
Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy


Abstract
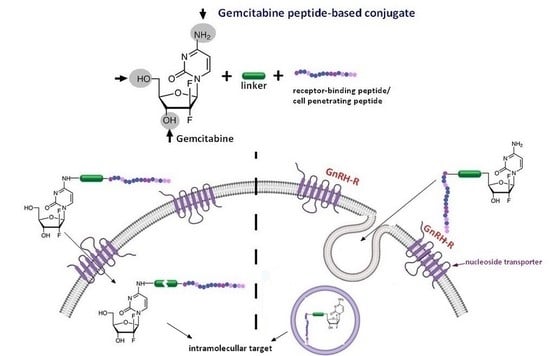
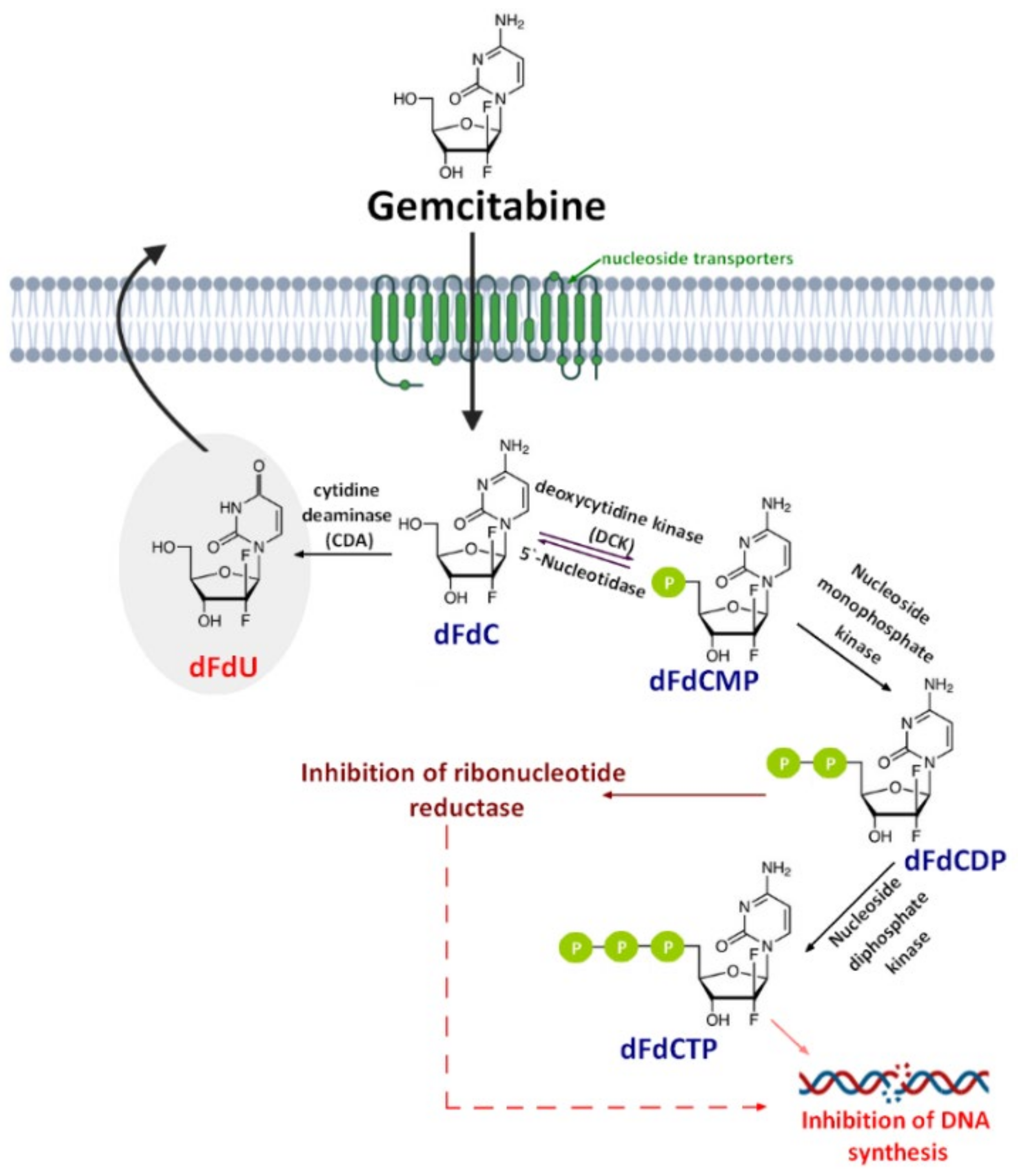
1. Introduction
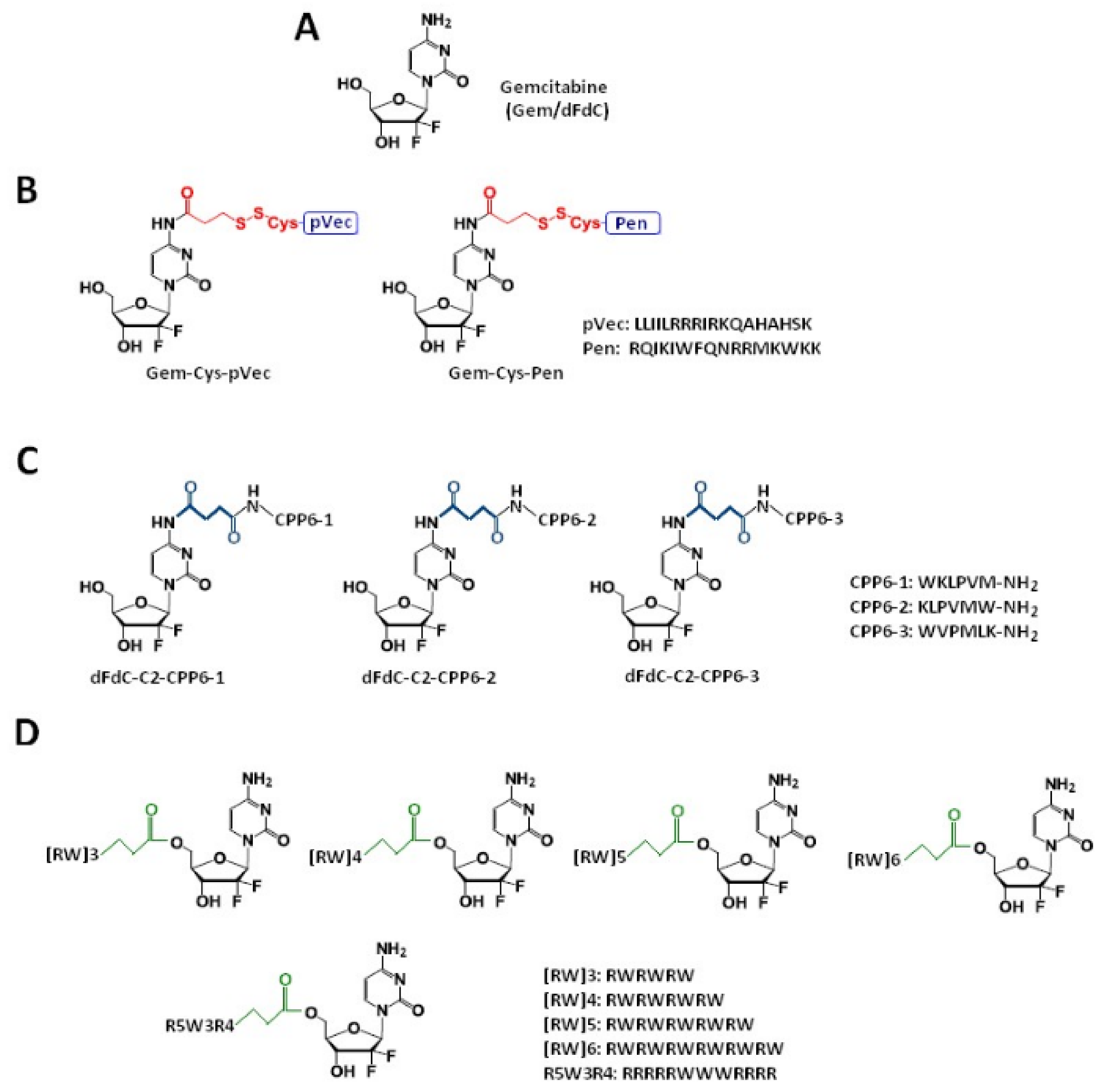
2. Gemcitabine Conjugated with Cell-Penetrating Peptides (CPPs)
2.1. Gemcitabine Conjugates with Arginine-Rich CPPs Modified at Tryptophan Residues
2.2. Gemcitabine Conjugated with Receptor-Binding Peptides
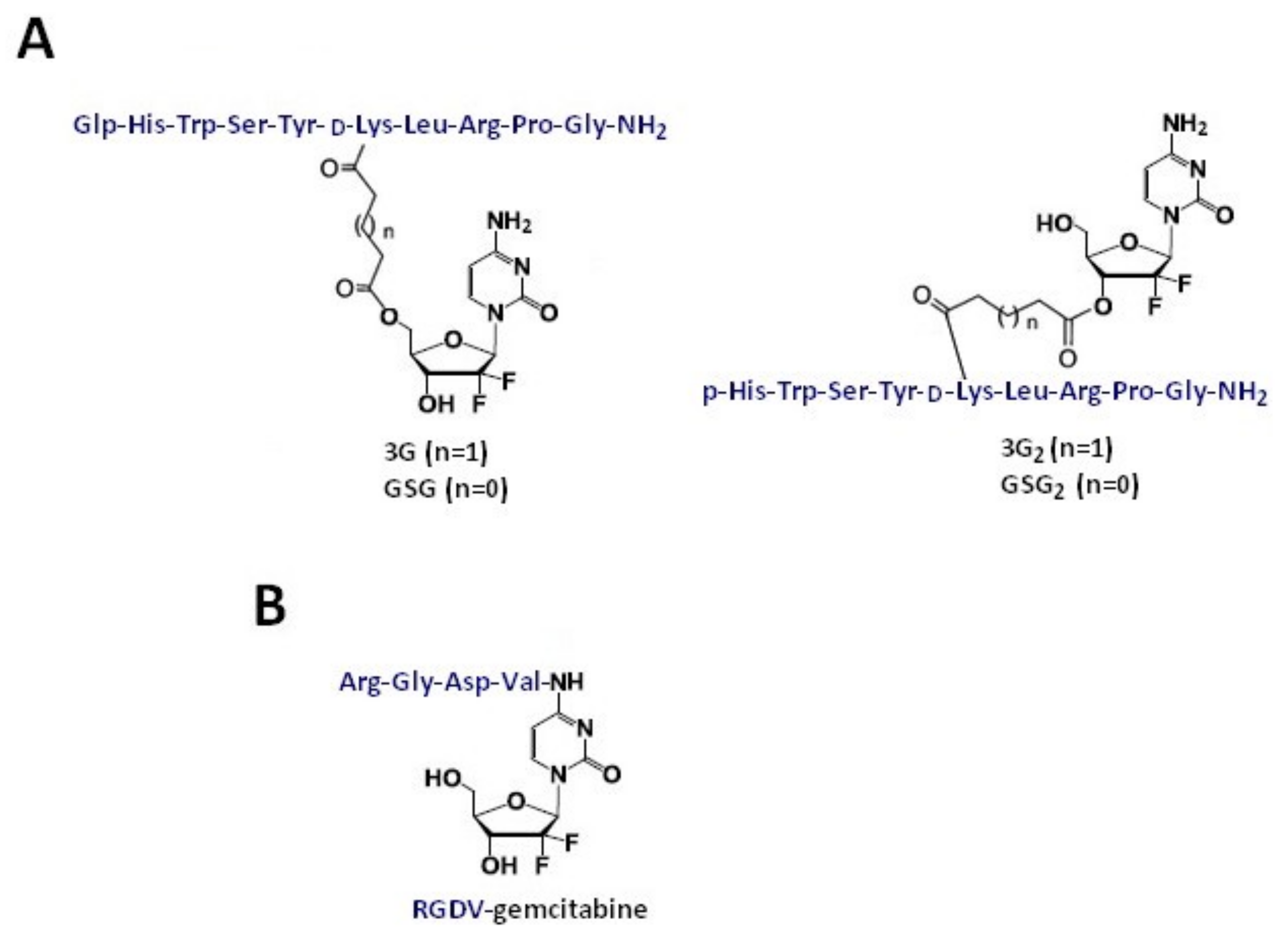
Gemcitabine-Succinate-GnRH Conjugates with Improved Metabolic Properties and Dual Mode of Efficacy
3. RGD Peptides-Gemcitabine Conjugates
3.1. Co-Administration of iRGD Peptide and Gemcitabine
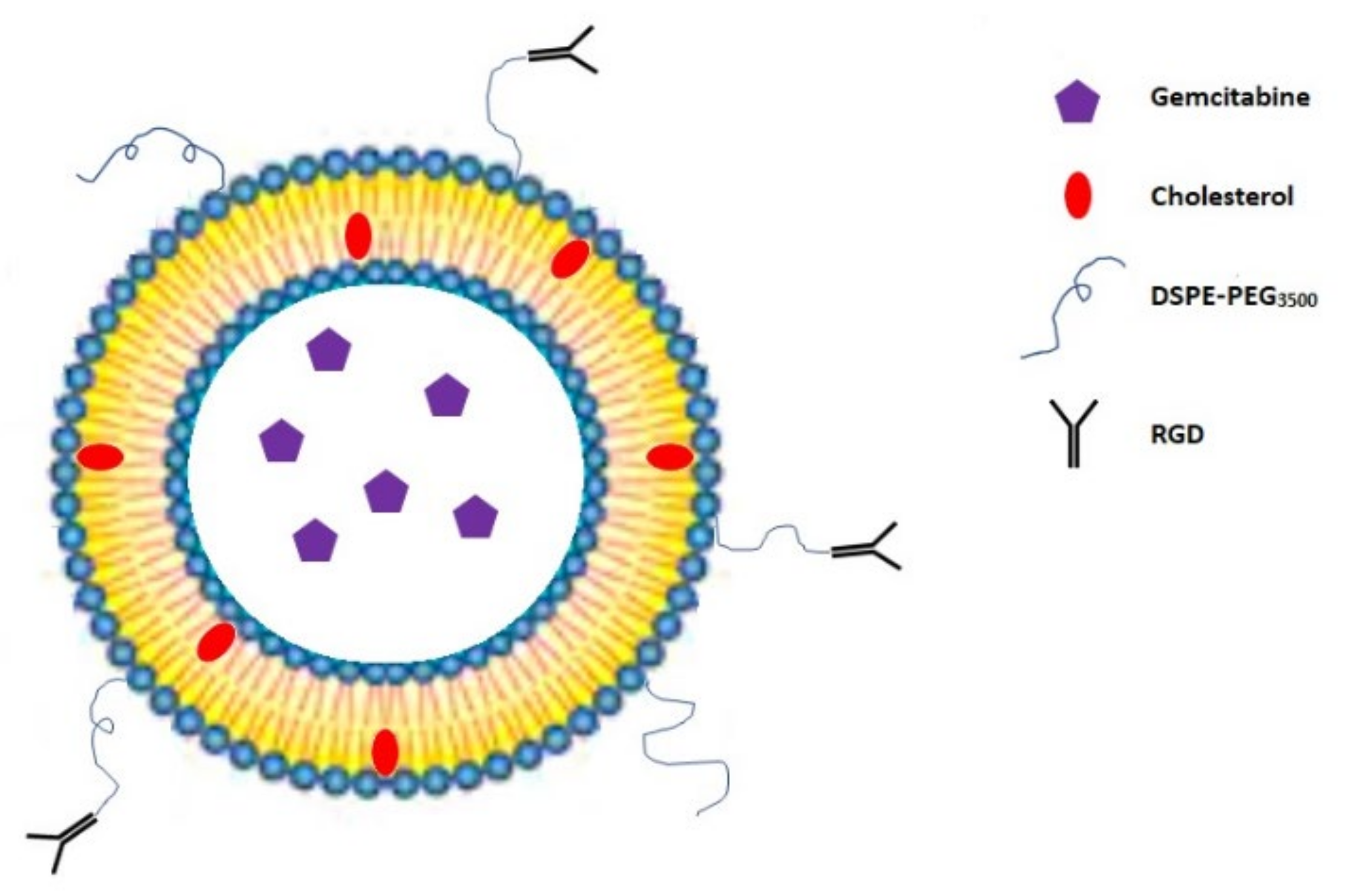
3.2. RGD Peptide-Gemcitabine-Loaded Nanocarriers
Co-Delivery of Gemcitabine and Paclitaxel in (&RGD&)-Modified Nanoparticles
3.3. Multifunctional Gemcitabine TPE-Gem-RGD Conjugate
3.4. RGDV-Gemcitabine Conjugate
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN, 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef]
- Duncan, R.; Vicent, M.J.; Greco, F.; Nicholson, R.I. Polymer-Drug Conjugates: Towards a Novel Approach for the Treatment of Endrocine-Related Cancer. Endocr. Relat. Cancer 2005, 12, 189–200. [Google Scholar] [CrossRef]
- Allen, T.M. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-Active Host Defense Peptides—Challenges and Perspectives for the Development of Novel Anticancer Drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef]
- Hirabayashi, H.; Nishikawa, M.; Takakura, Y.; Hashida, M. Development and pharmacokinetics of galactosylated poly-L-glutamic acid as a biodegradable carrier for liver-specific drug delivery. Pharm Res. 1996, 13, 880–884. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Drug Delivery and Release Systems for Targeted Tumor Therapy. J. Pept. Sci. 2015, 21, 186–200. [Google Scholar] [CrossRef]
- Haag, R.; Kratz, F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Nevozhay, D.; Kańska, U.; Budzyńska, R.; Boratyński, J. Current Status of Research on Conjugates and Related Drug Delivery Systems in the Treatment of Cancer and Other Diseases. Postepy Hig. Med. Dosw. 2007, 61, 350–360. [Google Scholar]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef]
- Majumdar, S.; Siahaan, T.J. Peptide-Mediated Targeted Drug Delivery. Med. Res. Rev. 2012, 32, 637–658. [Google Scholar] [CrossRef]
- Schreier, V.N.; Mezo, G.; Orbán, E.; Dürr, C.; Marquardt, A.; Manea, M. Synthesis, Enzymatic Stability and in Vitro Cytostatic Effect of Daunorubicin-GnRH-III Derivative Dimers. Bioorganic Med. Chem. Lett. 2013, 23, 2145–2150. [Google Scholar] [CrossRef]
- Leurs, U.; Mezo, G.; Orbán, E.; Ohlschläger, P.; Marquardt, A.; Manea, M. Design, Synthesis, in Vitro Stability and Cytostatic Effect of Multifunctional Anticancer Drug-Bioconjugates Containing GnRH-III as a Targeting Moiety. Biopolymers 2012, 98, 1–10. [Google Scholar] [CrossRef]
