
Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams


Abstract
1. Introduction
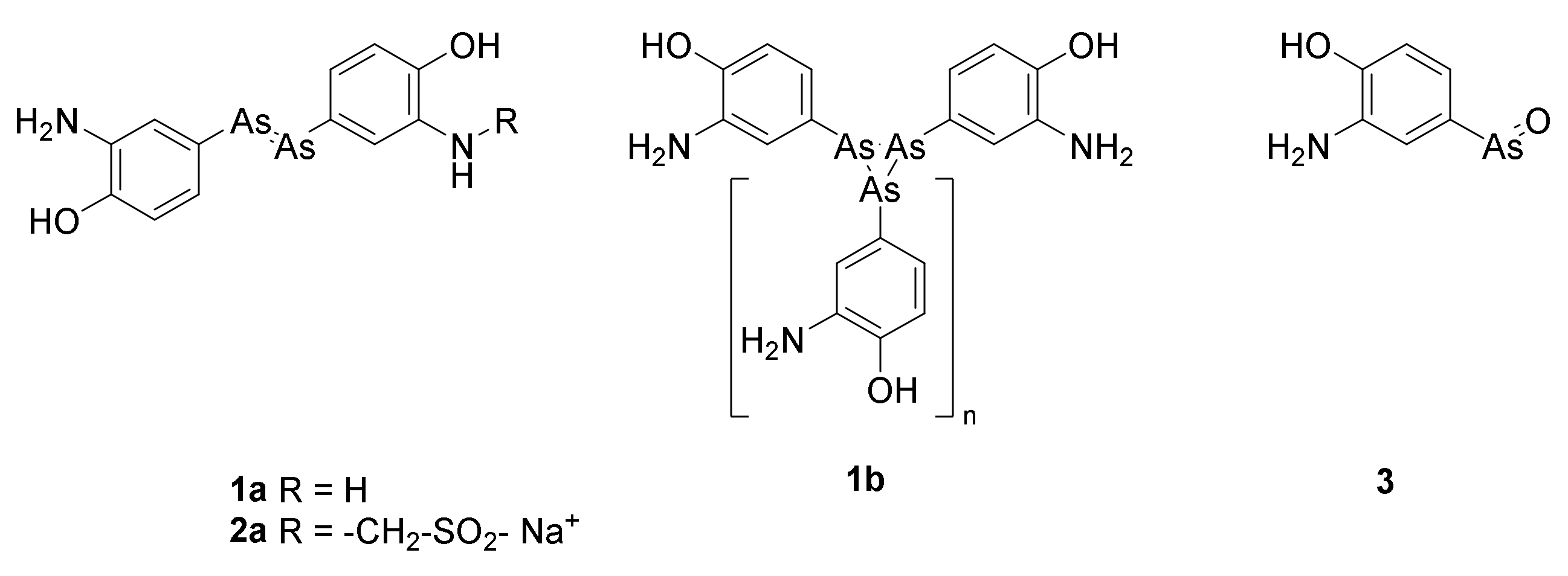
2. Arsphenamine (Salvarsan)
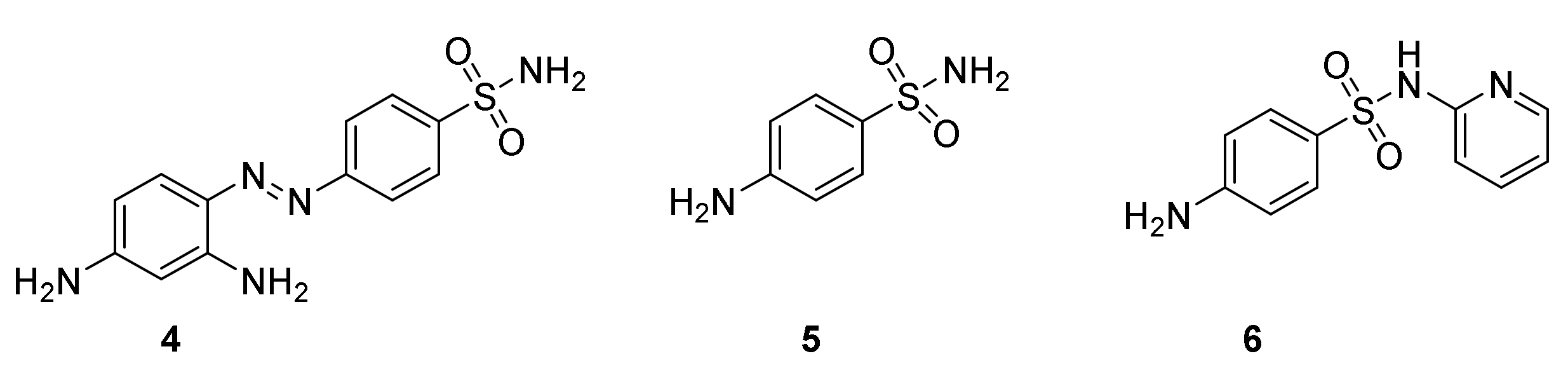
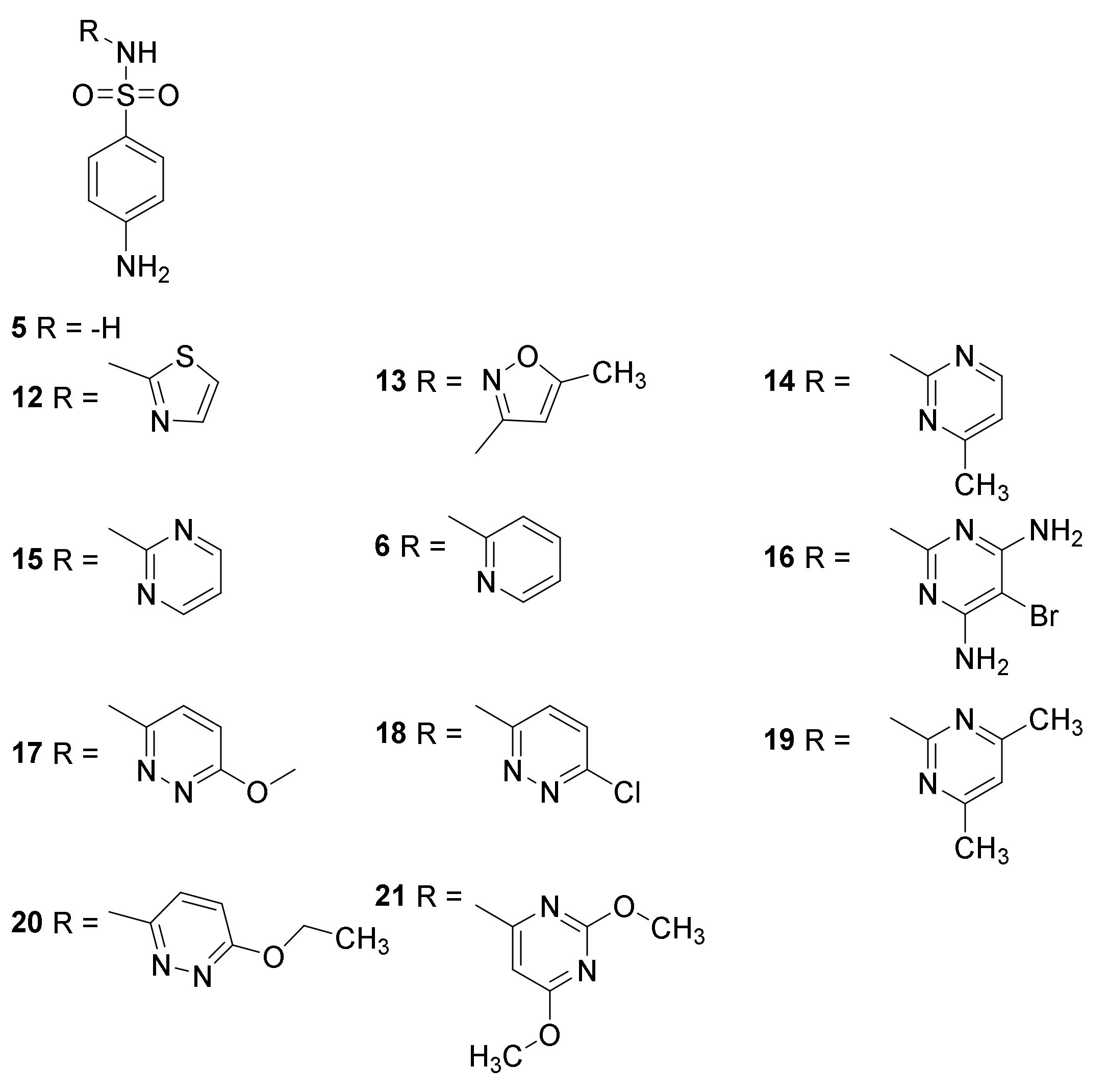
3. Sulfonamides
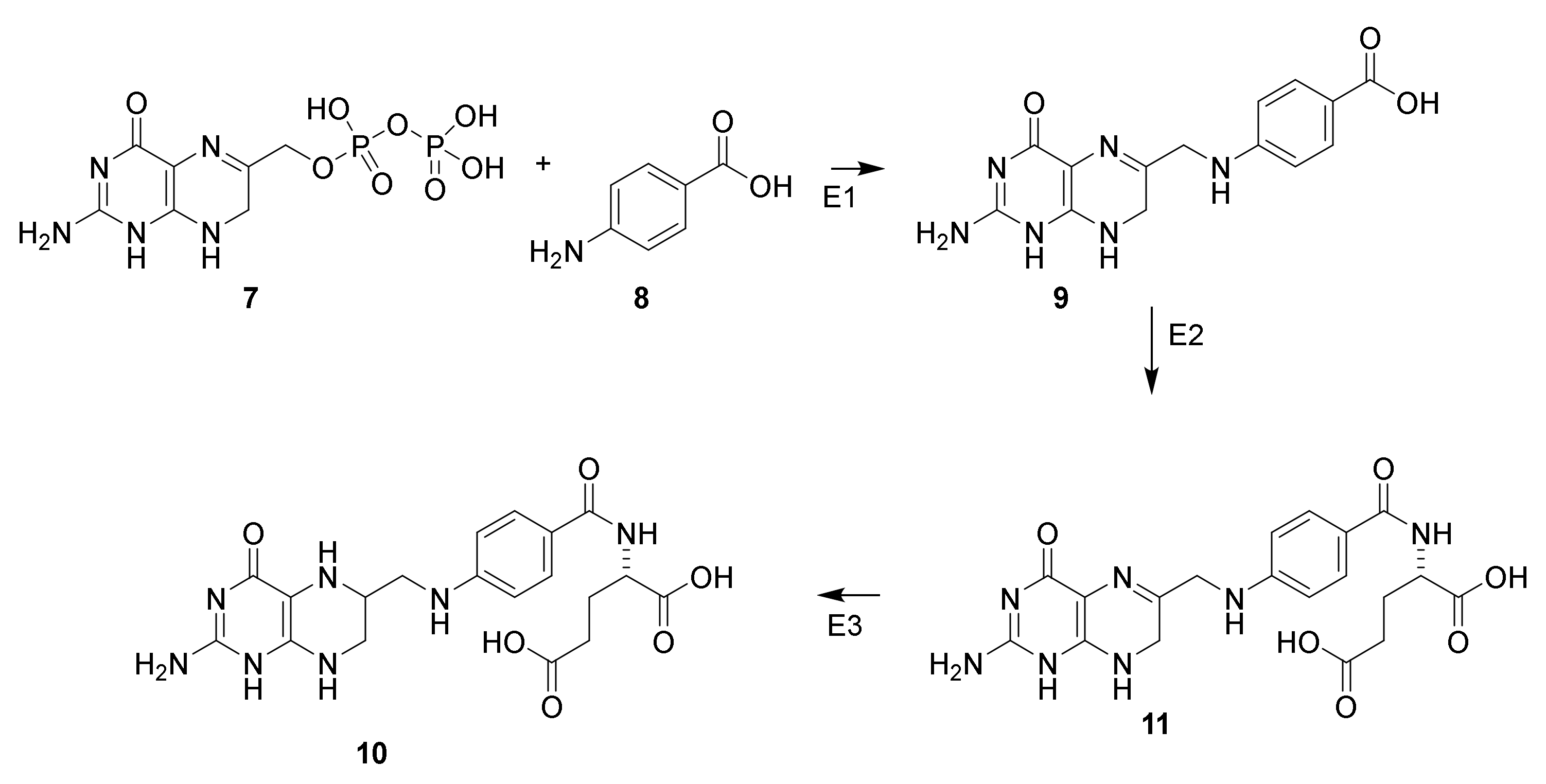
3.1. Discovery
3.2. Mechanism of Action
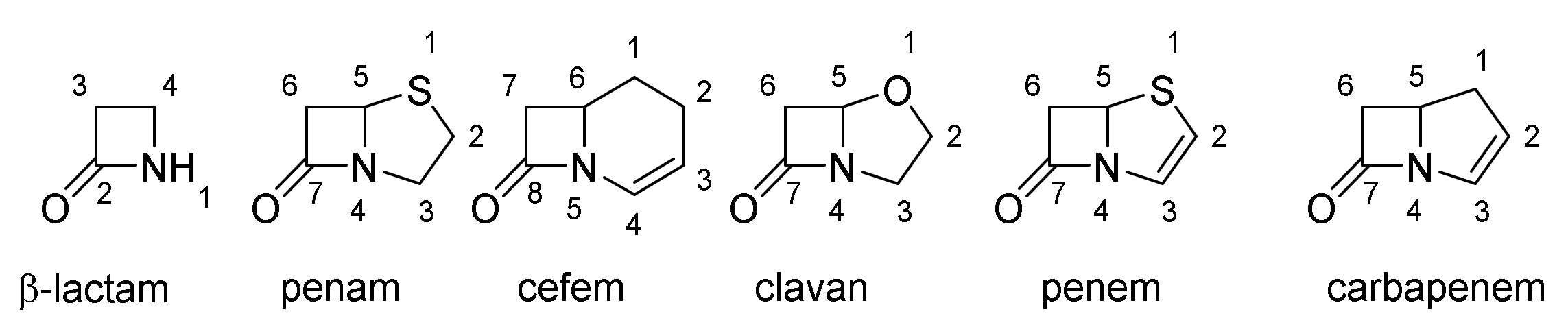
4. β-Lactams
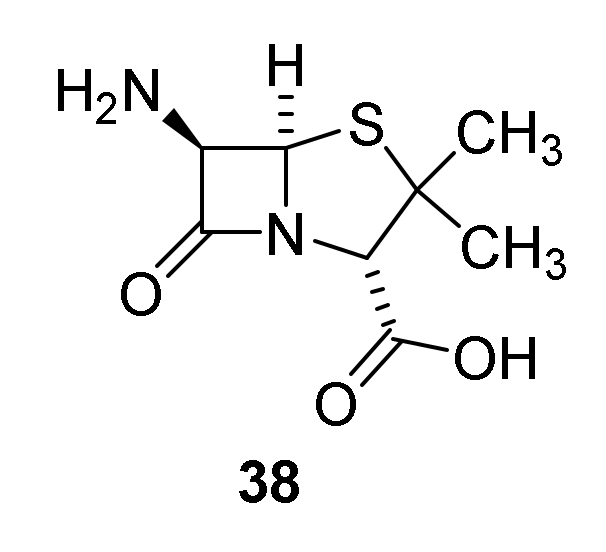
4.1. Discovery of the Penicillins
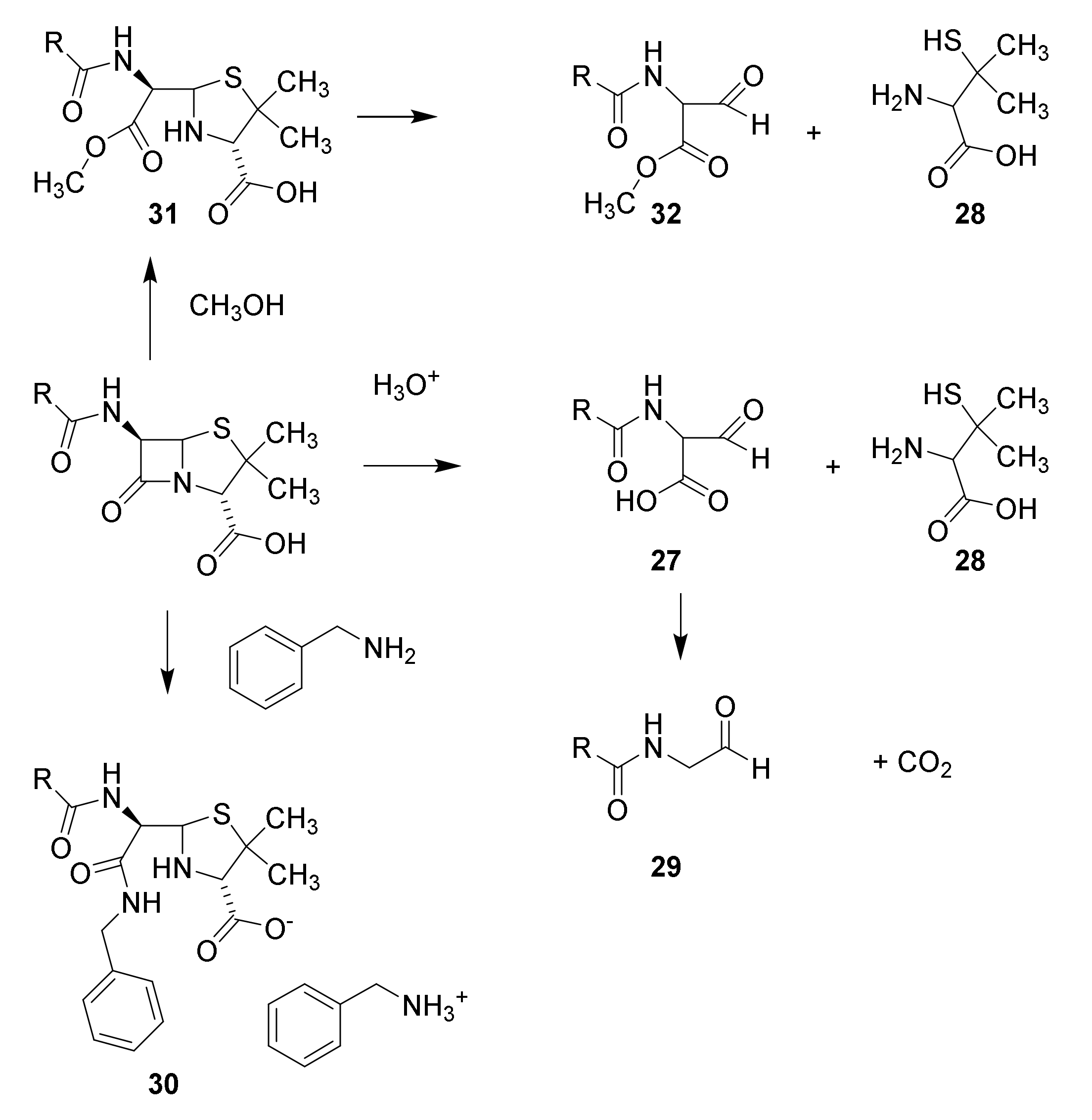
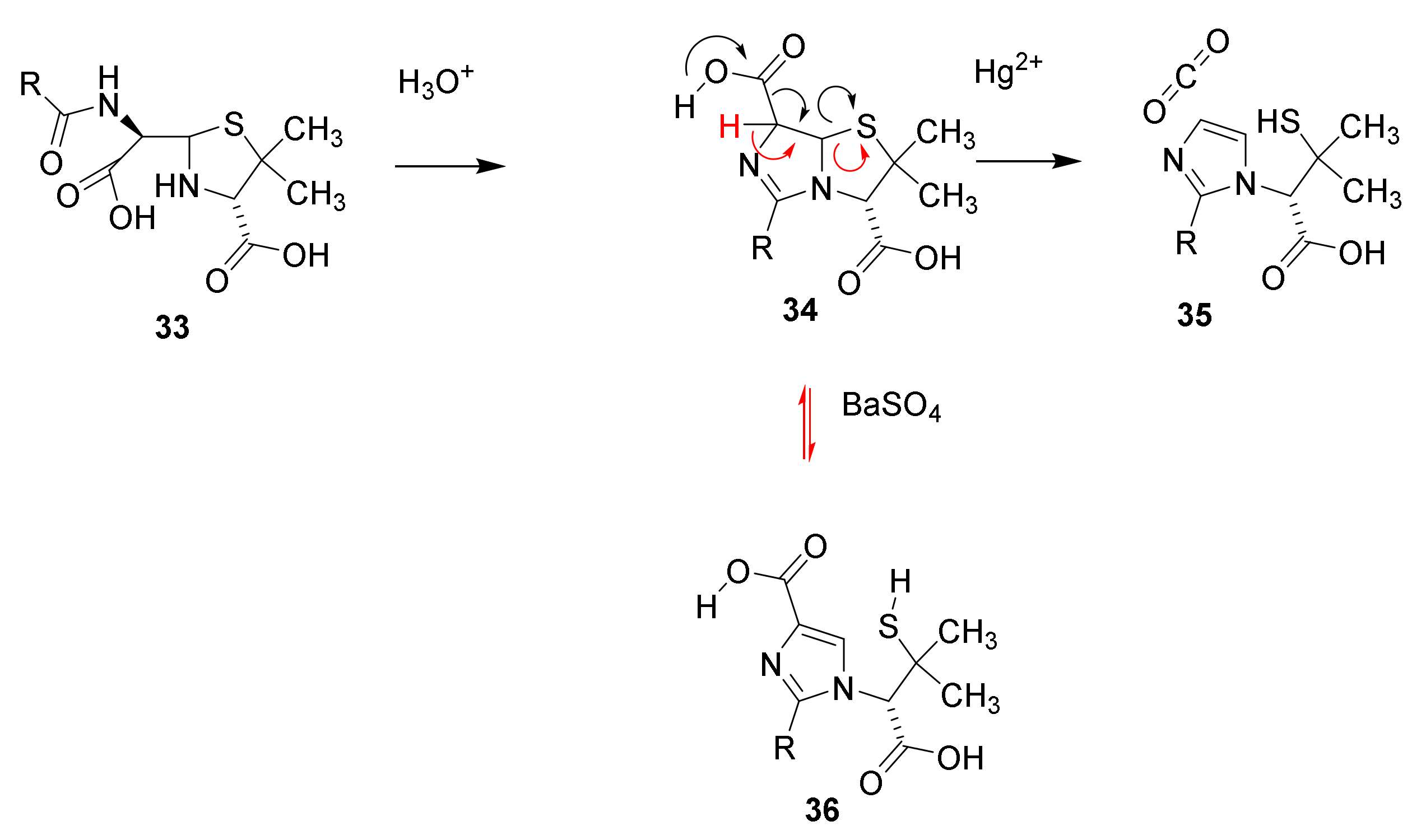
4.2. Structure Elucidation and Chemistry of Penicillin
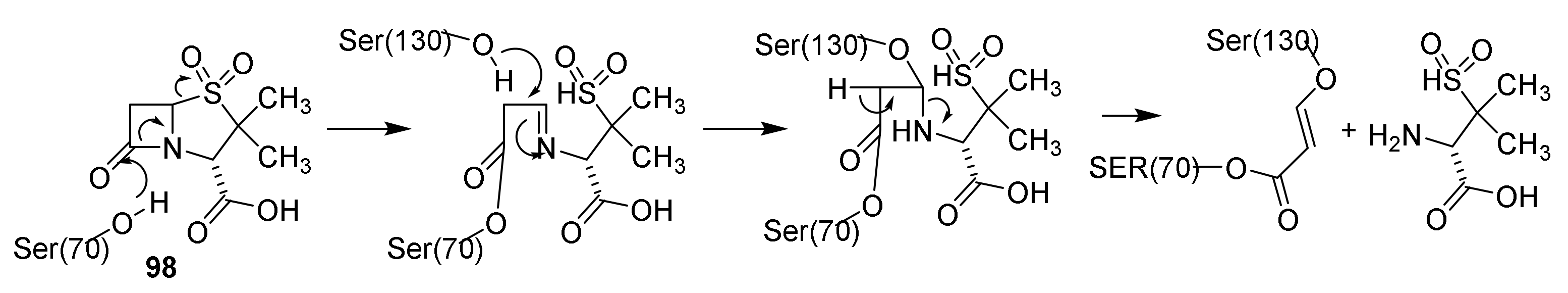
4.3. Mechanism of Action of Penicillins
4.4. Late Generations Penicillins
4.5. Penicillin Hypersensitivity
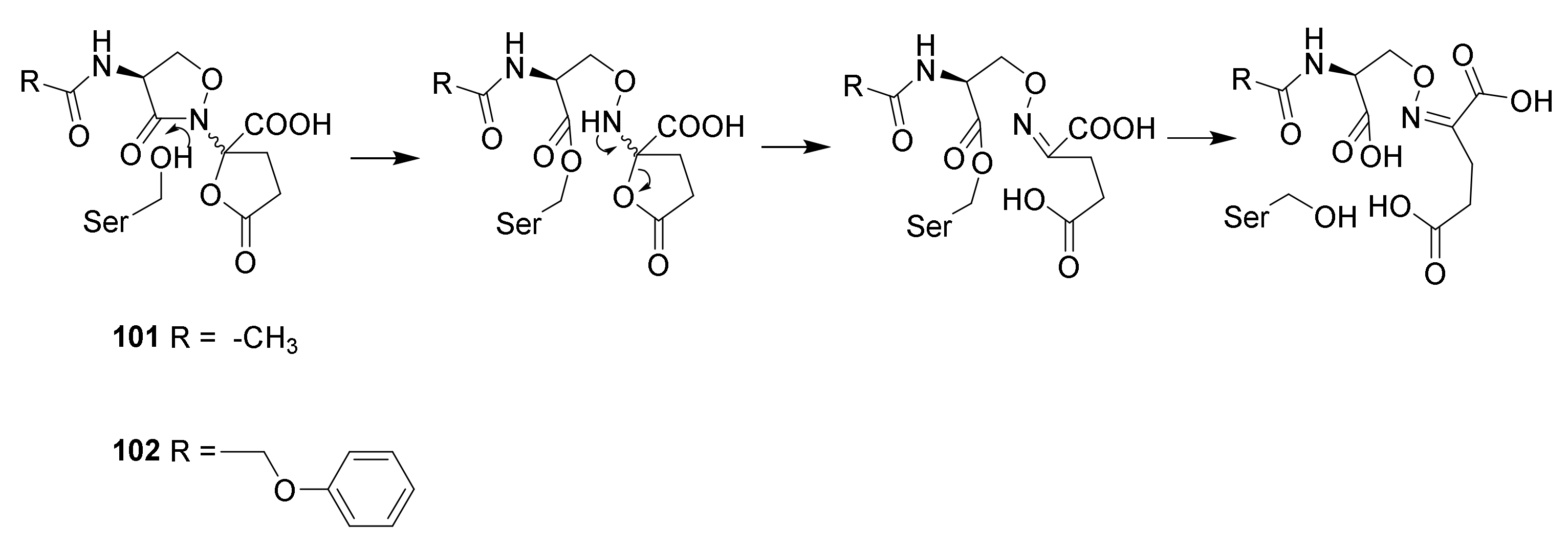
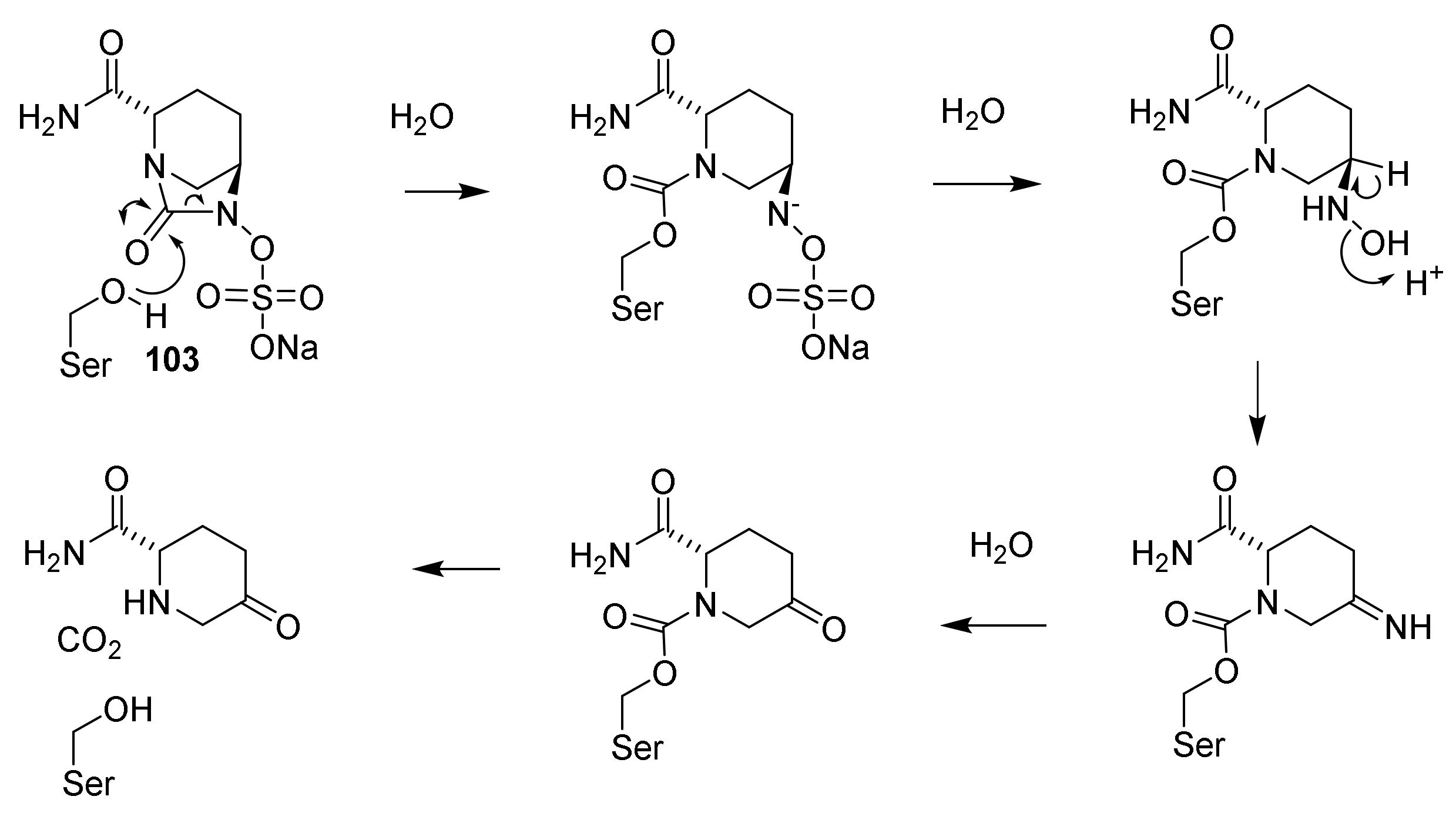
4.6. Penicillin Resistance
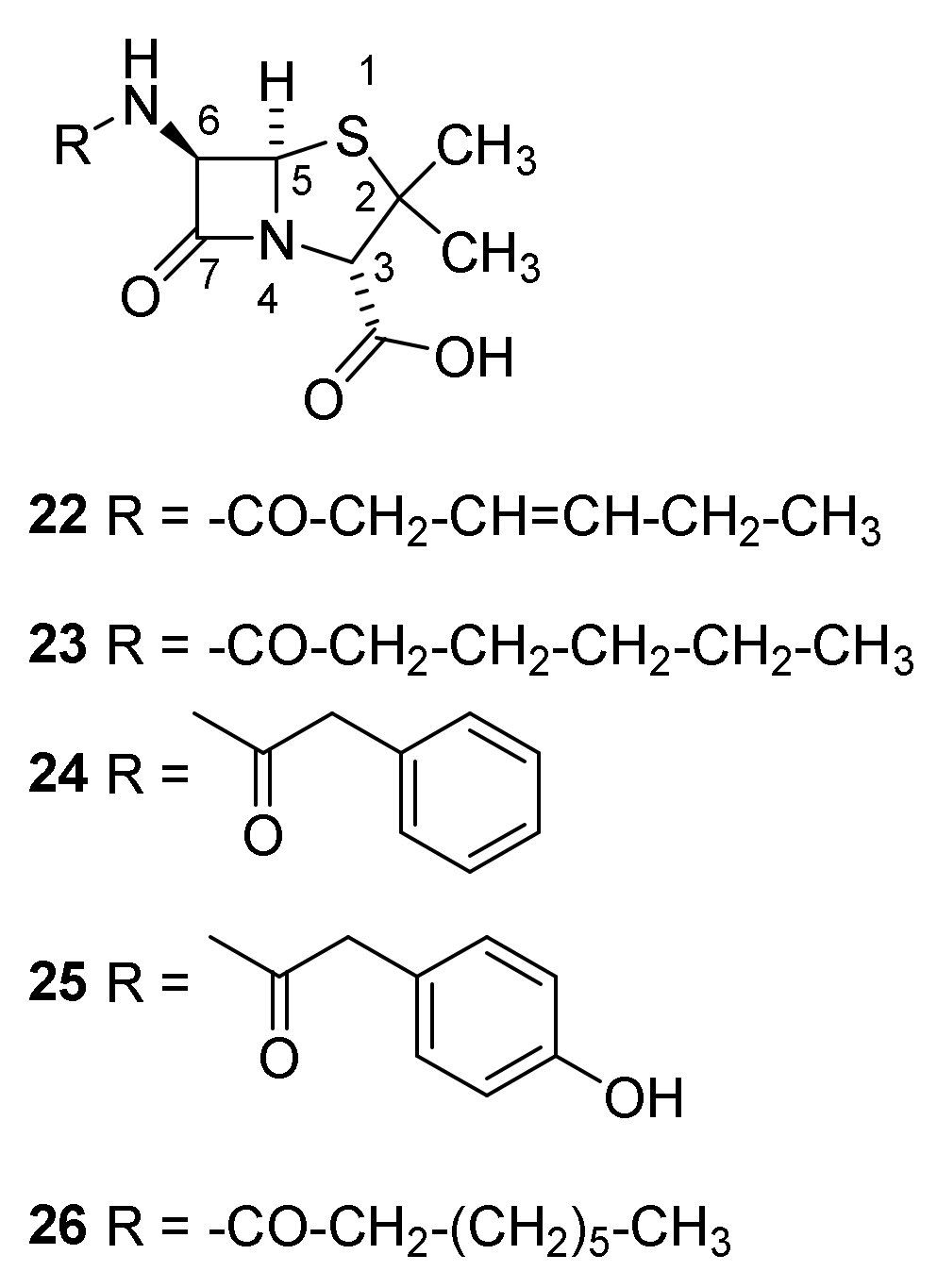
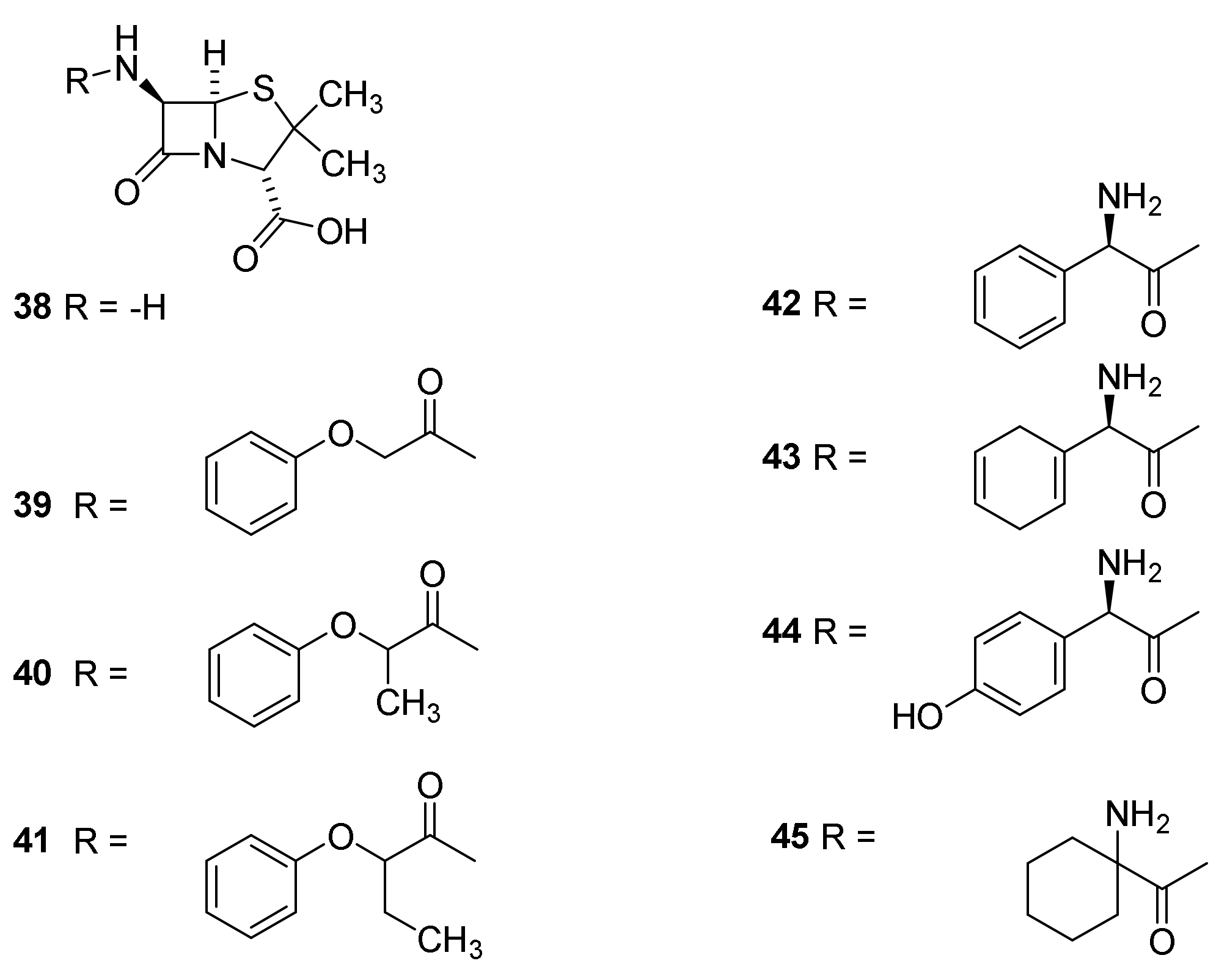
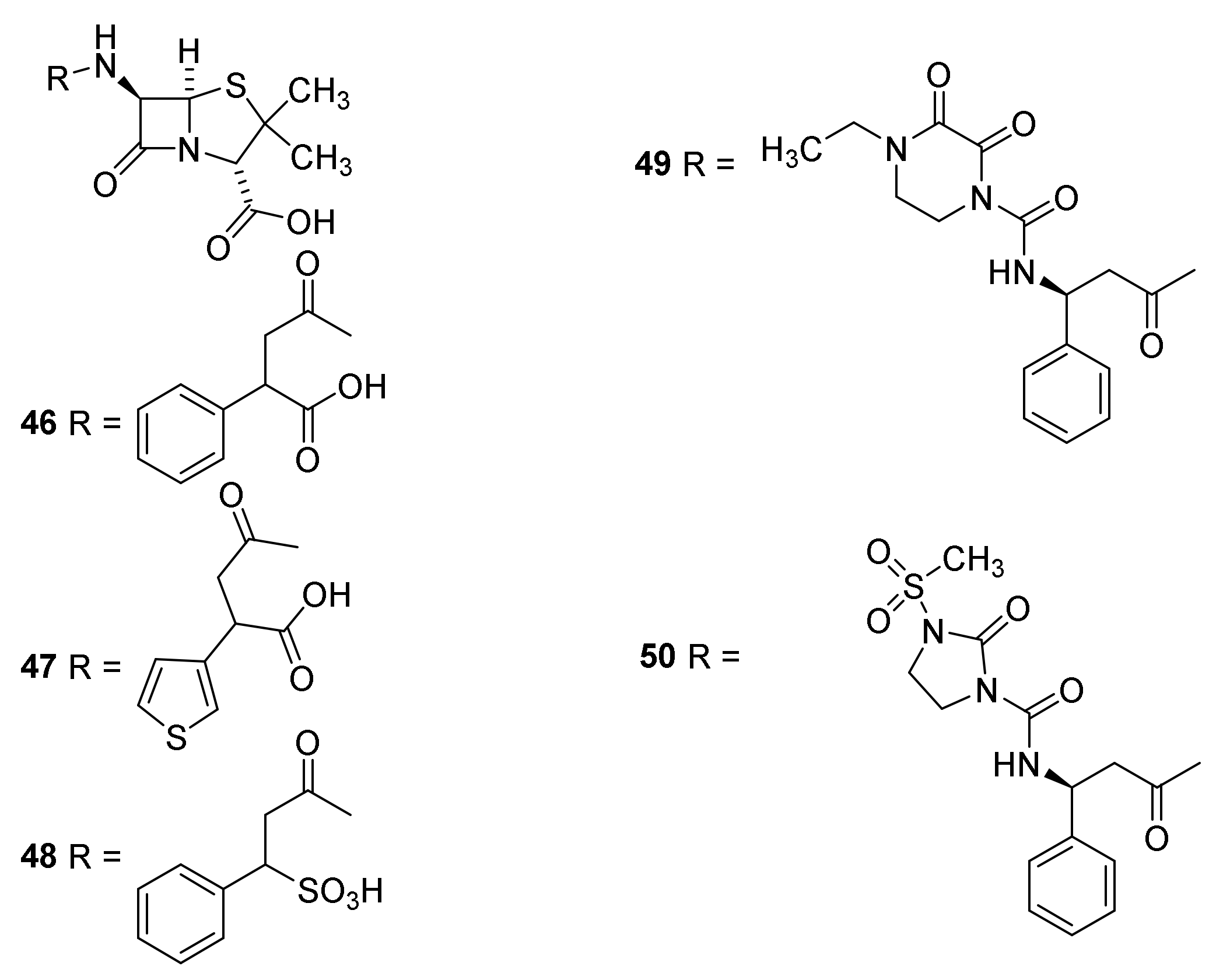
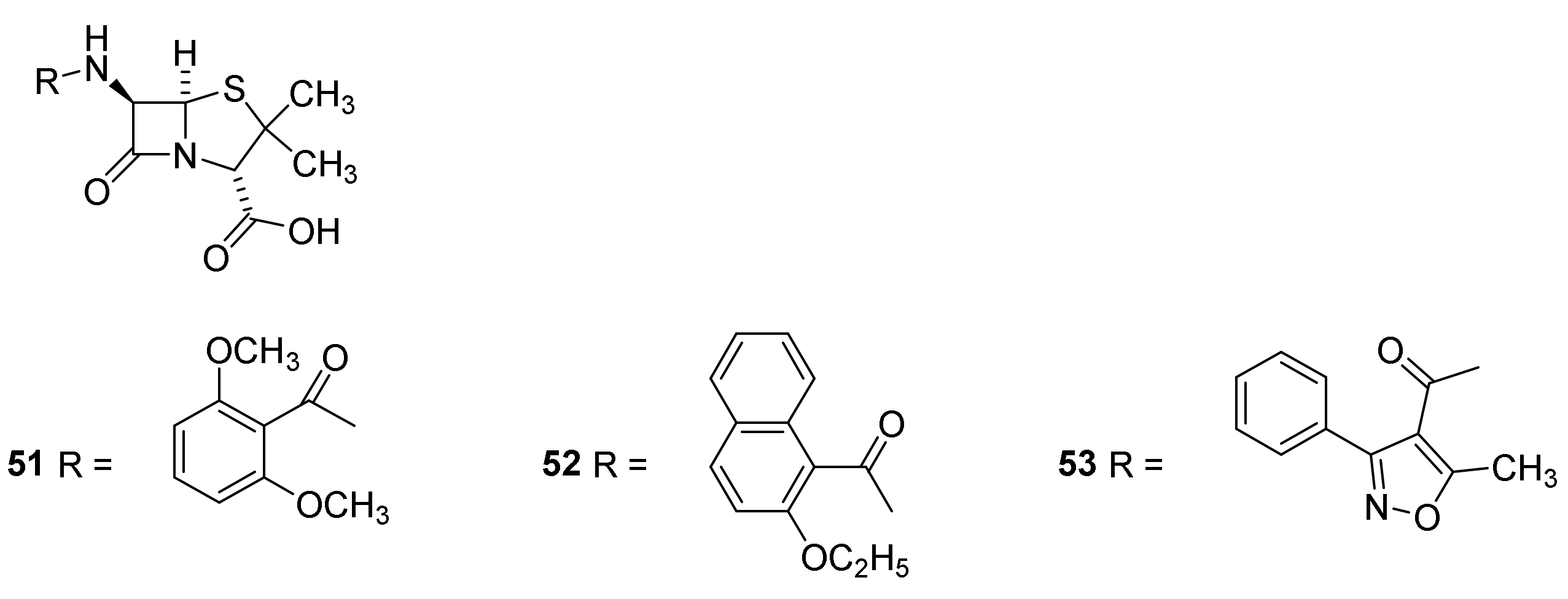
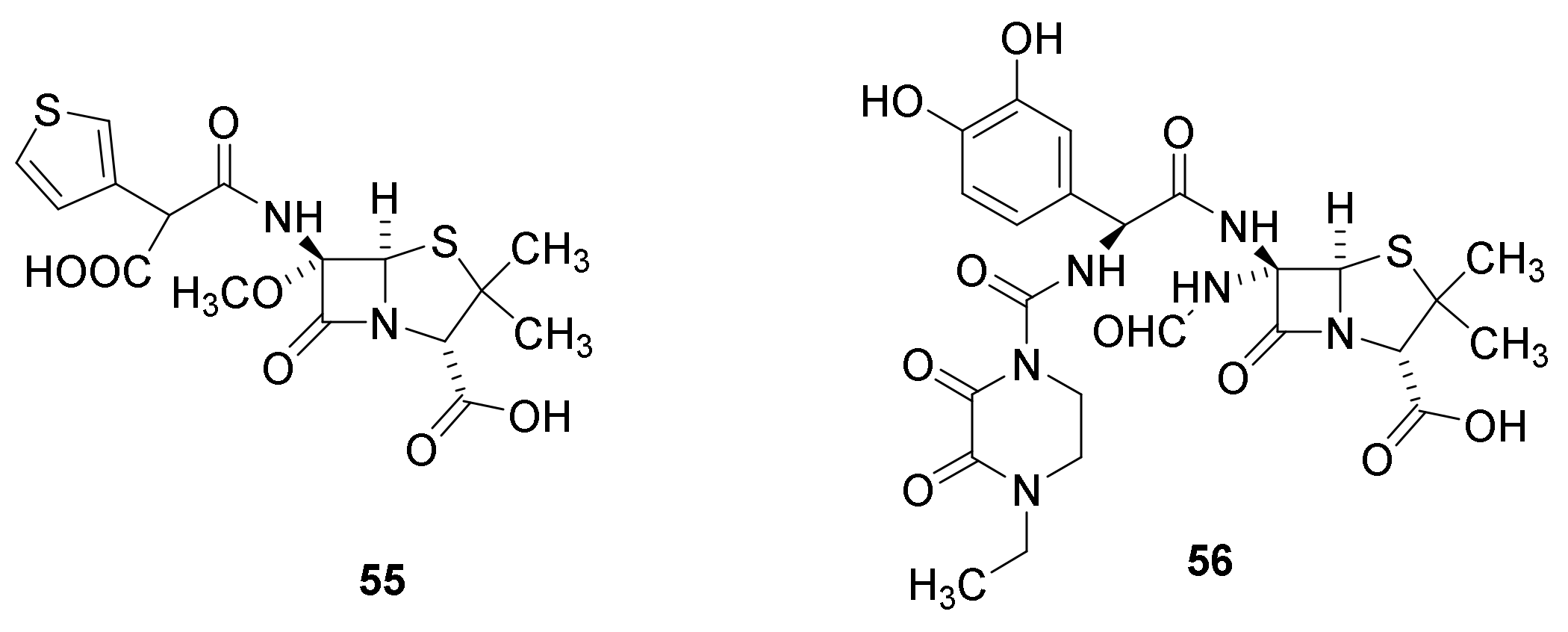
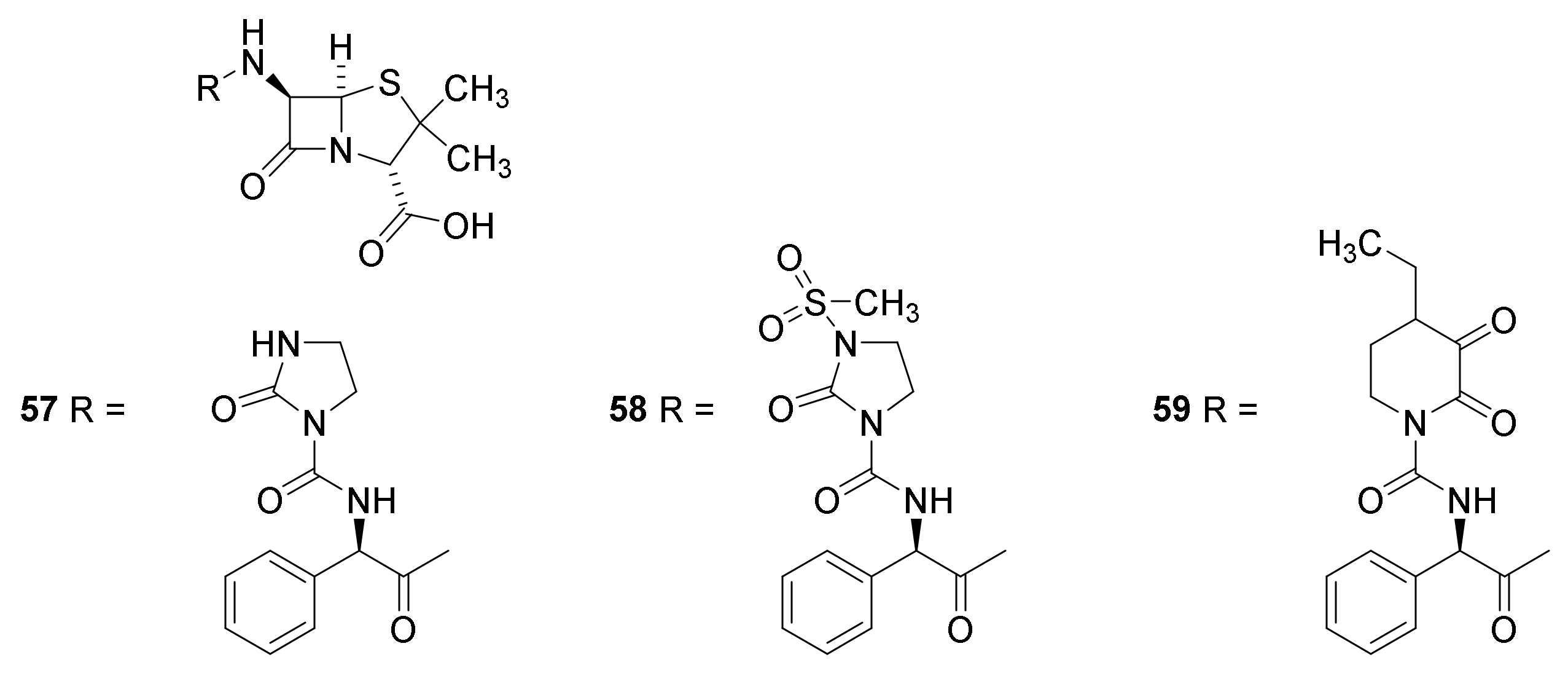
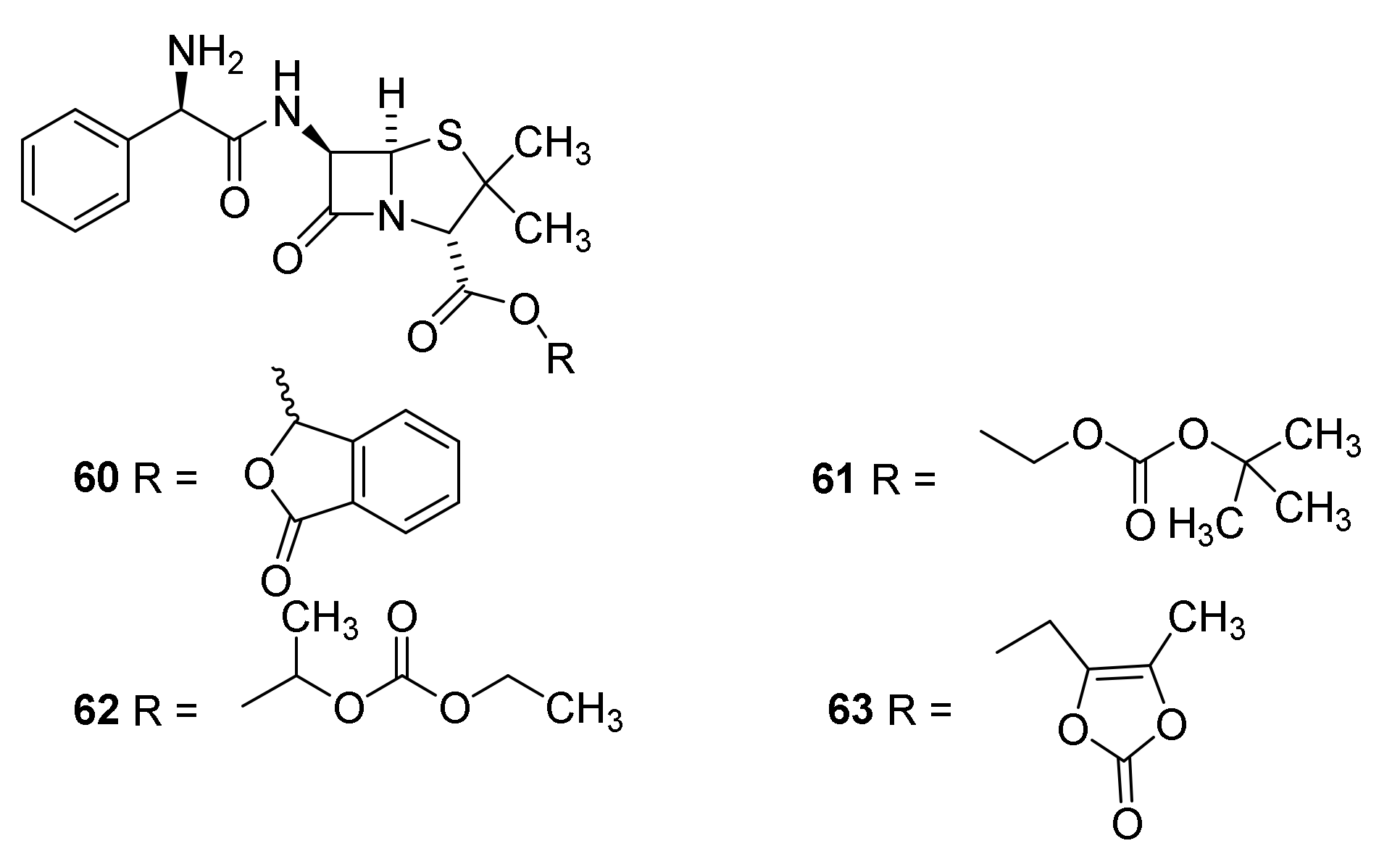
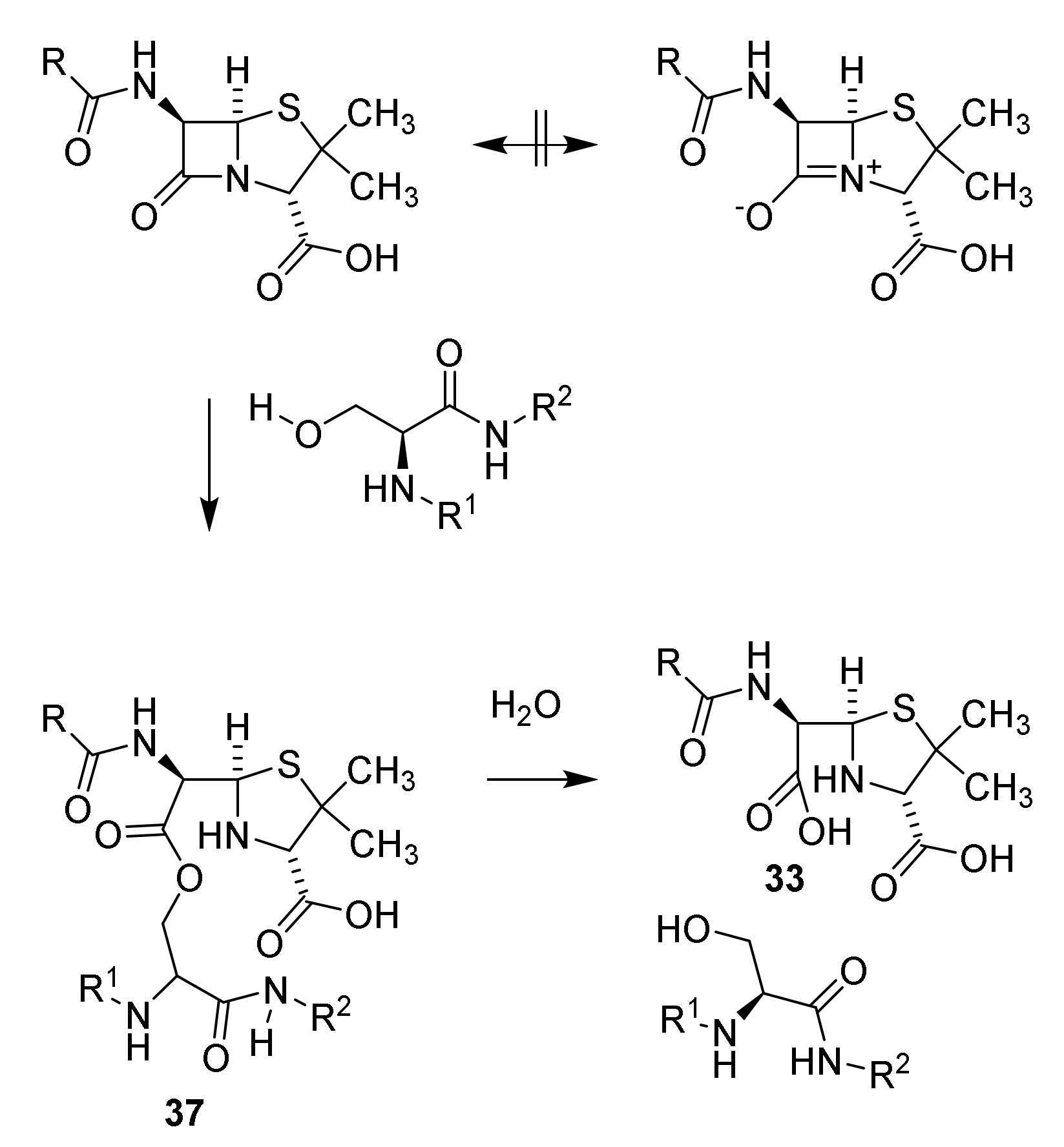
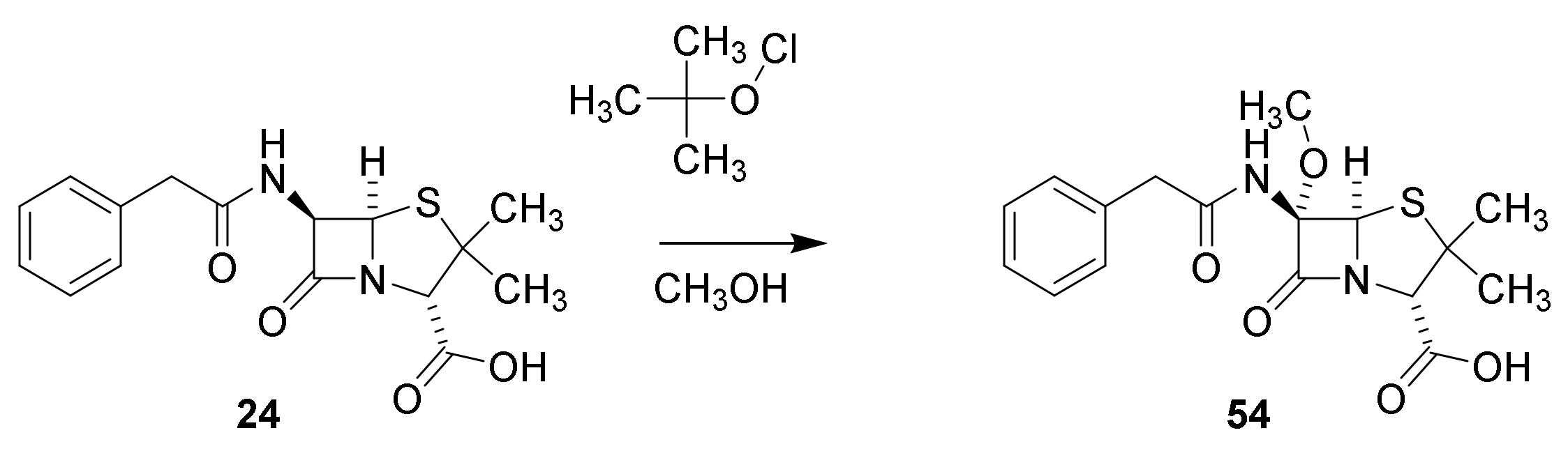
4.7. Penicillin Prodrugs
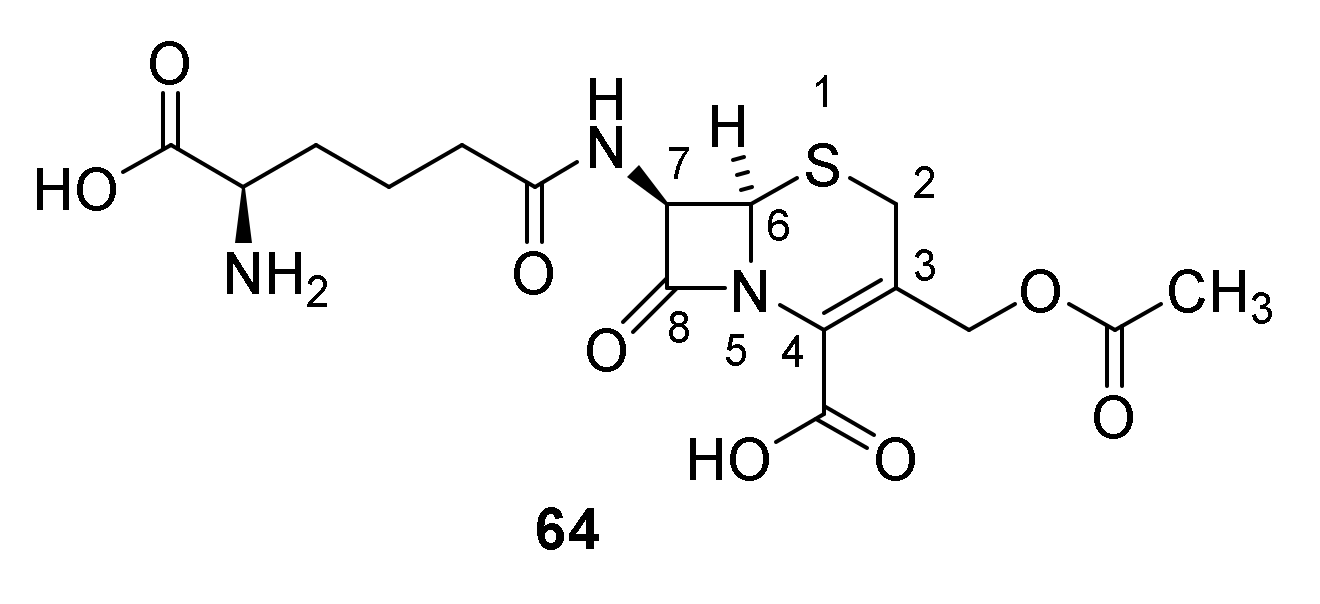
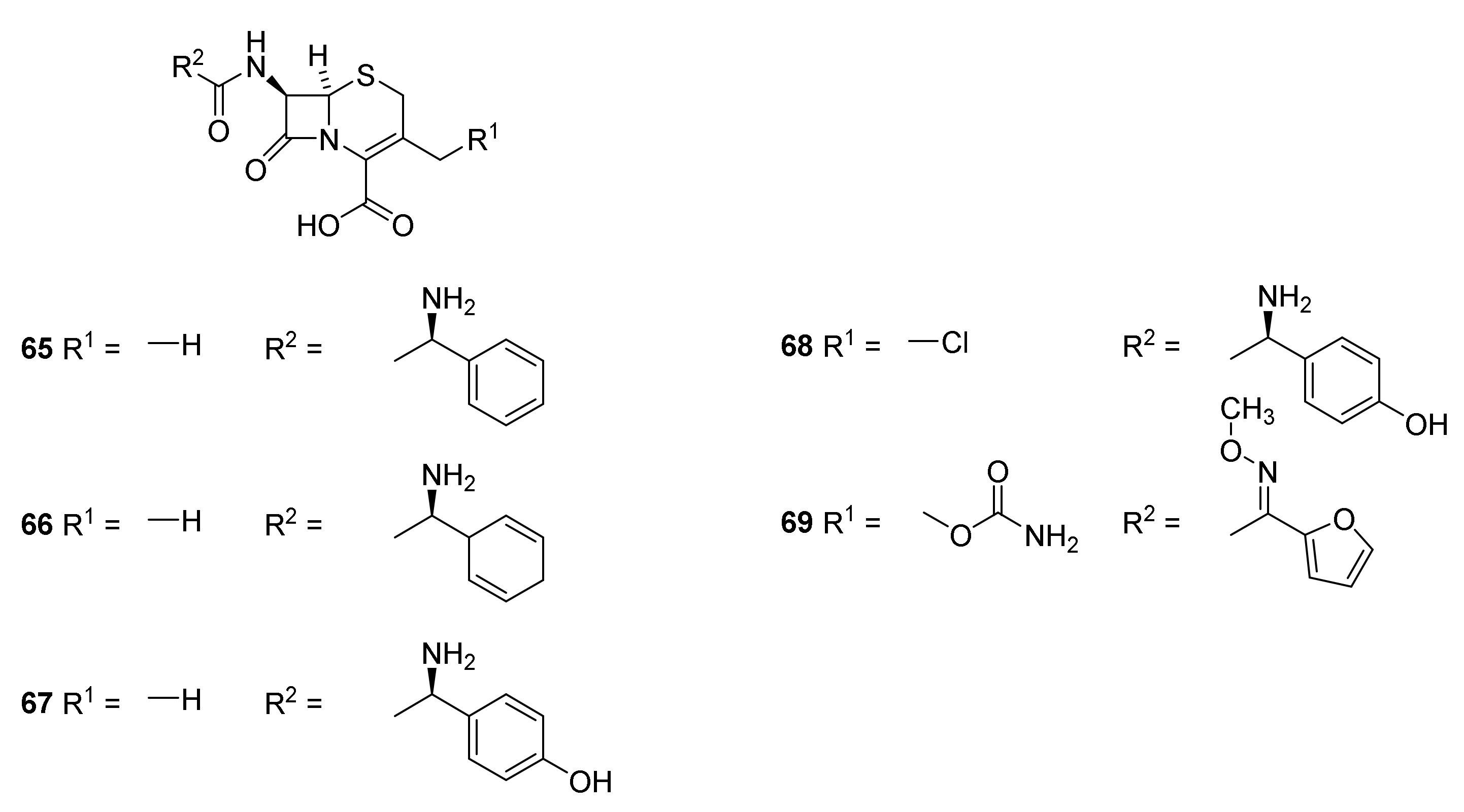
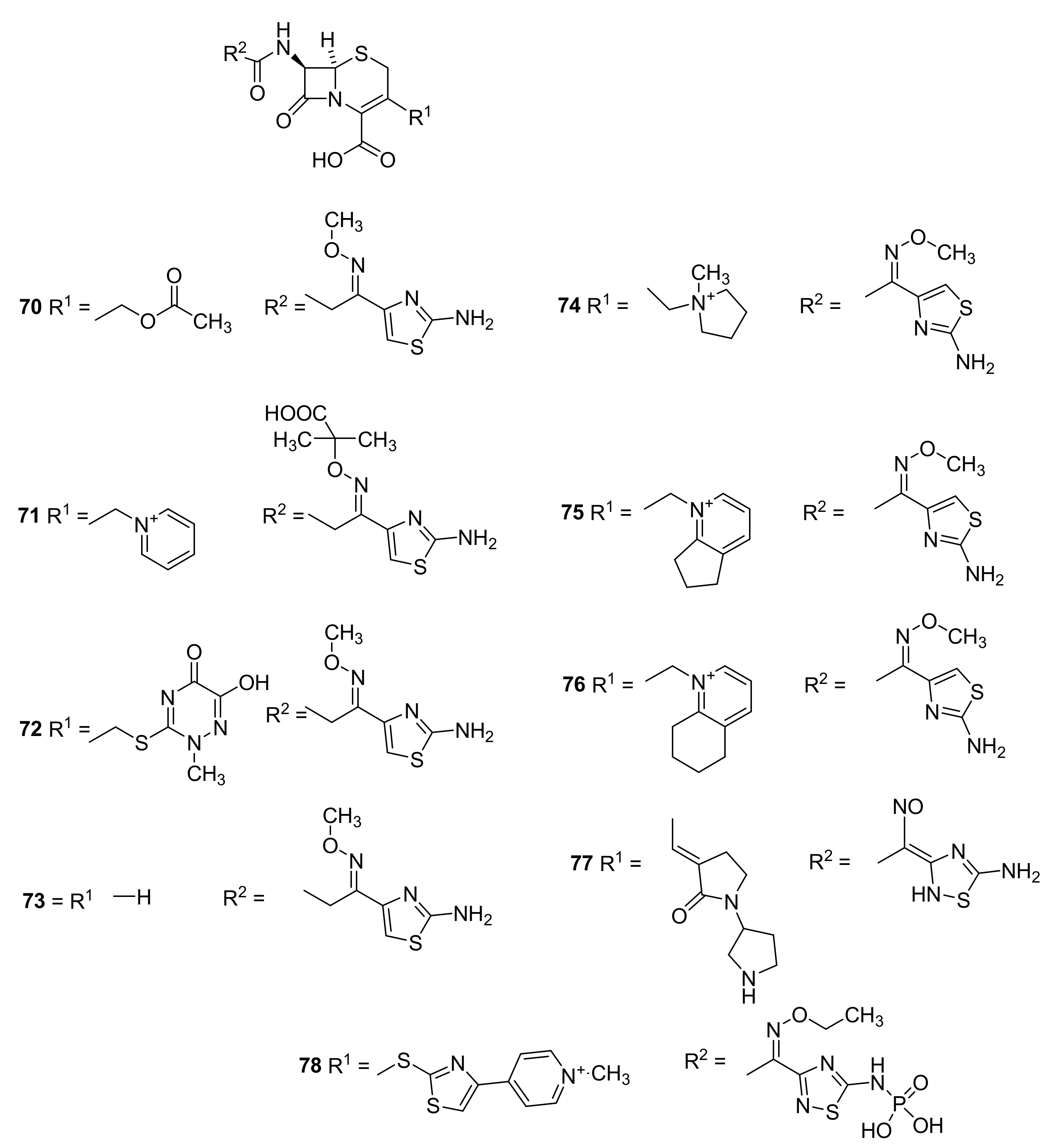
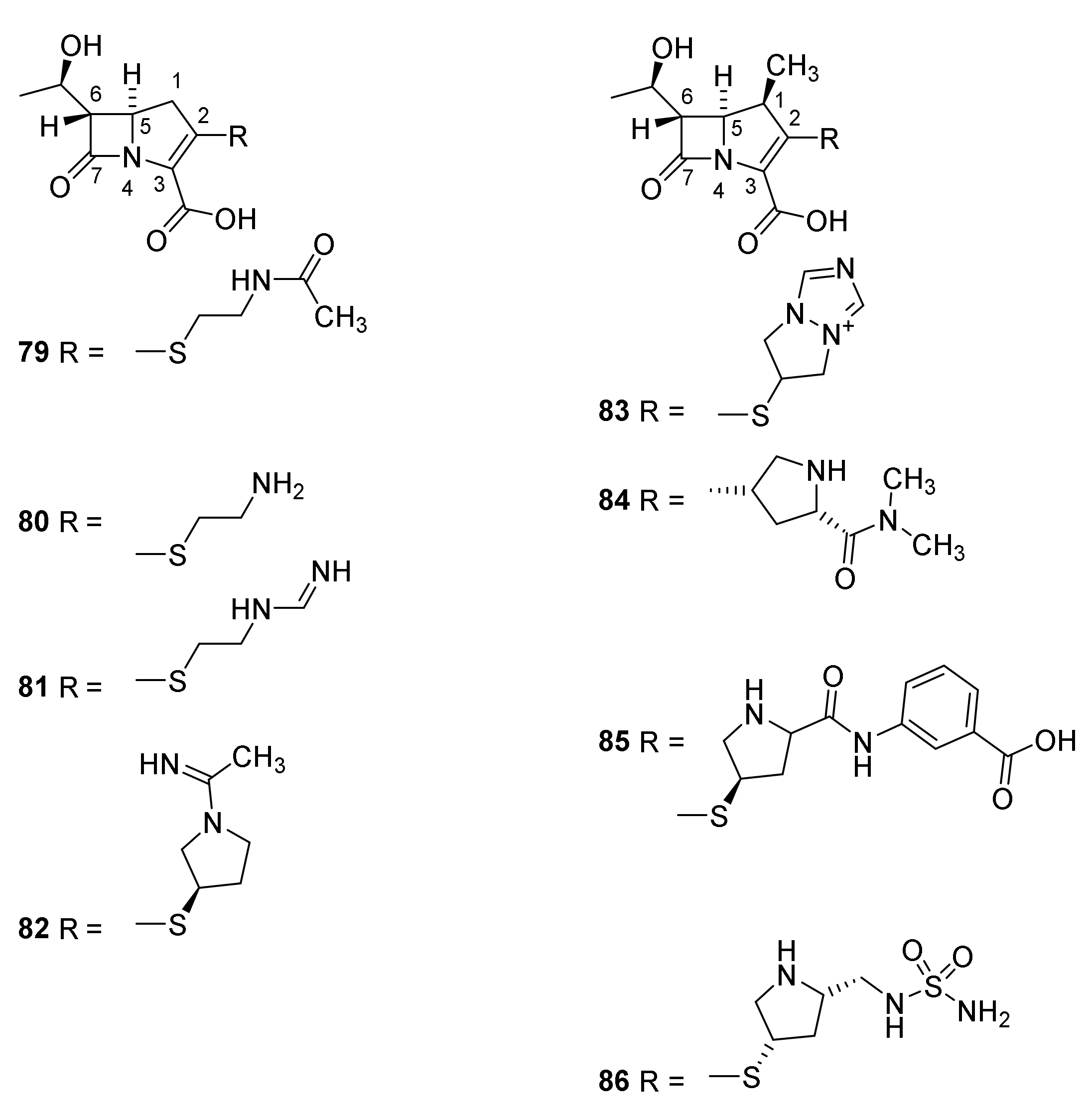
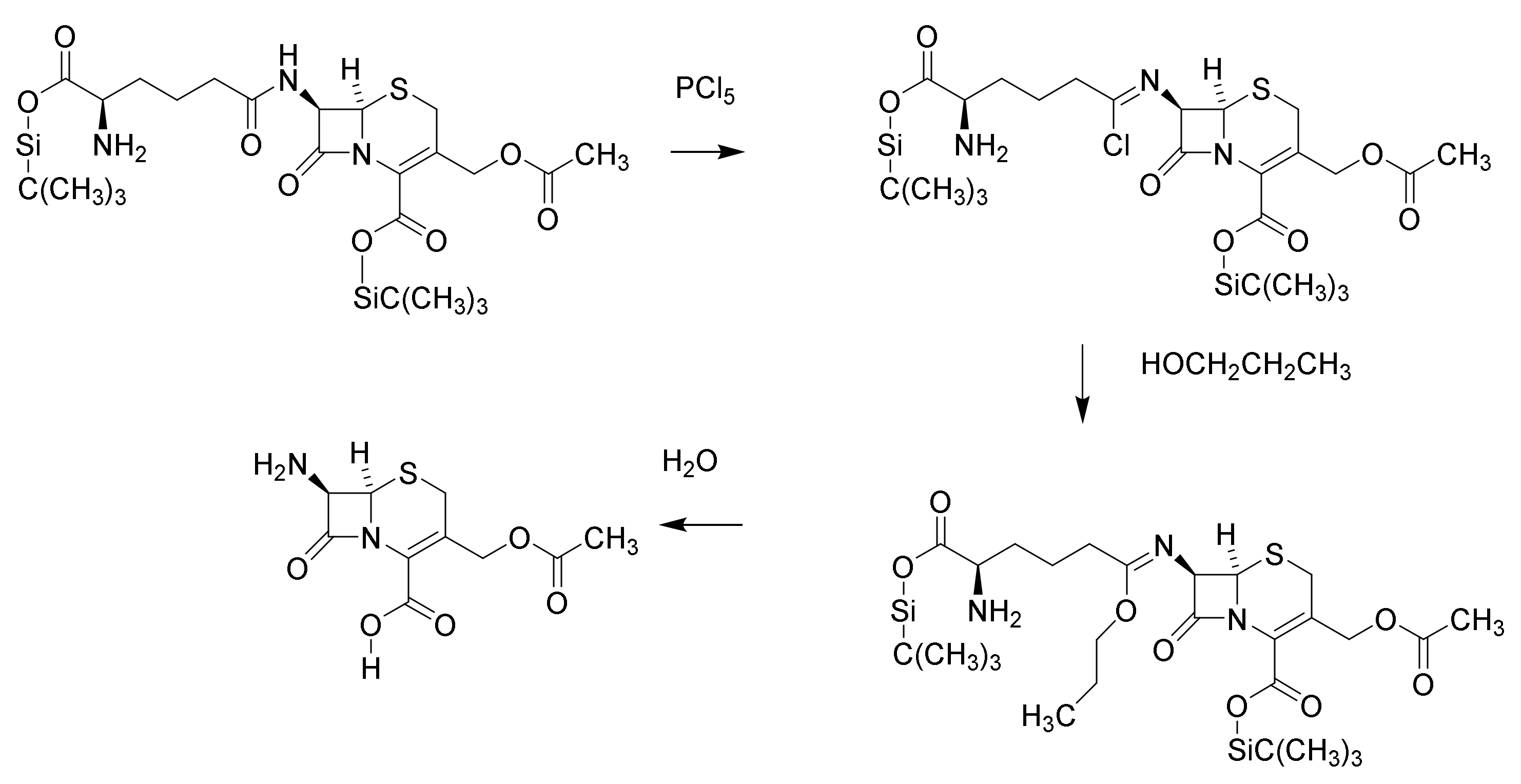
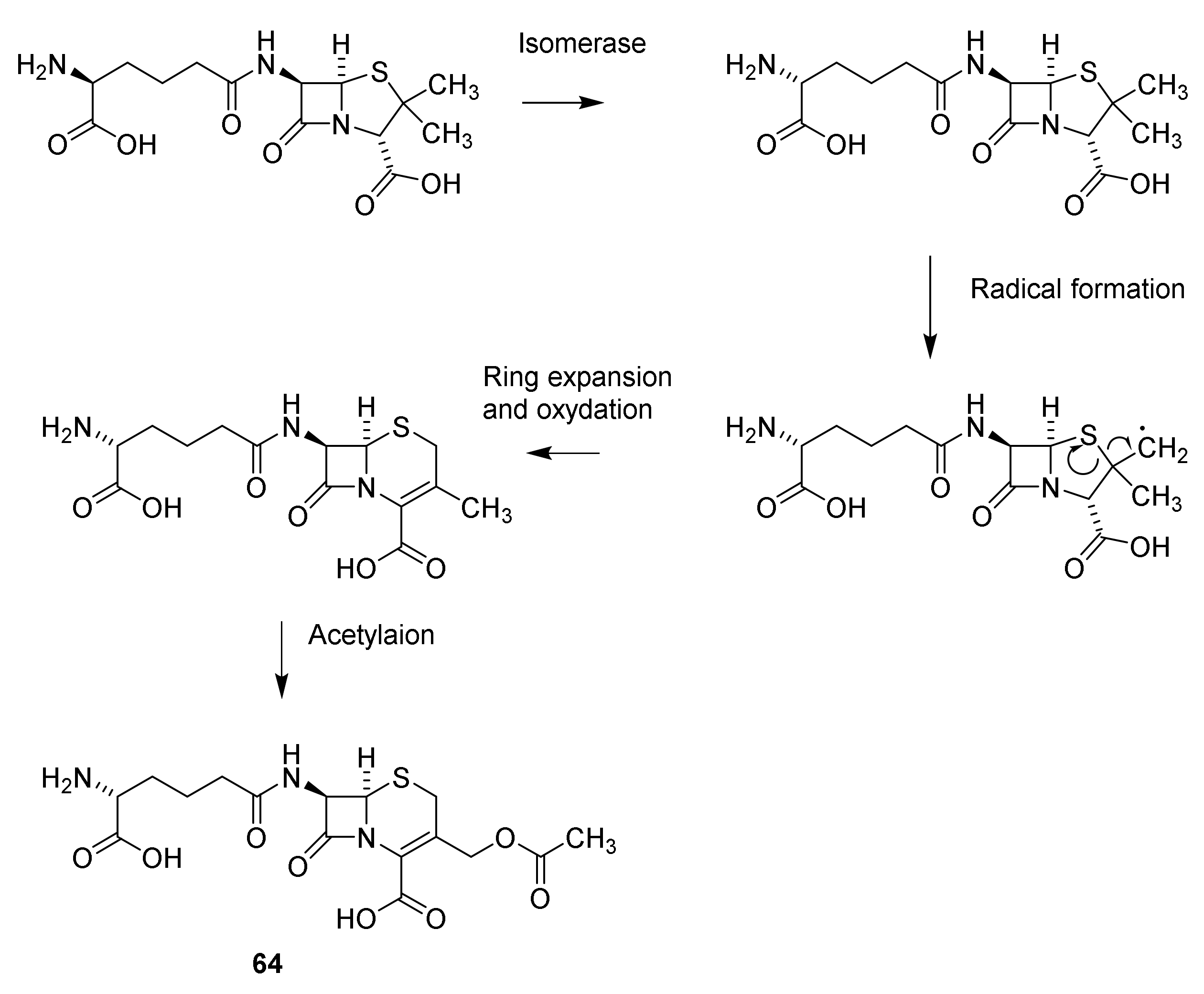
5. Cephalosporins
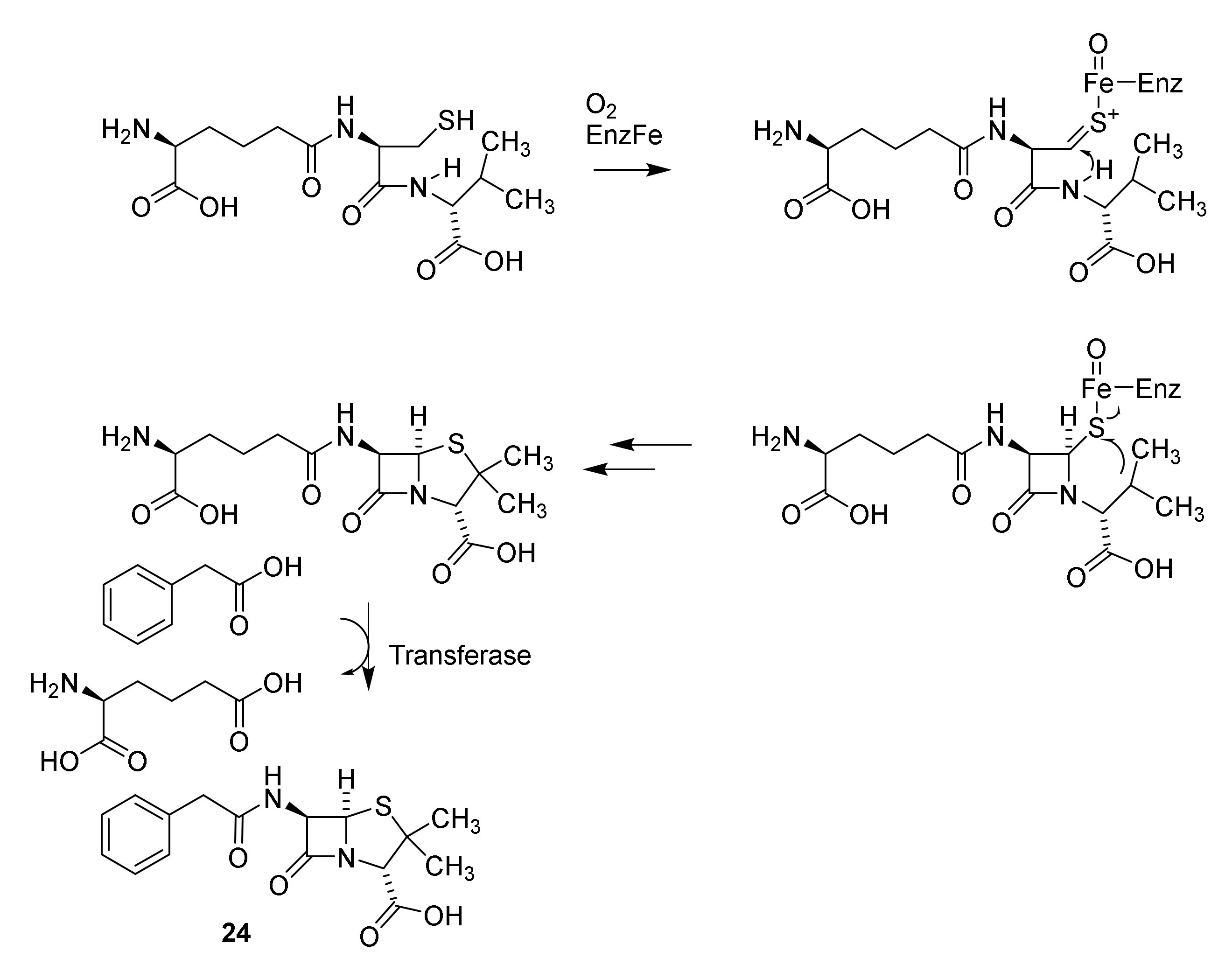
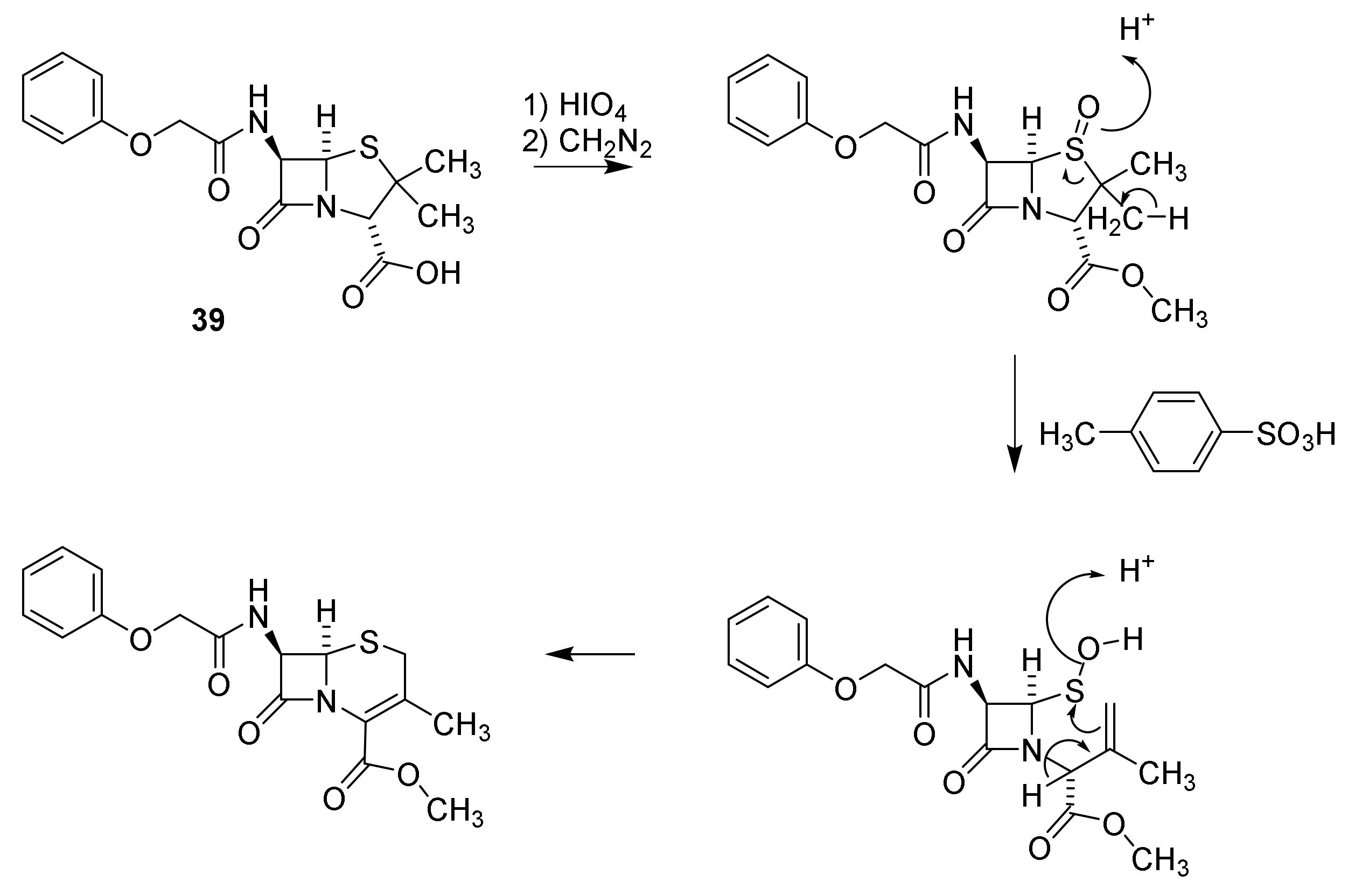
Discovery and Production of Cephalosporins
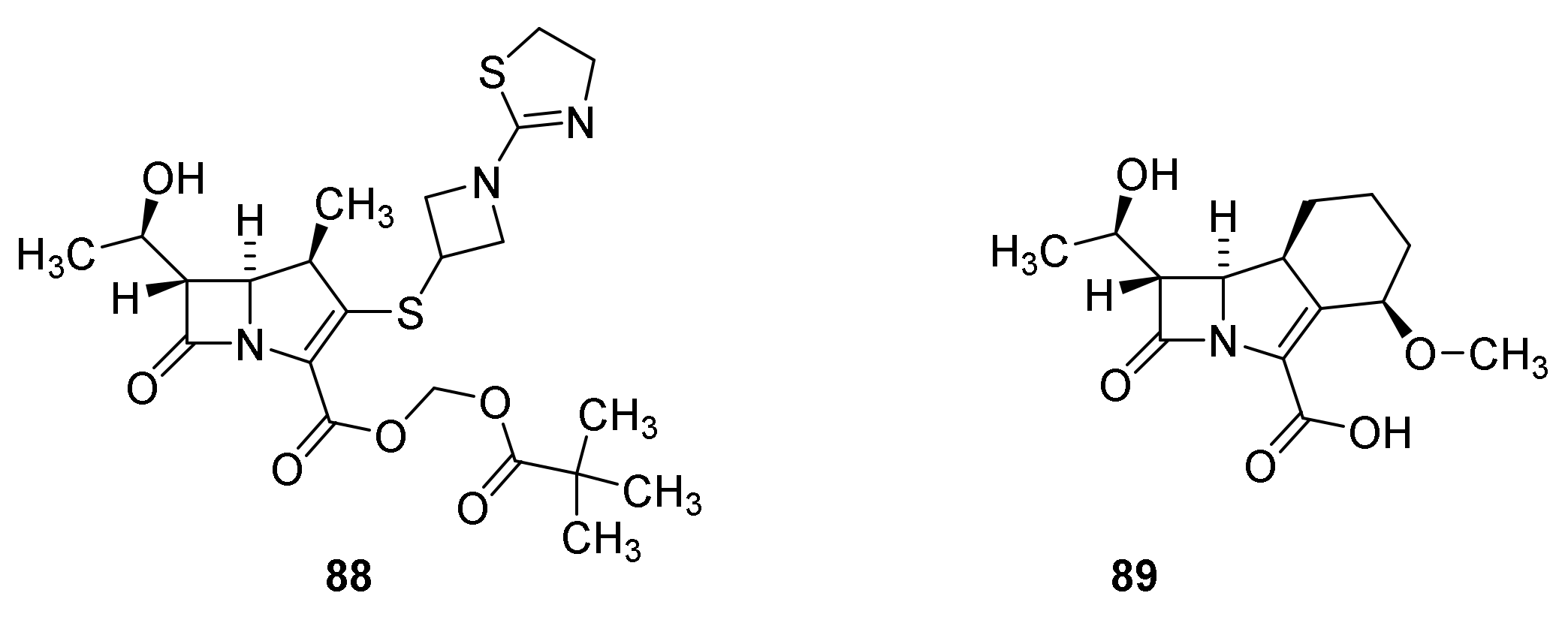
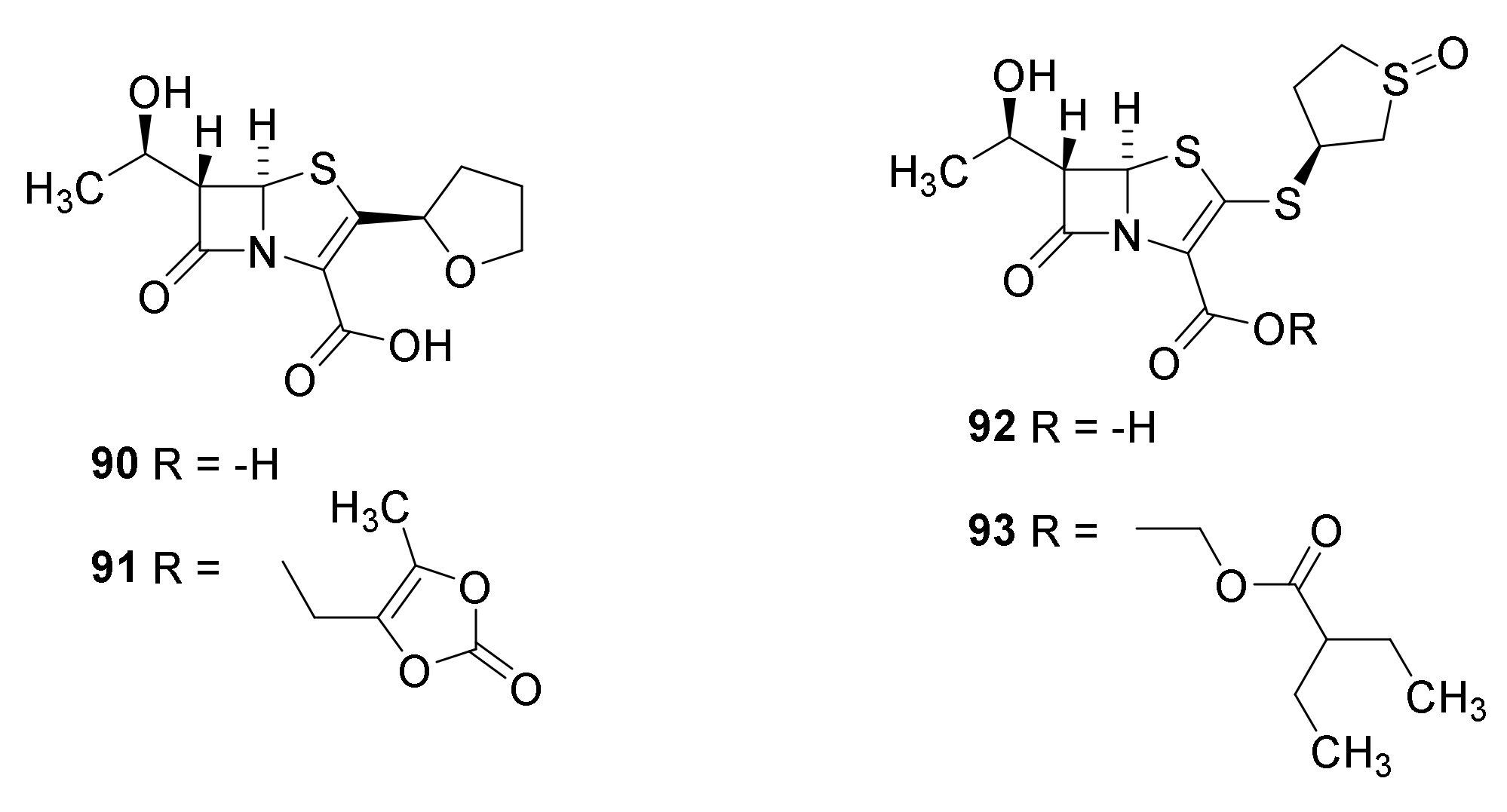
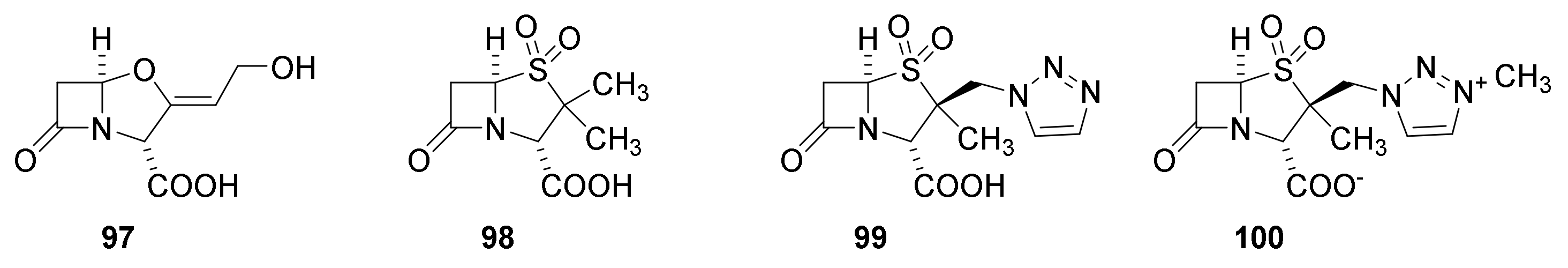
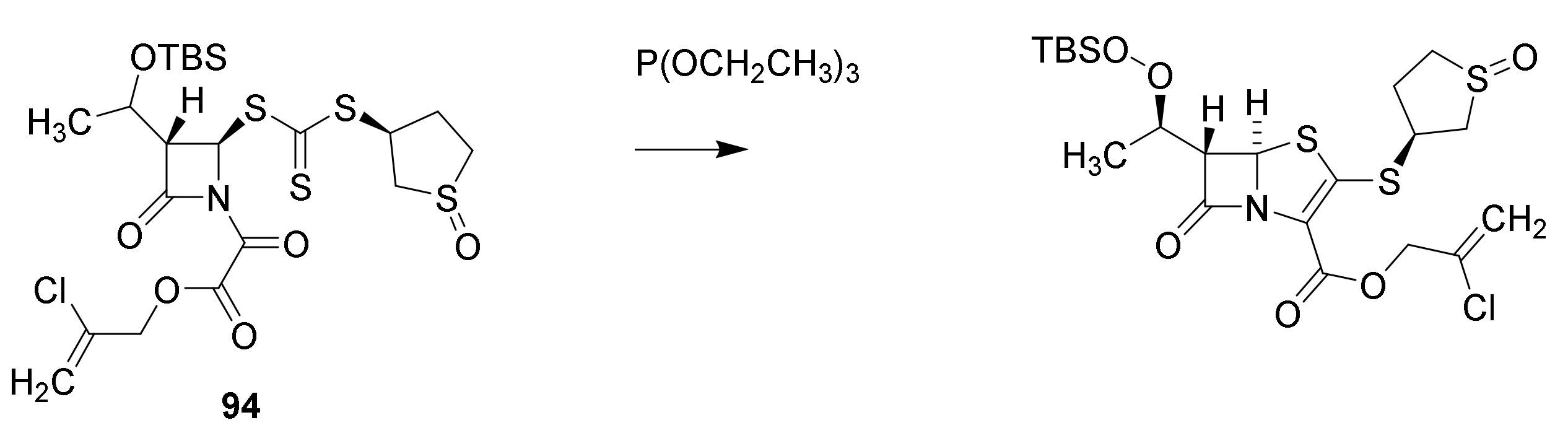
6. Carbapenems
7. Thiopenems
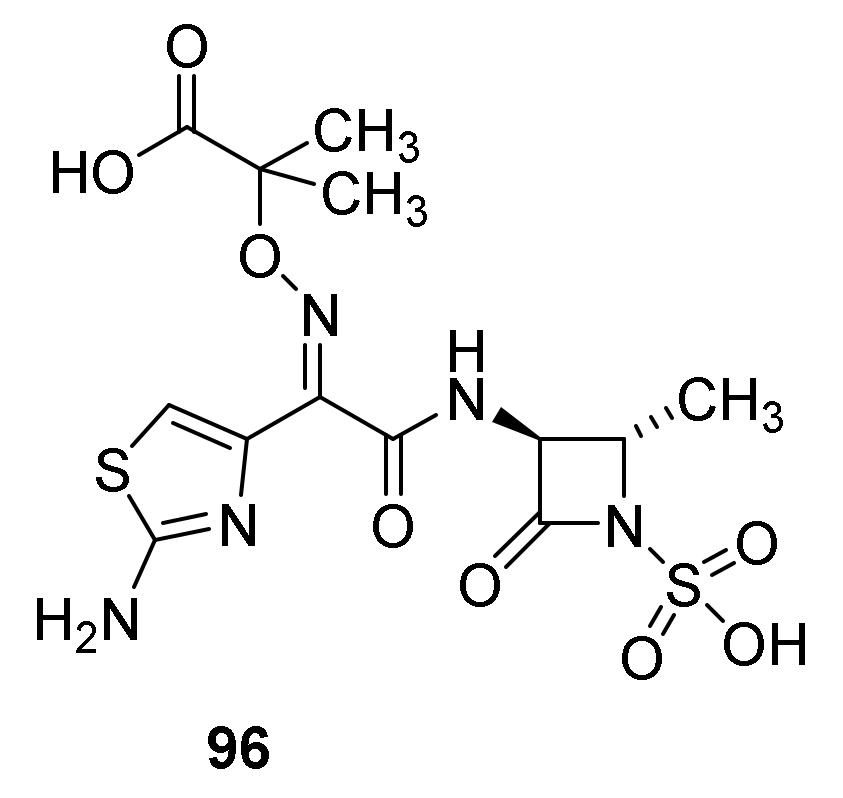
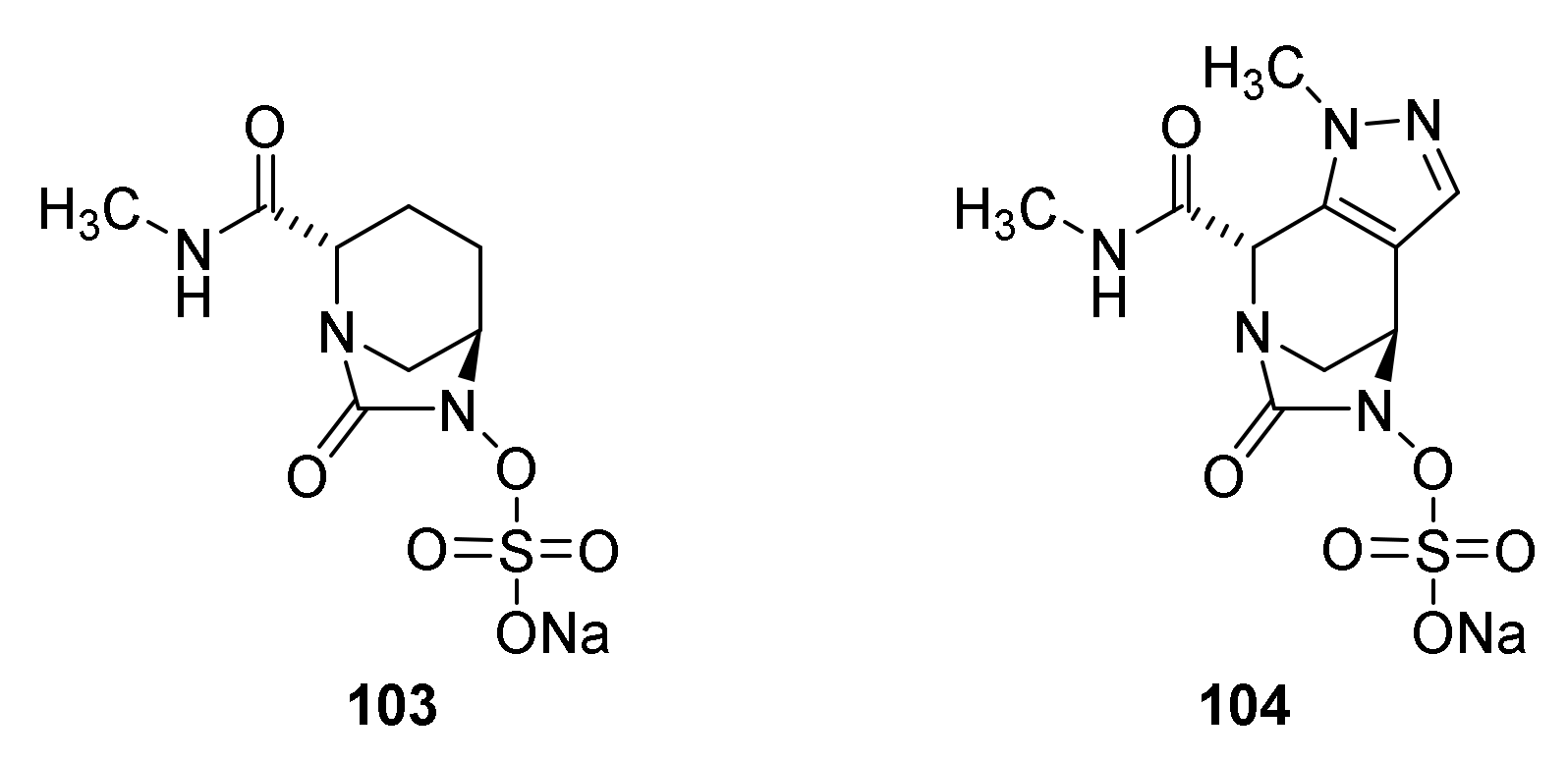
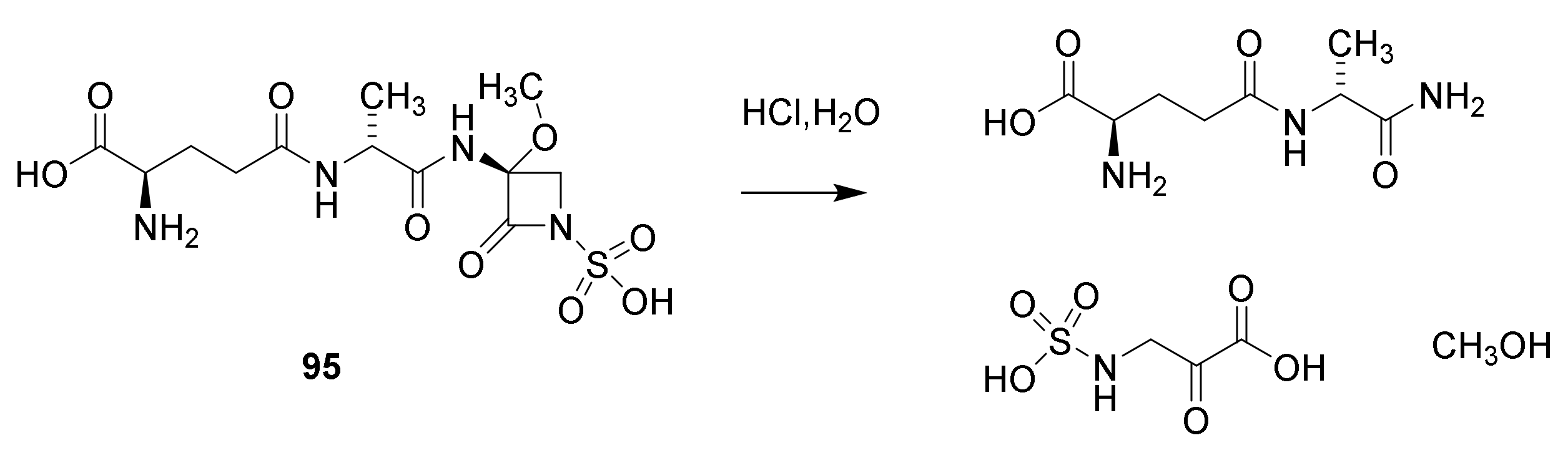
8. Monobactams
9. β-Lactamase Inhibitors
9.1. The β-Lactam Type of β-Lactamase Inhibitors
9.2. The Diazabicyclooctane Type of β-Lactamase Inhbitors
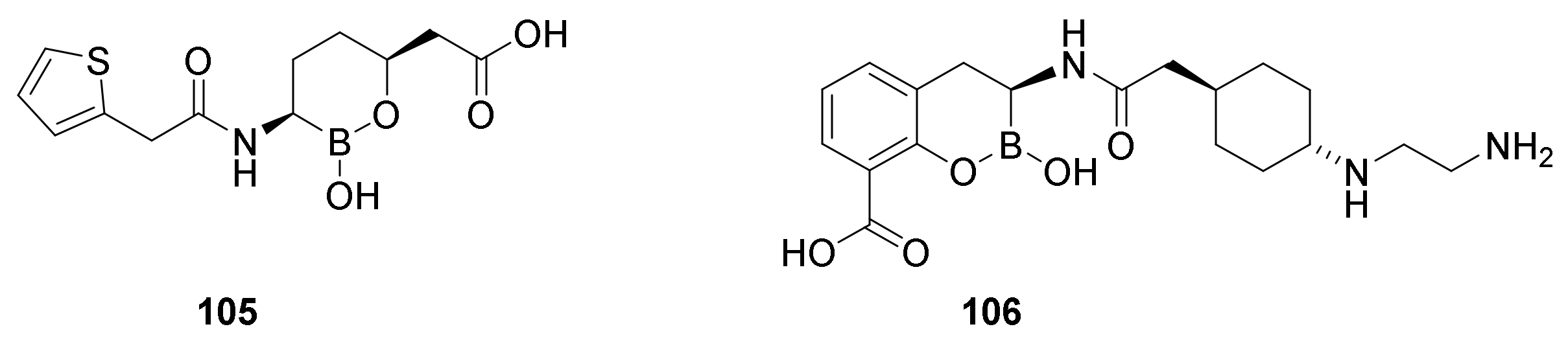
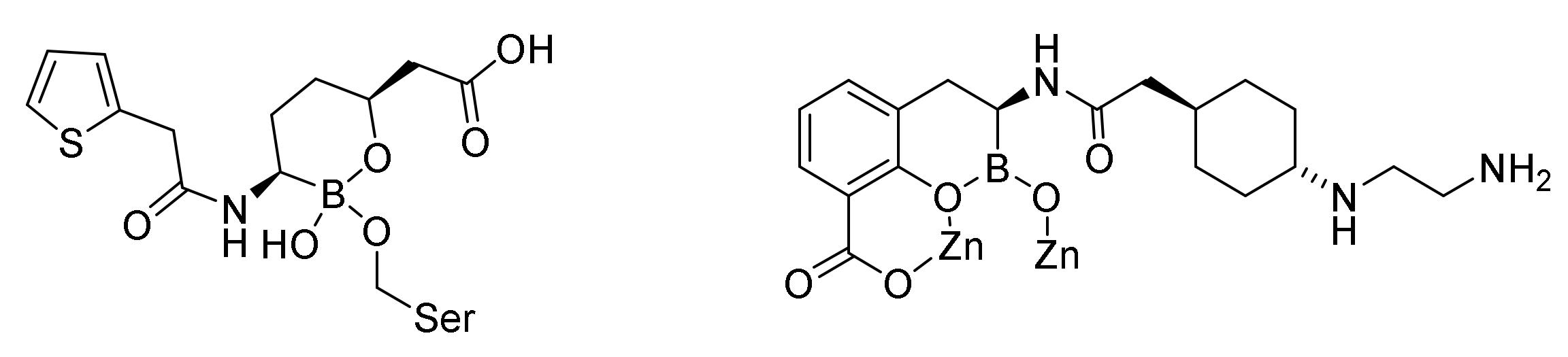
9.3. Boronic Acid β-Lactamase Inhibitors
10. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Koefoed, M. Befolkningsforholdene i Danmark; Statens Stistiske Buerau: Copenhagen, Denmark, 1905. [Google Scholar]
- Christensen, G. Døden Skifter Årsag; Dansk Sygeplejehistoriske Museum: Copenhagen, Denmark, 2017; p. 15. [Google Scholar]
- Singer, C.; Underwood, E.A. A Short History of Medicine, 2nd ed.; Clarendon Press: Oxford, UK, 1962. [Google Scholar]
- Slavkin, H.C. Biofilms, microbial ecology and Antoni van Leeuwenhoek. J. Am. Dent. Assoc. 1997, 128, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Müller, O.F. Animalcula Infusoria Fluviatilia er Marina; Mölleri, T.N., Ed.; Hauniæ: Copenhagen, Denmark, 1786. [Google Scholar]
- Pasteur, L. Étude sur le Vin, 2nd ed.; Librairie F. Savy: Paris, France, 1873. [Google Scholar]
- Warren, J.R. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273. [Google Scholar] [PubMed]
- Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273–1275. [Google Scholar]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1314. [Google Scholar] [CrossRef]
- Ciacci, C.; Mazzacca, G. The history of Helicobacter pylori: A reflection on the relationship between the medical community and industry. Dig. Liver Dis. 2006, 38, 778–780. [Google Scholar] [CrossRef]
