Abstract
Terpenylquinones are mixed biogenesis primary or secondary metabolites widespread in Nature with many biological activities, including the antineoplastic cytotoxicity, that have inspired this work. Here, we present a cytotoxic structure-activity relationship of several diterpenylhydroquinone (DTHQ) derivatives, obtained from the natural labdane diterpenoid myrceocommunic acid used as starting material. Different structural modifications, that changed the functionality and stereochemistry of the decalin, have been implemented on the bicyclic core through epoxidation, ozonolysis or decarboxylation, and through induction of biomimetic breaks and rearrangements of the diterpene skeleton. All the isomers generated were completely characterized by spectroscopic procedures. The resulting compounds have been tested in vitro on cultured cancer cells, showing their relevant antineoplastic cytotoxicity, with GI50 values in the μM and sub-μM range. The rearranged compound 8 showed the best cytotoxic results, with GI50 at the submicromolar range, retaining the cytotoxicity level of the parent compounds. In this report, the versatility of the labdane skeleton for chemical transformation and the interest to continue using structural modifications to obtain new bioactive compounds are demonstrated.
1. Introduction
Terpenyl-quinones/hydroquinones (TQs/THQs) are mixed biogenesis primary or secondary metabolites widespread in Nature. Examples of primary metabolites are vitamin K, tocopherols, ubiquinones or plastoquinones that play important roles in the electron transport chain or in photosynthesis [1]. As secondary metabolites, TQs/THQs have been isolated from different sources, particularly of marine origin such as avarol or avarone (Figure 1), and they often have a wide variety of biological activities, mainly antineoplastic and antioxidant properties, that have been related to their involvement in redox cycling and/or Michael-1,4-addition reactions [2,3].
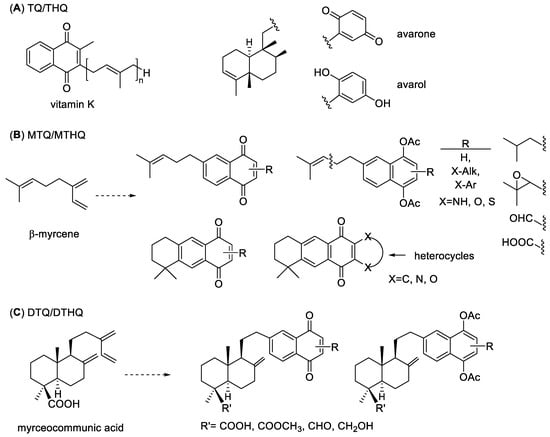
Figure 1.
Structures of representative terpenylquinones (TQ) and terpenylhydroquinones (THQ). (A) Examples of natural TQs and THQs. (B) General structure of monoterpenylquinones/hydroquinones (MTQ/MTHQs) derived from β-myrcene. (C) General structure of diterpenylquinones/hydroquinones (DTQ/DTHQs) derived from myrceocommunic acid.
Our research group has been involved in the design, synthesis and biological evaluation of several series of TQs/THQs, derived either from the monoterpene β-myrcene or the diterpenoid myrceocommunic acid, giving rise to two series of compounds named monoterpenylquinones (MTQs/MTHQs) and diterpenylquinones (DTQs/DTHQs) (Figure 1). We have prepared a large number of derivatives with variations in sizes and functionalities of both the quinone and the terpenoid moieties. Very interesting antineoplastic compounds were obtained from β-myrcene [4,5,6,7,8,9,10] and myrceocommunic acid [11,12,13,14], being the DTQs/DTHQs slightly more potent than the MTQs/MTHQs. About the quinone size, 1,4-naphthoquinone (NQ) and 1,4-anthracenequinone (AQ) analogues were more potent than the corresponding benzoquinones (BQs) and 9,10-AQs, the common moiety in clinically used anticancer drugs, without significant differences between the quinone functions and the corresponding acetylated hydroquinones. Particularly interesting have been the results of several DTHQ-cyclolignan hybrids against the osteosarcoma cell line MG-63, which is a very aggressive type of cancer [14].
In addition to the cytotoxicity assays, several of our TQs/THQs were evaluated as antifungal [10,15], antiviral [10,16] and anti-leishmanial [17], attaining promising results against yeasts, filamentous fungi and Leishmania infantum.
In our previous research, many MTQs/MTHQs have been obtained by transformation of the side chain derived from the monoterpenoid [5,6,9,10], however only a few examples dealt with modifications of the diterpenoid core, always keeping the labdane type decalin [11,12]. Now, we describe further chemical transformations performed at this decalin moiety in the DTHQ series, either at the A or B decalin rings, including isomerizations, decarboxylations or rearrangements, leading to derivatives with modified diterpenoid skeletons, which have been evaluated against four tumor cell lines, representative of common and multi-drug resistant cancer types affecting lung, colon, breast and skin, to analyze the influence of those modifications on their cytotoxicity.
2. Results and Discussion
2.1. Chemistry
Continuing with our research on the field of terpenylquinones and considering the previous interesting results, we decided to enlarge the structural diversity of our DTHQ derivatives by chemical transformations on the decalin core, taking advantage of the functional groups present on it, i.e., the double bond around C-8 and the carboxylic group at C-4, making use of reactions such as epoxidation, ozonolysis and decarboxylation, which were applied to the previously reported DTHQs 1–4.
The starting DTHQs 1 and 2 were prepared through a Diels-Alder cycloaddition between myrceocommunic acid or methyl myrceocommunate and p-benzoquinone (Scheme 1), following the procedure previously described by us [11,12]. Isomerization of the Δ8(17) double bond, in both 1 and 2 to the more stable endocyclic and tetrasubstituted Δ8 double bond, was achieved by treatment with aq 57% HI at 80 °C to yield the DTHQs 3 and 4 [13].
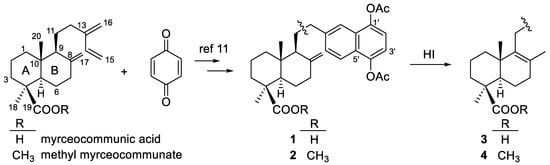
Scheme 1.
Synthesis of the starting DTHQs 1–4.
