
A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
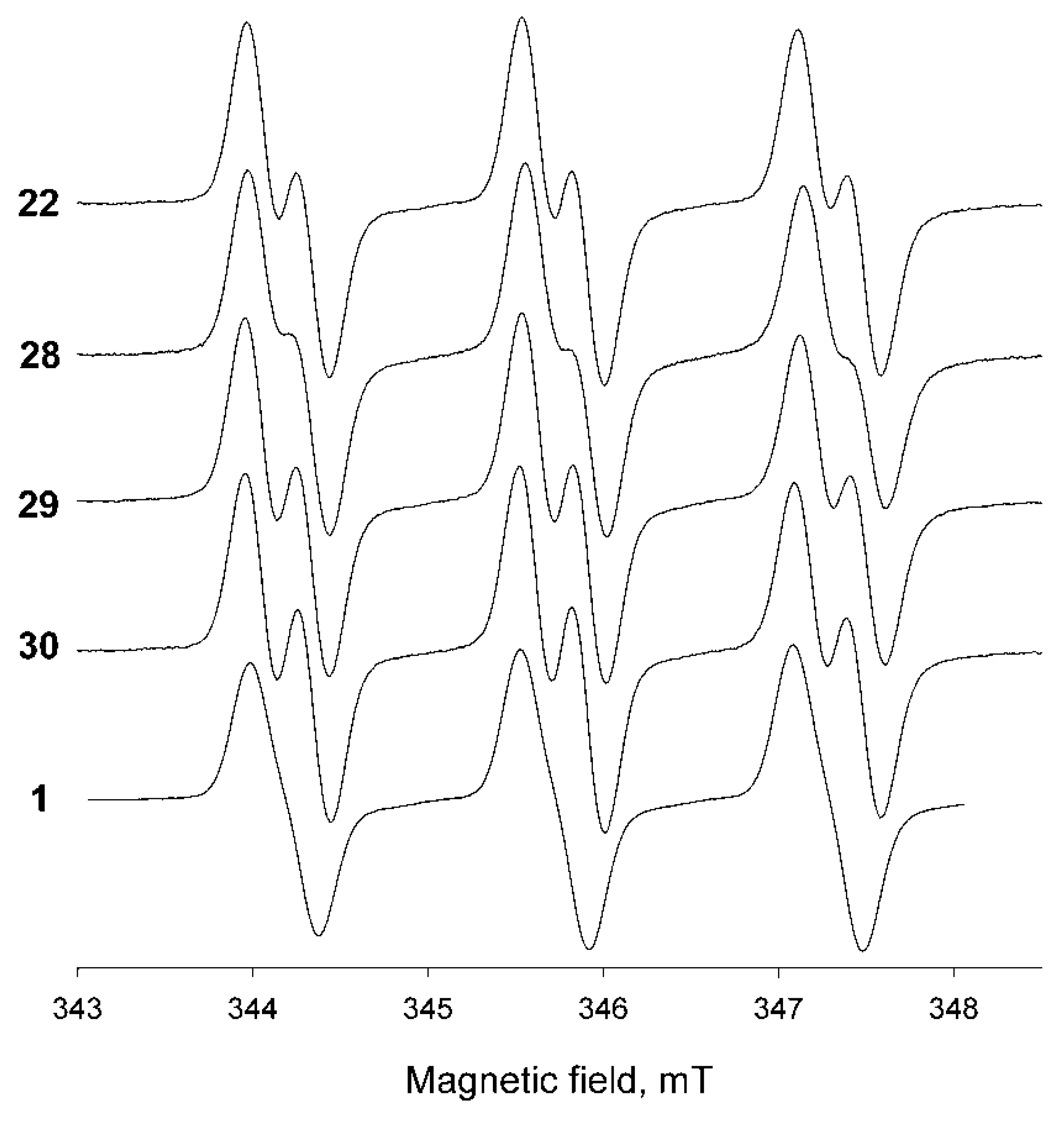
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of samples for 1H NMR from nitroxides
3.2. Synthesis
3.3. EPR Measurements and Kinetics
3.4. Animals
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hubbell, W.L.; López, C.J.; Altenbach, C.; Yang, Z. Technological advances in site-directed spin labelling. Curr. Opin. Struct. Biol. 2013, 23, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser, P.; Schmidt, M.J.; Drescher, M.; Summerer, D. Site-directed spin labeling of proteins for distance measurements in vitro and in cells. Org. Biomol. Chem. 2016, 14, 5468–5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endeward, B.; Marko, A.; Denysenkov, V.P.; Sigurdsson, S.T.; Prisner, T.F. Advanced EPR methods for studying conformational dynamics in nucleic acids. Methods Enzymol. 2015, 564, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-cell EPR: Progress towards structural studies inside cells. ChemBioChem 2019, 21, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Widder, P.; Schuck, J.; Summerer, D.; Drescher, M. Combining site-directed spin labeling in vivo and in-cell EPR distance determination. Phys. Chem. Chem. Phys. 2020, 22, 4875–4879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jugniot, N.; Duttagupta, I.; Rivot, A.; Massot, P.; Cardiet, C.; Pizzoccaro, A.; Jean, M.; Vanthuyne, N.; Franconi, J.-M.; Voisin, P.; et al. An elastase activity reporter for Electronic Paramagnetic Resonance (EPR) and Overhauser-enhanced Magnetic Resonance Imaging (OMRI) as a lineshifting nitroxide. Free. Radic. Biol. Med. 2018, 126, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Kocherginsky, N.; Swartz, H. Nitroxide Spin Labels—Reactions in Biology and Chemistry; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Marx, L.; Chiarelli, R.; Guiberteau, T.; Rassat, A. A comparative study of the reduction by ascorbate of 1,1,3,3-tetraethylisoindolin-2-yloxyl and of 1,1,3,3-tetramethylisoindolin-2-yloxyl. J. Chem. Soc. Perkin Trans. 1 2000, 2000, 1181–1182. [Google Scholar] [CrossRef]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radic. Res. 2014, 49, 78–85. [Google Scholar] [CrossRef]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of unnatural amino acids functionalized with sterically shielded pyrroline nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A bioresistant nitroxide spin label for in-cell EPR spectroscopy: In vitro and in oocytes protein structural dynamics studies. Angew. Chem. 2018, 130, 1380–1384. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. gem-Diethyl pyrroline nitroxide spin labels: Synthesis, EPR characterization, rotamer libraries and biocompatibility. ChemistryOpen 2019, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.A.; Polienko, Y.F.; Krumkacheva, O.A.; Strizhakov, R.K.; Gatilov, Y.V.; Grigor’ev, I.A.; Bagryanskaya, E.G. Synthesis of 2,5-bis(spirocyclohexane)-substituted Nitroxides of Pyrroline and Pyrrolidine series, including Thiol-specific spin label: An analogue of MTSSL with long relaxation time. J. Org. Chem. 2012, 77, 8016–8027. [Google Scholar] [CrossRef] [PubMed]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically shielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-bis(hydroxymethyl)-2,2,5,5-tetraethylpyrrolidin-1-oxyl via 1,3-dipolar cycloaddition of azomethine ylide to activated alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.A.; Khoroshunova, Y.V.; Kirilyuk, I.A. Method of Producing 2,2,5,5-Tetramethyl-3-Carboxypyrrolidine-1-Oxyl. RU Patent No. 2702331, 25 June 2019. [Google Scholar]