- Seitz, S.; Buchholz, S.; Schally, A.V.; Weber, F.; Klinkhammer-Schalke, M.; Inwald, E.C.; Perez, R.; Rick, F.G.; Szalontay, L.; Hohla, F.; et al. Triple Negative Breast Cancers Express Receptors for LHRH and Are Potential Therapeutic Targets for Cytotoxic LHRH-Analogs, AEZS 108 and AEZS 125. BMC Cancer 2014, 14, 847. [Google Scholar] [CrossRef]
- Liu, S.V.; Tsao-Wei, D.D.; Xiong, S.; Groshen, S.; Dorff, T.B.; Quinn, D.I.; Tai, Y.C.; Engel, J.; Hawes, D.; Schally, A.V.; et al. Phase I, Dose-Escalation Study of the Targeted Cytotoxic Lhrh Analog Aezs-108 in Patients with Castration- and Taxane-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 6277–6283. [Google Scholar] [CrossRef]
- Emons, G.; Gorchev, G.; Sehouli, J.; Wimberger, P.; Stähle, A.; Hanker, L.; Hilpert, F.; Sindermann, H.; Gründker, C.; Harter, P. Efficacy and Safety of AEZS-108 (INN: Zoptarelin Doxorubicin Acetate) an LHRH Agonist Linked to Doxorubicin in Women with Platinum Refractory or Resistant Ovarian Cancer Expressing LHRH Receptors: A Multicenter Phase II Trial of the Ago-Study Group (AGO G). Gynecol. Oncol. 2014, 133, 427–432. [Google Scholar] [CrossRef]
- Karampelas, T.; Skavatsou, E.; Argyros, O.; Fokas, D.; Tamvakopoulos, C. Gemcitabine Based Peptide Conjugate with Improved Metabolic Properties and Dual Mode of Efficacy. Mol. Pharm. 2017, 14, 674–685. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kamimura, H.; Tsuchiya, A.; Togashi, T.; Watanabe, K.; Seki, K.; Ohta, H.; Yoshida, T.; Takeda, K.; Kamimura, T. Clinical Efficacy of Intra-Arterial Pharmacokinetic Chemotherapy with 5-Fluorouracil, CDDP, Gemcitabine, and Angiotensin-II in Patients with Advanced Pancreatic Cancer. Hepatogastroenterology 2007, 54, 2378–2382. [Google Scholar]
- Reddy, L.; Couvreur, P. Novel Approaches to Deliver Gemcitabine to Cancers. Curr. Pharm. Des. 2008, 14, 1124–1137. [Google Scholar] [CrossRef]
- Lund, B.; Hansen, O.P.; Theilade, K.; Hansen, M.; Neijt, J.P. Phase II Study of Gemcitabine (2′,2′-Difluorodeoxycytidine) in Previously Treated Ovarian Cancer Patients. J. Natl. Cancer Inst. 1994, 86, 1530–1533. [Google Scholar] [CrossRef]
- Plunkett, W.; Huang, P.; Gandhi, V. Preclinical Characteristics of Gemcitabine. Anticancer Drugs 1995, 6, 7–13. [Google Scholar] [CrossRef]
- Hertel, L.W.; Boder, G.B.; Kroin, J.S.; Rinzel, S.M.; Poore, G.A.; Todd, G.C.; Grindey, G.B. Evaluation of the Antitumor Activity of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [Google Scholar]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17, 7–12. [Google Scholar] [CrossRef]
- Dyawanapelly, S.; Kumar, A.; Chourasia, M.K. Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy. Crit. Rev. Ther. Drug Carrier Syst. 2017, 34, 63–69. [Google Scholar] [CrossRef]
- Reid, J.M.; Qu, W.; Safgren, S.L.; Ames, M.M.; Krailo, M.D.; Seibel, N.L.; Kuttesch, J.; Holcenberg, J. Phase I Trial and Pharmacokinetics of Gemcitabine in Children with Advanced Solid Tumors. J. Clin. Oncol. 2004, 22, 2445–2451. [Google Scholar] [CrossRef]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef]
