc. for: C81H95N6O20 [M + H]+ m/z 1471.6596, measured 1471.6633.
2,3,5,6-tetra-(10-vindolinyl)-benzoquinone8, pink amorphous solid; melting point 280 °C; Rf = 0.47 (3%/97% MeOH/CH2Cl2); = -9.5 (c 0.0023, MeOH); UV (λmax MeOH): 229, 260, 310, 515; IR (KBr): 3608, 2957, 2879, 1738, 1614, 1500, 1449, 1372, 1244, 1040, 824, 746, 628 cm−1; 1H-NMR (500 MHz, CD3OD) δ = 7.01, 6.94, 6.92, 6.90, 6.82, 6.80, 6.87, 6.75, 6.68, 6.45, 6.40, 6.36, 6.34, 6.31 (4H, s, H-9), 6.28, 6.26, 6.22, 6.20, 6.17, 6.13, 6.12, 6.10 (4H, s, H-12), 5.85 (4H, m, H-14), 5.46, 5.42, 5.37, 5.36, 5.34 (3H, s, H-17), 5.28, 5.23, 5.12, 516 (4H, m, H-15), 3.87, 3.85, 3.83, 3.79, 3.76, 3.75 (24 H, s, C(11)OCH3, C(16)COOCH3), 3.76, 3.60 (4H, m, H-2), 3.50 (4H, m, Hβ-3), 3.47–3.45 (4H, m, Hβ-5), 2.89–2.73 (4H, m, Hα-3), 2.64, 2.59, 2.56,2.53 (4H, s, H-21), 2.67 (12H, s, H-1), 2.45–2.2.40, (4H, m, Hα-5), 2.30–2.06 (8H, m, H-6), 2.061 (12H, s, C(17)OCOCH3), 1.58–1.54 (4H, m, Ha-19),1.15–1.10 (3H, m, Hb-19), 0.57, 0.47, 0.41, 0.39, 0.30 (4H, overlap t, H-18); HRESI-MS calc. for: C106H125N8O26 [M + H]+ m/z 1925.8700, measured 1925.8702.
5.5. Computational Details
Considering methanol as an implicit solvent (CPCM [16,17]), the systems under study were optimized at the DFT (M06-2X [18]) level of theory and using the 6-311G** basis set within the restricted formalism. The program Gaussian-16 [19] was employed. For the transition states (TS), each geometry was characterized by the presence of a single imaginary vibrational frequency and IRC calculations have been performed to check that the TS connects the two desired minima. Reaction and activation free energies were obtained using the KiSThelP program [20].
Supplementary Materials
The following are available online. Figure S1: UV spectrum of compound 3, Figure S2: IR spectrum of compound 3, Figure S3: HRESI-MS spectrum of compound 3, Figure S4: 1H NMR spectrum of compound 3, Figure S5: 13C NMR spectrum of compound 3, Figure S6: UV spectrum of compound 4, Figure S7: IR spectrum of compound 4, Figure S8: HRESI-MS spectrum of compound 4, Figure S9: 1H NMR spectrum of compound 4, Figure S10: 13C NMR spectrum of compound 4, Figure S11: UV spectrum of compound 5, Figure S12: IR spectrum of compound 5, Figure S13: HRESI-MS spectrum of compound 5, Figure S14: 1H NMR spectrum of compound 5, Figure S15: 13C NMR spectrum of compound 5, Figure S16: UV spectrum of compounds 6, Figure S17: IR spectrum of compounds 6, Figure S18: 1H NMR spectrum of compounds 6, Figure S19: 13C NMR spectrum of compounds 6, Figure S20: UPLC profile of compound 6, Figure S21: UV spectrum of compounds 7, Figure S22: IR spectrum of compounds 7, Figure S23: HRESI-MS spectrum of compounds 7, Figure S24: 1H NMR spectrum of compounds 7, Figure S25: 13C NMR spectrum of compounds 7, Figure S26: UV spectrum of compounds 8, Figure S27: IR spectrum of compounds 8, Figure S28: HRESI-MS spectrum of compounds 8, Figure S29: 1H NMR spectrum of compounds 8, Figure S30: 13C NMR spectrum of compounds 8; Table S1: 13C NMR data for compounds 1, 3 (CDCl3), 4, 5 and 6a (CD3OD), Table S2: Energetics obtained at the DFT (M06-2X/6-311G**) level of theory, considering the methanol implicit solvent (CPCM). Values given in hartree.