- Taylor, L.H.; Latham, S.M.; Woolhouse, M.E. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Sigerist, H.E. The philosphy of hygiene. Bull. Inst. Hist. Med. 1933, 1, 323–331. [Google Scholar]
- Abellán, J. Water supply and sanitation services in modern Europe: Developments in 19th–20th centuries. In Proceedings of the XII International Congress of the Spanish Associations of Ecnonomic History, Salamanaca, Spain, 2–3 February 2017; University of Salamanaca: Salamanaca, Spain, 2017; pp. 1–17. [Google Scholar]
- Khalifa, M.; Bidaisee, S. The Importance of Clean Water. Sch. J. Appl. Sci. Res. 2018, 1, 17–20. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.; The International Natural Product Sciences Taskforce. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discovery 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Porras, G.; Chassagne, F.; Lyles, J.T.; Marquez, L.; Dettweiler, M.; Salam, A.M.; Samarakoon, T.; Shabih, S.; Farrokhi, D.R.; Quave, C.L. Ethnobotany and the Role of Plant Natural Products in Antibiotic Drug Discovery. Chem. Rev. 2021, 121, 3495–3560. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Riethmiller, S. From Atoxyl to Salvarsan: Searching for the magic bullet. Chemotherapy 2005, 51, 234–242. [Google Scholar] [CrossRef]
- Waksman, S.A. What is an antibiotic or an antibiotic substance? Mycologia 1947, 39, 565–569. [Google Scholar] [CrossRef]
- Watkins, W.J.; Plattner, J.J. Anti-infective drugs through medicinal chemistry: A 50-year retrospective. In 2015 Medicinal Chemistry Reviews; Desai, M.C., Ed.; American Chemical Society: Washington, DC, USA, 2015; Volume 50, pp. 242–282. [Google Scholar]
- Bassetti, M.; Poulakou, G.; Ruppe, E.; Bouza, E.; Van Hal, S.J.; Brink, A. Antimicrobial resistance in the next 30 years, humankind, bugs and drugs: A visionary approach. Intensive Care Med. 2017, 43, 1464–1475. [Google Scholar] [CrossRef]
- Marko, V. From Aspitin to Viagara; Springer Nature: Cham, Schwitzerland, 2020. [Google Scholar]
- Jones, H.W. Report of a series of cases of syphilis terated by Ehrlich’s arsenobenzol at the Walter Reed General Hospital Distict of Columbia. Boston Med. Surg. J. 1911, 164, 381–383. [Google Scholar] [CrossRef]
- Lloyd, N.C.; Morgan, H.W.; Nicholson, B.K.; Ronimus, R.S. The composition of Ehrlich’s salvarsan: Resolution of a century-old debate. Angew. Chem. Int. Ed. 2005, 44, 941–944. [Google Scholar] [CrossRef]
- Schwartz, R.S. Perspective: Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef]
- Christensen, S.B. Natrual Products that Changed Society. Biomedicines 2021, 9, 472. [Google Scholar] [CrossRef]
- Lesch, J.E. The First Miracle Drugs. How the Sulfa Drugs Transformed Medicine; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Domagk, G. Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Dtsch. Med. Wochenschr. 1935, 61, 250–253. [Google Scholar] [CrossRef]
- Klee, P.H.; Römer, H. Pontosil bei Streptokokkenkrankungen. Dtsch. Med. Wochenschr. 1935, 61, 253–255. [Google Scholar] [CrossRef]
- Ellis, H. The last year before the dawn of antibiotics. Br. J. Hosp. Med. 2009, 70, 475. [Google Scholar] [CrossRef] [PubMed]
- Trefouel, J.; Nitti, F.; Bovet, D. Activité du p-aminophénylsulfamide sur les infectións streptococciques enxpérimentales de la souris du lapin. C. R. Seances Soc. Biol. Ses. Fil. 1935, 120, 756–758. [Google Scholar]