- Lampp, L.; Morgenstern, U.; Merzweiler, K.; Imming, P.; Seidel, R.W. Synthesis and characterization of sterically and electrostatically shielded pyrrolidine nitroxide radicals. J. Mol. Struct. 2019, 1182, 87–94. [Google Scholar] [CrossRef]
- Baracz, N.M.; Hankovszky, O.H.; Sar, S.P.; Jerkovich, G.; Hideg, K. Synthesis of alkynyl-substituted pyrrolidin-1-yloxyl radicals from 1-pyrroline n-oxide nnitrones and alkynylmagnesium bromides. Synthesis 1996, 1996, 204–208. [Google Scholar] [CrossRef]
- Sár, C.P.; Jekö, J.; Fajer, P.; Hideg, K. Synthesis and reactions of new alkynyl substituted nitroxide radicals. Synthesis 1999, 1999, 1039–1045. [Google Scholar] [CrossRef]
- Sar, S.P.; Osz, E.; Jeko, J.; Hideg, K. Synthesis of spiro[pyrolidine-2,2-adamantane] nitrones and nitroxides. Synthesis 2005, 2005, 255–259. [Google Scholar] [CrossRef]
- Hideg, K.; Balog, M.; Abé, C.; Kálai, T.; Steinhoff, H.-J.; Jekő, J. Synthesis of new paramagnetic fatty acids and lipophilic spin labels. Synthesis 2007, 2007, 1663–1670. [Google Scholar] [CrossRef]
- Rey, P.; Ramasseul, R.; Rassat, A. A useful protecting group in the preparation of amino-nitroxides. J. Chem. Soc. Chem. Commun. 1976, 3, 83–84. [Google Scholar] [CrossRef]
- Sosnovsky, G.; Cai, Z. A study of the Favorskii rearrangement with 3-bromo-4-oxo-2,2,6,6-tetramethylpiperidine-1-oxyl. J. Org. Chem. 1995, 60, 3414–3418. [Google Scholar] [CrossRef]
- Polienko, Y.F.; Kuprikova, N.M.; Parkhomenko, D.A.; Gatilov, Y.V.; Chernyak, E.I.; Kirilyuk, I.A. Synthesis of 2,5-bis(spirocyclohexane)-substituted nitroxides: New spin labeling agents. Tetrahedron 2020, 81, 131915. [Google Scholar] [CrossRef]
- Yan’shole, V.V.; Kirilyuk, I.A.; Grigor’ev, I.A.; Morozov, S.V.; Tsentalovich, Y.P. Antioxidative properties of nitroxyl radicals and hydroxyamines in reactions with triplet and deaminated kynurenine. Russ. Chem. Bull. 2010, 59, 66–74. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Morris, J.C.; Fairfull-Smith, K.E.; Graingerb, R.S.; Bottle, S.E. A novel protecting group methodology for syntheses using nitroxides. Chem. Commun. 2013, 49, 10382–10384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murayama, K.; Yoshioka, T. Studies on stable free radicals. III. Reactions of stable nitroxide radicals with s-radicals derived from benzenethiols and thiamine. Bull. Chem. Soc. Jpn. 1969, 42, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Bálint, J.; Kiss, V.; Egri, G.; Kálai, T.; Demeter, Á.; Balog, M.; Fogassya, E.; Hideg, K. Kinetic resolution of 1-oxyl-3-hydroxymethyl-2,2,5,5-tetramethylpyrrolidine derivatives by lipase-catalyzed enantiomer selective acylation. Tetrahedron Asymmetry 2004, 15, 671–679. [Google Scholar] [CrossRef]