- Grunewald, R.; Kantarjian, H.; Du, M.; Faucher, K.; Tarassoff, P.; Plunkett, W. Gemcitabine in Leukemia: A Phase I Clinical, Plasma, and Cellular Pharmacology Study. J. Clin. Oncol. 1992, 10, 406–413. [Google Scholar] [CrossRef]
- Immordino, M.L.; Brusa, P.; Rocco, F.; Arpicco, S.; Ceruti, M.; Cattel, L. Preparation, Characterization, Cytotoxicity and Pharmacokinetics of Liposomes Containing Lipophilic Gemcitabine Prodrugs. J. Control. Release 2004, 100, 331–346. [Google Scholar] [CrossRef]
- Alvarellos, M.L.; Lamba, J.; Sangkuhl, K.; Thorn, C.F.; Wang, L.; Klein, D.J.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Gemcitabine Pathway. Pharmacogenet. Genom. 2014, 24, 564–574. [Google Scholar] [CrossRef]
- Kroep, J.; Van Moorsel, C.; Veerman, G.; Voorn, D.; Schultz, R.; Worzalla, J.; Tanzer, L.; Merriman, R.; Pinedo, H.; Peters, G. Role of Deoxycytidine Kinase (DCK), Thymidine Kinase 2 (TK2), and Deoxycytidine Deaminase (DCDA) in the Antitumor Activity of Gemcitabine (DFdC). In Purine and Pyrimidine Metabolism in Man IX; Springer: Boston, MA, USA, 1998; pp. 657–660. [Google Scholar]
- Correia, C.; Xavier, C.P.R.; Duarte, D.; Ferreira, A.; Moreira, S.; Vasconcelos, M.H.; Vale, N. Development of Potent CPP6-Gemcitabine Conjugates against Human Prostate Cancer Cell Line (PC-3). RSC Med. Chem. 2020, 11, 268–273. [Google Scholar] [CrossRef]
- Moysan, E.; Bastiat, G.; Benoit, J.P. Gemcitabine versus Modified Gemcitabine: A Review of Several Promising Chemical Modifications. Mol. Pharm. 2013, 10, 430–444. [Google Scholar] [CrossRef] [PubMed]
- May, J.P.; Ernsting, M.J.; Undzys, E.; Li, S.D. Thermosensitive Liposomes for the Delivery of Gemcitabine and Oxaliplatin to Tumors. Mol. Pharm. 2013, 10, 4499–4508. [Google Scholar] [CrossRef]
- Grazia Calvagno, M.; Celia, C.; Paolino, D.; Cosco, D.; Iannone, M.; Castelli, F.; Doldo, P.; Fresta, M. Effects of Lipid Composition and Preparation Conditions on Physical-Chemical Properties, Technological Parameters and In Vitro Biological Activity of Gemcitabine-Loaded Liposomes. Curr. Drug Deliv. 2006, 4, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Bersani, S.; Vila-Caballer, M.; Brazzale, C.; Barattin, M.; Salmaso, S. PH-Sensitive Stearoyl-PEG-Poly(Methacryloyl Sulfadimethoxine) Decorated Liposomes for the Delivery of Gemcitabine to Cancer Cells. Eur. J. Pharm. Biopharm. 2014, 88, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Qian, W.P.; Wang, L.; Wang, Y.A.; Staley, C.A.; Satpathy, M.; Nie, S.; Mao, H.; Yang, L. Theranostic Nanoparticles with Controlled Release of Gemcitabine for Targeted Therapy and MRI of Pancreatic Cancer. ACS Nano 2013, 7, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, J.E.N.; Valizadeh, H.; Hamishehkar, H. Solid Lipid Nanoparticles as Efficient Drug and Gene Delivery Systems: Recent Breakthroughs. Adv. Pharm. Bull. 2015, 5, 151–159. [Google Scholar] [CrossRef]