- Colebrook, L.; Buttle, G.A.H.; O’Meara, R.A.Q. The mode of action of p-aminobenzenesulfonamide and Prontosil in hæmolytic streptococcal infections. Lancet 1936, 228, 1323–1326. [Google Scholar] [CrossRef]
- Fuller, A.T. Is p-aminobenzenesulphonamide the active agent in prontosil therapy? Lancet 1937, 229, 194–198. [Google Scholar] [CrossRef]
- Loudon, I. Maternal Care and Maternal Mortality in Britain, 1935–1950. In Death in Childbirth: An International Study of Maternal Care and Maternal Mortality 1800–1950; Loudon, I., Ed.; Oxford University Press: Oxford, UK, 1992; pp. 255–270. [Google Scholar]
- Fildes, P. A rational approach to research in chemotherapy. Lancet 1940, 238, 955–957. [Google Scholar] [CrossRef]
- Azevedo-Barbosa, H.; Dias, D.F.; Franco, L.L.; Hawkes, J.A.; Carvalho, D.T. From Antibacterial to Antitumour Agents: A Brief Review on The Chemical and Medicinal Aspects of Sulfonamides. Mini-Rev. Med. Chem. 2020, 20, 2052–2066. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products, 3rd ed.; John Wiley and Sons Ltd.: Chicester, UK, 2009. [Google Scholar]
- Lima, L.M.; Silva, B.N.M.d.; Barbosa, G.; Barreiro, E.J. β-Lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef]
- Palavecino, E.L. Clinical, Epidemiologic, and Laboratory Aspects of Methicillin-Resistant Staphylococcus aureus Infections. Cutting-Edge Technologies and Advancements. In Methicillin-Resistant Staphylococcus aureus (MRSA) Protocols, 3rd ed.; Ji, Y., Ed.; Humana Press: New York, NY, USA, 2020; pp. 1–28. [Google Scholar]
- Fleming, A. The antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Zaffiri, L.; Gardner, J.; Toledo-Pereyra, L.H. History of antibiotics. From salvarsan to cephalosporins. J. Investig. Surg. 2012, 25, 67–77. [Google Scholar] [CrossRef]
- Nayler, J.H.C. Early discoveries in the penicillin series. Trends Biochem. Sci. 1991, 16, 195–197. [Google Scholar] [CrossRef]
- Petri, W.A. Penicillins, cephalosporins, and other beta-lactam antibiotics. In Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 12th ed.; Brunton, L.L., Ed.; McGraw Hill Medicinal: New York, NY, USA, 2011; pp. 1477–1504. [Google Scholar]
- Burton, G.; Osborne, N.F.; Pearson, M.; Southgate, R. The b-Lactam Antibiotics. In Burger’s Medicinal Chemistry and Drug Discovery; Wolff, M.E., Ed.; John Wiley & Sons: New York, NY, USA, 1997; Volume 4, pp. 277–363. [Google Scholar]
- Pitts, C.R.; Lectka, T. Chemical Synthesis of β-Lactams: Asymmetric Catalysis and Other Recent Advances. Chem. Rev. 2014, 114, 7930–7953. [Google Scholar] [CrossRef]
- Schrøder, P. Fra Løvens Kemiske Fabrik til Leo Pharma A/S. Theriaca 2005, 36, 27–118. [Google Scholar]
- Chemistry of Penicilin. Nature 1945, 156, 766–767. [CrossRef]
- Council T.C.o.M.R.a.T.M.R. Chemistry of Penicillin. Science 1945, 102, 627–629. [Google Scholar]
- Nicolaou, K.C.; Snyder, S.A. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef]
- Hodgkin, D.C. X-ray analysis of the structure of penicillin. Adv. Sci. 1949, 6, 85–89. [Google Scholar]
- Woodward, R.B. Penems and related substances. Philos. Trans. R. Soc. Lond. Ser. B 1980, 289, 239–250. [Google Scholar]
- Fisher, J.F.; Mobashery, S. b-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev. 2021, 121, 3412–3463. [Google Scholar] [CrossRef]