- Úr, G.; Kálai, T.; Balog, M.; Bognár, B.; Gulyás-Fekete, G.; Hideg, K. Synthesis of new pyrroline nitroxides with ethynyl functional group. Synth. Commun. 2015, 45, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Barratt, M.D.; Davies, A.P.; Evans, M.T.A. Maleimide and Isomaleimide Pyrrolidine-Nitroxide Spin Labels. Eur. J. Biochem. 1971, 24, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Bacic, G.; Pavićević, A.; Peyrot, F. In vivo evaluation of different alterations of redox status by studying pharmacokinetics of nitroxides using magnetic resonance techniques. Redox Biol. 2015, 8, 226–242. [Google Scholar] [CrossRef] [Green Version]
- Kosem, N.; Naganuma, T.; Ichikawa, K.; Morales, N.P.; Yasukawa, K.; Hyodo, F.; Yamada, K.; Utsumi, H. Whole-body kinetic image of a redox probe in mice using overhauser-enhanced MRI. Free Radic. Biol. Med. 2012, 53, 328–336. [Google Scholar] [CrossRef]
- Davis, R.M.; Mitchell, J.B.; Krishna, M.C. Nitroxides as cancer imaging agents. Anti-Cancer Agents Med. Chem. 2011, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.-I.; Hyodo, F.; Matsumoto, A.; Koretsky, A.P.; Sowers, A.L.; Mitchell, J.B.; Krishna, M.C. High-resolution mapping of tumor redox status by magnetic resonance imaging using nitroxides as redox-sensitive contrast agents. Clin. Cancer Res. 2006, 12, 2455–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, G.M.; Burks, S.R.; Kohr, M.J.; Kao, J.P.Y. Synthesis and biological testing of aminoxyls designed for long-term retention by living cells. Org. Biomol. Chem. 2005, 3, 645–648. [Google Scholar] [CrossRef]
- Legenzov, E.A.; Muralidharan, S.; Woodcock, L.B.; Eaton, G.R.; Eaton, S.S.; Rosen, G.M.; Kao, J.P.Y. Designing molecular probes to prolong intracellular retention: Application to nitroxide spin probes. Bioconjugate Chem. 2016, 27, 2923–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Smith, R.A.J. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted Triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.; Shibata, S.; Ishii, I.; Zlateva, G.; Zhelev, Z.; Aoki, I.; Bakalova, R. Imaging of redox-imbalance and oxidative stress in kidney in vivo, induced by dietary cholesterol. Biotechnol. Biotechnol. Equip. 2019, 33, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalova, S.I.; Kirilyuk, I.A.; Dikalova, A.E. Antihypertensive effect of mitochondria-targeted proxyl nitroxides. Redox Biol. 2014, 4, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.D.B.; Funk, R.S.; Rajewski, R.A.; Krise, J.P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 2009, 1, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.M.; Sowers, A.L.; DeGraff, W.; Bernardo, M.; Thetford, A.; Krishna, M.C.; Mitchell, J.B. A novel nitroxide is an effective brain redox imaging contrast agent and in vivo radioprotector. Free Radic. Biol. Med. 2011, 51, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobko, A.A.; Kirilyuk, I.A.; Gritsan, N.P.; Polovyanenko, D.N.; Grigor’ev, I.A.; Khramtsov, V.V.; Bagryanskaya, E.G. EPR and quantum chemical studies of the pH-sensitive imidazoline and imidazolidine nitroxides with bulky substituents. Appl. Magn. Reson. 2010, 39, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and properties. Molecules 2020, 25, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwama, M.; Amano, A.; Shimokado, K.; Maruyama, N.; Ishigami, A. Ascorbic acid levels in various tissues, plasma and urine of mice during aging. J. Nutr. Sci. Vitaminol. 2012, 58, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.E.; Meister, A. Dynamic state of glutathione in blood plasma. J. Biol. Chem. 1980, 255, 9530–9533. [Google Scholar] [CrossRef]