2.1.1. Epoxidation and Rearrangement Reactions
It is well known that the Lewis acid-catalyzed rearrangement of epoxides is an efficient tool to obtain new compounds through carbocation rearrangements [18]. In fact, in our previous work [11], the epoxidation of the Δ8(17) double bond, followed by rearrangement promoted by BF3·Et2O, gave the corresponding aldehyde at C-17, with improved cytotoxicity respecting DTHQ 2. Here in this work, in order to increase the structural diversity of our DTHQs, we explore the rearrangement products of the endocyclic Δ8 double bond epoxidation.
Treatment of 4 with MCPBA afforded a mixture of the two possible epoxides 5 and 6 in a 2:1 ratio. Both isomers were separated by column chromatography, characterized through their NMR spectra and treated separately with BF3·Et2O (Scheme 2).
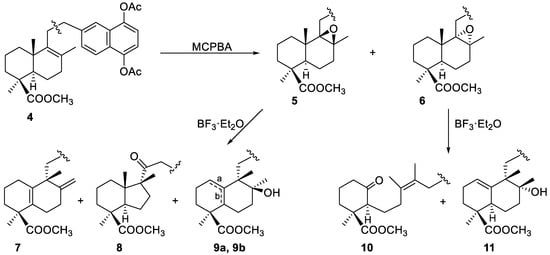
Scheme 2.
Epoxidation of 4 and rearrangement of epoxides 5 and 6 promoted by BF3·Et2O.
Reaction of the β-epoxide 5 with the Lewis acid at −78 °C gave compounds 7, 8 and the isomers 9a and 9b. The 1H- and 13C-NMR spectra of DTHQ 7 indicated the presence of the exocyclic double bond at C-8, similar to that present in DTHQs 1 and 2, whereas the spectrum of 7 didn’t have the characteristic signal of the axial methyl group of a labdane type decalin. In fact, such signal moved from around 0.5 ppm to 1.19 ppm; this fact, together with the 13C-NMR data and the molecular formula C31H36O6 by HRMS, indicated us that the labdane decalin was rearranged to an halimane type decalin [19] with a tetrasubstituted Δ5(10) double bond. The configuration at C-9 was deduced from the proposed epoxide rearrangement mechanism, probably via an unconcerted mechanism [19], that involved 1,2-shift of the axial methyl group at C-10 as illustrated by pathway “I” in Scheme 3.
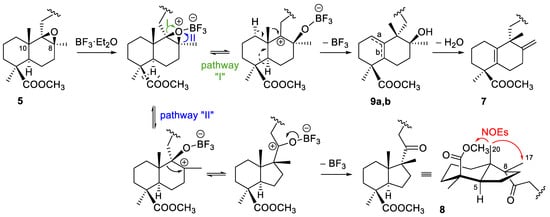
Scheme 3.
Proposed mechanism for the epoxide 5 rearrangement leading to compounds 7–9.
The 1H and 13C-NMR spectra of DTHQ 8 showed signals for a ketone function and three methyl groups on aliphatic quaternary carbons. Several spectroscopic experiments (NOE, HMBC and HMQC and HRMS data) confirmed the contraction of the ring by migration of the chain following the pathway “II” shown in Scheme 3.
Compounds 9a and 9b showed to be regioisomers that practically coeluted in the CC, and only a small amount of 9a was purified. They had a tertiary hydroxyl group and very similar spectroscopic properties, being the main difference in the 1H-NMR spectra, the presence or absence of an olefinic proton signal at 5.52 ppm. NOE, HMBC and HMQC experiments performed on 9a allowed to locate the double bond at the Δ1(10) position and to assign the S configuration for C-8; consequently, the double bond in 9b should be placed in position Δ5(10) and this compound would be the precursor of 7 through loss of a water molecule (Scheme 3).
Rearrangement of the α-epoxide 6 in the same conditions yielded a reaction product from which the ketone 10 and the alcohol 11 were isolated by CC (Scheme 2). The NMR data of 10 indicated the presence of a ketone (210.0 ppm) and two methyl groups on a double bond (1.62 and 1.69 ppm). HRMS, HMBC and HMQC experiments corroborated the opening of the B-ring of the decalin, and its degradation can be explained from the proposed rearrangement mechanism shown in Scheme 4. The Z configuration for the double bond was proposed on the basis of this mechanism and literature data for the 13C NMR chemical shift of methyl groups on double bonds [20].
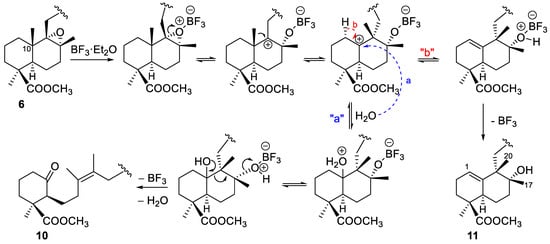
Scheme 4.
Proposed mechanism for the epoxide 6 rearrangement leading to ketoester 10 and hydroxyester 11.
Compound 11 had spectra data very similar to those of compound 9a, with simple differences in the chemical shifts of the C-17 and C-20 methyl groups, thus indicating that 11 should be the C-8 epimer of 9a.
2.1.2. Ozonolysis
Ozonolysis is a well-known reaction for oxidative cleavage of carbon-carbon double bonds to get carbonyl compounds under reductive or oxidative conditions [21]. It has been widely used for structure elucidation of organic compounds, but also to get fragments with carbonyl groups. Interestingly, if the double bond is within a ring, a chain containing two carbonyls is obtained.
In the work described here, DTHQs 1 and 2 have an exocyclic double bond and its ozonolysis under reductive work-up conditions gave respectively the ketones 12 and 13, both having a 17-norlabdane decalin core (Scheme 5). However, DTHQ 4 has an endocyclic tetrasubstituted double bond and its ozonolysis afforded the 8,9-secolabdane diketone 14 and the epoxide 6 in a 1:1.5 ratio (Scheme 5). The formation of epoxides is frequent during ozonolysis of sterically hindered double bonds [22] as it happens in the case of 4.
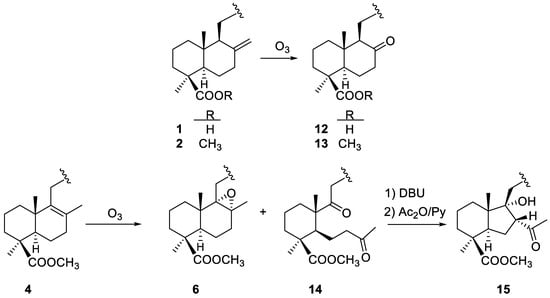
Scheme 5.