- Aggarwal, S.; Gupta, S.; Pabla, D.; Murthy, R.S.R. Gemcitabine-Loaded PLGA-PEG Immunonanoparticles for Targeted Chemotherapy of Pancreatic Cancer. Cancer Nanotechnol. 2013, 4, 145–157. [Google Scholar] [CrossRef]
- Chitkara, D.; Mittal, A.; Behrman, S.W.; Kumar, N.; Mahato, R.I. Self-Assembling, Amphiphilic Polymer-Gemcitabine Conjugate Shows Enhanced Antitumor Efficacy against Human Pancreatic Adenocarcinoma. Bioconjug. Chem. 2013, 24, 1161–1173. [Google Scholar] [CrossRef]
- Vale, N.; Ferreira, A.; Fernandes, I.; Alves, C.; Araújo, M.J.; Mateus, N.; Gomes, P. Gemcitabine Anti-Proliferative Activity Significantly Enhanced upon Conjugation with Cell-Penetrating Peptides. Bioorganic Med. Chem. Lett. 2017, 27, 2898–2901. [Google Scholar] [CrossRef]
- Zakeri-Milani, P.; Farkhani, S.M.; Shirani, A.; Mohammadi, S.; Mojarrad, J.S.; Akbari, J.; Valizadeh, H. Cellular Uptake and Anti-Tumor Activity of Gemcitabine Conjugated with New Amphiphilic Cell Penetrating Peptides. EXCLI J. 2017, 16, 650–662. [Google Scholar] [PubMed]
- Karampelas, T.; Argyros, O.; Sayyad, N.; Spyridaki, K.; Pappas, C.; Morgan, K.; Kolios, G.; Millar, R.P.; Liapakis, G.; Tzakos, A.G.; et al. GnRH-Gemcitabine Conjugates for the Treatment of Androgen-Independent Prostate Cancer: Pharmacokinetic Enhancements Combined with Targeted Drug Delivery. Bioconjug. Chem. 2014, 25, 813–823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Akashi, Y.; Oda, T.; Ohara, Y.; Miyamoto, R.; Kurokawa, T.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Satake, M.; Ohkohchi, N. Anticancer Effects of Gemcitabine Are Enhanced by Co-Administered IRGD Peptide in Murine Pancreatic Cancer Models That Overexpressed Neuropilin-1. Br. J. Cancer 2014, 110, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol. Pharm. 2016, 13, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Geng, C.; Jiang, L.; Sun, J.; Chen, B.; Zhou, Y.; Yang, B.; Lu, H. Encapsulation of Gemcitabine in RGD-Modified Nanoliposomes Improves Breast Cancer Inhibitory Activity. Pharm. Dev. Technol. 2020, 25, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Song, Y.; Di, Y.; He, H.; Fu, D.; Jin, C. Enhanced Tumor Targeting of CRGD Peptide-Conjugated Albumin Nanoparticles in the BxPC-3 Cell Line. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, P.; Zou, Q.; Li, X.; Fu, J.; Luo, Y.; Liang, X.; Jin, Y. Co-Delivery of Gemcitabine and Paclitaxel in CRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules 2018, 23, 2906. [Google Scholar] [CrossRef]
- Han, H.; Jin, Q.; Wang, Y.; Chen, Y.; Ji, J. The Rational Design of a Gemcitabine Prodrug with AIE-Based Intracellular Light-up Characteristics for Selective Suppression of Pancreatic Cancer Cells. Chem. Commun. 2015, 51, 17435–17438. [Google Scholar] [CrossRef]
- Liu, W.; Mao, Y.; Zhang, X.; Wang, Y.; Wu, J.; Zhao, S.; Peng, S.; Zhao, M. RGDV-Modified Gemcitabine: A Nano-Medicine Capable of Prolonging Half-Life, Overcoming Resistance and Eliminating Bone Marrow Toxicity of Gemcitabine. Int. J. Nanomed. 2019, 14, 7263–7279. [Google Scholar] [CrossRef]
