
Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases

 , ,
, , 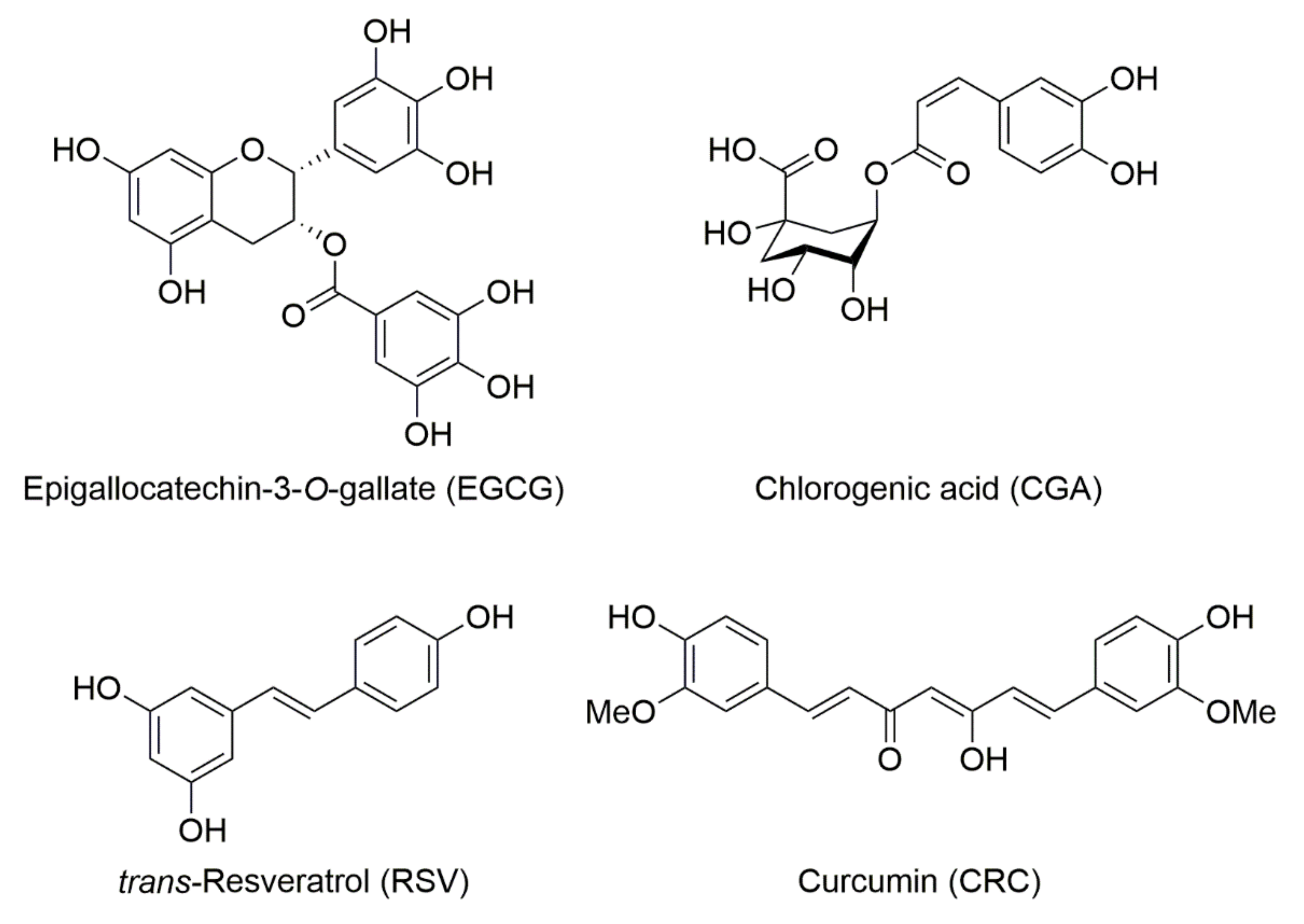{kind=link}
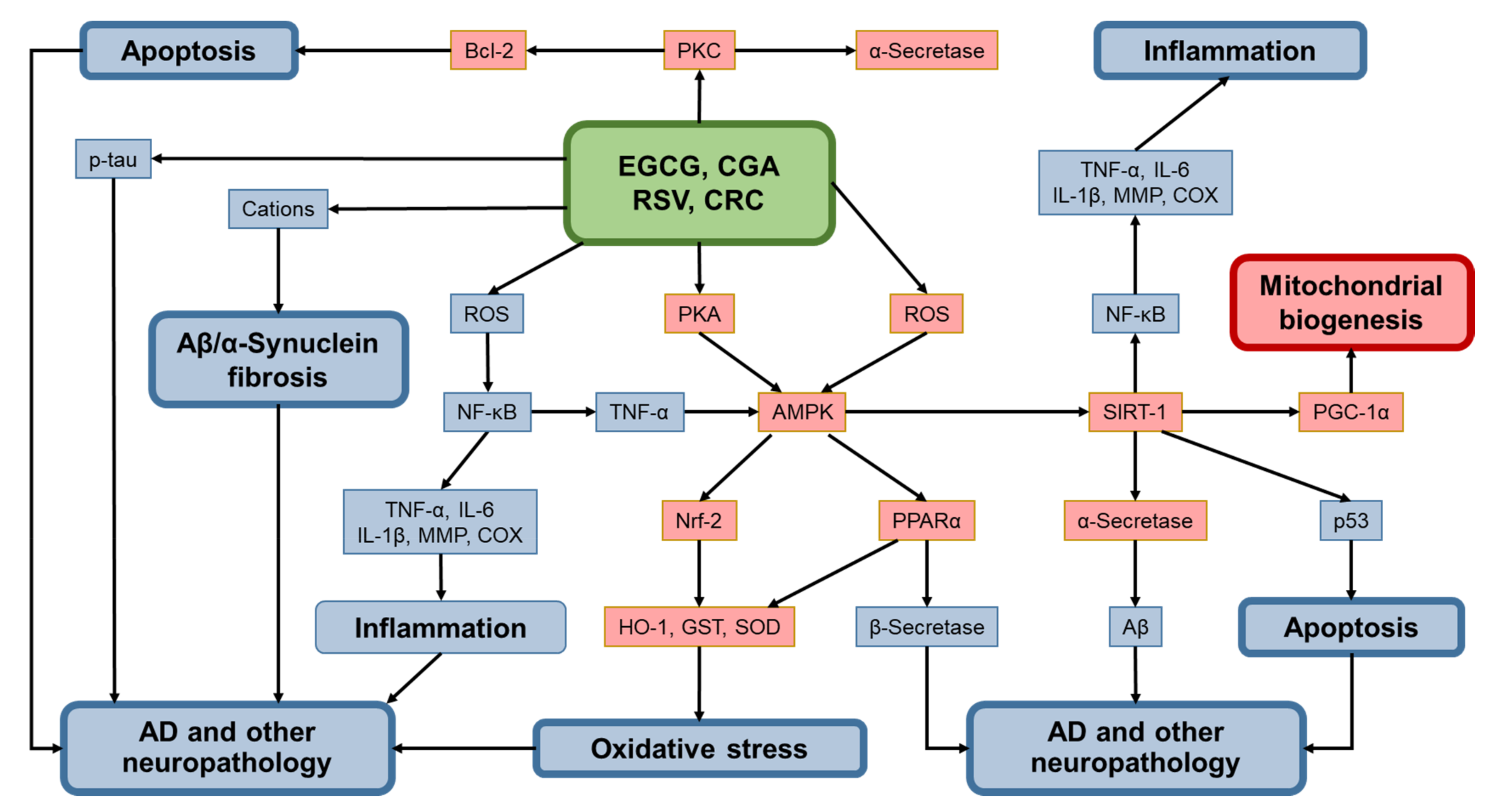{kind=link}
Abstract
1. Introduction
2. Alzheimer’s Disease (AD)
2.1. Effects of Tea/EGCG on AD
2.1.1. Human Studies of Tea/EGCG on AD
2.1.2. Basic Studies of Tea/EGCG on AD
2.2. Effects of Coffee/CGA on AD
2.2.1. Human Studies of Coffee/CGA on AD
2.2.2. Basic Studies of Coffee/CGA on AD
2.3. Human Studies of Wine/RSV
2.3.1. Human Studies of Wine/RSV on AD
2.3.2. Basic Studies of Wine/RSV on AD
2.4. Studies of Curry/CRC on AD
2.4.1. Human Studies of Curry/CRC on AD
2.4.2. Basic Studies of CRC on AD and Action Mechanism
3. Parkinson’s Disease (PD)
3.1. Effects of Tea/EGCG on PD
3.1.1. Human Studies of Effects of Tea/EGCG on PD
3.1.2. Basic Studies of Effects of TEA/EGCG on PD and Action Mechanism
3.2. Effects of Coffee/CGA on PD
3.2.1. Human Studies of Effects of Coffee/CGA on PD
3.2.2. Basic Studies of Effects of Coffee/CGA on PD and Action Mechanism
3.3. Effects of Wine/RSV on PD
3.3.1. Human Studies of Effects of Wine/RSV on PD
3.3.2. Basic Studies of Effects of RSV on PD and Action Mechanism
3.4. Effects of CRC on PD
3.4.1. Human Studies of Effects of CRC on PD
3.4.2. Basic Studies of Effects of CRC on PD and Action Mechanism
4. Other NDDs and Healthy Subjects
4.1. Effects of Tea/EGCG
4.1.1. Human Studies of Tea/EGCG on Other NDDs and Healthy Subjects
4.1.2. Basic Studies of Tea/EGCG on Other NDDs and Action Mechanism
4.2. Effects of Coffee/CGA on Other NDDs and Healthy Subjects
4.2.1. Human Studies of Effects of Coffee/CGA on other NDD and Healthy Subjects
4.2.2. Basic Studies of CGA on Other NDDs and Action Mechanism
4.3. Effects of Wine/RSV on Other NDDs
4.3.1. Human Studies of Effects of Wine/RSV on Other NDDs
4.3.2. Basic Studies of RSV’s Effects on Other NDDs and Action Mechanism
4.4. CRC’s Effects on Others NDDs and Healthy Subjects
4.4.1. Human Studies of CRC’s Effects on Other NDDs and Healthy Subjects
4.4.2. Basic Studies of CRC on Other NDDs and Action Mechanism
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Tanabe, H.; Pervin, M.; Goto, S.; Isemura, M.; Nakamura, Y. Beneficial Effects of Plant Polyphenols on Obesity. Obes. Control Ther. 2017, 4, 1–16. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant Polyphenols as Dietary Antioxidants in Human Health and Disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, S.; Ohishi, T.; Miyoshi, N.; Oishi, Y.; Nakamura, Y.; Isemura, M. Anti-Cancer Effects of Green Tea Epigallocatchin-3-Gallate and Coffee Chlorogenic Acid. Molecules 2020, 25, 4553. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, E.; Incocciati, A.; Palombarini, F.; Boffi, A.; Bonamore, A.; Macone, A. Ethylchloroformate Derivatization for GC–MS Analysis of Resveratrol Isomers in Red Wine. Molecules 2020, 25, 4603. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, S.; Saito, K.; Miyoshi, N.; Ohishi, T.; Oishi, Y.; Miyoshi, M.; Nakamura, Y. Anti-Cancer Effects of Green Tea by Either Anti- or Pro-Oxidative Mechanisms. Asian Pac. J. Cancer Prev. 2016, 17, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Pervin, M.; Goto, S.; Isemura, M.; Nakamura, Y. Beneficial Effects of Tea and the Green Tea Catechin Epigallocatechin-3-Gallate on Obesity. Molecules 2016, 21, 1305. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Potential Neuroprotective Properties of Epigallocatechin-3-Gallate (EGCG). Nutr. J. 2015, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.-W.; Chen, Y.; Huang, X.; Zhou, F.; Wang, W.; Xian, B.; Zhang, X.; Masliah, E.; Chen, Q.; et al. Appoptosin Is a Novel Pro-Apoptotic Protein and Mediates Cell Death in Neurodegeneration. J. Neurosci. 2012, 32, 15565–15576. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.M.; Katsouri, L.; Sastre, M. Modulation of Inflammation in Transgenic Models of Alzheimer’s Disease. J. Neuroinflamm. 2014, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, S.L.; Stab, B.; Casas, Z.; Sutachan, J.J.; Samudio, I.; Gonzalez, J.; Gonzalo, L.; Capani, F.; Morales, L.; Barreto, G.E. Effects of Natural Antioxidants in Neurodegenerative Disease. Nutr. Neurosci. 2012, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.; Mandelkow, E.; Holtzman, D. Deciphering Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP Processing in Alzheimer’s Disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jin, X.; Yan, J.; Jin, Y.; Yu, W.; Wu, H.; Xu, S. Prevalence of Dementia, Cognitive Status and Associated Risk Factors among Elderly of Zhejiang Province, China in 2014. Age Ageing 2016, 45, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Melo van Lent, D.; Wolfsgruber, S.; Weinhold, L.; Kleineidam, L.; Bickel, H.; Scherer, M.; Eisele, M.; van den Bussche, H.; Wiese, B.; et al. Prospective Associations between Single Foods, Alzheimer’s Dementia and Memory Decline in the Elderly. Nutrients 2018, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Kwak, S.M.; Myung, S.-K. Caffeine Intake from Coffee or Tea and Cognitive Disorders: A Meta-Analysis of Observational Studies. Neuroepidemiology 2015, 44, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chong, M.-S.; Lim, W.-S.; Lee, T.-S.; Kua, E.-H.; Ng, T.-P. Tea for Alzheimer Prevention. J. Prev. Alzheimer’s Dis. 2015, 2, 136–141. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Liu, M.-Y.; Wang, S.; Tang, Q.-S.; Yao, W.-F.; Zhao, H.-S.; Wei, M.-J. Research on EGCG Improving the Degenerative Changes of the Brain in AD Model Mice Induced with Chemical Drugs. J. Chin. Med. Mater. 2012, 35, 1641–1644. [Google Scholar] [PubMed]