Ozonolysis and further reactions of DTHQs 1, 2 and 4.
Diketone 14 was submitted to further intramolecular aldolic condensation in the presence of DBU giving rise to the rearranged labdane derivative 15 (Scheme 5). Under basic conditions, a partial saponification of the aromatic acetates was observed, so the reaction product was acetylated to restore the naphthohydroquinone diacetate moiety of the starting DTHQs. Both the secodecalin and the rearranged labdane derivative were characterized by spectroscopic comparison with the natural products chapecoderins A and B, which have the same labdane skeletons without the carboxylic function at C-19 [23].
2.1.3. Decarboxylation Reactions
The last type of transformations performed on the DTHQs take advantage of the presence of the carboxylic group, which allows to get derivatives modified at the A-ring of the decalin. We obtained several 19-nor-DTHQs from 1 and 3 by decarboxylation with lead tetraacetate (LTA) [24]. Thus, the oxidative decarboxylation of 1 with LTA and cupric acetate in toluene-pyridine gave a 5:7:10 mixture (as deduced from the C-20 methyl signal the 1H-NMR spectrum) of the three unsaturated derivatives 16a–c respectively, and the triacetate 17 [25] (Scheme 6). When this reaction product was treated with BF3·Et2O, the triacetate was transformed into the olefins 16a–c. The three isomers 16a–c were practically inseparable by CC, even using silica gel impregnated with silver nitrate, and only a small amount of the 16a, which has two exocyclic methylenes, was separated. Consequently, the mixture 16a–c was treated with HI to attempt the isomerization to the more stable Δ4 and Δ8 endocyclic double bonds. However, the isomerization was not complete and a mixture of 18b and 18c was obtained (Scheme 6). To force the isomerization towards 18c, the mixture 18b,c was treated with iodine in toluene for longer time, however, a complex reaction product was obtained from which the rearranged aromatic derivative 19 was separated and characterized (Scheme 6). The isomer 18c was also obtained after treatment of DTHQ 3 with LTA followed by reaction with iodine for 3 h (Scheme 6).
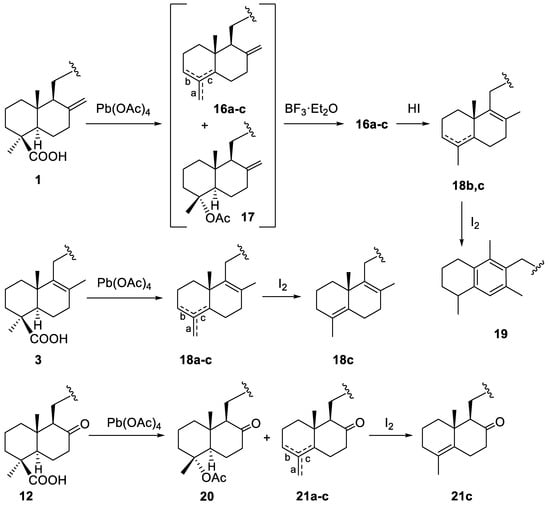
Scheme 6.
Decarboxylations and further reactions of DTHQs 1, 3 and 12.
The oxidative decarboxylation was also applied to DTHQ 12 to afford the ketoacetate 20 and the isomers 21a–c (Scheme 3). When the mixture 21a–c was treated with iodine, the isomer 21c, having the most stable tetrasubstituted double bond, was isolated and characterized.
The structure of compound 19 was deduced from the analysis of its spectroscopic properties. Among the most characteristic signals in its 1H-NMR spectrum, those corresponding to three methyl groups stood out, one doublet at 1.30 ppm and two singlets at 2.35 and 2.23 ppm, the latter assignable to methyls attached to an aromatic ring. Additional spectroscopic experiments (13C-NMR, HRMS, NOE, HMBC and HMQC) confirmed the structure of 19, whose formation can be explained by the proposed mechanism shown in Scheme 7, in which the observed NOEs for 19 are shown with blue arrows, although the absolute configuration at C-4 could not be determined. Similar rearrangements towards B-ring aromatization have also been described for other natural labdanes [26].
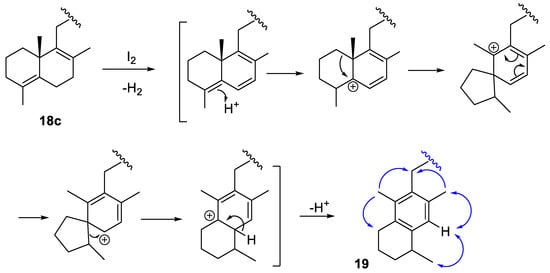
Scheme 7.
Proposed mechanism for the formation of compound 19. The blue arrows represent the experimentally observed NOEs.
2.2. Biological Evaluation
Cell killing ability of the synthetized DTHQ derivatives was conducted against a panel of three human tumor cell lines with high incidence in the population [27], including non-small cell lung carcinoma (A-549), colon adenocarcinoma (HT-29), and multi-drug-resistant breast carcinoma (MDA-MB-231). The in vitro antiproliferative activity was assessed using the colorimetric SRB (sulforhodamine B) method [28] and the common anticancer naphthacenequinone drug doxorubicin was included in the assays as reference. The GI50 (μM) values (concentration that causes 50% growth inhibition), for all the tested compounds are shown in Table 1. Additionally, compounds 1, 2, 8, and 21a were also assayed on malignant skin melanoma SK-MEL-28 cells with GI50 values of 0.24, 0.12, 0.54 and 1.66 μM respectively.

Table 1.
Antineoplastic cytotoxicity (GI50 values in μM) of DTHQs 1–21 against A-549 (lung), HT-29 (colorectal) and MB-231 (breast) tumor cell lines (nt: not tested).
From these results, it can be stated that the compounds assayed were fairly cytotoxic for neoplastic cells. Indeed, some of them showed GI50 values similar to those of doxorubicin. In general, the presence of oxygenated functions, the opening, contraction or rearrangement of the decalin moiety gave compounds that kept or slightly decreased the potency of the lead compounds 1 and 2.