Author Contributions
G.M. conceived the project, analyzed the data, wrote and edited the manuscript. S.A. performed the experiments and analyzed the data. R.L. analyzed the data and gave amplitude to the symmetry considerations. E.H. devised and performed the calculations and discussed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
S.A. was supported by a grant from the Pakistan Higher Education Commission. Travel grants from ICCBS allowed G.M. to visit ICCBS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Full NMR data are available upon request from S.A. or G.M.
Acknowledgments
Atta-ur-Rahman, patron-in-chief of ICCBS and I. M. Choudhary, director of ICCBS, are gratefully acknowledged for their constant interest in and support for this work. We wish to particularly thank Atia-Tul-Wahab, who took care of the biological evaluation of all compounds, and the technical staff of ICCBS for all physical measurements. Professor Marco Ciufolini (UBC, Vancouver) kindly suggested the use of copper and took part in the discussions. The article was critically read and edited by J. D. Connolly (U of Glasgow), who is gratefully thanked. This work could not have been achieved without the generous gift of vindoline from Minakem, SAS. Thanks are given to the MaSCA (Maison de la Simulation de Champagne-Ardenne, France) and to the CRIANN computational center (Rouen, France, http://www.criann.fr) for computing facilities.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
On the representation of the adducts. A full and complete representation of the structures limited to the C, N, O and to a few selected H atoms is certainly the best choice for the simple adduct 3. However, when it comes to the larger adducts, these 2D representations of tridimensional edifices lack legibility. Figure A1 gives an example of these drawings for mono-adduct 3 and for di-adduct 6a1.