- Walker, J.M.; Klakotskaia, D.; Ajit, D.; Weisman, G.A.; Wood, W.G.; Sun, G.Y.; Serfozo, P.; Simonyi, A.; Schachtman, T.R. Beneficial Effects of Dietary EGCG and Voluntary Exercise on Behavior in an Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2015, 44, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green Tea Epigallocatechin-3-Gallate (EGCG) Reduces β-Amyloid Mediated Cognitive Impairment and Modulates Tau Pathology in Alzheimer Transgenic Mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Liu, M.; Zhong, X.; Yao, W.; Xiao, Q.; Wen, Q.; Yang, B.; Wei, M. Epigallocatechin Gallate Reduces Amyloid β-Induced Neurotoxicity via Inhibiting Endoplasmic Reticulum Stress-Mediated Apoptosis. Mol. Nutr. Food Res. 2018, 62, 1700890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, C.; Jiang, Y.; Wang, S.; Wu, X.; Wang, K. PPARγ Coactivator-1α (PGC-1α) Protects Neuroblastoma Cells against Amyloid-Beta (Aβ) Induced Cell Death and Neuroinflammation via NF-ΚB Pathway. BMC Neurosci. 2017, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (−)-Epigallocatechin-3-Gallate Ameliorates Memory Impairment and Rescues the Abnormal Synaptic Protein Levels in the Frontal Cortex and Hippocampus in a Mouse Model of Alzheimer’s Disease. Neuroreport 2017, 28, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Ramis, M.R.; Sarubbo, F.; Tejada, S.; Jiménez, M.; Esteban, S.; Miralles, A.; Moranta, D. Chronic Polyphenon-60 or Catechin Treatments Increase Brain Monoamines Syntheses and Hippocampal SIRT1 LEVELS Improving Cognition in Aged Rats. Nutrients 2020, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Tanabe, H.; Suzuki, T.; Saeki, K.; Hara, Y. Applications of a Standardized Green Tea Catechin Preparation for Viral Warts and Human Papilloma Virus-Related and Unrelated Cancers. Molecules 2020, 25, 2588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, Q.; Chen, J.-Y.; OuYang, D.; Lu, J.-H. The Pharmacological Activity of Epigallocatechin-3-Gallate (EGCG) on Alzheimer’s Disease Animal Model: A Systematic Review. Phytomedicine 2020, 79, 153316. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green Tea (-)-Epigallocatechin-3-Gallate Inhibits β-Amyloid-Induced Cognitive Dysfunction through Modification of Secretase Activity via Inhibition of ERK and NF-ΚB Pathways in Mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B.H. Neuroprotection and Neurorescue against Aβ Toxicity and PKC-dependent Release of Non-amyloidogenic Soluble Precursor Protein by Green Tea Polyphenol (-)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Shytle, D.N.; Mori, T.; Huayan Hou, S.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; Hardy, J.; et al. Green Tea Epigallocatechin-3-Gallate (EGCG) Modulates Amyloid Precursor Protein Cleavage and Reduces Cerebral Amyloidosis in Alzheimer Transgenic Mice. J. Neurosci. 2005, 25, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.; Kennedy, O.J.; Roderick, P.; Fallowfield, J.A.; Hayes, P.C.; Parkes, J. Coffee Consumption and Health: Umbrella Review of Meta-Analyses of Multiple Health Outcomes. BMJ 2017, j5024. [Google Scholar] [CrossRef] [PubMed]
- Wasim, S.; Kukkar, V.; Awad, V.M.; Sakhamuru, S.; Malik, B.H. Neuroprotective and Neurodegenerative Aspects of Coffee and Its Active Ingredients in View of Scientific Literature. Cureus 2020. [Google Scholar] [CrossRef] [PubMed]
- Quintana, J.L.B.; Allam, M.F.; Del Castillo, A.S.; Navajas, R.F.-C. Alzheimer’s Disease and Coffee: A Quantitative Review. Neurol. Res. 2007, 29, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-P.; Wu, Y.-F.; Cheng, H.-Y.; Xia, T.; Ding, H.; Wang, H.; Wang, Z.-M.; Xu, Y. Habitual Coffee Consumption and Risk of Cognitive Decline/Dementia: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Nutrition 2016, 32, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Zhao, Q.-F.; Li, J.-Q.; Wang, J.; Yu, J.-T. Meta-Analysis of Modifiable Risk Factors for Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sun, D.; He, Y. Coffee Intake and the Incident Risk of Cognitive Disorders: A Dose–Response Meta-Analysis of Nine Prospective Cohort Studies. Clin. Nutr. 2017, 36, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Jung, G.; Lee, H.N.; Sohn, B.K.; Lee, J.-Y.; Kim, Y.K.; et al. Coffee Intake and Decreased Amyloid Pathology in Human Brain. Transl. Psychiatry 2019, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Gelber, R.P.; Petrovitch, H.; Masaki, K.H.; Ross, G.W.; White, L.R. Coffee Intake in Midlife and Risk of Dementia and Its Neuropathologic Correlates. J. Alzheimer’s Dis. 2011, 23, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Orsini, N. Coffee Consumption and Risk of Dementia and Alzheimer’s Disease: A Dose-Response Meta-Analysis of Prospective Studies. Nutrients 2018, 10, 1501. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.K.; Leung, G.M.; Schooling, C.M. Habitual Coffee Consumption and Risk of Type 2 Diabetes, Ischemic Heart Disease, Depression and Alzheimer’s Disease: A Mendelian Randomization Study. Sci. Rep. 2016, 6, 36500. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Yamamoto, M.; Misawa, K.; Nishimura, H.; Misawa, K.; Ota, N.; Shimotoyodome, A. Coffee Polyphenols Prevent Cognitive Dysfunction and Suppress Amyloid β Plaques in APP/PS2 Transgenic Mouse. Neurosci. Res. 2020, 154, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Misawa, K.; Nishimura, H.; Hirata, T.; Yamamoto, M.; Ota, N. 5-Caffeoylquinic Acid Ameliorates Cognitive Decline and Reduces Aβ Deposition by Modulating Aβ Clearance Pathways in APP/PS2 Transgenic Mice. Nutrients 2020, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Sun, F.; Wang, Y.; Kang, J.; Zhang, S.; Li, H. CGA Restrains the Apoptosis of Aβ 25-35 -Induced Hippocampal Neurons. Int. J. Neurosci. 2020, 130, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Ciaramelli, C.; Palmioli, A.; De Luigi, A.; Colombo, L.; Sala, G.; Riva, C.; Zoia, C.P.; Salmona, M.; Airoldi, C. NMR-Driven Identification of Anti-Amyloidogenic Compounds in Green and Roasted Coffee Extracts. Food Chem. 2018, 252, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Miyamae, Y.; Shigemori, H.; Isoda, H. Neuroprotective Effect of 3,5-Di-O-Caffeoylquinic Acid on SH-SY5Y Cells and Senescence-Accelerated-Prone Mice 8 through the up-Regulation of Phosphoglycerate Kinase-1. Neuroscience 2010, 169, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Akinyemi, A.J.; Ademiluyi, A.O.; Adefegha, S.A. Comparative Study on the Inhibitory Effect of Caffeic and Chlorogenic Acids on Key Enzymes Linked to Alzheimer’s Disease and Some Pro-Oxidant Induced Oxidative Stress in Rats’ Brain-In Vitro. Neurochem. Res. 2013, 38, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.; Şener, B.; Choudhary, M.; Khalid, A. Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activity of Some Turkish Medicinal Plants. J. Ethnopharmacol. 2004, 91, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Yadav, E.; Singh, D.; Debnath, B.; Rathee, P.; Yadav, P.; Verma, A. Molecular Docking and Cognitive Impairment Attenuating Effect of Phenolic Compound Rich Fraction of Trianthema Portulacastrum in Scopolamine Induced Alzheimer’s Disease Like Condition. Neurochem. Res. 2019, 44, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.-H.; Lee, H.-K.; Kim, J.-A.; Hong, S.-I.; Kim, H.-C.; Jo, T.-H.; Park, Y.-I.; Lee, C.-K.; Kim, Y.-B.; Lee, S.-Y.; et al. Neuroprotective Effects of Chlorogenic Acid on Scopolamine-Induced Amnesia via Anti-Acetylcholinesterase and Anti-Oxidative Activities in Mice. Eur. J. Pharmacol. 2010, 649, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hu, J.; Li, J.; Yang, Z.; Xin, X.; Wang, J.; Ding, J.; Geng, M. Effect of Acidic Oligosaccharide Sugar Chain on Scopolamine-Induced Memory Impairment in Rats and Its Related Mechanisms. Neurosci. Lett. 2005, 374, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Kakio, S.; Nakazawa, Y.; Kobata, K.; Funakoshi-Tago, M.; Suzuki, T.; Tamura, H. Roasted Coffee Reduces β-Amyloid Production by Increasing Proteasomal β-Secretase Degradation in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2018, 62, 1800238. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Chang, C.-Q.; Ma, F.-Y.; Yu, C.-L. Modulating Effects of Chlorogenic Acid on Lipids and Glucose Metabolism and Expression of Hepatic Peroxisome Proliferator-Activated Receptor-α in Golden Hamsters Fed on High Fat Diet. Biomed. Environ. Sci. 2009, 22, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.; Dartigues, J.; Lafont, S.; Letenneur, L.; Commenges, D.; Salamon, R. Wine Consumption and Dementia in the Elderly: A Prospective Community Study in the Bordeaux Area. Rev. Neurol. 1997, 153, 185–192. [Google Scholar] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol Regulates Neuro-Inflammation and Induces Adaptive Immunity in Alzheimer’s Disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Freyssin, A.; Page, G.; Fauconneau, B.; Rioux Bilan, A. Natural Stilbenes Effects in Animal Models of Alzheimer’s Disease. Neural. Regen. Res. 2020, 15, 843. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-Activated Protein Kinase Signaling Activation by Resveratrol Modulates Amyloid-β Peptide Metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary Resveratrol Prevents Alzheimer’s Markers and Increases Life Span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Ruscica, M.; Banach, M. Resveratrol and Cognitive Decline: A Clinician Perspective. Arch. Med. Sci. 2019, 15, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds That Extend Longevity Are Protective in Neurodegenerative Diseases and Provide a Novel Treatment Strategy for These Devastating Disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef] [PubMed]
- Al-Edresi, S.; Alsalahat, I.; Freeman, S.; Aojula, H.; Penny, J. Resveratrol-Mediated Cleavage of Amyloid Β1–42 Peptide: Potential Relevance to Alzheimer’s Disease. Neurobiol. Aging 2020, 94, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Koukoulitsa, C.; Villalonga-Barber, C.; Csonka, R.; Alexi, X.; Leonis, G.; Dellis, D.; Hamelink, E.; Belda, O.; Steele, B.R.; Micha-Screttas, M.; et al. Biological and Computational Evaluation of Resveratrol Inhibitors against Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2016, 31, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, G.; Liang, Z.; Sheng, S.; Shi, Y.; Peng, L.; Wang, Y.; Wang, F.; Zhang, X. Resveratrol Improves Cognition and Decreases Amyloid Plaque Formation in Tg6799 Mice. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-Y.; Dong, Q.-X.; Zhu, J.; Sun, X.; Zhang, L.-F.; Qiu, M.; Yu, X.-L.; Liu, R.-T. Resveratrol Rescues Tau-Induced Cognitive Deficits and Neuropathology in a Mouse Model of Tauopathy. Curr. Alzheimer Res. 2019, 16, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.-J.; Ratka, A. Telomere Shortening and Alzheimer’s Disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Zatta, P. Resveratrol and Alzheimerâ€TMs Disease: Message in a Bottle on Red Wine and Cognition. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, F.; Watanabe, T.; Hasegawa, S.; Hoshi, T.; Higami, Y.; Tanuma, S. The Effect of Resveratrol on the Werner Syndrome RecQ Helicase Gene and Telomerase Activity. Curr. Aging Sci. 2011, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 Contributes to Telomere Maintenance and Augments Global Homologous Recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Smythe, G.; Braidy, N.; Münch, G.; Sachdev, P. The Role of Polyphenols in the Modulation of Sirtuins and Other Pathways Involved in Alzheimer’s Disease. Ageing Res. Rev. 2013, 12, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, M.; Chandra, V.; Kamboh, M.I.; Johnston, J.M.; Dodge, H.H.; Thelma, B.K.; Juyal, R.C.; Pandav, R.; Belle, S.H.; DeKosky, S.T. Apolipoprotein E Polymorphism and Alzheimer Disease. Arch. Neurol. 2000, 57, 824. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Pandav, R.; Dodge, H.H.; Johnston, J.M.; Belle, S.H.; DeKosky, S.T.; Ganguli, M. Incidence of Alzheimer’s Disease in a Rural Community in India: The Indo-US Study. Neurology 2001, 57, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Takahashi, Y.; Amakusa, Y.; Tanno, Y.; Tuji, Y.; Niwa, H.; Murakami, N.; Krishna, U. Effects of Turmeric on Alzheimer′s Disease with Behavioral and Psychological Symptoms of Dementia. AYU 2012, 33, 499. [Google Scholar] [CrossRef] [PubMed]
- DiSilvestro, R.A.; Joseph, E.; Zhao, S.; Bomser, J. Diverse Effects of a Low Dose Supplement of Lipidated Curcumin in Healthy Middle Aged People. Nutr. J. 2012, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-Week Randomized, Double Blind, Placebo-Controlled Study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients With Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Mahalakshmi, A.M.; Ray, B.; Tuladhar, S.; Hediyal, T.A.; Manthiannem, E.; Padamati, J.; Chandra, R.; Chidambaram, S.B.; Sakharkar, M.K. Benefits of Curcumin in Brain Disorders. BioFactors 2019, 45, 666–689. [Google Scholar] [CrossRef] [PubMed]