- Gupta, A.; Mandal, D.; Ahmadibeni, Y.; Parang, K.; Bothun, G. Hydrophobicity Drives the Cellular Uptake of Short Cationic Peptide Ligands. Eur. Biophys. J. 2011, 40, 727–736. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Mandal, D.; Tiwari, R.K.; Guo, L.; Lu, W.; Parang, K. Cyclic Peptide-Capped Gold Nanoparticles as Drug Delivery Systems. Mol. Pharm. 2013, 10, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Prochiantz, J.A. Penetratin Story: An Overview. Methods Mol. Biol. 2015, 1324, 29–37. [Google Scholar] [PubMed]
- Nan, Y.H.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Antimicrobial Activity, Bactericidal Mechanism and LPS-Neutralizing Activity of the Cell-Penetrating Peptide PVEC and Its Analogs. J. Pept. Sci. 2011, 17, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Lapa, R.; Vale, N. Combination of Gemcitabine with Cell-Penetrating Peptides: A Pharmacokinetic Approach Using in Silico Tools. Biomolecules 2019, 9, 693. [Google Scholar] [CrossRef]
- Gomez, J.A.; Chen, J.; Ngo, J.; Hajkova, D.; Yeh, I.J.; Gama, V.; Miyagi, M.; Matsuyama, S. Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity. Pharmaceuticals 2010, 3, 3594–3613. [Google Scholar] [CrossRef]
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C.L. Arginine-Rich Cell-Penetrating Peptides. FEBS Lett. 2010, 584, 1806–1813. [Google Scholar] [CrossRef]
- Rydberg, H.A.; Matson, M.; Åmand, H.L.; Esbjörner, E.K.; Nordén, B. Effects of Tryptophan Content and Backbone Spacing on Uptake Efficiency of Cell-Penetrating Peptides. Biophys. J. 2012, 102, 487a3. [Google Scholar] [CrossRef]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The Preference of Tryptophan for Membrane Interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C.; Williams, K.A.; Deber, C.M.; Reithmeier, R.A. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J. Mol. Biol. 1993, 229, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Membrane-Proteins: From Sequence to Structure. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Reithmeier, R.A.F. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 1995, 5, 491–500. [Google Scholar] [CrossRef]
- Cavallaro, G.; Mariano, L.; Salmaso, S.; Caliceti, P.; Gaetano, G. Folate-Mediated Targeting of Polymeric Conjugates of Gemcitabine. Int. J. Pharm. 2006, 307, 258–269. [Google Scholar] [CrossRef]
- Pili, B.; Bourgaux, C.; Amenitsch, H.; Keller, G.; Lepêtre-Mouelhi, S.; Desmaële, D.; Couvreur, P.; Ollivon, M. Interaction of a New Anticancer Prodrug, Gemcitabine-Squalene, with a Model Membrane. Coupled DSC and XRD Study. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1522–1532. [Google Scholar] [CrossRef]
- Sheldrake, H.M.; Patterson, L.H. Function and Antagonism of β: Integrins in the Development of Cancer Therapy. Curr. Cancer Drug Targets 2009, 9, 519–540. [Google Scholar] [CrossRef]