There were no differences in potency between compounds with exocyclic or endocyclic double bond in the B-ring of the decalin core (compounds 1–4). The presence of a ketone in such ring led to less potent compounds (12, 13 and 21 vs. 1 and 2), while DTHQ 12, with a free carboxylic group, was the less potent compound of the series on the three cell lines. Interestingly, the α-epoxide 6 was four times more potent than the β-epoxide 5, what is in accordance with previous results obtained with the Δ8(17) epoxides [11]. Those analogues with a halimane type decalin and a hydroxyl group at C-8 (9 and 11), were less potent than their precursors, though keeping their cytotoxicity values in the micromolar range. The same happened with DTHQs 10 and 14, which lacked the decalin B-ring. Several differences were observed for DTHQs 8 and 15, which had an indane system, being 8 as potent as its precursors, while 15 was fairly less potent. Such a remarkable difference, of more than one order of magnitude, would be accounted for by an intramolecular hydrogen bonding between the carbonyl and the hydroxyl group attached to the cyclopentane ring in 15, which would prevent potential interactions with hypothetical targets, unlike the free ketone in 8. The presence of the ketone out of the bicyclic core in 8 may also have a positive impact on activity, since other decalin compounds with ketone groups located on the ring, such as 12, 13 or 21, resulted less potent than the indanyl ketone 8.
The decarboxylated analogues were, in general, less potent than the parent compounds; however, DTHQ 18 had the same GI50 value than its precursor without differences between the a–c isomers, and DTHQs 17 and 20, with an acetoxy group at C-4, retained the submicromolar cytotoxicity.
Finally, in terms of cytotoxic potency, these results indicated that a varied range of functionalization in the B-ring was tolerated and that the ester function in the A-ring seemed to be important for the antineoplastic cytotoxicity.
3. Materials and Methods
3.1. Chemistry
NMR experiments were recorded on a Bruker WP 200 SY (Bruker GmbH, Karlsruhe, Germany) (200 and 50.3 MHz for 1H and 13C) or Bruker Avance 400DRX (Bruker GmbH, Karlsruhe, Germany) (400 and 100 MHz) spectrometers in CDCl3 using TMS as internal reference. Chemical shift (δ) values are expressed in ppm followed by multiplicity and coupling constants (J) in Hz. Optical rotations were recorded on a Perkin-Elmer 241 polarimeter (Perkin-Elmer GmbH, Uberlingen, Germany) in CHCl3 solution. UV spectra were obtained on a Hitachi 100–60 spectrophotometer (Hitachi High-Technologies Corporation, Tokyo, Japan) in ethanol solution, and λmax are given in nm. IR spectra were obtained on a Nicolet Impact 410 spectrophotometer (Nicolet Instrument Corporation, Madison, WI, USA) in NaCl film. HRMS were run in a VG TS-250 spectrometer (VG Instruments, Manchester, UK) working at 70 eV, using electrospray ionization (ESI) or fast atom bombardment (FAB). Elemental analysis (C, H, N) were obtained with a PERKIN-ELMER 2400 CHN (Perkin-Elmer GmbH, Uberlingen, Germany). Solvents and reagents were purified by standard procedures as necessary. Column chromatography (CC) purifications were performed using silica gel 60 (40–63 mm, 230–400 mesh, Merck) and TLC was carried out on silica gel 60 F245 (Merck, 0.25 mm thick). NMR assignments are included in the Supplementary material: Tables S1–S20.
Diterpenylnaphthohydroquinone diacetates 1, 2 and 4 were obtained by means of previously described procedures [11,12]. Other compounds were prepared as follows.
DTHQ 3. To a 10−2 M solution of 1 (185 mg, 0.38 mmol) in toluene (37 mL) aq HI 57% (0.86 mL) was added. The mixture was stirred at 80 °C for 10 min. Then, ethyl acetate was added and the organic layer was washed with aq satd NaHCO3 and brine, dried over Na2SO4, filtered and evaporated off yielding the Δ8 product 3 (182 mg, 0.37 mmol, 98%): IR cm−1 (film): 2930, 1765, 1690, 1450, 1365, 1200, 1180, 1050, 820, 735. Anal. Calcd. for C30H36O6: C,73.15; H, 7.37. Found C, 73.12%; H, 7.36%. RMN 1H: Table 2. RMN 13C: Table 3.
Table 2.
1H-NMR data for compounds 3,5–13 (δ ppm (J in Hz)).
Table 3.
13C-NMR data for compounds 3,5–13 (δ ppm).
DTHQ epoxides 5 and 6. Compound 4 (178 mg, 0.35 mmol) was dissolved in dichloromethane and MCPBA (485 mg, 1.55 mmol) and NaHCO3 (528 mg) were added. The mixture was kept at room temperature for 3 h. Then dichloromethane was added, followed by 10% aqueous Na2S2O3 until the oxidant was eliminated. The organic layer was washed with brine and dried over anhydrous Na2SO4, and the solvent evaporated off. The reaction product was purified by CC (CHCl3/EtOAc 9:1) to obtain: 65 mg (35%) of epoxide 5, 42 mg (23%) of mixture of 5 and 6 and 28 mg (15%) of epoxide 6. Compound 5: [α = +50.9 (c, 0.97%); IR cm−1 (film): 2945, 1764, 1717, 1462, 1433, 1366, 1262, 1203, 1182, 1053, 891, 825; UV λmax (ε): 240 (12300), 287 (5500). HRMS-FAB: calcd for C31H39O7 [M + H]+ 523.2696; found 523.2742 m/z. RMN 1H: Table 2. RMN 13C: Table 3. Compound 6: [α = +25.1 (c, 1.0%); IR cm−1 (film):2920, 2850, 1765, 1720, 1610, 1465, 1365, 1200, 1180, 1050, 890, 825, 735; HRMS-FAB: calcd for C31H39O7 [M + H]+ 523.2696 u; found 523.2698 m/z; RMN 1H: Table 2. RMN 13C: Table 3.