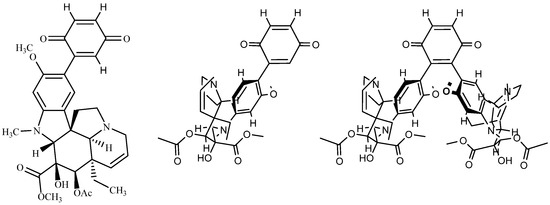
Figure A1.
Two representations of mono adduct 3 and of di-adduct 6a1.
Figure A1.
Two representations of mono adduct 3 and of di-adduct 6a1.
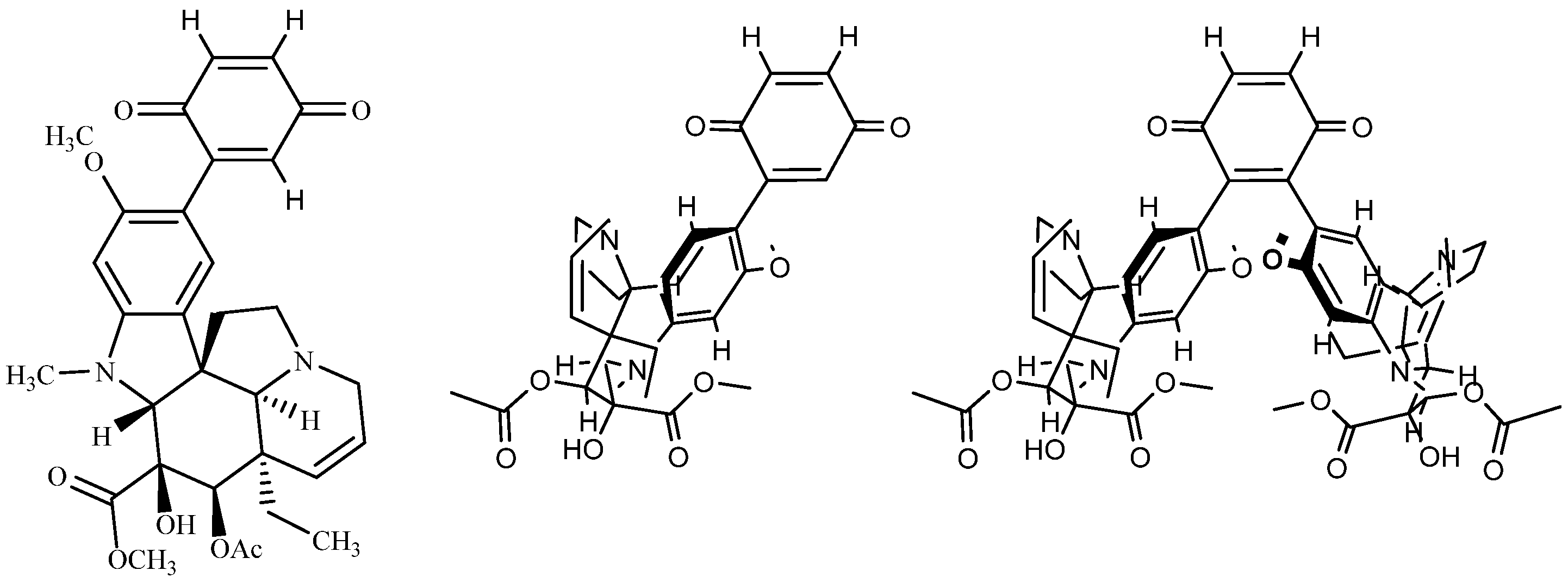
To sort out the gross structures of the di-tri- and tetra-adducts, we chose to write V for vindoline but this ignored the chirality of V. The most intuitive representations consist of drawings where some of the elements of the aromatic ring of vindoline are present and the fact that the OMe group is above or below the plane of the benzoquinone is per se an indication of the chirality (Figure A2). One of the drawbacks of these representations is the difficulty in locating the real elements of chirality of the vindoline and this is reason we propose an alternative in which a “rod” stands for the vindoline with H-2 as a label of chirality.
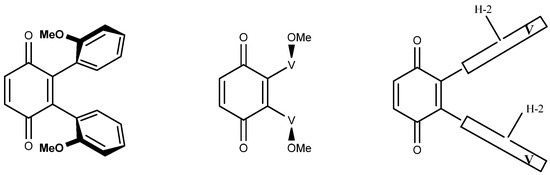
Figure A2.
Three simplified representations of di-adduct 6b.
Figure A2.
Three simplified representations of di-adduct 6b.
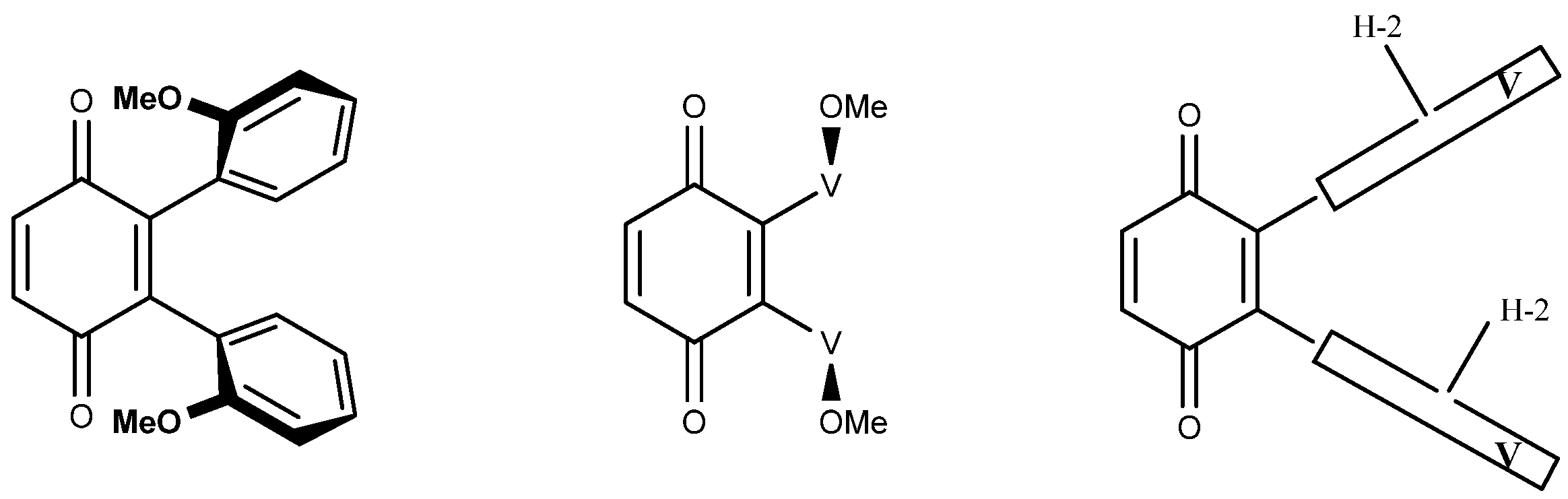
This latter was found to be of convenience in the more complex tetra-adducts and it helped decide the number of possible isomers (Figure A3). There is only one possibility with the four H-2 outside or inside (two isomers). In each of these, whichever H-2 is turned inside/outside or the other way around gives a single possibility because the four H-2 are “equivalent”, giving two more isomers. There are three possibilities with two H-2 inside and two outside: one on the same side and two on opposite sides. This accounts for the seven isomers described above.