- Kadonosono, T.; Yamano, A.; Goto, T.; Tsubaki, T.; Niibori, M.; Kuchimaru, T.; Kizaka-Kondoh, S. Cell Penetrating Peptides Improve Tumor Delivery of Cargos through Neuropilin-1-Dependent Extravasation. J. Control. Release 2015, 201, 14–21. [Google Scholar] [CrossRef]
- De, G.; Ko, J.K.; Tan, T.; Zhu, H.; Li, H.; Ma, J. Amphipathic Tail-Anchoring Peptide Is a Promising Therapeutic Agent for Prostate Cancer Treatment. Oncotarget 2014, 5, 7734–7747. [Google Scholar] [CrossRef][Green Version]
- Li, Z.; Cho, C. Development of Peptides as Potential Drugs for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1180–1189. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, Q.; Yang, J.; Wang, H.; Xu, J.; Zheng, J. IRGD as a Tumor-Penetrating Peptide for Cancer Therapy (Review). Mol. Med. Rep. 2017, 15, 2925–2930. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled Multimeric Cyclic RGD Peptides as Integrin Αvβ3 Targeted Radiotracers for Tumor Imaging. Mol. Pharm. 2006, 3, 472–487. [Google Scholar] [CrossRef]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-Based Strategies to Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Prakash Karmali, P.; Ramana Kotamraju, V.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of Tumor-Targeted Delivery of Macromolecular Drugs, Including the EPR Effect in Solid Tumor and Clinical Overview of the Prototype Polymeric Drug SMANCS. J. Control. Release 2001, 74, 47–61. [Google Scholar] [CrossRef]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and Nanoparticles: Nanosized Vehicles for Drug Delivery in Cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active Targeting Schemes for Nanoparticle Systems in Cancer Therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-Based Strategies for Selective Delivery of Therapeutics and Imaging Agents to the Tumour Vasculature. Drug Resist. Update 2005, 8, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R.; Amberger, A.; Saks, V.; Grimm, M. Changes in Mitochondrial Redox State, Membrane Potential and Calcium Precede Mitochondrial Dysfunction in Doxorubicin-Induced Cell Death. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS Stress in Cancer Cells and Therapeutic Implications. Drug Resist. Update 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Alva, A.; Slovin, S.; Daignault, S.; Carducci, M.; DiPaola, R.; Pienta, K.; Agus, D.; Cooney, K.; Chen, A.; Smith, D.C.; et al. Phase II Study of Cilengitide (EMD 121974, NSC 707544) in Patients with Non-Metastatic Castration Resistant Prostate Cancer, NCI-6735. A Study by the DOD/PCF Prostate Cancer Clinical Trials Consortium. Investig. New Drugs 2012, 30, 749–757. [Google Scholar] [CrossRef]
- Tang, Z.; Feng, W.; Yang, Y.; Wang, Q. Gemcitabine-Loaded RGD Modified Liposome for Ovarian Cancer: Preparation, Characterization and Pharmacodynamic Studies. Drug Des. Dev. Ther. 2019, 13, 3281–3290. [Google Scholar] [CrossRef]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The Relationship between Bone Metastasis from Human Breast Cancer and Integrin Αvβ3 Expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar]
