
Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments

 ,
, 
Abstract
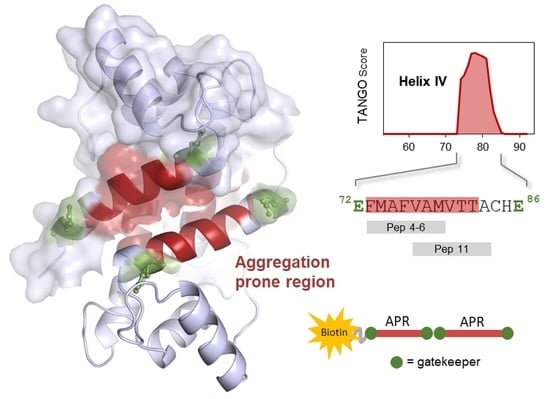
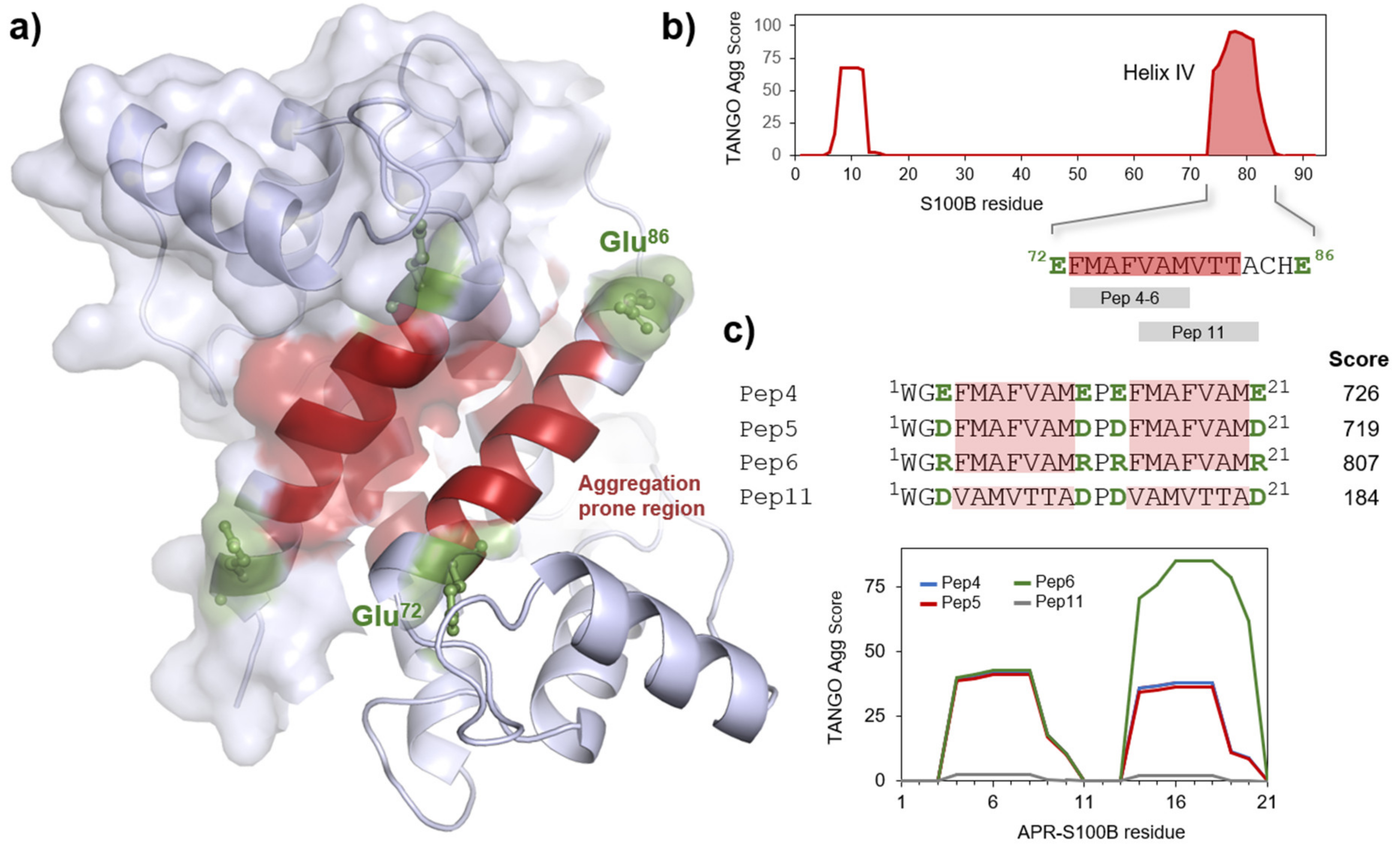
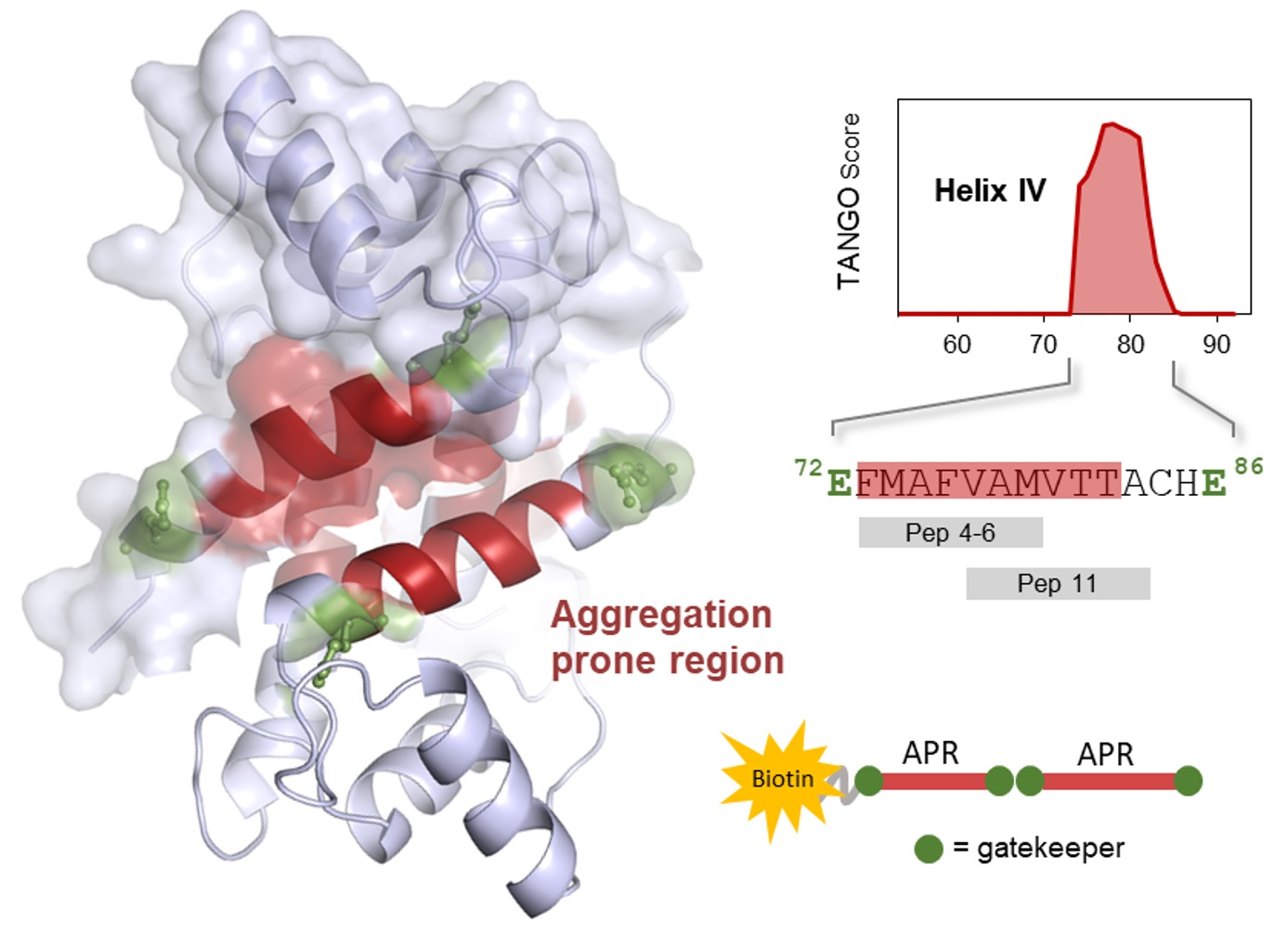{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. S100B-Derived APR Peptides: Design and Synthesis
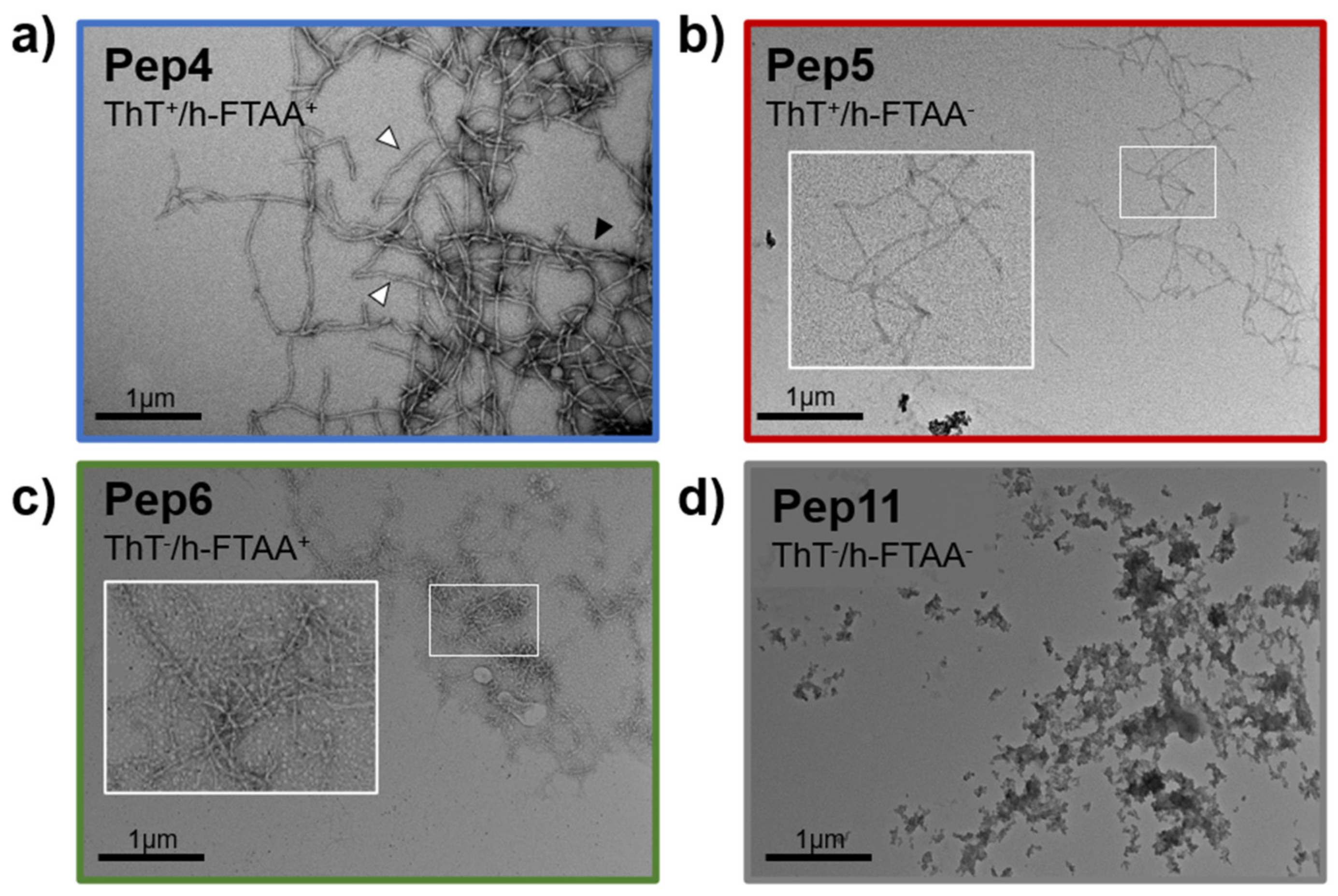
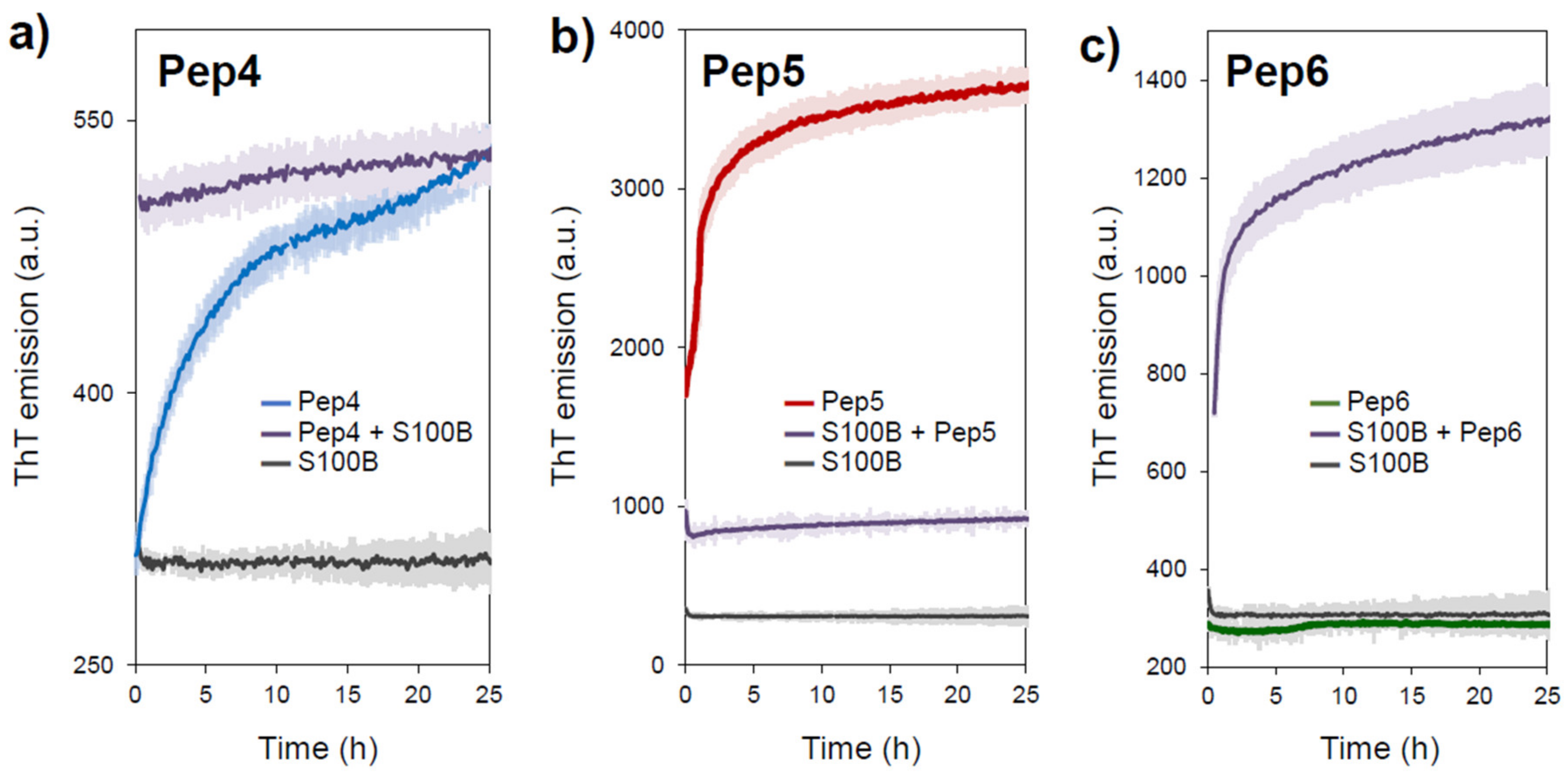
2.2. Aggregation Propensity of S100B-Derived APR Peptides
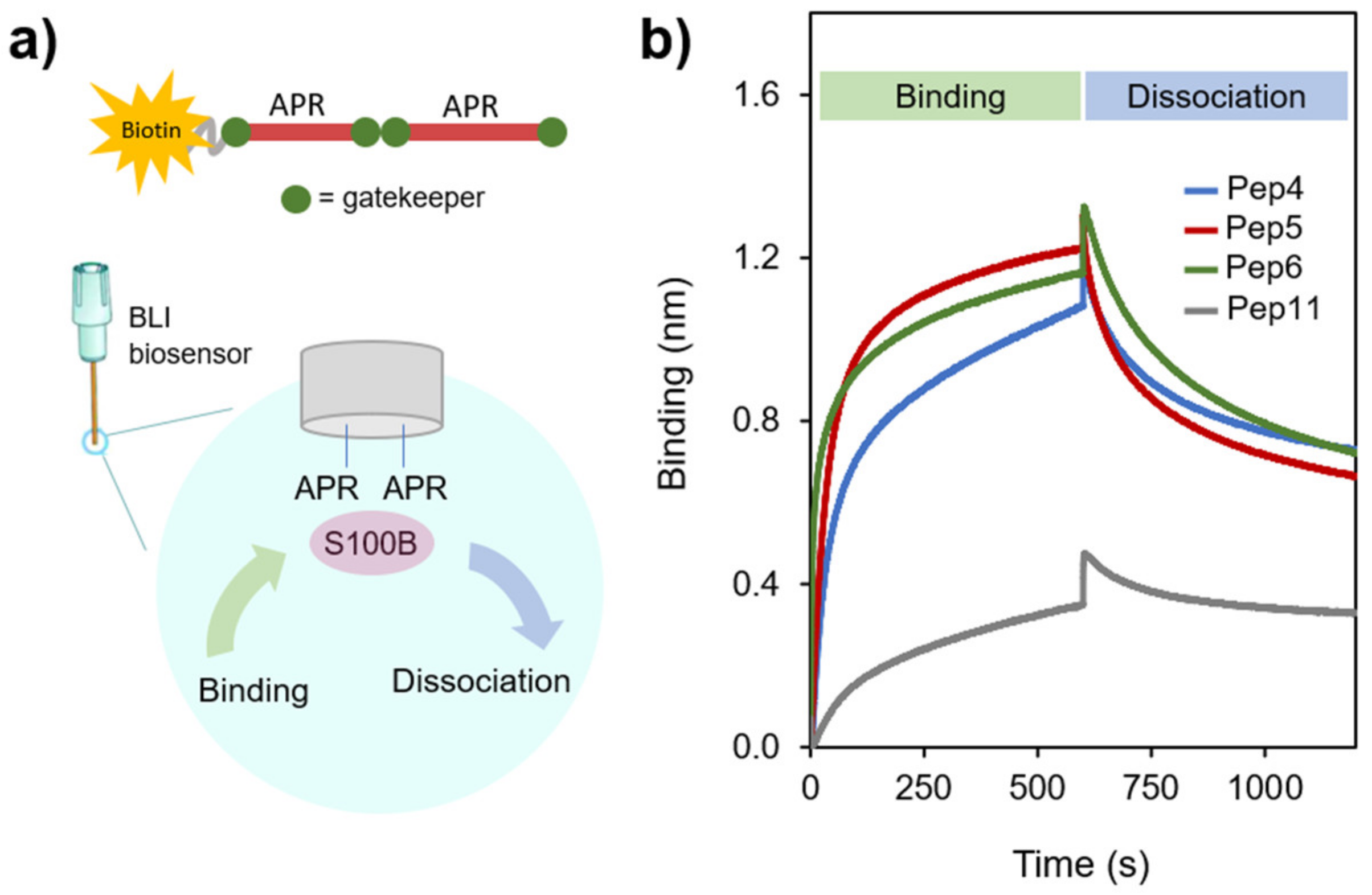
2.3. APR-Peptides Bind to S100B, which Modulates Their Aggregation Propensity
3. Materials and Methods
3.1. S100B Expression and Purification
3.2. Peptide Design and Synthesis
3.3. Fluorescence Spectroscopy
3.4. Transmission Electron Microscopy (TEM)
3.5. Bio-Layer Interferometry (BLITZ)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AD | Alzheimer’s Disease |
| Aβ | Amyloid-β Peptide |
| APR | Aggregation Prone Region |
| ThT | Thioflavin T |
| RAGE | Receptor Advanced Glycation End Product |
References