- Voulgaropoulou, S.D.; van Amelsvoort, T.A.M.J.; Prickaerts, J.; Vingerhoets, C. The Effect of Curcumin on Cognition in Alzheimer’s Disease and Healthy Aging: A Systematic Review of Pre-Clinical and Clinical Studies. Brain Res. 2019, 1725, 146476. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, D.; Taguchi, H.; Yamamoto, A.; Shirai, N.; Hirao, K.; Tooyama, I. Curcuminoid Binds to Amyloid-Β1-42 Oligomer and Fibril. J. Alzheimer’s Dis. 2011, 24, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Kotia, V.; Ghosh, D.; Mohite, G.M.; Kumar, A.; Maji, S.K. Curcumin Modulates α-Synuclein Aggregation and Toxicity. ACS Chem. Neurosci. 2013, 4, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Mohorko, N.; Repovš, G.; Popović, M.; Kovacs, G.G.; Bresjanac, M. Curcumin Labeling of Neuronal Fibrillar Tau Inclusions in Human Brain Samples. J. Neuropathol. Exp. Neurol. 2010, 69, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s?-Amyloid Fibrils in Vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Palanivelu, K. The Effect of Curcumin (Turmeric) on Alzheimer′s Disease: An Overview. Ann. Indian Acad. Neurol. 2008, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin Labels Amyloid Pathology in Vivo, Disrupts Existing Plaques, and Partially Restores Distorted Neurites in an Alzheimer Mouse Model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Teter, B.; Frautschy, S.A. Neuroprotective Effects of Curcumin. In The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease; Springer: Boston, MA, USA, 2007; pp. 197–212. [Google Scholar]
- Huang, H.-C.; Xu, K.; Jiang, Z.-F. Curcumin-Mediated Neuroprotection Against Amyloid-β-Induced Mitochondrial Dysfunction Involves the Inhibition of GSK-3β. J. Alzheimer’s Dis. 2012, 32, 981–996. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Dai, X.; Xiao, N.; Wu, X.; Wei, Z.; Fang, W.; Zhu, Y.; Zhang, J.; Chen, X. Curcumin Ameliorates Memory Decline via Inhibiting BACE1 Expression and β-Amyloid Pathology in 5 × FAD Transgenic Mice. Mol. Neurobiol. 2017, 54, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in Vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.-P.; Maiti, P.; et al. Curcumin Suppresses Soluble Tau Dimers and Corrects Molecular Chaperone, Synaptic, and Behavioral Deficits in Aged Human Tau Transgenic Mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate via c-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin Structure-Function, Bioavailability, and Efficacy in Models of Neuroinflammation and Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The Curry Spice Curcumin Reduces Oxidative Damage and Amyloid Pathology in an Alzheimer Transgenic Mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular Chaperone Dysfunction in Neurodegenerative Diseases and Effects of Curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, M.; Gyengesi, E.; Münch, G. Curcumin and Apigenin—Novel and Promising Therapeutics against Chronic Neuroinflammation in Alzheimer′s Disease. Neural Regen. Res. 2015, 10, 1181. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Park, B.; Goel, A.; Aggarwal, B.B. Epigenetic Changes Induced by Curcumin and Other Natural Compounds. Genes Nutr. 2011, 6, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; McClure, D.; Jimenez, L.A.; Megson, I.L.; Rahman, I. Curcumin Induces Glutathione Biosynthesis and Inhibits NF-ΚB Activation and Interleukin-8 Release in Alveolar Epithelial Cells: Mechanism of Free Radical Scavenging Activity. Antioxid. Redox Signal. 2005, 7, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Morasso, C.; Stivaktakis, P.; Pandini, C.; Tinelli, V.; Tsatsakis, A.; Prosperi, D.; Hickey, M.; Corsi, F.; Cereda, C. Curcumin Formulations and Trials: What’s New in Neurological Diseases. Molecules 2020, 25, 5389. [Google Scholar] [CrossRef] [PubMed]
- Boulos, C.; Yaghi, N.; El Hayeck, R.; Heraoui, G.N.; Fakhoury-Sayegh, N. Nutritional Risk Factors, Microbiota and Parkinson’s Disease: What Is the Current Evidence? Nutrients 2019, 11, 1896. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyake, Y.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Intake of Japanese and Chinese Teas Reduces Risk of Parkinson’s Disease. Parkinsonism Relat. Disord. 2011, 17, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Kandinov, B.; Giladi, N.; Korczyn, A.D. Smoking and Tea Consumption Delay Onset of Parkinson’s Disease. Parkinsonism Relat. Disord. 2009, 15, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Barranco Quintana, J.L.; Allam, M.F.; Del Castillo, A.S.; Navajas, R.F.-C. Parkinson’s Disease and Tea: A Quantitative Review. J. Am. Coll. Nutr. 2009, 28, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, S. Dose-Response Meta-Analysis on Coffee, Tea and Caffeine Consumption with Risk of Parkinson’s Disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Checkoway, H.; Powers, K.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D. Parkinson’s Disease Risks Associated with Cigarette Smoking, Alcohol Consumption, and Caffeine Intake. Am. J. Epidemiol. 2002, 155, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.-K.; Tan, C.; Fook-Chong, S.M.C.; Lum, S.Y.; Chai, A.; Chung, H.; Shen, H.; Zhao, Y.; Teoh, M.L.; Yih, Y.; et al. Dose-Dependent Protective Effect of Coffee, Tea, and Smoking in Parkinson’s Disease: A Study in Ethnic Chinese. J. Neurol. Sci. 2003, 216, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Bidel, S.; Jousilahti, P.; Antikainen, R.; Tuomilehto, J. Coffee and Tea Consumption and the Risk of Parkinson’s Disease. Mov. Disord. 2007, 22, 2242–2248. [Google Scholar] [CrossRef] [PubMed]
