
Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces


Abstract
:
1. Introduction
2. Results and Discussion
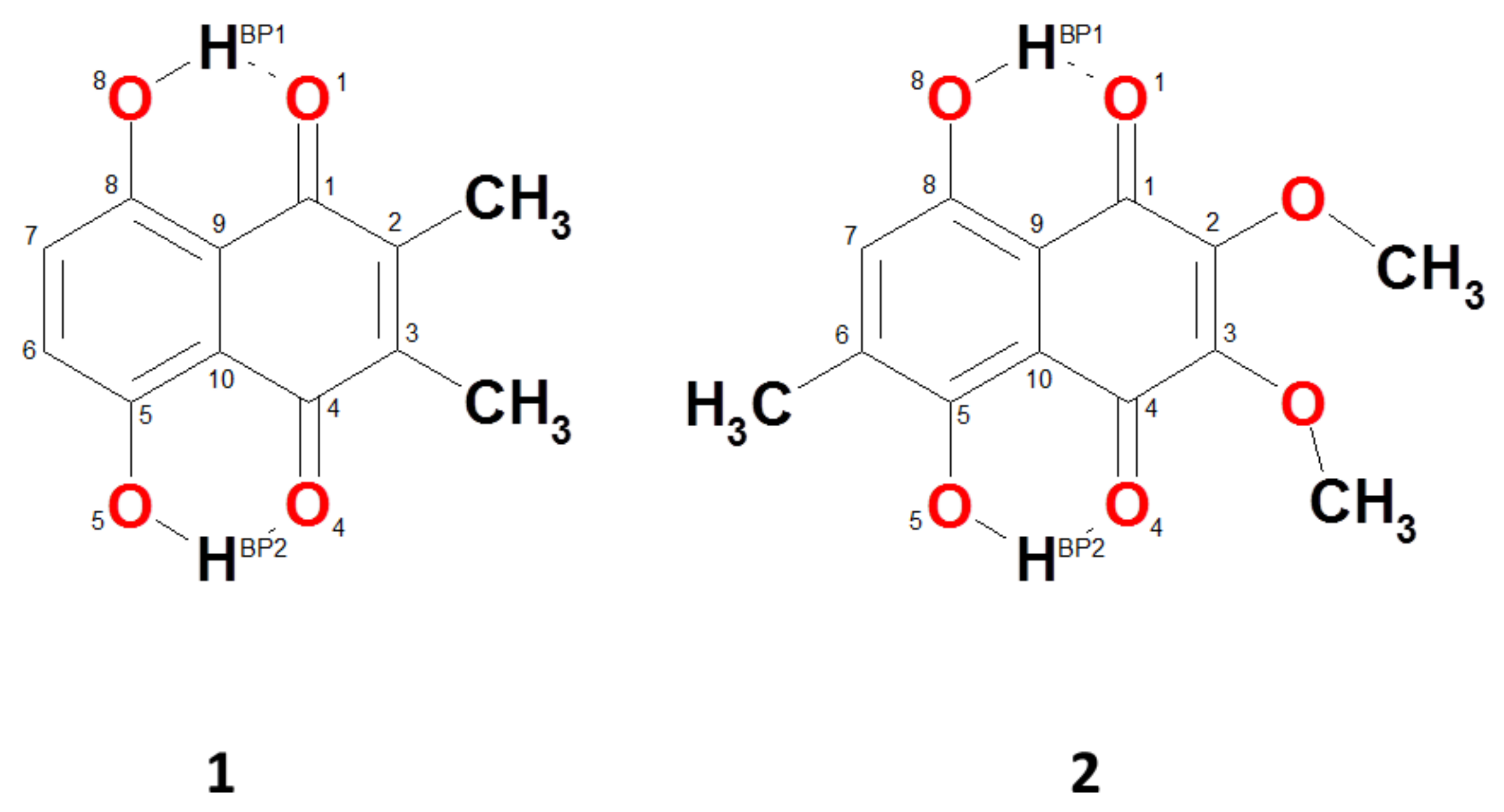
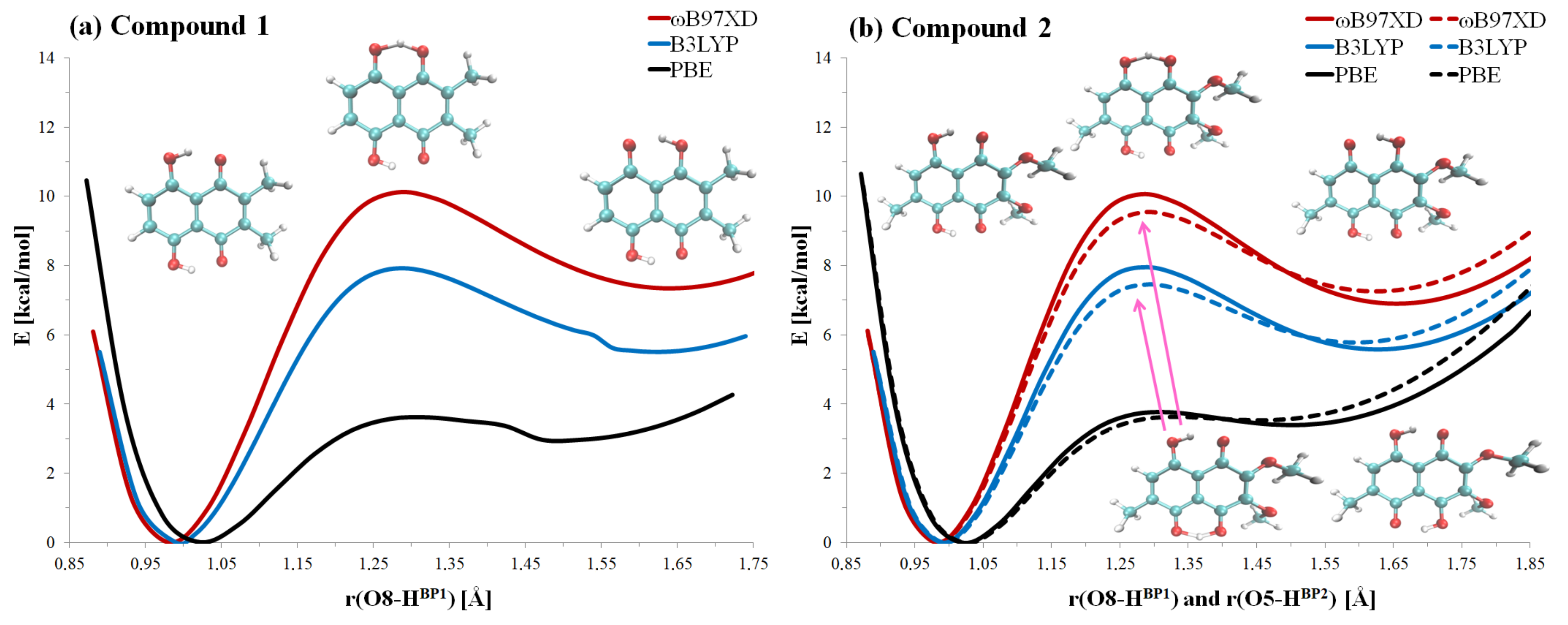
2.1. Geometric and Electronic Structure Description of Naphthazarin Derivatives Monomers with Special Emphasis on Intramolecular Hydrogen Bonds
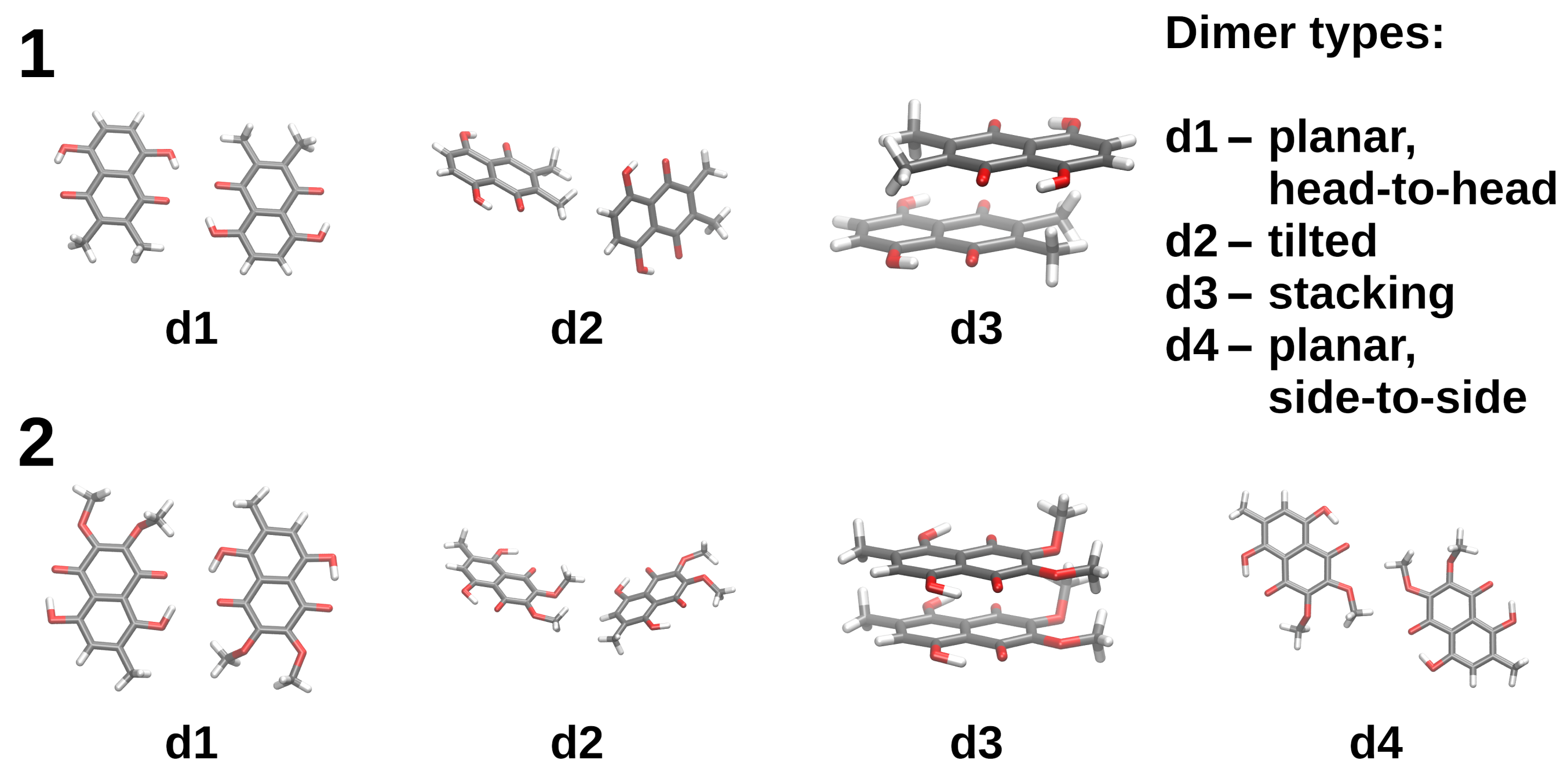
2.2. Intermolecular Forces in Naphthazarin Derivatives Dimers Based on Symmetry-Adapted Perturbation Theory (SAPT)
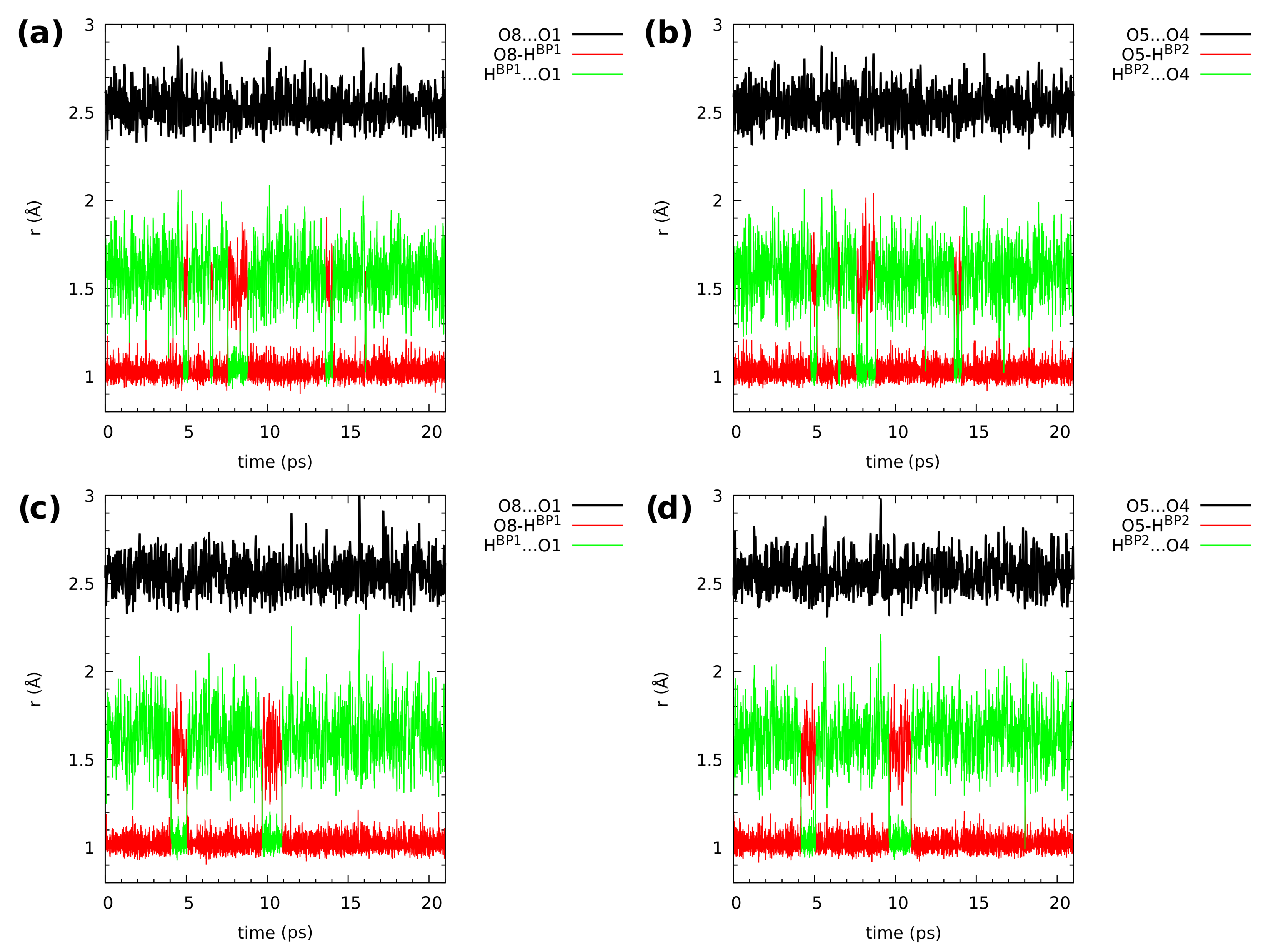
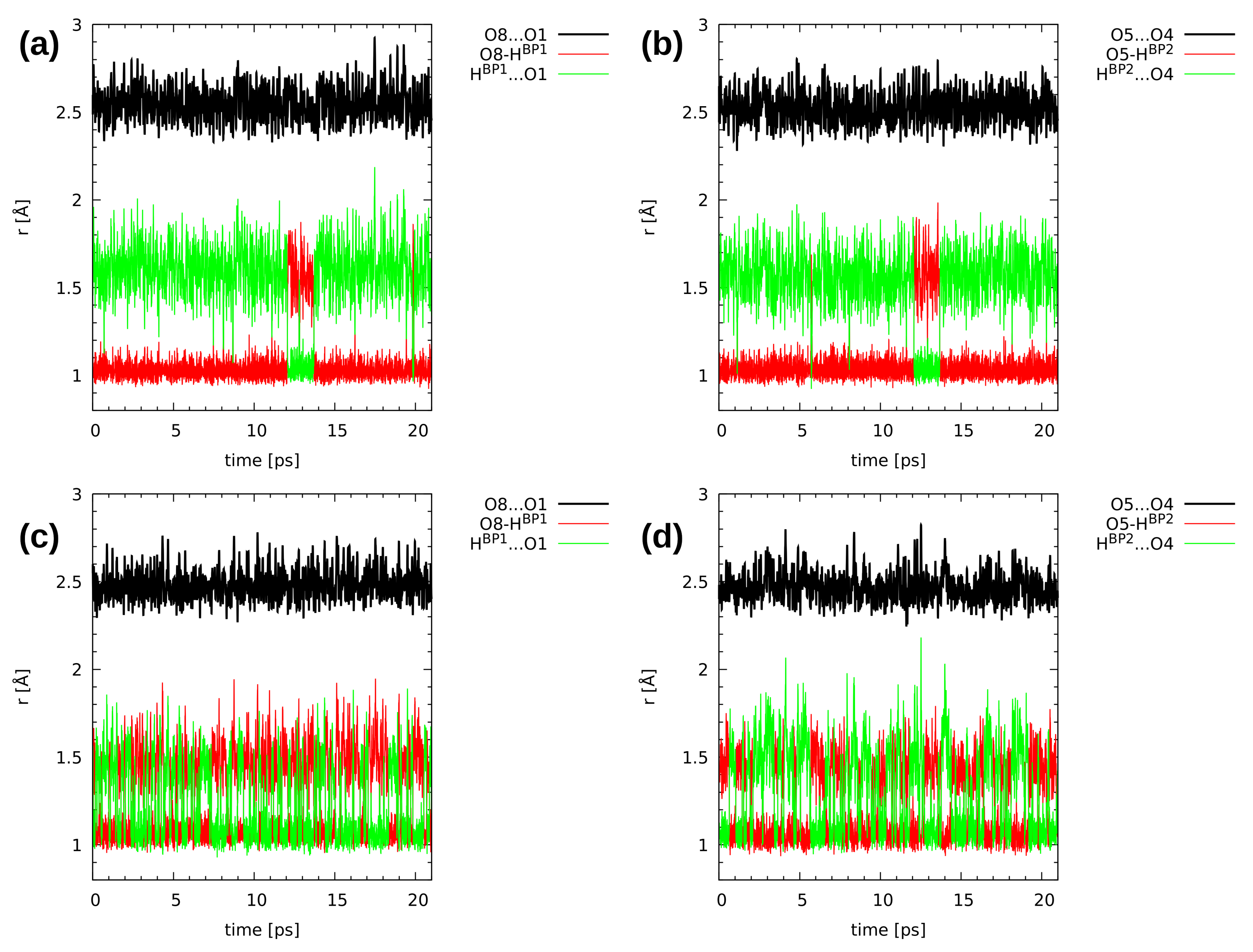
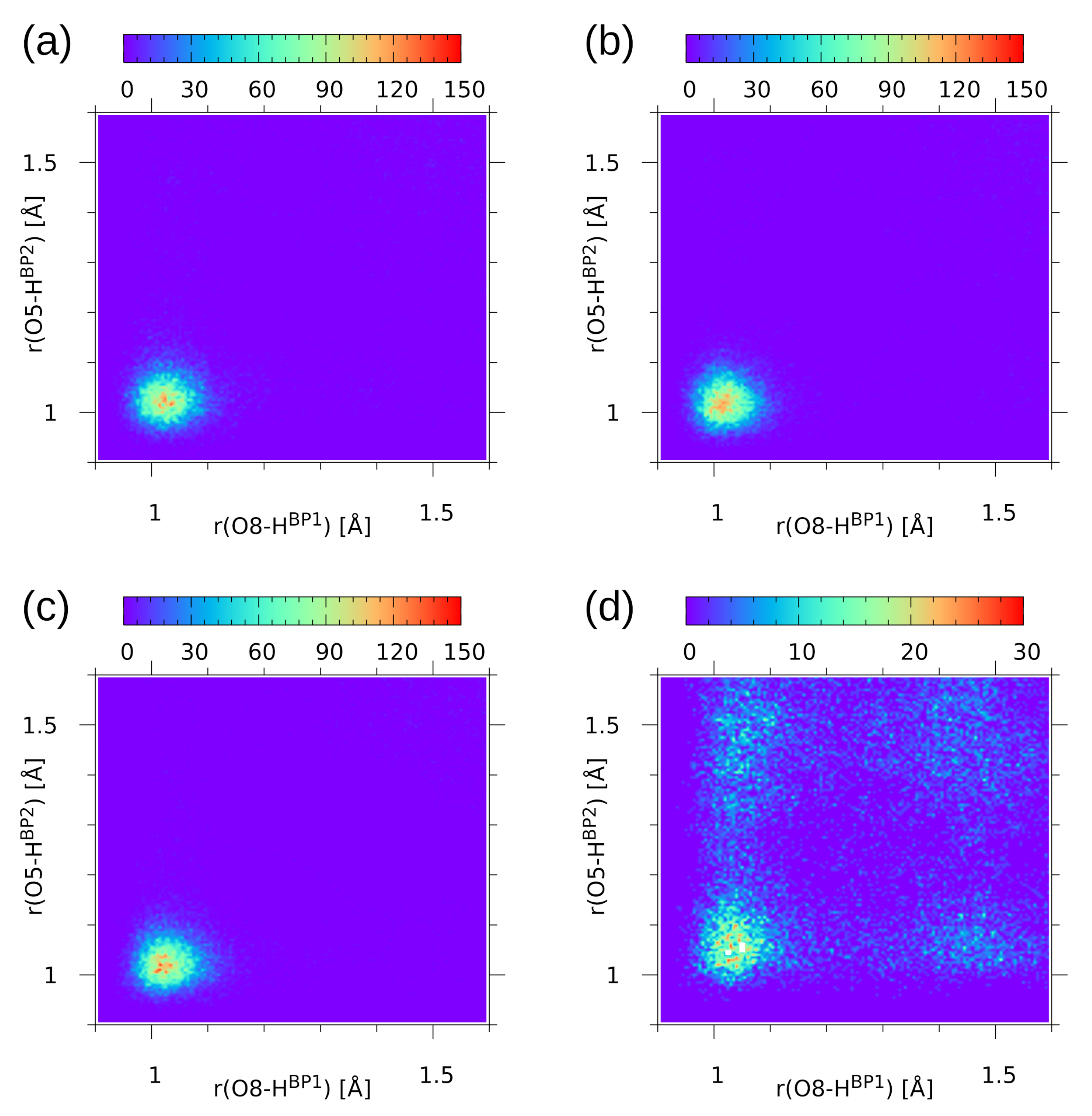
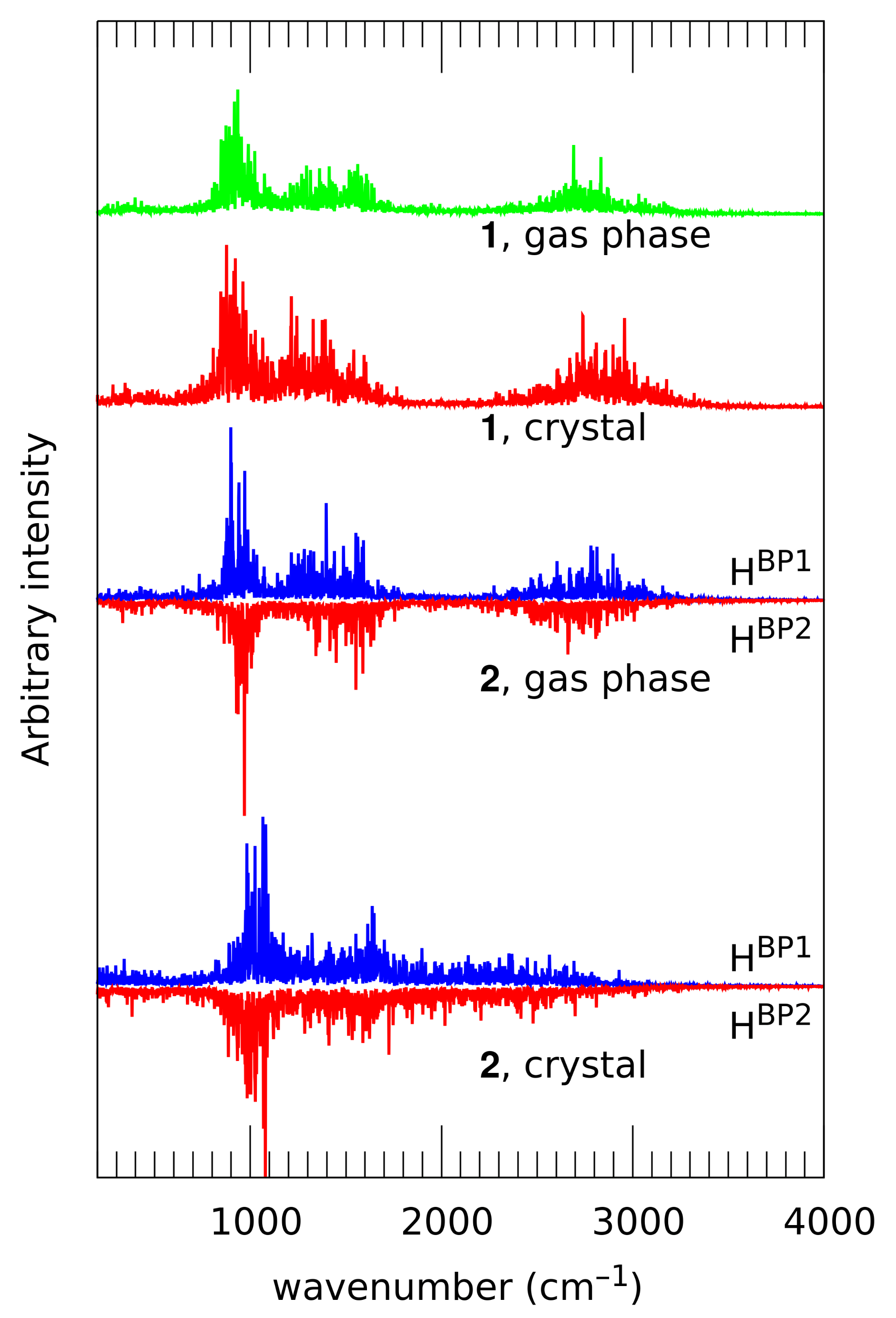
2.3. First-Principle Molecular Dynamics (FPMD) in the Gas and Crystalline Phases
3. Computational Methodology
3.1. Static Models on the Basis of Density Functional Theory (DFT)
3.2. An Application of Symmetry-Adapted Perturbation Theory (SAPT) to Dimers
3.3. Car-Parrinello Molecular Dynamics in the Gas Phase and Solid State