Rearranged DTHQs 7, 8 and 9. To a solution of epoxide 5 (62 mg, 0.12 mmol) in dry dichloromethane, cooled at −78 °C, BF3-Et2O (55 μL, 0.43 mmol) was added under inert atmosphere. The mixture was stirred at this temperature for 3 h. Then the solvent was removed, and the crude dissolved in EtOAc, washed with brine, dried over Na2SO4, and the solvent evaporated off. The reaction crude was chromatographed to yield: 6 mg (9%, eluent: Hex/EtOAc, 9:1) of 7, 24 mg (38%, eluent: Hex/EtOAc, 9:1) of 8, 7 mg (11%, eluent: Hex/EtOAc, 7:3) of 9a and 17 mg (27%, eluent: Hex/EtOAc, 7:3) of 9a,b. Compound 7: IR cm−1 (film): 2935, 1765, 1725, 1610, 1450, 1365, 1200, 1050, 1010, 895, 825, 735; HRMS-FAB: calcd for C31H37O6 [M + H]+ 505.2590; found 505.2623 m/z. RMN 1H: Table 2. RMN 13C: Table 3. Compound 8: [α = +19.7 (c, 0.97%); IR cm−1 (film): 2495, 1765, 1720, 1625, 1610, 1450, 1365, 1200, 1050, 1005, 895, 825, 735; HRMS-FAB: calcd for C31H39O7 [M+H]+ 523.2696; found 523.2700 m/z. RMN 1H: Table 2. RMN 13C: Table 3. HMQC and HMBC experiments: Table S1. Compound 9a: IR cm−1 (film): 3515, 2935, 1765, 1610, 1450, 1365, 1265, 1200, 1120, 1010, 915, 820, 730; HRMS-FAB: calcd for C31H37O6 [M+H]+ 505.2590; found 505.2623 m/z. RMN 1H: Table 2. RMN 13C: Table 3. HMQC and HMBC experiments: Table S2.
Rearranged DTHQs 10 and 11. To a solution of epoxide 6 (89 mg, 0.17 mmol) in dry dichloromethane, cooled at −78 °C, BF3-Et2O (78 μL, 0.61 mmol) was added under inert atmosphere. The mixture was stirred at this temperature for 3 h. Then the solvent was removed, and the crude dissolved in EtOAc, washed with brine, dried over Na2SO4, and the solvent evaporated off. The reaction crude was chromatographed to yield: 25 mg (28%, eluent: Hex/EtOAc, 7:3) of 10 and 29 mg (32%, eluent: Hex/EtOAc, 6:4) of 11. Compound 10: [α = +2.8 (c, 0.65%); IR cm−1 (film): 2948, 1765, 1727, 1608, 1462, 1433, 1366, 1020, 1182, 1053, 897, 825, 735. HRMS-FAB: calcd for C31H39O7 [M + H]+: 523.2696; found 523.2674 m/z. Anal. Calcd. for C31H38O7: C,71.24; H, 7.33. Found C, 71.19%; H, 6.90%. RMN 1H: Table 2. RMN 13C: Table 3. HMQC and HMBC experiments: Table S3. Compound 11: [α = −62.4 (c, 0.65%); IR cm−1 (film): 3536, 29,36, 1765, 1728, 1609, 1462, 1433, 1365, 1203, 1182, 1054, 1016, 939, 899, 736. UV λmax (ε): 240 (12600), 286 (4600). HRMS-ESI: calcd for C31H38O7Na [M + Na]+: 545.2509; found 545.2528 m/z. RMN 1H: Table 2. RMN 13C: Table 3. HMQC and HMBC experiment: Table S4.
General procedure for ozonolysis. nor-DTHQ 12. A solution of compound 1 (340 g, 0.69 mmol) in dry CH2Cl2 (6 mL) was cooled to –78 °C, and ozone was bubbled (30 nL/h, 0.6 amperes) through the mixture for 10 min. Then the reaction mixture was purged with oxygen for 10 min and argon for additional 5 min to remove the ozone excess. Then, dimethyl sulfide (1.5 mL, 19 mmol) was added to the mixture and stirred for 6 h. The solvent was distilled off, the residue was dissolved in EtOAc and washed with brine. The organic layer was dried over anhydrous Na2SO4 and evaporated off to give a reaction product that was purified by CC on silica-gel (eluent: Hex/EtOAc, 7:3) to yield 12 (227 mg, 66%). IR cm−1 (film): 2950, 1765, 1705, 1695, 1610, 1450, 1365, 1200, 1050, 1010, 915, 895, 735. RMN 1H: Table 2. RMN 13C: Table 3. HRMS-FAB: calcd for C29H35O7 [M + H]+: 495.2383; found 495.2377.
nor-DTHQ 13. Starting from 2 (220 mg, 0.44 mmol) and following the above procedure for ozonolysis, the reaction product yielded 13 (212 mg, 96%). IR cm−1 (film): 2935, 1765, 1720, 1710, 1610, 1450, 1365, 1200, 1050, 825, 735. HRMS-FAB: calcd for C30H37O7 [M + H]+: 509.2539; found 509.2602 m/z. RMN 1H: Table 2. RMN 13C: Table 3.
seco-DTHQ 14. Starting from 4 (171 mg, 0.34 mmol) and following the above procedure for ozonolysis, the reaction product was chromatographed to yield 6 (74 mg, 40%, eluent: Hex/EtOAc, 7:3) and 14 (49 mg, 27%, eluent: Hex/EtOAc, 8:3). [α = +19.0 (c, 0.97%); IR cm−1 (film): 2945, 1764, 1717, 1462, 1433, 1366, 1262, 1203, 1182, 1053, 891, 825. UV λmax (ε): 240 (12300), 289 (4500). HRMS-FAB: calcd for C31H39O8 [M + H]+: 539.2645; found 539.2618 m/z. RMN 1H: Table 4. RMN 13C: Table 5. HMQC and HMBC experiments: Table S5.
Table 4.
1H-NMR data for compounds 14–21 (δ ppm (J in Hz)).
Table 5.
13C-NMR data for compounds 14–21 (δ ppm).
Rearranged DTHQ 15. To a stirred solution of 14 (59 mg, 0.11 mmol) in dry toluene (3 mL) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (27 μL, 0.16 mmol). The reaction mixture was stirred for 6 h at 60 °C under nitrogen atmosphere. The reaction was quenched by addition of 2 M HCl and extracted with ethyl acetate. The combined organic layer was washed with brine and dried over anhydrous Na2SO4. The reaction product was acetylated with acetic anhydride in pyridine and then purified by column chromatography (eluent: Hex/EtOAc, 7:3) to yield unreacted 14 (45%) and 15 (30 mg, 51%). [α = +8.6 (c, 1.0%); IR cm−1 (film): 3421, 2932, 1764, 1721, 1687, 1609, 1461, 1433, 1365, 1020, 1181, 1053, 896, 825. UV λmax (ε): 240 (12300), 285 (5900). HRMS-FAB: calcd for C31H39O8 [M + H]+: 539.2645; found 539.2642 m/z. RMN 1H: Table 4. RMN 13C: Table 5. HMQC and HMBC experiment: Table S6.