- Furger, K.A.; Allan, A.L.; Wilson, S.M.; Hota, C.; Vantyghem, S.A.; Postenka, C.O.; Al-Katib, W.; Chambers, A.F.; Tuck, A.B. Β3 Integrin Expression Increases Breast Carcinoma Cell Responsiveness to the Malignancy-Enhancing Effects of Osteopontin. Mol. Cancer Res. 2003, 1, 810–819. [Google Scholar]
- Vellon, L.; Menendez, J.A.; Liu, H.; Lupu, R. Up-Regulation of Αvβ3 Integrin Expression Is a Novel Molecular Response to Chemotherapy-Induced Cell Damage in a Heregulin-Dependent Manner. Differentiation 2007, 75, 819–830. [Google Scholar] [CrossRef]
- Park, Y.H.; Jung, K.H.; Im, S.A.; Sohn, J.H.; Ro, J.; Ahn, J.H.; Kim, S.B.; Nam, B.H.; Oh, D.Y.; Han, S.W.; et al. Phase III, Multicenter, Randomized Trial of Maintenance Chemotherapy versus Observation in Patients with Metastatic Breast Cancer after Achieving Disease Control with Six Cycles of Gemcitabine plus Paclitaxel as First-Line Chemotherapy: KCSG-BR07-02. J. Clin. Oncol. 2013, 31, 1732–1739. [Google Scholar] [CrossRef]
- Borsoi, C.; Leonard, F.; Lee, Y.; Zaid, M.; Elganainy, D.; Alexander, J.F.; Kai, M.; Liu, Y.T.; Kang, Y.; Liu, X.; et al. Gemcitabine Enhances the Transport of Nanovector-Albumin-Bound Paclitaxel in Gemcitabine-Resistant Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2017, 403, 296–304. [Google Scholar] [CrossRef]
- Yu, D.M.; Li, W.; Zhang, Y.; Zhang, B. Anti-Tumor Efficiency of Paclitaxel and DNA When Co-Delivered by PH Responsive Ligand Modified Nanocarriers for Breast Cancer Treatment. Biomed. Pharmacother. 2016, 83, 1428–1435. [Google Scholar] [CrossRef]
- Noh, I.; Kim, H.O.; Choi, J.; Choi, Y.; Lee, D.K.; Huh, Y.M.; Haam, S. Co-Delivery of Paclitaxel and Gemcitabine via CD44-Targeting Nanocarriers as a Prodrug with Synergistic Antitumor Activity against Human Biliary Cancer. Biomaterials 2015, 53, 763–774. [Google Scholar] [CrossRef]
- Bai, K.B.; Láng, O.; Orbán, E.; Szabó, R.; Köhidai, L.; Hudecz, F.; Mezö, G. Design, Synthesis, and in Vitro Activity of Novel Drug Delivery Systems Containing Tuftsin Derivatives and Methotrexate. Bioconjug. Chem. 2008, 19, 2260–2269. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugate | Type of Linker | Target (Cell Line) | Results | Ref. |
|---|---|---|---|---|
| Gemcitabine Conjugate with Cell-penetrating Peptides (CPPs) | ||||
| Gem-Cys-pVec, Gem-Cys-Pen | Disulfide bridge | Three human cancer cell lines: MKN-28 (human gastric cancer), Caco-2 (heterogeneous human epithelial colorectal adenocarcinoma), and HT-29 (human colon adenocarcinoma) | Longer half-life: 9.6 days for Gem-Cys-Pen) and 42 h for Gem-Cys-pVec MKN-28, Caco-2 and HT-29 IC50 < 50 µM *, gemcitabine > 100 µM | [41] |
| dFdC-C2-CPP6-1, dFdC-C2-CPP6-2, dFdC-C2-CPP6-3 | Succinyl spacer | Human pancreatic adenocarcinoma (BxPC-3), human breast adenocarcinoma (MCF-7), and human prostate adenocarcinoma (PC-3) cancer cell lines | IC50:15 ± 0.6 nM for dFdC-C2-CPP6-1; 14 ± 0.4 nM for dFdC-C2-CPP6-3 and 74 ± 6.1 nM for gemcitabine alone in the PC-3 cell line | [32] |