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019. [Google Scholar] [CrossRef]
- McKnight, L.E.; Raman, E.P.; Bezawada, P.; Kudrimoti, S.; Wilder, P.T.; Hartman, K.G.; Godoy-Ruiz, R.; Toth, E.A.; Coop, A.; MacKerell, A.D.; et al. Structure-Based Discovery of a Novel Pentamidine-Related Inhibitor of the Calcium-Binding Protein S100B. ACS Med. Chem. Lett. 2012, 3, 975–979. [Google Scholar] [CrossRef]
- Markowitz, J.; Mackerell, A.D., Jr.; Carrier, F.; Charpentier, T.H.; Weber, D.J. Design of Inhibitors for S100BCurr. Top. Med. Chem. 2005, 5, 1093–1108. [Google Scholar] [CrossRef]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef]
- Cho, C.C.; Chou, R.H.; Yu, C. Pentamidine blocks the interaction between mutant S100A5 and RAGE V domain and inhibits the RAGE signaling pathway. Biochem. Biophys. Res. Commun. 2016, 477, 188–194. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Arendash, G.W.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 2010, 58, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martinez, J.; Botelho, H.M.; Göbl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The neuronal S100B protein is a calcium-tuned suppressor of amyloid-beta aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Figueira, A.J.; Carapeto, A.; Rodrigues, M.S.; Cardoso, I.; Gomes, C.M. The S100B alarmin is a dual-function chaperone suppressing Aβ oligomerization through combined zinc chelation and inhibition of protein aggregation. ACS Chem. Neurosci. 2020, 11, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Serrano, L.; Schymkowitz, J.W. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 2006, 355, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Beerten, J.; Schymkowitz, J.; Rousseau, F. Aggregation prone regions and gatekeeping residues in protein sequences. Curr. Top. Med. Chem. 2012, 12, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Betti, C.; Vanhoutte, I.; Coutuer, S.; De Rycke, R.; Mishev, K.; Vuylsteke, M.; Aesaert, S.; Rombaut, D.; Gallardo, R.; De Smet, F.; et al. Sequence-Specific Protein Aggregation Generates Defined Protein Knockdowns in Plants. Plant Physiol. 2016, 171, 773–787. [Google Scholar] [CrossRef]