General procedure for decarboxylation with lead tetracetate. nor-DTHQs 16 and 17. A solution of 1 (256 mg, 0.52 mmol) and pyridine (1.3 mL, 16 mmol) in dry toluene (10 mL) was heated at 145 °C for 1 h. Then lead tetraacetate (299 mg, 0.67 mmol) and copper diacetate (104 mg, 0.52 mmol) were added and kept stirred at the same temperature for additional 4.5 h. The cooled mixture was evaporated and redissolved in EtOAc. The organic layer was washed consecutively with 2 M HCl and brine and dried over anhydrous Na2SO4. The reaction product was purified by column chromatography to yield 16a–c (109 mg, 47%, eluent: Hex/EtOAc, 95:5) and 17 (53 mg, 20%, eluent: Hex/EtOAc, 9:1). RMN 1H: Table S14. RMN 13C: Table S15. The mixture 16a–c was chromatographed over silica gel impregnated with 20% silver nitrate, using Hex/EtOAc, 7:3 as eluent, to yield 16a–c (63 mg, 47%, eluent:) and 16a (21 mg. Compound 16a: [α = +10.5 (c, 0.54%); IR cm−1 (film): 3400, 2929, 2849, 1766, 1737, 1720, 1643, 1461, 1440, 1366, 1020, 1181, 1053, 890. UV λmax (ε): 240 (5700), 284 (2100). HRMS-FAB: calcd for C29H35O4 [M + H]+: 447.2535; found 447.2516 m/z. RMN 1H: Table 4. RMN 13C: Table 5. Compound 17: [α = +11.3 (578), (c, 0.99%) IR cm−1 (film): 3078, 2935, 2870, 1766, 1724, 1641, 1608, 1432, 1366, 1252, 1020, 1181, 1053, 1013, 893, 826, 736. UV λmax (ε): 240 (11400), 285 (4800). RMN 1H: Table 4. RMN 13C: Table 5.
nor-DTHQ 18. Starting from 3 (250 mg, 0.51 mmol) and following the above procedure for decarboxylation, the CC of the reaction product over AgNO3 impregned silicagel (eluent: Hex/EtOAc, 9:1), yielded 18a (23 mg, 10%), a 1:1:1 mixture of 18a–c (64 mg, 28%) and 18c (25 mg, 11%). Compound 18a: [α = +62.1 (c, 0.40%); IR cm−1 (film): 2930, 1765, 1610, 1450, 1365, 1200, 1180, 1050, 1010, 890, 820, 735. HRMS-FAB: calcd for C29H35O4 [M + H]+: 447.2535; found 447.2563 m/z. RMN 1H: Table 4. RMN 13C: Table 5. Compound 18c: [α = +99.7 (c, 0.76%); IR cm−1 (film): 2930, 1765, 1610, 1450, 1365, 1200, 1180, 1050, 1010, 895, 825, 735. HRMS-FAB: calcd for C29H35O4 [M + H]+: 447.2535; found 447.2542 m/z. RMN 1H: Table 4. RMN 13C: Table 5.
The mixture 18a–c was dissolved in toluene and iodine (10 mg) was added and stirred at 95 °C for 3 h. The cooled mixture was evaporated and redissolved in EtOAc. The organic layer was washed consecutively with aq sat sodium thiosulphate (Na2S2O3) and brine and dried over anhydrous Na2SO4, to yield 18c (96%).
Treatment of the mixture 16a–c with aq HI 57% (following the procedure described above to obtain 3), yield a 1:1 mixture of 18b and 18c (66%) after CC of the reaction product, eluted with Hex/EtOAc 95:5.
nor-DTHQ 19. The mixture 18b–c (116 mg, 0.26 mmol) was dissolved in toluene (7 mL) and iodine (44 mg, 0.17 mmol) was added and stirred at 95 °C for 24 h. The cooled mixture was evaporated and redissolved in EtOAc. The organic layer was washed consecutively with aq sat Na2S2O3 and brine and dried over anhydrous Na2SO4, to yield a reaction crude that was purified by CC to yield 19 (37 mg, 32%, eluent: Hex/EtOAc, 95:5). [α = −1.7 (c, 0.9%); IR cm−1 (film): 2926, 2855, 1766, 1608, 1461, 1431, 1364, 1201, 1180, 1054, 1007, 894, 823. UV λmax (ε): 241 (15100), 283 (4900). MS (m/z): 444 (M)+(10). RMN 1H: Table 4. RMN 13C: Table 5. HMQC experiment: Table S7.
dinor-DTHQs 20 and 21. Starting from 12 (230 mg, 0.46 mmol) and following the above procedure for decarboxylation, the CC of the reaction product yielded 21a (7 mg, 3%, eluent: Hex/EtOAc, 95:5), 21a–c (85 mg, 41%, eluent: Hex/EtOAc, 95:5) and 20 (15 mg, 7%, eluent: Hex/EtOAc, 9:1). Compound 20: [α = −16.5 (c, 0.96%); IR cm−1 (film): 2940, 1765, 1725, 1710, 1365, 1250, 1200, 1180, 1050, 1010. HRMS-ESI: calcd for C30H36O7Na [M + Na]+: 531.2353; found 531.2346 m/z. RMN 1H: Table 4. RMN 13C: Table 5. Compound 21a: [α = −1.6 (c, 0.67%); IR cm−1 (film): 2936, 2865, 1765, 1707, 1643, 1608, 1433, 1366, 1202, 1182, 1053, 1009, 895, 827, 735. UV λmax (ε): 240 (8800), 285 (4000). HRMS-FAB: calcd for C28H33O5 [M + H]+: 449.2328; found 449.2330 m/z. RMN 1H: Table 4. RMN 13C: Table 5.
The mixture 21a–c (85 mg, 0.17 mmol) was dissolved in toluene and iodine (20 mg) was added and stirred at 95 °C for 3 h. The cooled mixture was evaporated and redissolved in EtOAc. The organic layer was washed consecutively with aq sat Na2S2O3 and brine, and dried over anhydrous Na2SO4. CC of the reaction product yielded 21c (27 mg, 32%, eluent: Hex/EtOAc, 9:1). Compound 21c: IR cm−1 (film): 2935, 1765, 1705, 1600, 1430, 1365, 1200, 1180, 1050, 1005, 900, 825. HRMS-FAB: calcd for C28H33O5 [M + H]+: 449.2328 u; found 449.2342 m/z. RMN 1H: Table 4. RMN 13C: Table 5.