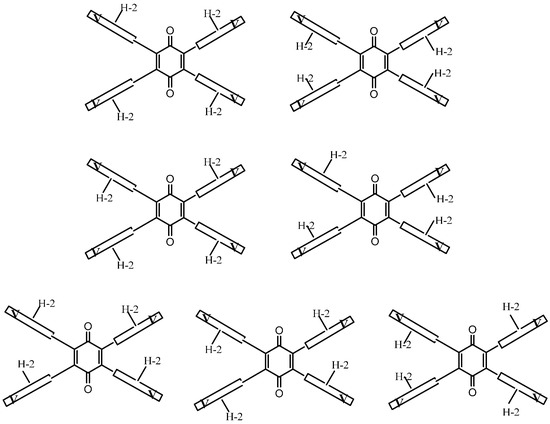
Figure A3.
Representation of the seven isomers of tetra-adduct 8.
Figure A3.
Representation of the seven isomers of tetra-adduct 8.
Appendix B
NMR assignments: compound 5 as an example. At the request of one of the reviewers, this appendix was written to detail the NMR assignments for compound 5; this, of course can be extended to other compounds in the series. As a preliminary remark, we considered that the vindoline moiety present in compound 5 was unaffected by the reaction conditions since all functionalities of the molecule could be observed in the spectra. A single aromatic proton was missing from vindoline, suggesting that the reaction occurred on nucleophilic position C-10 of vindoline (see above Scheme 2 for numbering). The HSQC experiment displayed correlations for eight methines, three methylenes and five methyl groups. The methylene on the ethyl side chain was identified through couplings with CH3-18 (COSY and HSQC) and the methylenes C-3 and C-5 were distinguished through their direct couplings with H-3 and H-5. Worthy of note is the fact that this assignment, probably the most delicate in the 13C NMR spectrum of vindoline was solved almost 50 years ago by Wenkert et al. [21]. Assignment of the methyl and methine resonances was straightforwardly deduced from their chemical environment except for the aromatic, which required used of the HMBC experiment. As said in the above part, the 1H NMR spectrum of 5 showed three singlets at low field and the two vindoline protons (H-9 and H-12) were assigned by the observation of couplings in the HMBC experiment with C-7 (in turn coupling with H-5 and H-6). The remaining singlet was assigned to the quinone protons, which were equivalent for symmetry reasons. The quaternary aromatic carbon C-11 (159.5) and C-13 (155.1) were assigned by HMBC correlations with OCH3 and NCH3 protons respectively, while C-10 (112.6) was detected owing to a 3J coupling with the quinone H-3′.