- Bednarska, N.G.; van Eldere, J.; Gallardo, R.; Ganesan, A.; Ramakers, M.; Vogel, I.; Baatsen, P.; Staes, A.; Goethals, M.; Hammarström, P.; et al. Protein aggregation as an antibiotic design strategy. Mol. Microbiol. 2016, 99, 849–865. [Google Scholar] [CrossRef]
- Yanamandra, K.; Alexeyev, O.; Zamotin, V.; Srivastava, V.; Shchukarev, A.; Brorsson, A.C.; Tartaglia, G.G.; Vogl, T.; Kayed, R.; Wingsle, G.; et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS ONE 2009, 4, e5562. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Botelho, H.M.; Leal, S.S.; Cardoso, I.; Fritz, G.; Gomes, C.M. Intrinsically disordered and aggregation prone regions underlie beta-aggregation in S100 proteins. PLoS ONE 2013, 8, e76629. [Google Scholar] [CrossRef]
- Martinez, J.; Cristovao, J.S.; Sanchez, R.; Gasset, M.; Gomes, C.M. Preparation of Amyloidogenic Aggregates from EF-Hand beta-Parvalbumin and S100 Proteins. Methods Mol. Biol. 2018, 1779, 167–179. [Google Scholar]
- Carvalho, S.B.; Cardoso, I.; Botelho, H.M.; Yanamandra, K.; Fritz, G.; Gomes, C.M.; Morozova-Roche, L.A. Chapter 18—Structural Heterogeneity and Bioimaging of S100 Amyloid Assemblies. In Bio-Nanoimaging; Uversky, V.N., Lyubchenko, Y.L., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 197–212. [Google Scholar]
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Beerten, J.; Jonckheere, W.; Rudyak, S.; Xu, J.; Wilkinson, H.; De Smet, F.; Schymkowitz, J.; Rousseau, F. Aggregation gatekeepers modulate protein homeostasis of aggregating sequences and affect bacterial fitness. Protein Eng. Des. Sel. 2012, 25, 357–366. [Google Scholar] [CrossRef] [PubMed]
- De Baets, G.; Van Durme, J.; Rousseau, F.; Schymkowitz, J. A Genome-Wide Sequence–Structure Analysis Suggests Aggregation Gatekeepers Constitute an Evolutionary Constrained Functional Class. J. Mol. Biol. 2014, 426, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System; Version 