- Wiesner, R.J.; Ruegg, J.C.; Morano, I. Counting target molecules by exponential polymerase chain reaction: Copy number of mitochondrial DNA in rat tissues. Biochem. Biophys. Res. Commun. 1992, 183, 553–559. [Google Scholar] [CrossRef]
- Soltoff, S.P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Babic, N.; Orio, M.; Peyrot, F. Unexpected rapid aerobic transformation of 2,2,6,6-tetraethyl-4-oxo(piperidin-1-yloxyl) radical by cytochrome P450 in the presence of NADPH: Evidence against a simple reduction of the nitroxide moiety to the hydroxylamine. Free Radic. Biol. Med. 2020, 156, 144–156. [Google Scholar] [CrossRef]
- Hedlund, E.; Gustafsson, J.A.; Warner, M. Cytochrome P450 in the brain: A review. Curr. Drug Metab. 2001, 2, 245–263. [Google Scholar] [CrossRef]
- Knights, K.M.; Rowland, A.; Miners, J.O. Renal drug metabolism in humans: The potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br. J. Clin. Pharmacol. 2013, 76, 587–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosbie, S.J.; Blain, P.G.; Williams, F.M. An investigation into the role of rat skeletal muscle as a site for xenobiotic metabolism using microsomes and isolated cells. Hum. Exp. Toxicol. 1997, 16, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Mitochondrial P450s. Chem. Biol. Interact. 2006, 163, 86–93. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitroxide | aN, mT | aH, mT | Kp | kred × 104, M−1 s−1 |
|---|---|---|---|---|
| 22 | 1.57 | 0.28 | 5.5 | 4.1 ± 0.2 |
| 28 | 1.60 | 0.27 | 13 | 2.4 ± 0.4 |
| 29 | 1.58 | 0.29 | 70 | 2.5 ± 0.4 |
| 30 | 1.56 | 0.30 | 30 | 2.2 ± 0.5 |
| 1 | 1.56 | 0.20 | 270 | 3.3 + 0.5 12.5 ± 0.3 [45] 20 ± 10 [9] ≤10 [14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules 2021, 26, 5761. https://doi.org/10.3390/molecules26195761
Dobrynin SA, Usatov MS, Zhurko IF, Morozov DA, Polienko YF, Glazachev YI, Parkhomenko DA, Tyumentsev MA, Gatilov YV, Chernyak EI, et al. A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules. 2021; 26(19):5761. https://doi.org/10.3390/molecules26195761
Chicago/Turabian StyleDobrynin, Sergey A., Mikhail S. Usatov, Irina F. Zhurko, Denis A. Morozov, Yuliya F. Polienko, Yurii I. Glazachev, Dmitriy A. Parkhomenko, Mikhail A. Tyumentsev, Yuri V. Gatilov, Elena I. Chernyak, and et al. 2021. "A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series" Molecules 26, no. 19: 5761. https://doi.org/10.3390/molecules26195761
APA StyleDobrynin, S. A., Usatov, M. S., Zhurko, I. F., Morozov, D. A., Polienko, Y. F., Glazachev, Y. I., Parkhomenko, D. A., Tyumentsev, M. A., Gatilov, Y. V., Chernyak, E. I., Bagryanskaya, E. G., & Kirilyuk, I. A. (2021). A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules, 26(19), 5761. https://doi.org/10.3390/molecules26195761