3.2. Biological Evaluation
A colorimetric type of assay using sulforhodamine B (SRB) reaction has been adapted for a quantitative measurement of cell growth and viability, following a previously described method [28]. This assay employs 96 well cell culture microplates of 9 mm diameter. Cell lines are obtained from American Type Culture Collection (ATCC) derived from different human cancer types. Cells are maintained in RPMI 1640 10% Fetal Bovine Serum (FBS), supplemented with 0.1 g/L penicillin and 0.1 g/L streptomycin sulfate and then incubated at 37 °C, 5% CO2 and 98% humidity. For the experiments, cells were harvested from subconfluent cultures using trypsin and resuspended in fresh medium before plating.
Cells were seeded in 96 well microtiter plates, at 5 × 103 cells per well in aliquots of 195 μL of RPMI medium, and they were allowed to attach to the plate surface by growing in drug free medium for 18 h. Afterward, samples were added in aliquots of 5 μL in a ranging from 10 to 10−8 μg/mL, dissolved in DMSO:EtOH:Phosphate Buffered Saline (PBS) (0.5:0.5:99). After 72 h exposure, the antitumor effect was measured by the SRB methodology: cells were fixed by adding 50 μL of cold 50% (wt/vol) trichloroacetic acid (TCA) and incubating for 60 min at 4 °C. Plates were washed with deionised water and dried; 100 μL of SRB solution (0.4% wt/vol in 1% acetic acid) was added to each microtiter well and incubated for 10 min at room temperature. Unbound SRB was removed by washing with 1% acetic acid. Plates are air-dried and bound stain was solubilized with tris(hydroxymethyl)aminomethane (Tris) buffer. Optical densities (OD) were read on an automated spectrophotometric plate reader at a single wavelength of 490 nm. Data analyses were generated automatically by LIMS implementation. Using control OD values (C), test OD values (T) and time zero OD values (T0), the drug concentration that causes 50% Growth Inhibition (GI50 value) was calculated from the equation: 100 × [(T − T0)/(C − T0)] = 50. Each value represents the mean from triplicate determinations.
4. Conclusions
In this work, we reported several modifications of the decalin core of DTHQs obtained from the natural labdanic diterpenoid myrceocommunic acid. With the aim to enlarge the structural diversity and the number of DTHQ derivatives, we have carried out certain chemical transformations into the natural bicyclic diterpene core, such as epoxidation, ozonolysis or decarboxylation, taking advantage of the functional groups present on it, that are, the double bond around C-8 and the carboxylic group at C-4. Considering this, we have obtained a collection of novel compounds that included some derivatives rearranged towards different diterpenic skeletons as halimane, norlabdane or secoditerpene analogues.
These chemical approaches illustrate the versatility of the labdane ring and show the complex reactivity of the system. Despite the chemical complexity of some rearrangements, most of the final molecules obtained, were thoroughly characterized and their cytotoxicity tested on common solid tumor cell lines. According to the GI50 values obtained from the in vitro evaluation, all the compounds tested maintained their cytotoxicity in the µM level, showing no changes in potency when the doble bound in the decalin B-ring was isomerized, but the conservation of the terpenic bicyclic system, either decalin or indane, in these molecules seems to be important. Additionally, oxygenated functions such as the ester group in the A-ring appears to be a main point for the activity of this family of compounds, without forgetting that its cytotoxicity would also be associated with the hydroquinonic part of the molecule, which is identical in all the compounds. These results lay the foundations for the introduction of further functional changes in the decalin moiety that would make possible to obtain more potent and effective antineoplastic compounds.
Supplementary Materials
The following are available online, Tables S1–S7: Correlations and assignments for compounds 8, 9a, 10, 11, 14, 15 and 19. Figures S1–S28: NMR spectra for compounds 3, 5–21.
Author Contributions
Design and synthesis Á.P.H., P.C., M.L.R., J.M.M.d.C., P.A.G. and M.Á.C.; Biological assay A.F.; original draft preparation Á.P.H., P.A.G. and M.Á.C.; writing—review and editing A.S.F., J.M.M.d.C. and M.Á.C.; supervision, project administration and funding acquisition A.S.F. and M.Á.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish MINECO (CTQ2015-68175-R, AGL2016-79813-C2-2-R), Junta de Castilla y Leoń (SA076P20) and ISCIII-RICET Network (RD16/00270018) cofinanced by the Fondo Social Europeo of the European Union (Fondos FEDER—EU) and USAL (Financiación GIR). The APC was funded by MDPI.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of most of the compounds prepared are available from the authors.