- Rahman, M.; Pyle, D.J.; Bisz, E.; Dziuk, B.; Ejsmont, K.; Lalancette, R.; Wang, Q.; Chen, H.; Szostak, R.; Szostak, M. Evaluation of Cyclic Amides as Activating Groups in N−C Bond Cross-Coupling: Discovery of N-Acyl-δ-valerolactams as Effective Twisted Amide Precursors for Cross-Coupling Reactions. J. Org. Chem. 2021, 86, 10455–10466. [Google Scholar] [CrossRef]
- Lejon, S.; Ellis, J.; Valegard, K. The Last Step in Cephalosporin C Formation Revealed: Crystal Structures of Deacetylcephalosporin C Acetyltransferase from Acremonium chrysogenum in Complexes with Reaction Intermediates. J. Mol. Biol. 2008, 377, 935–944. [Google Scholar] [CrossRef]
- Rolinson, G.N. Forty years of β-lactam research. J. Antimicrob. Chemother. 1998, 41, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, F.R.; Doyle, F.P.; Nayler, J.H.C.; Rolinson, G.N. Synthesis of penicillin-6-aminopenicillanic acid in penicillin fermentations. Nature 1959, 183, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Nayler, J.H.C. Semi-synthetic approaches to novel penicillins. Trends Biochem. Sci. 1991, 16, 234–237. [Google Scholar] [CrossRef]
- Wang, D.Y.; Abboud, M.I.; Markoulides, M.S.; Brem, J.; Schofield, C.J. The road to avibactam: The first clinically useful non-β-lactam working somewhat like a β-lactam. Future Med. Chem. 2016, 8, 1063–1084. [Google Scholar] [CrossRef]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins; a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1133–1141. [Google Scholar] [CrossRef]
- Oliva, M.; Dideberg, O.; Field, M.J. Understanding the acylation mechanisms of active-site serine penicillin-recognizing proteins: A molecular dynamics simulation study. Proteins Struct. Funct. Genet. 2003, 53, 88–100. [Google Scholar] [CrossRef]
- Boehme, E.H.W.; Applegate, H.E.; Toeplitz, B.; Dolfini, J.E.; Gougoutas, J.Z. 6-Methyl penicillins and 7-methyl cephalosporins. J. Amer. Chem. Soc. 1971, 93, 4324–4326. [Google Scholar] [CrossRef]
- Sakaguchi, K.i.; Murao, S. A new enzyme, penicillin-amidase. Preliminary. Nippon Nogei Kagaku Kaishi 1950, 23, 411. [Google Scholar] [CrossRef]
- Williamson, G.M.; Morrison, J.K.; Stevens, K.J. A new synthetic penicillin PA-248. Lancet 1961, 1, 847–850. [Google Scholar] [CrossRef]
- Nagley, M. Clinical use of a new synthetic penicillin: PA-248. Lancet 1961, 1, 851. [Google Scholar] [CrossRef]
- Peter, G. Streptococcal pharyngitis: Current therapy and criteria for evaluation of new agents. Clin. Infect. Dis. 1992, 14 (Suppl. S2), S218–S223. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for Emerging Pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Rolinson, G.N.; Geddes, A.M. The 50th anniversary of the discovery of 6-aminopenicillanic acid (6-APA). Int. J. Antimicrob. Agents 2007, 29, 3–8. [Google Scholar] [CrossRef]
- Nikaido, H. Antibiotic resistance caused by Gram-negative multidrug efflux pumps. Clin. Infect. Dis. 1998, 27 (Suppl. S1), S32–S41. [Google Scholar] [CrossRef]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Ogawara, H. Self-resistance in Streptomyces, with special reference to β-lactam antibiotics. Molecules 2016, 21, 605. [Google Scholar] [CrossRef]
- Anderson, E.S.; Datta, N. Resistance to Penicillins and its transfer in enterobacteriaceae. Lancet 1965, 1, 407–409. [Google Scholar] [CrossRef]