| [RW]3-Gem[RW]4-Gem [RW]5-Gem [RW]6-Gem R5W3R4-Gem | Succinyl spacer | A549 cell line | Conjugates display increased toxicity compared with the free drug | [42] |
| Gemcitabine Conjugate with Receptor-Binding Peptides | ||||
| Gemcitabine-succinate-GnRH (GSG) | Ester linkages (four or five carbons) glutaric or succinyl spacer | Prostate cancer (CaP) cell lines (DU145 and PC3) Xenograft animal model Bone marrow cells derived from male C57BL/6 mice | IC50: 308 ± 170 nM for GSG and 231 ± 34 nM for gemcitabine GSG efficacy was achieved with a significantly lower dose when compared with gemcitabine (approximately 25 times) IC90: 41.4 ± 13.3 nM for GSG vs. 20.9 ± 8.5 nM for gemcitabine; IC50: 24.3 ± 6.4 nM vs. 12.1 ± 6.7 nM, respectively | [42] [43] [18] |
| Internalized-RGD ** (iRGD) peptide with gemcitabine | None, the mixture of gemcitabine and iRGD | Five mouse pancreatic cancer cell xenograft models: AsPC-1, BxPC-3, Capan-1, MIA PaCa-2, SUIT-2 | iRGD peptide demonstrated a substantial booster accumulation effect of drugs in the mouse pancreatic cancer models with high NRP1 expression | [44] |
| RGD peptide-gemcitabine-loaded nanocarriers | Drug encapsulation | Human ovarian cancer cell line SKOV-3, human breast adenocarcinoma cell line MDA-MB-231 and MCF-7, and human pancreatic cancer cell line BxPC-3 | IC50: 0.1 μg/mL for cRGD-Gem-HSA-NP, 0.28 μg/mL for gemcitabine, 0.38 μg/mL for Gem-C14, and 0.42 μg/mL for Gem-HSA-NP | [45,46,47,48] |
| Multifunctional gemcitabine prodrug TPE-Gem-RGD | GFLG *** tetrapeptide, disulfide bond | Human pancreatic cancer cell line BxPC-3 | RGD-targeted TPE-Gem-RGD prodrug was inhibiting the proliferation of pancreatic cancer cells more efficiently | [49] |
| RGDV-gemcitabine conjugate | Directly connected via amide bond | Cell lines: MCF-7, HCT-8, A549, 95D, and HepG2 | The IC50 values of gemcitabine and RGDV-gemcitabine show no significant difference; half-life of RGDV-Gem is 17-fold higher than for gemcitabine alone, no kidney toxicity, no liver toxicity, no marrow toxicity, and no drug resistance. The minimal effective dose and activity of RGDV-gemcitabine are 100-fold lower and 10-fold higher than that of gemcitabine, respectively | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawryłkiewicz, A.; Ptaszyńska, N. Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy. Molecules 2021, 26, 364. https://doi.org/10.3390/molecules26020364
Hawryłkiewicz A, Ptaszyńska N. Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy. Molecules. 2021; 26(2):364. https://doi.org/10.3390/molecules26020364
Chicago/Turabian StyleHawryłkiewicz, Aleksandra, and Natalia Ptaszyńska. 2021. "Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy" Molecules 26, no. 2: 364. https://doi.org/10.3390/molecules26020364
APA StyleHawryłkiewicz, A., & Ptaszyńska, N. (2021). Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy. Molecules, 26(2), 364. https://doi.org/10.3390/molecules26020364