- Hosseini tabatabaei, N. Non-Genetic Factors Associated with the Risk of Parkinson’s Disease in Iranian Patients. Funct. Neurol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ding, D.; Wang, J.; Zhao, Q.; Meng, H.; Li, H.; Gao, Y.-T.; Shu, X.-O.; Tanner, C.M.; Hong, Z.; et al. Parkinson’s Disease Research in a Prospective Cohort in China. Parkinsonism Relat. Disord. 2015, 21, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.C.; Koh, W.-P.; Yuan, J.-M.; Wang, R.; Au, W.-L.; Tan, J.H.; Tan, E.-K.; Yu, M.C. Differential Effects of Black versus Green Tea on Risk of Parkinson’s Disease in the Singapore Chinese Health Study. Am. J. Epidemiol. 2007, 167, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bandres-Ciga, S.; Kim, J.; Heilbron, K.; Kia, D.; Hemani, G.; Xue, A.; Lawlor, D.A.; Smith, G.D.; Duran, R.; et al. The Parkinson’s Disease Mendelian Randomization Research Portal. Mov. Disord. 2019, 34, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.H.; Mandel, S. Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate Prevents N-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Dopaminergic Neurodegeneration. J. Neurochem. 2001, 78, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.M.; Jeong-Ja, O.; Jeon, B.S. Inhibition of Inducible Nitric Oxide Synthase Expression and Cell Death by (−)-Epigallocatechin-3-Gallate, a Green Tea Catechin, in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease. J. Clin. Neurosci. 2010, 17, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhu, M.; Liang, Z. (-)-Epigallocatechin-3-Gallate Modulates Peripheral Immunity in the MPTP-Induced Mouse Model of Parkinson’s Disease. Mol. Med. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-H.; Guan, M.S.H.; Koh, C.; Ouyang, X.; Yu, F.; Tan, E.-K.; O’Neill, S.P.; Zhang, X.; Chung, J.; Lim, K.-L. AMP Kinase Activation Mitigates Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson’s Disease. J. Neurosci. 2012, 32, 14311–14317. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-H.; Lill, C.M.; Lee, P.-C.; Hansen, J.; Lassen, C.F.; Bertram, L.; Greene, N.; Sinsheimer, J.S.; Ritz, B. Gene-Environment Interaction in Parkinson’s Disease: Coffee, ADORA2A, and CYP1A2. Neuroepidemiology 2016, 47, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A Meta-Analysis of Coffee Drinking, Cigarette Smoking, and the Risk of Parkinson’s Disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective Study of Caffeine Consumption and Risk of Parkinson’s Disease in Men and Women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W. Association of Coffee and Caffeine Intake With the Risk of Parkinson Disease. JAMA 2000, 283, 2674. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.D.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Smoking, Alcohol, and Coffee Consumption Preceding Parkinson’s Disease: A Case-Control Study. Neurology 2000, 55, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Gigante, A.F.; Asabella, A.N.; Iliceto, G.; Martino, T.; Ferrari, C.; Defazio, G.; Rubini, G. Chronic Coffee Consumption and Striatal DAT-SPECT Findings in Parkinson’s Disease. Neurol. Sci. 2018, 39, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Grosso, G.; Godos, J.; Galvano, F.; Giovannucci, E.L. Coffee, Caffeine, and Health Outcomes: An Umbrella Review. Annu. Rev. Nutr. 2017, 37, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Sääksjärvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Männistö, S. Prospective Study of Coffee Consumption and Risk of Parkinson’s Disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D.; Luchsinger, J.A.; Tang, M.X.; Mayeux, R. Parkinsonian Signs in Older People: Prevalence and Associations with Smoking and Coffee. Neurology 2003, 61, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Morano, A.; Jiménez-Jiménez, F.J.; Molina, J.A.; Antolín, M.A. Risk-Factors for Parkinson’s Disease: Case-Control Study in the Province of Cáceres, Spain. Acta Neurol. Scand. 1994, 89, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, M.; Nakaso, K.; Kusumoto, C.; Katano, S.; Tajima, N.; Yamashita, A.; Zushi, T.; Ito, S.; Matsura, T. Cytoprotective Effect of Chlorogenic Acid against α-Synuclein-Related Toxicity in Catecholaminergic PC12 Cells. J. Clin. Biochem. Nutr. 2012, 51, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of Chlorogenic Acid Supplementation in MPTP-Intoxicated Mouse. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective Effect of Chlorogenic Acid on Mitochondrial Dysfunction-Mediated Apoptotic Death of DA Neurons in a Parkinsonian Mouse Model. Oxid. Med. Cell. Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of Enteric Environmental Modification by Coffee Components on Neurodegeneration in Rotenone-Treated Mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Dong, S.-Y.; Cui, X.-X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.-H.; Tan, E.-K.; Zhao, W.-J.; Wu, Y.-C. Resveratrol Alleviates MPTP-Induced Motor Impairments and Pathological Changes by Autophagic Degradation of α-Synuclein via SIRT1-Deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wu, Q.; Lu, Y.-F.; Gong, Q.-H.; Shi, J.-S. Neuroprotective Effect of Resveratrol on 6-OHDA-Induced Parkinson’s Disease in Rats. Eur. J. Pharmacol. 2008, 600, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.K.; Juvekar, A.R. Neuroprotective Effect of Curcumin as Evinced by Abrogation of Rotenone-Induced Motor Deficits, Oxidative and Mitochondrial Dysfunctions in Mouse Model of Parkinson’s Disease. Pharmacol. Biochem. Behav. 2016, 150–151, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nehru, B. Curcumin Affords Neuroprotection and Inhibits α-Synuclein Aggregation in Lipopolysaccharide-Induced Parkinson’s Disease Model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Mythri, R.B.; Srinivas Bharath, M.M. Curcumin: A Potential Neuroprotective Agent in Parkinson’s Disease. Curr. Pharm. Des. 2012, 18, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Tong, Y.; Guo, M. Iron Chelators as Potential Therapeutic Agents for Parkinsons Disease. Curr. Bioact. Compd. 2008, 4, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.; Juvekar, A. Kinetics of Inhibition of Monoamine Oxidase Using Curcumin and Ellagic Acid. Pharmacogn. Mag. 2016, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Choi, J.H.; Yoo, D.Y.; Kim, W.; Jung, H.Y.; Kim, J.W.; Yoo, M.; Lee, S.; Kim, C.J.; Yoon, Y.S.; et al. Effects of Curcumin (Curcuma Longa) on Learning and Spatial Memory as Well as Cell Proliferation and Neuroblast Differentiation in Adult and Aged Mice by Upregulating Brain-Derived Neurotrophic Factor and CREB Signaling. J. Med. Food 2014, 17, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Elangovan, N.; Manigandan, K.; Singh, S.; Shukla, S. CNB-001 a Novel Curcumin Derivative, Guards Dopamine Neurons in MPTP Model of Parkinson’s Disease. Biomed. Res. Int. 2014, 2014, 236182. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, H.; Ma, J.-F.; Tan, Y.-Y.; Xiao, Q.; Ding, J.-Q.; Chen, S.-D. Curcumin Inhibition of JNKs Prevents Dopaminergic Neuronal Loss in a Mouse Model of Parkinson’s Disease through Suppressing Mitochondria Dysfunction. Transl. Neurodegener. 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Polito, C.; Cai, Z.-Y.; Shi, Y.-L.; Li, X.-M.; Yang, R.; Shi, M.; Li, Q.-S.; Ma, S.-C.; Xiang, L.-P.; Wang, K.-R.; et al. Association of Tea Consumption with Risk of Alzheimer’s Disease and Anti-Beta-Amyloid Effects of Tea. Nutrients 2018, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green Tea Consumption and Cognitive Function: A Cross-Sectional Study from the Tsurugaya Project. Am. J. Clin. Nutr. 2006, 83, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.-P.; Feng, L.; Niti, M.; Kua, E.-H.; Yap, K.-B. Tea Consumption and Cognitive Impairment and Decline in Older Chinese Adults. Am. J. Clin. Nutr. 2008, 88, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Shinohara, M.; Yuki, S.; Dohmoto, C.; Ikeda, Y.; Samuraki, M.; Iwasa, K.; Yokogawa, M.; Asai, K.; Komai, K.; Nakamura, H.; et al. Consumption of Green Tea, but Not Black Tea or Coffee, Is Associated with Reduced Risk of Cognitive Decline. PLoS ONE 2014, 9, e96013. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gwee, X.; Kua, E.-H.; Ng, T.-P. Cognitive Function and Tea Consumption in Community Dwelling Older Chinese in Singapore. J. Nutr. Health Aging 2010, 14, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.-J.; He, C.-H.; Li, S.; Zhang, S.-Y.; Duan, S.-Y.; Sun, H.-P.; Shen, Y.-P.; Xu, Y.; Yin, J.-Y.; Pan, C.-W. Tea Consumption Is Associated with Cognitive Impairment in Older Chinese Adults. Aging Ment. Health 2018, 22, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Scholey, A.; Downey, L.A.; Ciorciari, J.; Pipingas, A.; Nolidin, K.; Finn, M.; Wines, M.; Catchlove, S.; Terrens, A.; Barlow, E.; et al. Acute Neurocognitive Effects of Epigallocatechin Gallate (EGCG). Appetite 2012, 58, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Dietz, C.; Dekker, M.; Piqueras-Fiszman, B. An Intervention Study on the Effect of Matcha Tea, in Drink and Snack Bar Formats, on Mood and Cognitive Performance. Food Res. Int. 2017, 99, 72–83. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and Efficacy of Cognitive Training plus Epigallocatechin-3-Gallate in Young Adults with Down’s Syndrome (TESDAD): A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Watanabe, Y.; Nakamura, K.; Sanpei, K.; Wakasugi, M.; Yokoseki, A.; Onodera, O.; Ikeuchi, T.; Kuwano, R.; Momotsu, T.; et al. Modifiable Factors Associated with Cognitive Impairment in 1,143 Japanese Outpatients: The Project in Sado for Total Health (PROST). Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Yamada, H.; Takuma, N.; Kawasaki, Y.; Harada, S.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Effects of Green Tea Consumption on Cognitive Dysfunction in an Elderly Population: A Randomized Placebo-Controlled Study. Nutr. J. 2015, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Xiao, Y.; Ying, X.; Li, S.; Zhai, Y.; Shang, X.; Li, F.; Wang, X.; He, F.; Lin, J. Correction: Tea Consumption and Cognitive Impairment: A Cross-Sectional Study among Chinese Elderly. PLoS ONE 2015, 10, e0140739. [Google Scholar] [CrossRef] [PubMed]
- Wightman, E.L.; Haskell, C.F.; Forster, J.S.; Veasey, R.C.; Kennedy, D.O. Epigallocatechin Gallate, Cerebral Blood Flow Parameters, Cognitive Performance and Mood in Healthy Humans: A Double-Blind, Placebo-Controlled, Crossover Investigation. Hum. Psychopharmacol. Clin. Exp. 2012, 27, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Baros, S.; Valko, M. Redox Active Metal-Induced Oxidative Stress in Biological Systems. Transit. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B.H. Cell Signaling Pathways in the Neuroprotective Actions of the Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate: Implications for Neurodegenerative Diseases. J. Neurochem. 2004, 88, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-B.; Zhou, L.; Wang, Y.-Z.; Wang, X.; Zhou, Y.; Ho, W.-Z.; Li, J.-L. Neuroprotective Activity of (-)-Epigallocatechin Gallate against Lipopolysaccharide-Mediated Cytotoxicity. J. Immunol. Res. 2016, 2016, 4962351. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; de Sola, S.; Farré, M.; Xicota, L.; Cuenca-Royo, A.; Rodriguez, J.; León, A.; Langohr, K.; Gomis-González, M.; Hernandez, G.; et al. A Phase 1, Randomized Double-Blind, Placebo Controlled Trial to Evaluate Safety and Efficacy of Epigallocatechin-3-Gallate and Cognitive Training in Adults with Fragile X Syndrome. Clin. Nutr. 2020, 39, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Liu, M.; Zhong, X.; Yao, W.; Wei, M. Increased BBB Permeability Contributes to EGCG-Caused Cognitive Function Improvement in Natural Aging Rats: Pharmacokinetic and Distribution Analyses. Acta Pharmacol. Sin. 2019, 40, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood Brain Barrier Permeability of (−)-Epigallocatechin Gallate, Its Proliferation-Enhancing Activity of Human Neuroblastoma SH-SY5Y Cells, and Its Preventive Effect on Age-Related Cognitive Dysfunction in Mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Coffee and Tea Drinking and the Risk of Late-Life Dementia: A Population-Based CAIDE Study. J. Alzheimer’s Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Cropley, V.; Croft, R.; Silber, B.; Neale, C.; Scholey, A.; Stough, C.; Schmitt, J. Does Coffee Enriched with Chlorogenic Acids Improve Mood and Cognition after Acute Administration in Healthy Elderly? A Pilot Study. Psychopharmacology 2012, 219, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Camfield, D.A.; Silber, B.Y.; Scholey, A.B.; Nolidin, K.; Goh, A.; Stough, C. A Randomised Placebo-Controlled Trial to Differentiate the Acute Cognitive and Mood Effects of Chlorogenic Acid from Decaffeinated Coffee. PLoS ONE 2013, 8, e82897. [Google Scholar] [CrossRef] [PubMed]