- Ding, Y.; Li, Z.; Xu, C.; Qin, W.; Wu, Q.; Wang, X.; Cheng, X.; Li, L.; Huang, W. Fluorogenic Probes/Inhibitors of β-Lactamase and their Applications in Drug-Resistant Bacteria. Angew. Chem. Int. Ed. 2021, 60, 24–40. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. Ser. B 1980, 289, 321–331. [Google Scholar]
- Datta, N.; Kontomichalou, P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 1965, 208, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Cullmann, W.; Dick, W. Induction potency of various beta-lactam derivatives in gram-negative rods. Chemotherapy 1989, 35, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.T.; Everett, M. Designing Inhibitors of β-Lactamase Enzymes to Overcome Carbapenem Resistance in Gram-Negative Bacteria. Acc. Chem. Res. 2021, 54, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- De Araujo Viana Marques, D.; Machado, S.E.F.; Ebinuma, V.C.S.; de Albuquerque Lima Duarte, C.; Converti, A.; Porto, A.L.F. Production of β-lactamase inhibitors by Streptomyces species. Antibiotics 2018, 7, 61. [Google Scholar] [CrossRef]
- Doyle, F.P.; Hardy, K.; Nayler, J.H.C.; Soulal, M.J.; Stove, E.R.; Waddington, H.R.J. Derivatives of 6-aminopenicillanic acid. III. 2,6-Dialkoxybenzoyl derivatives. J. Chem. Soc. 1962, 1453–1458. [Google Scholar] [CrossRef]
- Moriyama, Y.; Ishikane, M.; Mezaki, K.; Ohmagari, N. Comparison of penicillins (penicillin G and ampicillin) and cefazolin as a definitive therapy against penicillin-susceptible Staphylococcus aureus (PSSA) bacteremia in Japan: A retrospective cohort study. J. Infect. Chemother. 2020, 26, 358–362. [Google Scholar] [CrossRef]
- Sanders, C.C.; Sanders, W.E., Jr. Beta-Lactam resistance in gram-negative bacteria: Global trends and clinical impact. Clin. Infect. Dis. 1992, 15, 824–839. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Urban, F.J.; Cooper, R.D.; Jose, F.L. Direct 6-methoxylation of penicillin derivatives. A convenient pathway to substituted -lactam antibiotics. J. Am. Chem. Soc. 1973, 95, 2401–2403. [Google Scholar] [CrossRef]
- Basker, M.J.; Edmondson, R.A.; Knott, S.J.; Ponsford, R.J.; Slocombe, B.; White, S.J. In vitro antibacterial properties of BRL 36650, a novel 6α-substituted penicillin. Antimicrob. Agents Chemother. 1984, 26, 734–740. [Google Scholar] [CrossRef]
- Brown, D.G.; Wobst, H.J. A Decade of FDA-Approved Drugs (2010–2019): Trends and Future Directions. J. Med. Chem. 2021, 64, 2312–2338. [Google Scholar] [CrossRef]
- Murakami, M.; Takahashi, K.; Isaka, I.; Kawahara, N.; Horiguchi, H.; Murakami, Y.; Sakamoto, N.; Ozasa, T.; Kashiwagi, T.; Nakano, K.; et al. New Oxymethylester Derivatives of Penicillin and Cephalosporin. German Patent DE2228670A1, 11 January 1973. [Google Scholar]
- Roholt, K.; Nielsen, B.; Kristensen, E. Clinical pharmacology of pivampicillin. Antimicrob. Agents Chemother. 1974, 6, 563–571. [Google Scholar] [CrossRef][Green Version]
- Rose, S.J.; Stokes, T.C.; Patel, S.; Cooper, M.B.; Betteridge, D.J.; Payne, J.E. Carnitine deficiency associated with long-term pivampicillin treatment: The effect of a replacement therapy regime. Postgrad. Med. J. 1992, 68, 932–934. [Google Scholar] [CrossRef][Green Version]
- Clayton, J.P.; Cole, M.; Elson, S.W.; Ferres, H. BRL.8988 (Talampicillin), a well-absorbed oral form of ampicillin. Antimicrob. Agents Chemother. 1974, 5, 670–671. [Google Scholar] [CrossRef]
- Tan, J.S.; Salstrom, S.J. Bacampicillin, ampicillin, cephalothin and cephapirin levels in human blood and interstitial fluid. Antimicrob. Agents Chemother. 1979, 15, 510–512. [Google Scholar] [CrossRef]