1.8.; Schrodinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Klingstedt, T.; Aslund, A.; Simon, R.A.; Johansson, L.B.; Mason, J.J.; Nystrom, S.; Hammarstrom, P.; Nilsson, K.P. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org. Biomol. Chem. 2011, 9, 8356–8370. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.; Witte, K.; Wartchow, C.; Choo, S.; Yao, D.; Persson, H.; Wei, J.; Li, P.; Heidecker, B.; Ma, W.; et al. Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen. 2009, 12, 791–800. [Google Scholar] [CrossRef]
- Ostendorp, T.; Leclerc, E.; Galichet, A.; Koch, M.; Demling, N.; Weigle, B.; Heizmann, C.W.; Kroneck, P.M.H.; Fritz, G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007, 26, 3868–3878. [Google Scholar] [CrossRef]
- Thulin, E.; Kesvatera, T.; Linse, S. Molecular determinants of S100B oligomer formation. PLoS ONE 2011, 6, e14768. [Google Scholar] [CrossRef]
- Fields, C.G.; Lloyd, D.H.; Macdonald, R.L.; Otteson, K.M.; Noble, R.L. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept Res. 1991, 4, 95–101. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cristóvão, J.S.; Romão, M.A.; Gallardo, R.; Schymkowitz, J.; Rousseau, F.; Gomes, C.M. Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments. Molecules 2021, 26, 440. https://doi.org/10.3390/molecules26020440
Cristóvão JS, Romão MA, Gallardo R, Schymkowitz J, Rousseau F, Gomes CM. Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments. Molecules. 2021; 26(2):440. https://doi.org/10.3390/molecules26020440
Chicago/Turabian StyleCristóvão, Joana S., Mariana A. Romão, Rodrigo Gallardo, Joost Schymkowitz, Frederic Rousseau, and Cláudio M. Gomes. 2021. "Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments" Molecules 26, no. 2: 440. https://doi.org/10.3390/molecules26020440
APA StyleCristóvão, J. S., Romão, M. A., Gallardo, R., Schymkowitz, J., Rousseau, F., & Gomes, C. M. (2021). Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments. Molecules, 26(2), 440. https://doi.org/10.3390/molecules26020440