References
- Gorman, M.; Neuss, N.; Biemann, K. Vinca alkaloids. X. The structure of vindoline. J. Am. Chem. Soc. 1962, 84, 1058–1059. [Google Scholar] [CrossRef]
- Passarella, D.; Giardini, A.; Peretto, B.; Fontana, G.; Sacchetti, A.; Silvani, A.; Ronchi, C.; Cappelletti, G.; Cartelli, D.; Borlake, J.; et al. Inhibitors of tubulin polymerization: Synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg. Med. Chem. 2008, 16, 6269–6285. [Google Scholar] [CrossRef]
- Keglevich, A.; Dányi, L.; Rieder, A.; Horváth, D.; Szigetvári, A.; Dékány, M.; Szántay, C., Jr.; Dhahir Latif, A.; Hunyadi, A.; Zupkó, I.; et al. Synthesis and Cytotoxic Activity of New Vindoline Derivatives Coupled to Natural and Synthetic Pharmacophores. Molecules 2020, 25, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley-Mason, J. Total synthesis of (±)-16-hydroxydihydrocleavamines and partial synthesis of demethoxycarbonyldeoxyvinblastine. J. Chem. Soc. Chem. Commun. 1967, 20, 1048–1049. [Google Scholar] [CrossRef]
- Atta-ur-Rahman. Partial synthesis of D-15, 20-anhydrovinblastine. Pak. J. Sci. Ind. Res. 1971, 14, 487–488. [Google Scholar]
- Potier, P.; Langlois, N.; Langlois, Y.; Guéritte, F. Partial synthesis of vinblastine-type alkaloids. J. Chem. Soc. Chem. Commun. 1975, 16, 670–671. [Google Scholar] [CrossRef]
- El-Sayed, A.; Handy, G.A.; Cordell, G.A. Catharanthus alkaloids. Confirming structural evidences and antineoplastic activity of the bisindole alkaloids leurosine N-oxide, roseadine and vindolicine from Catharanthus roseus. J. Nat. Prod. 1983, 46, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Tiong, S.H.; Looi, C.Y.; Arya, A.; Wong, F.W.; Hazni, H.; Mustafa, M.R.; Awang, K. Vindogentianine, a hypoglycemic alkaloid from Catharanthus roseus (L) G. Don (Apocynaceae). Fitoterapia 2015, 102, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Chaudhary, M.I. Bannucine, a new dihydroindole from Catharanthus roseus (L) G. Don. J. Chem. Soc. Perkin Trans. 1986, 1, 923–926. [Google Scholar] [CrossRef]
- Morrison, M.; Steele, W.; Danner, D. The Reaction of Benzoquinone with Amines and Proteins. Arch. Biochem. Biophys. 1969, 134, 515–523. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Fedoseyenko, D.; Mohapatra, P.P.; Steel, P.J. Reactions of p-Benzoquinone with S-Nucleophiles. Synthesis 2008, 5, 778–785. [Google Scholar] [CrossRef]
- Jiang, J.-H.; Boominathan, S.S.K.; Hu, W.-P.; Chen, C.-H.; Vandavasi, J.K.; Lin, Y.-T.; Wang, J.-J. Sequential, one-pot access to arylated benzoquinones/naphtoquinones from phenols/naphtols. Eur. J. Org. Chem. 2016, 2016, 2284–2289. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Liu, L.; Chen, Y.-J.; Wang, D.; Li, C.-J. Synthesis of aryl-substituted 1,4-benzoquinone via water-promoted and In(OTf)3-catalyzed in situ conjugate addition-dehydrogenation of aromatic compounds to 1,4-benzoquinone in water. Adv. Synth. Catal. 2006, 348, 229–235. [Google Scholar] [CrossRef]
- Jonasson, C.; Karstens, W.F.J.; Hiemstra, H.; Bäckvall, J.E. A Palladium(II)-catalyzed intramolecular 1,2-oxidation of allenes involving nitrogen nucleophiles. Tetrahedron Lett. 2000, 41, 1619–1622. [Google Scholar] [CrossRef]
- Escolastico, C.; Santa Maria, M.D.; Claramunt, R.M.; Jimeno, M.L.; Alkorta, I.; Foces-Foces, C.; Cano, F.H.; Elguero, J. Imidazole and benzimidazole addition to quinones. Formation of meso and d,l isomers and crystal structure of the d,l isomer of 2,3-bis(benzimidazol-l’-yl)-1,4-dihydroxybenzene. Tetrahedron 1994, 50, 12489–12510. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.0; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Canneaux, S.; Bohr, F.; Henon, E. KiSThelP: A Program to Predict Thermodynamic Properties and Rate Constants from Quantum Chemistry Results. J. Comput. Chem. 2014, 35, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wenkert, E.; Cochran, D.W.; Hagaman, E.W.; Schell, F.M.; Neuss, N.; Katner, A.S.; Potier, P.; Kan, C.; Plat, M.; Koch, M.; et al. Carbon-13 nuclear magnetic resonance spectroscopy of naturally occurring substances. XIX. Aspidosperma alkaloids. J. Am. Chem. Soc. 1973, 95, 4990–4995. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).