- Saitou, K.; Ochiai, R.; Kozuma, K.; Sato, H.; Koikeda, T.; Osaki, N.; Katsuragi, Y. Effect of Chlorogenic Acids on Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2018, 10, 1337. [Google Scholar] [CrossRef] [PubMed]
- Hoelzl, C.; Knasmüller, S.; Wagner, K.-H.; Elbling, L.; Huber, W.; Kager, N.; Ferk, F.; Ehrlich, V.; Nersesyan, A.; Neubauer, O.; et al. Instant Coffee with High Chlorogenic Acid Levels Protects Humans against Oxidative Damage of Macromolecules. Mol. Nutr. Food Res. 2010, 54, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, S.; Sun, J.; Li, Y.; Zhang, D. Association of Coffee, Decaffeinated Coffee and Caffeine Intake from Coffee with Cognitive Performance in Older Adults: National Health and Nutrition Examination Survey (NHANES) 2011–2014. Nutrients 2020, 12, 840. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, B.M.; Buijsse, B.; Tijhuis, M.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Coffee Consumption Is Inversely Associated with Cognitive Decline in Elderly European Men: The FINE Study. Eur. J. Clin. Nutr. 2007, 61, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Panza, F.; Imbimbo, B.P.; D’Introno, A.; Galluzzo, L.; Gandin, C.; Misciagna, G.; Guerra, V.; Osella, A.; Baldereschi, M.; et al. Coffee Consumption Habits and the Risk of Mild Cognitive Impairment: The Italian Longitudinal Study on Aging. J. Alzheimer’s Dis. 2015, 47, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Kuriki, K.; Otsuka, R.; Kato, Y.; Nishita, Y.; Tange, C.; Tomida, M.; Imai, T.; Ando, F.; Shimokata, H. Green Tea and Coffee Intake and Risk of Cognitive Decline in Older Adults: The National Institute for Longevity Sciences, Longitudinal Study of Aging. Public Health Nutr. 2020, 23, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Shim, J.; Kim, H.W.; Kim, J.; Jang, Y.J.; Yang, H.; Park, J.; Choi, S.H.; Yoon, J.H.; et al. Caffeinated Coffee, Decaffeinated Coffee, and the Phenolic Phytochemical Chlorogenic Acid up-Regulate NQO1 Expression and Prevent H2O2-Induced Apoptosis in Primary Cortical Neurons. Neurochem. Int. 2012, 60, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Jang, Y.J.; Hwang, M.K.; Kang, N.J.; Lee, K.W.; Lee, H.J. Attenuation of Oxidative Neuronal Cell Death by Coffee Phenolic Phytochemicals. Mutat. Res. Mol. Mech. Mutagen. 2009, 661, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Shimazawa, M.; Mishima, S.; Hara, H. Water Extract of Propolis and Its Main Constituents, Caffeoylquinic Acid Derivatives, Exert Neuroprotective Effects via Antioxidant Actions. Life Sci. 2007, 80, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Qi, R.; Zhang, J.; Wang, Z.; Wang, H.; Hu, C.; Zhao, Y.; Bie, M.; Wang, Y.; Fu, Y.; et al. Chlorogenic Acid Inhibits LPS-Induced Microglial Activation and Improves Survival of Dopaminergic Neurons. Brain Res. Bull. 2012, 88, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, J.-S.; Jang, H.-J.; Kim, S.-M.; Chang, M.S.; Park, S.H.; Kim, K.S.; Bae, J.; Park, J.-W.; Lee, B.; et al. Chlorogenic Acid Ameliorates Brain Damage and Edema by Inhibiting Matrix Metalloproteinase-2 and 9 in a Rat Model of Focal Cerebral Ischemia. Eur. J. Pharmacol. 2012, 689, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Basli, A.; Soulet, S.; Chaher, N.; Mérillon, J.-M.; Chibane, M.; Monti, J.-P.; Richard, T. Wine Polyphenols: Potential Agents in Neuroprotection. Oxid. Med. Cell. Longev. 2012, 2012, 805762. [Google Scholar] [CrossRef] [PubMed]
- Marx, W.; Kelly, J.T.; Marshall, S.; Cutajar, J.; Annois, B.; Pipingas, A.; Tierney, A.; Itsiopoulos, C. Effect of Resveratrol Supplementation on Cognitive Performance and Mood in Adults: A Systematic Literature Review and Meta-Analysis of Randomized Controlled Trials. Nutr. Rev. 2018, 76, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Mehlig, K.; Skoog, I.; Guo, X.; Schutze, M.; Gustafson, D.; Waern, M.; Ostling, S.; Bjorkelund, C.; Lissner, L. Alcoholic Beverages and Incidence of Dementia: 34-Year Follow-up of the Prospective Population Study of Women in Goteborg. Am. J. Epidemiol. 2007, 167, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Arntzen, K.A.; Schirmer, H.; Wilsgaard, T.; Mathiesen, E.B. Moderate Wine Consumption Is Associated with Better Cognitive Test Results: A 7 Year Follow up of 5033 Subjects in the Tromsø Study. Acta Neurol. Scand. 2010, 122, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.V.; Kerti, L.; Margulies, D.S.; Floel, A. Effects of Resveratrol on Memory Performance, Hippocampal Functional Connectivity, and Glucose Metabolism in Healthy Older Adults. J. Neurosci. 2014, 34, 7862–7870. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.; Howe, P.; Wong, R. Effects of Resveratrol on Cognitive Performance, Mood and Cerebrovascular Function in Post-Menopausal Women; A 14-Week Randomised Placebo-Controlled Intervention Trial. Nutrients 2017, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Thaung Zaw, J.J.; Howe, P.R.; Wong, R.H. Long-Term Effects of Resveratrol on Cognition, Cerebrovascular Function and Cardio-Metabolic Markers in Postmenopausal Women: A 24-Month Randomised, Double-Blind, Placebo-Controlled, Crossover Study. Clin. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.O.; Wightman, E.L.; Reay, J.L.; Lietz, G.; Okello, E.J.; Wilde, A.; Haskell, C.F. Effects of Resveratrol on Cerebral Blood Flow Variables and Cognitive Performance in Humans: A Double-Blind, Placebo-Controlled, Crossover Investigation. Am. J. Clin. Nutr. 2010, 91, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A Randomized, Double-Blind, Placebo-Controlled Trial of Resveratrol for Alzheimer Disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuw, R.; Kevers, C.; Pincemail, J.; Defraigne, J.O.; Dommes, J. Antioxidant Capacity and Phenolic Composition of Red Wines from Various Grape Varieties: Specificity of Pinot Noir. J. Food Compos. Anal. 2014, 36, 40–50. [Google Scholar] [CrossRef]