- Omata, H.; Komiya, M.; Akimoto, Y.; Kaneko, K.; Fujii, A. Ampicillin concentrations in human serum and oral tissues following a single oral administration of lenampicillin. Chemotherapy 1994, 42, 172–176. [Google Scholar]
- Abraham, E.P.; Loder, P.B. Cephalosporin C. In Cephalosporins and Penicillins; Flynn, E.H., Ed.; Princeton University Press: London, UK, 1972; pp. 1–26. [Google Scholar]
- Newton, G.G.F.; Abraham, E.P. Cephalosporin C, a new antibiotic containing sulfur and D-α-aminoadipic acid. Nature 1955, 175, 548. [Google Scholar] [CrossRef]
- Abraham, E.P.; Newton, G.G.F. Structure of cephalosporin C. Biochem. J. 1961, 79, 377–393. [Google Scholar] [CrossRef]
- Hodgkin, D.C.; Maslen, E.N. The X-ray analysis of the structure of cephalosporin C. Biochem. J. 1961, 79, 393–402. [Google Scholar] [CrossRef]
- Morin, R.B.; Jackson, B.G.; Mueller, R.A.; Lavagnino, E.R.; Scanlon, W.B.; Andrews, S.L. Chemistry of cephalosporin antibiotics. III. Chemical correlation of penicillin and cephalosporin antibiotics. J. Am. Chem. Soc. 1963, 85, 1896–1897. [Google Scholar] [CrossRef]
- Lucinova, E.; Welward, L.; Frimm, R.; Kosalko, R.; Sokol, M. 7-Aminocephalosporanic Acid. Czech Patent CS193181B1, 31 October 1979. [Google Scholar]
- Hardianto, D.; Royani, J.I.; Safarrida, A. Cephalosporin C acylase from microbes for one-step enzymatic transformation of cephalosporin C to 7-aminocephalosporanic acid. J. Pure Appl. Microbiol. 2016, 10, 2495–2499. [Google Scholar] [CrossRef]
- Masoud, M.S.; Ali, A.E.; Nasr, N.M. Chemistry, classification, pharmacokinetics, clinical uses and analysis of beta lactam antibiotics: A review. J. Chem. Pharm. Res. 2014, 6, 28–58. [Google Scholar]
- Mehta, D.; Sharma, A.K. Cephalosporins: A Review on Imperative Class of Antibiotics. Inventi Rapid Mol. Pharmacol. 2016, 2016, 17877. [Google Scholar]
- Ge, Y.; Maynard, D.; Rickert, D.E. Comparative pharmacokinetics of ceftaroline in rats, rabbits, and monkeys following a single intravenous or intramuscular injection. Antimicrob. Agents Chemother. 2010, 54, 912–914. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef]
- Kropp, H.; Sundelof, J.G.; Hajdu, R.; Kahan, F.M. Metabolism of thienamycin and related carbapenem antibiotics by the renal dipeptidase, dehydropeptidase-I. Antimicrob. Agents Chemother. 1982, 22, 62–70. [Google Scholar] [CrossRef]
- Breilh, D.; Texier-Maugein, J.; Allaouchiche, B.; Saux, M.-C.; Boselli, E. Carbapenems. J. Chemother. 2013, 25, 1–17. [Google Scholar] [CrossRef]
- Fukasawa, M.; Sumita, Y.; Harabe, E.T.; Tanio, T.; Nouda, H.; Kohzuki, T.; Okuda, T.; Matsumura, H.; Sunagawa, M. Stability of meropenem and effect of 1β-methyl substitution on its stability in the presence of renal dehydropeptidase I. Antimicrob. Agents Chemother. 1992, 36, 1577–1579. [Google Scholar] [CrossRef] [PubMed]
- Bouffard, F.A.; Johnston, D.B.R.; Christensen, B.G. Thienamycin total synthesis. 1. Synthesis of azetidinone precursors of (±)-thienamycin and its stereoisomers. J. Org. Chem. 1980, 45, 1130–1135. [Google Scholar] [CrossRef]
- Schmitt, S.M.; Johnston, D.B.R.; Christensen, B.G. Thienamycin total synthesis. 2. Model studies-synthesis of a simple 2-(alkylthio)carbapen-2-em. J. Org. Chem. 1980, 45, 1135–1142. [Google Scholar] [CrossRef]