References
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from marine invertebrates: Structures, occurrence and ecological implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef]
- García, P.A.; Hernández, A.P.; San Feliciano, A.; Castro, M.A. Bioactive prenyl- and terpenyl-quinones/hydroquinones of marine origin. Mar. Drugs 2018, 16, 292. [Google Scholar]
- Sunassee, S.N.; Davies-Coleman, M.T. Cytotoxic and antioxidant marine prenylated quinones and hydroquinones. Nat. Prod. Rep. 2012, 29, 513–535. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Gordaliza, M.; Castro, A.; Mahiques, M.M.; San Feliciano, A.; Garcia-Gravalos, M.D. Further antineoplastic terpenylquinones and terpenylhydroquinones. Bioorg. Med. Chem. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Reinoso, P.; Miguel del Corral, J.M.; Castro, M.A.; Gordaliza, M.; Gupta, M.P.; Solis, P.; San Feliciano, A. Cytotoxic-antineoplastic activity of hydroquinone derivatives. Eur. J. Med. Chem. 2002, 37, 177–182. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Ojeda, C.; Escobar, J.; Gallardo, C.; Miguel del Corral, J.M.; Castro, A.; Cuevas, C.; San Feliciano, A. Synthesis, characterisation and cytotoxicity of chloro derivatives of prenylnaphthohydroquinone. Bioorg. Med. Chem. 2005, 13, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Castro, M.A.; Gordaliza, M.; Martin, M.L.; Gualberto, S.A.; Gamito, A.M.; Cuevas, C.; San Feliciano, A. Synthesis and cytotoxicity of new aminoterpenylquinones. Bioorg. Med. Chem. 2005, 13, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Garcia, P.A.; Gamito, A.M.; Gualberto, S.A.; Batista, R.; San Feliciano, A. A novel synthetic route to cytotoxic 1,4-anthraquinones from 1,4-benzoquinones. Synthesis 2005, 19, 3202–3208. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Ojeda, C.; Miguel del Corral, J.M.; Castro, M.A.; Mollinedo, F.; San Feliciano, A. Synthesis and evaluation as antitumor agents of 1,4-naphthohydroquinone derivatives conjugated with amino acids and purines. Arch. Pharm. 2013, 346, 882–890. [Google Scholar] [CrossRef]
- Castro, M.A.; Gamito, A.M.; Tangarife-Castano, V.; Roa-Linares, V.; Miguel del Corral, J.M.; Mesa-Arango, A.C.; Betancur-Galvis, L.; Francesch, A.M.; San Feliciano, A. New 1,4-anthracenedione derivatives with fused heterocyclic rings: Synthesis and biological evaluation. RSC Adv. 2015, 5, 1244–1261. [Google Scholar] [CrossRef]
- Miguel del Corral, J.M.; Gordaliza, M.; Castro, M.A.; Mahiques, M.M.; Chamorro, P.; Molinari, A.; Garcia-Gravalos, M.D.; Broughton, H.B.; San Feliciano, A. New selective cytotoxic diterpenylquinones and diterpenylhydroquinones. J. Med. Chem. 2001, 44, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Castro, M.A.; Rodriguez, M.L.; Chamorro, P.; Cuevas, C.; San Feliciano, A. New cytotoxic diterpenylnaphthohydroquinone derivatives obtained from a natural diterpenoid. Bioorg. Med. Chem. 2007, 15, 5760–5774. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Rodriguez, M.L.; San Feliciano, A. An easy route to pentacyclic terpenylquinones. Tetrahedron Lett. 2012, 53, 519–521. [Google Scholar] [CrossRef]
- Hernandez, A.P.; Diez, P.; Garcia, P.A.; Miguel del Corral, J.M.; Perez-Andres, M.; Diez, D.; San Feliciano, A.; Fuentes, M.; Castro, M.A. New hybrids derived from podophyllic aldehyde and diterpenylhydroquinones with selectivity toward osteosarcoma cells. ACS Med. Chem. Lett. 2018, 9, 328–333. [Google Scholar] [CrossRef]
- Castro, M.A.; Gamito, A.M.; Tangarife-Castatno, V.; Zapata, B.; Miguel del Corral, J.M.; Mesa-Arango, A.C.; Betancur-Galvis, L.; San Feliciano, A. Synthesis and antifungal activity of terpenyl-1,4-naphthoquinone and 1,4-anthracenedione derivatives. Eur. J. Med. Chem. 2013, 67, 19–27. [Google Scholar] [CrossRef]
- Roa-Linares, V.C.; Miranda-Brand, Y.; Tangarife-Castatno, V.; Ochoa, R.; Garcia, P.A.; Castro, M.A.; Betancur-Galvis, L.; San Feliciano, A. Anti-herpetic, anti-Dengue and antineoplastic activities of simple and heterocycle-fused derivatives of terpenyl-1,4-naphthoquinone and 1,4-anthraquinone. Molecules 2019, 24, 1279. [Google Scholar] [CrossRef]
- Pérez-Pertejo, Y.; Escudero-Martínez, J.M.; Reguera, R.M.; Balaña-Fouce, R.; Garcia, P.A.; Jambrina, P.G.; San Feliciano, A.; Castro, M.A. Antileishmanial activity of terpenylquinones on Leishmania infantum and their effects on Leishmania topoisomerase IB. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 70–79. [Google Scholar] [CrossRef]
- El Haib, A.; Benharref, A.; Parrès-Maynadié, S.; Manoury, E.; Daran, J.C.; Urrutigoïty, M.; Gouygou, M. Molecular rearrangemente of epoxide derived from sesquitepenes by Lewis acid catalysis. Tetrahedron Assymmetry 2010, 21, 1272–1277. [Google Scholar] [CrossRef]
- Geroge, J.H.; McArdle, M.; Baldwin, J.E.; Adlington, R.M. Biomimetic rearrangements of simplified labdane diterpenoids. Tetrahedron 2010, 66, 6321–6330. [Google Scholar] [CrossRef]
- Mohanraj, S.; Herz, W. Photosensitized oxygenation of labda-8(17),12-diene, labda-8(17), 13-diene, and the biformenes. Synthesis of pumiloxide. J. Org. Chem. 1981, 46, 1362–1366. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanism, and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Bailey, P.S.; Hwang, H.H.; Chiang, C.-Y. Mechanisms of epoxidation during ozonation of carbon-carbon double bonds. J. Org. Chem. 1985, 50, 231–234. [Google Scholar] [CrossRef]
- Hagiwara, H.; Takeuchi, F.; Nozawa, M.; Hoshi, T.; Suzuki, T. The first total synthesis and determination of the absolute configuration of chapecoderin A, B and C. Tetrahedron 2004, 60, 1983–1989. [Google Scholar] [CrossRef]
- Yang, Z.; Kitano, Y.; Chiba, K.; Shibata, N.; Kurokawa, H.; Doi, Y.; Arakawa, Y.; Tada, M. Synthesis of variously oxidized abietane diterpenes and their antibacterial activities against MRSA and VRE. Bioorg. Med. Chem. 2001, 9, 347–356. [Google Scholar] [CrossRef]
- Fernández-Mateos, A.; Ferrero-Barrueco, O.; De Pascual-Teresa, J.; Rubio-González, R. Synthesis of (-)4,8β-dimethyl testolactone from (+)O-15-methyl isoagathate. Tetrahedron 1991, 47, 4375–4382. [Google Scholar] [CrossRef]
- Marcos, I.S.; Basabe, P.; Laderas, M.; Díez, D.; Jorge, A.; Rodilla, J.M.; Moro, R.F.; Lithgow, A.M.; Barata, I.G.; Urones, J.G. Side-chain migration reactions and ring B aromatization in labdanes: Scope and limitations. Synthesis of isogregenedane type tetrahydronaphthalenic diterpenes. Tetrahedron 2003, 59, 2333–2343. [Google Scholar] [CrossRef]
- National Cancer Institute. Common Cancer Types. Available online: https://www.cancer.gov/types/common-cancers (accessed on 8 January 2021).
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Waren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).