- Rossetti, F.; Jouin, A.; Jourdes, M.; Teissedre, P.-L.; Foligni, R.; Longo, E.; Boselli, E. Impact of Different Stoppers on the Composition of Red and Rosé Lagrein, Schiava (Vernatsch) and Merlot Wines Stored in Bottle. Molecules 2020, 25, 4276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Wang, Y.; Wang, G.; Mao, L.; Zhang, D.; Wang, J. Effects of Resveratrol on Learning and Memory in Rats with Vascular Dementia. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, H.; Xu, G.; Yang, Y.; Xu, R.; Liu, Q.; Xue, X.; Liu, J.; Wang, H. Resveratrol Prevents Cognitive Impairment in Type 2 Diabetic Mice by Upregulating Nrf2 Expression and Transcriptional Level. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Gocmez, S.S.; Gacar, N.; Utkan, T.; Gacar, G.; Scarpace, P.J.; Tumer, N. Protective Effects of Resveratrol on Aging-Induced Cognitive Impairment in Rats. Neurobiol. Learn. Mem. 2016, 131, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Ménard, C.; Quirion, R. Neuroprotective Action of Resveratrol. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, N.; Zhao, L.; Tong, L.; Kawano, H.; Yan, H.-J.; Li, H.-P. Protective Effect of Resveratrol against Nigrostriatal Pathway Injury in Striatum via JNK Pathway. Brain Res. 2017, 1654, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yao, M.; Wei, L.; Ge, S. Obesity Caused by a High-Fat Diet Regulates the Sirt1/PGC-1α/FNDC5/BDNF Pathway to Exacerbate Isoflurane-Induced Postoperative Cognitive Dysfunction in Older Mice. Nutr. Neurosci. 2020, 23, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.-P.; Chiam, P.-C.; Lee, T.; Chua, H.-C.; Lim, L.; Kua, E.-H. Curry Consumption and Cognitive Function in the Elderly. Am. J. Epidemiol. 2006, 164, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Goozee, K.G.; Shah, T.M.; Sohrabi, H.R.; Rainey-Smith, S.R.; Brown, B.; Verdile, G.; Martins, R.N. Examining the Potential Clinical Value of Curcumin in the Prevention and Diagnosis of Alzheimer’s Disease. Br. J. Nutr. 2016, 115, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.H.; Pipingas, A.; Scholey, A.B. Investigation of the Effects of Solid Lipid Curcumin on Cognition and Mood in a Healthy Older Population. J. Psychopharmacol. 2015, 29, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.-P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Kucukgoncu, S.; Guloksuz, S.; Tek, C. Effects of Curcumin on Cognitive Functioning and Inflammatory State in Schizophrenia. J. Clin. Psychopharmacol. 2019, 39, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and Cognition: A Randomised, Placebo-Controlled, Double-Blind Study of Community-Dwelling Older Adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Qi, S.S.; Zhou, P.; Cui, H.R.; Chen, S.X.; Dai, K.Y.; Tang, M.L. Neurobiological and Pharmacological Validity of Curcumin in Ameliorating Memory Performance of Senescence-Accelerated Mice. Pharmacol. Biochem. Behav. 2013, 105, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zeng, Q.; Mitchell, E.S.; Xiu, J.; Duan, Y.; Li, C.; Tiwari, J.K.; Hu, Y.; Cao, X.; Zhao, Z. Curcumin Enhances Neurogenesis and Cognition in Aged Rats: Implications for Transcriptional Interactions Related to Growth and Synaptic Plasticity. PLoS ONE 2012, 7, e31211. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Ma, J.; Wang, Y.; Huang, Z.; Zou, Q.; Cai, Z.; Tang, Y. Curcumin Prevents Neuroinflammation by Inducing Microglia to Transform into the M2-Phenotype via CaMKKβ-Dependent Activation of the AMP-Activated Protein Kinase Signal Pathway. Curr. Alzheimer Res. 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer Prevention by Tea: Animal Studies, Molecular Mechanisms and Human Relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical Antioxidants as Novel Neuroprotective Agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health Benefits of Resveratrol: Evidence from Clinical Studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Leem, E.; Lee, J.M.; Kim, S.R. Control of Reactive Oxygen Species for the Prevention of Parkinson’s Disease: The Possible Application of Flavonoids. Antioxidants 2020, 9, 583. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 Activation as a Novel Mechanism Underlying the Prevention of Alzheimer Disease Amyloid Neuropathology by Calorie Restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, J.; Sheng, F.; Li, P. Attenuation of Palmitic Acid–Induced Lipotoxicity by Chlorogenic Acid through Activation of SIRT1 in Hepatocytes. Mol. Nutr. Food Res. 2019, 63, 1801432. [Google Scholar] [CrossRef] [PubMed]
- Baghbaderani, S.; Hashemi, M.; Ebrahimi-Ghiri, M.; Zarrindast, M.-R.; Nasehi, M.; Entezari, M. Curcumin Prevents Cognitive Deficits in the Bile Duct Ligated Rats. Psychopharmacology 2020, 237, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Quon, M.J.; Kim, J. New Insights into the Mechanisms of Polyphenols beyond Antioxidant Properties; Lessons from the Green Tea Polyphenol, Epigallocatechin 3-Gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.W.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor Necrosis Factor α-Induced Skeletal Muscle Insulin Resistance Involves Suppression of AMP-Kinase Signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukutomi, R.; Ohishi, T.; Koyama, Y.; Pervin, M.; Nakamura, Y.; Isemura, M. Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases. Molecules 2021, 26, 415. https://doi.org/10.3390/molecules26020415
Fukutomi R, Ohishi T, Koyama Y, Pervin M, Nakamura Y, Isemura M. Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases. Molecules. 2021; 26(2):415. https://doi.org/10.3390/molecules26020415
Chicago/Turabian StyleFukutomi, Ryuuta, Tomokazu Ohishi, Yu Koyama, Monira Pervin, Yoriyuki Nakamura, and Mamoru Isemura. 2021. "Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases" Molecules 26, no. 2: 415. https://doi.org/10.3390/molecules26020415
APA StyleFukutomi, R., Ohishi, T., Koyama, Y., Pervin, M., Nakamura, Y., & Isemura, M. (2021). Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases. Molecules, 26(2), 415. https://doi.org/10.3390/molecules26020415