- Schmitt, S.M.; Johnston, D.B.R.; Christensen, B.G. Thienamycin total synthesis. 3. Total synthesis of (±)-thienamycin and (±)-8-epithienamycin. J. Org. Chem. 1980, 45, 1142–1148. [Google Scholar] [CrossRef]
- Kametani, T.; Huang, S.-P.; Yokohama, S.; Suzuki, Y.; Ihara, M. Studies on the syntheses of heterocyclic compounds. 800. A formal total synthesis of (±)-thienamycin and a (±)-decysteaminylthienamycin derivative. J. Am. Chem. Soc. 1980, 102, 2060–2065. [Google Scholar] [CrossRef]
- Berks, A.H. Preparations of two pivotal intermediates for the synthesis of 1-β-methyl carbapenem antibiotics. Tetrahedron 1996, 52, 331–375. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Brahim, I.; Hisham, N.; Aladdin, R.; Mohammed, H.; Bahaaeldin, A. Recent updates of carbapenem antibiotics. Eur. J. Med. Chem. 2017, 131, 185–195. [Google Scholar] [CrossRef]
- Cotroneo, N.; Rubio, A.; Critchley, I.A.; Pillar, C.; Pucci, M.J. In vitro and in vivo characterization of tebipenem, an oral carbapenem. Antimicrob. Agents Chemother. 2020, 64, e02240-19. [Google Scholar] [CrossRef]
- Han, H.; Liu, J.; Hu, L.; Wu, Y. Synthesis of Faropenem. Zhongguo Yiyao Gongye Zazhi 2001, 32, 339–341. [Google Scholar]
- Volkmann, R.A.; Kelbaugh, P.R.; Nason, D.M.; Jasys, V.J. 2-Thioalkyl penems: An efficient synthesis of sulopenem, a (5R,6S)-6-(1(R)-hydroxyethyl)-2-[(cis-1-oxo-3-thiolanyl)thio]-2-penem antibacterial. J. Org. Chem. 1992, 57, 4352–4361. [Google Scholar] [CrossRef]
- Woodcock, J.M.; Andrews, J.M.; Brenwald, N.P.; Ashby, J.P.; Wise, R. The in-vitro activity of faropenem, a novel oral penem. J. Antimicrob. Chemother. 1997, 39, 35–43. [Google Scholar] [CrossRef][Green Version]
- Schurek, K.N.; Wiebe, R.; Karlowsky, J.A.; Rubinstein, E.; Hoban, D.J.; Zhanel, G.G. Faropenem: Review of a new oral penem. Expert Rev. Anti-Infect. Ther. 2007, 5, 185–198. [Google Scholar] [CrossRef]
- Imada, A.; Kitano, K.; Kintaka, K.; Muroi, M.; Asai, M. Sulfazecin and isosulfazecin, novel β-lactam antibiotics of bacterial origin. Nature 1981, 289, 590–591. [Google Scholar] [CrossRef]
- Sykes, R.B.; Bonner, D.P.; Bush, K.; Georgopapadakou, N.H.; Wells, J.S. Monobactams-monocyclic β-lactam antibiotics produced by bacteria. J. Antimicrob. Chemother. 1981, 8, 1–16. [Google Scholar] [CrossRef]
- Ramsey, C.; MacGowan, A.P. A review of the pharmacokinetics and pharmacodynamics of aztreonam. J. Antimicrob. Chemother. 2016, 71, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Jalde, S.S.; Choi, H.K. Recent advances in the development of β-lactamase inhibitors. J. Microbiol. 2020, 58, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Yahav, D.; Giske, C.G.; Graematniece, A.; Abodakpi, H.; Tam, V.H.; Leibovici, L. New β-Lactam-β-Lactamase Inhibitor Combinations. Clin. Microbiol. Rev. 2021, 34, e00115. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.G.; Butterworth, D.; Cole, M.; Hanscomb, G.; Hood, J.D.; Reading, C.; Rolinson, G.N. Naturally occurring β-lactamase inhibitors with antibacterial activity. J. Antibiot. 1976, 29, 668–669. [Google Scholar] [CrossRef]
- Howarth, T.T.; Brown, A.G.; King, T.J. Clavulanic acid, a novel β-lactam isolated from Streptomyces clavuligerus; X-ray crystal structure analysis. J. Chem. Soc. Chem. Commun. 1976, 266–267. [Google Scholar] [CrossRef]
- Buynak, J.D. Understanding the longevity of the β-lactam antibiotics and of antibiotic/β-lactamase inhibitor combinations. Biochem. Pharmacol. 2006, 71, 930–940. [Google Scholar] [CrossRef]
- Harada, S.; Tsubotani, S.; Hida, T.; Ono, H.; Okazaki, H. Structure of lactivicin, an antibiotic having a new nucleus and similar biological activities to β-lactam antibiotics. Tetrahedron Lett. 1986, 27, 6229–6232. [Google Scholar] [CrossRef]
- Nozaki, Y.; Katayama, N.; Ono, H.; Tsubotani, S.; Harada, S.; Okazaki, H.; Nakao, Y. Binding of a non-β-lactam antibiotic to penicillin-binding proteins. Nature 1987, 325, 179–180. [Google Scholar] [CrossRef]
- Holzgrabe, U. Lactivicin—An antibiotic against penicillin-resistant pneumococci. Pharm. Unserer Zeit 2007, 36, 421–423. [Google Scholar] [CrossRef]
- Vazquez, J.A.; Gonzalez Patzan, L.D.; Stricklin, D.; Duttaroy, D.D.; Kreidly, Z.; Lipka, J.; Sable, C. Efficacy and safety of ceftazidime-avibactam versus imipenem-cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: Results of a prospective, investigator-blinded, randomized study. Curr. Med. Res. Opin. 2012, 28, 1921–1931. [Google Scholar] [CrossRef]
- Vazquez-Ucha, J.C.; Rodriguez, D.; Lasarte-Monterrubio, C.; Lence, E.; Arca-Suarez, J.; Maneiro, M.; Gato, E.; Perez, A.; Martinez-Guitian, M.; Juan, C.; et al. 6-Halopyridylmethylidene Penicillin-Based Sulfones Efficiently Inactivate the Natural Resistance of Pseudomonas aeruginosa to β-Lactam Antibiotics. J. Med. Chem. 2021, 64, 6310–6328. [Google Scholar] [CrossRef]
- Dhillon, S. Meropenem/Vaborbactam: A Review in Complicated Urinary Tract Infections. Drugs 2018, 78, 1259–1270. [Google Scholar] [CrossRef]
- Morandi, S.; Morandi, F.; Caselli, E.; Shoichet, B.K.; Prati, F. Structure-based optimization of cephalothin-analogue boronic acids as β-lactamase inhibitors. Bioorg. Med. Chem. 2008, 16, 1195–1205. [Google Scholar] [CrossRef]
- Barriere, S.L. Clinical, economic and societal impact of antibiotic resistance. Expert Opin. Pharmacother. 2015, 16, 151–153. [Google Scholar] [CrossRef]
- Roberts, R.R.; Hota, B.; Ahmad, I.; Scott, R.D., 2nd; Foster, S.D.; Abbasi, F.; Schabowski, S.; Kampe, L.M.; Ciavarella, G.G.; Supino, M.; et al. Hospital and societal costs of antimicrobial-resistant infections in a Chicago teaching hospital: Implications for antibiotic stewardship. Clin. Infect. Dis. 2009, 49, 1175–1184. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K., Jr.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. 10 × ′20 Progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E. Antibiotics in late clinical development. Biochem. Pharmacol. 2017, 133, 152–163. [Google Scholar] [CrossRef]
- Remy, V.; Largeron, N.; Quilici, S.; Carroll, S. The economic value of vaccination: Why prevention is wealth. J. Mark. Access Health Policy 2015, 3, 1. [Google Scholar] [CrossRef]








































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christensen, S.B. Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams. Molecules 2021, 26, 6057. https://doi.org/10.3390/molecules26196057
Christensen SB. Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams. Molecules. 2021; 26(19):6057. https://doi.org/10.3390/molecules26196057
Chicago/Turabian StyleChristensen, Søren Brøgger. 2021. "Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams" Molecules 26, no. 19: 6057. https://doi.org/10.3390/molecules26196057
APA StyleChristensen, S. B. (2021). Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams. Molecules, 26(19), 6057. https://doi.org/10.3390/molecules26196057