3.4. Estimation of the Nuclear Quantum Effects on the Structural Properties in the Gas Phase and Solid State
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| CPMD | Car–Parrinello Molecular Dynamics |
| PT | Proton Transfer |
| AIM | Atoms In Molecules |
| SAPT | Symmetry-Adapted Perturbation Theory |
| NQE | Nuclear Quantum Effects |
| RAHB | Resonance-Assisted Hydrogen Bond |
| PES | Potential Energy Surface |
| MP2 | Møller–Plesset second-order perturbation theory |
| CCSD | Coupled Clusters with Singles and Doubles |
| BCP | Bond Critical Point |
| RCP | Ring Critical Point |
| BSSE | Basis Set Superposition Error |
| FPMD | First-Principle Molecular Dynamics |
| PBCs | Periodic Boundary Conditions |
References and Note
- Belkova, N.V.; Shubina, E.S.; Epstein, L.M. Diverse World of Unconventional Hydrogen Bonds. Acc. Chem. Res. 2005, 38, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Steiner, T. Other weak and non-conventional hydrogen bonds. In The Weak Hydrogen Bond; Oxford University Press: Oxford, UK, 2001; pp. 122–292. [Google Scholar] [CrossRef]
- Honacker, C.; Kappelt, B.; Jabłoński, M.; Hepp, A.; Layh, M.; Rogel, F.; Uhl, W. Aluminium Functionalized Germanes: Intramolecular Activation of Ge-H Bonds, Formation of a Dihydrogen Bond and Facile Hydrogermylation of Unsaturated Substrates. Eur. J. Inorg. Chem. 2019, 2019, 3287–3300. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef]
- Hobza, P.; Zahradník, R.; Müller-Dethlefs, K. The World of Non-Covalent Interactions: 2006. Collect. Czechoslov. Chem. Commun. 2006, 71, 443–531. [Google Scholar] [CrossRef] [Green Version]
- Hobza, P.; Řezáč, J. Introduction: Noncovalent Interactions. Chem. Rev. 2016, 116, 4911–4912. [Google Scholar] [CrossRef] [Green Version]
- Puzzarini, C.; Spada, L.; Alessandrini, S.; Barone, V. The challenge of non-covalent interactions: Theory meets experiment for reconciling accuracy and interpretation. J. Phys. Condens. Matter 2020, 32, 343002. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Competition between Intra and Intermolecular Triel Bonds. Complexes between Naphthalene Derivatives and Neutral or Anionic Lewis Bases. Molecules 2020, 25, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: London, UK, 1992. [Google Scholar]
- Jeffrey, G.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Jeffrey, G. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [Green Version]
- Trylska, J. The role of hydrogen bonding in the enzymatic reaction catalyzed by HIV-1 protease. Protein Sci. 2004, 13, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Simón, L.; Goodman, J.M. Enzyme Catalysis by Hydrogen Bonds: The Balance between Transition State Binding and Substrate Binding in Oxyanion Holes. J. Org. Chem. 2009, 75, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-Bond and Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef] [PubMed]
- Filarowski, A.; Koll, A.; Sobczyk, L. Intramolecular Hydrogen Bonding in o-hydroxy Aryl Schiff Bases. Curr. Org. Chem. 2009, 13, 172–193. [Google Scholar] [CrossRef]
- Kwocz, A.; Panek, J.J.; Jezierska, A.; Hetmanczyk, Ł.; Pawlukojć, A.; Kochel, A.; Lipkowski, P.; Filarowski, A. A molecular roundabout: Triple cyclically arranged hydrogen bonds in light of experiment and theory. New J. Chem. 2018, 42, 19467–19477. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Liao, L.; Ma, Q.; Zhang, Z.; Fan, G. Stabilization of an intramolecular hydrogen-bond block in an s-triazine insensitive high-energy material. New J. Chem. 2019, 43, 10675–10679. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Marchenko, A.V.; Pozharskii, A.F.; Filarowski, A.; Spiridonova, D.V. Combination of “Buttressing” and “Clothespin” Effects for Reaching the Shortest NHN Hydrogen Bond in Proton Sponge Cations. J. Org. Chem. 2021, 86, 3637–3647. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Harata, K.; Fujiwara, M.; Ohbu, K. Molecular Arrangement and Intermolecular Hydrogen Bonding in Crystals of Methyl 6-O-Acyl-d-glycopyranosides. Langmuir 1996, 12, 636–640. [Google Scholar] [CrossRef]
- Vella-Zarb, L.; Braga, D.; Orpen, A.G.; Baisch, U. The influence of hydrogen bonding on the planar arrangement of melamine in crystal structures of its solvates, cocrystals and salts. CrystEngComm 2014, 16, 8147. [Google Scholar] [CrossRef]
- Mes, T.; Smulders, M.M.J.; Palmans, A.R.A.; Meijer, E.W. Hydrogen-Bond Engineering in Supramolecular Polymers: Polarity Influence on the Self-Assembly of Benzene-1,3,5-tricarboxamides. Macromolecules 2010, 43, 1981–1991. [Google Scholar] [CrossRef]
- Sikder, A.; Ghosh, S. Hydrogen-bonding regulated assembly of molecular and macromolecular amphiphiles. Mater. Chem. Front. 2019, 3, 2602–2616. [Google Scholar] [CrossRef]
- Han, K.L.; Zhao, G.J. (Eds.) Hydrogen Bonding and Transfer in the Excited State; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the .beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O-H-O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Mautner, M.M.N. The Ionic Hydrogen Bond. Chem. Rev. 2005, 105, 213–284. [Google Scholar] [CrossRef] [Green Version]
- Gilli, G.; Gilli, P. Towards an unified hydrogen-bond theory. J. Mol. Struct. 2000, 552, 1–15. [Google Scholar] [CrossRef]
- Jesus, A.J.L.; Redinha, J.S. Charge-Assisted Intramolecular Hydrogen Bonds in Disubstituted Cyclohexane Derivatives. J. Phys. Chem. A 2011, 115, 14069–14077. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S.; Stuchebrukhov, A.A. Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev. 2010, 110, 6939–6960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemin, D.; Zúñiga, J.; Requena, A.; Céron-Carrasco, J.P. Assessing the Importance of Proton Transfer Reactions in DNA. Acc. Chem. Res. 2014, 47, 2467–2474. [Google Scholar] [CrossRef]
- Ishikita, H.; Saito, K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc. Interface 2014, 11, 20130518. [Google Scholar] [CrossRef]
- Jana, S.; Dalapati, S.; Guchhait, N. Proton Transfer Assisted Charge Transfer Phenomena in Photochromic Schiff Bases and Effect of –NEt2 Groups to the Anil Schiff Bases. J. Phys. Chem. A 2012, 116, 10948–10958. [Google Scholar] [CrossRef]
- Panek, J.J.; Filarowski, A.; Jezierska-Mazzarello, A. Impact of proton transfer phenomena on the electronic structure of model Schiff bases: An AIM/NBO/ELF study. J. Chem. Phys. 2013, 139, 154312. [Google Scholar] [CrossRef]
- Joshi, H.C.; Antonov, L. Excited-State Intramolecular Proton Transfer: A Short Introductory Review. Molecules 2021, 26, 1475. [Google Scholar] [CrossRef] [PubMed]
- Stuchebrukhov, A.A. Mechanisms of proton transfer in proteins: Localized charge transfer versus delocalized soliton transfer. Phys. Rev. E 2009, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, S.; Wyatt, D.L.; Crumrine, D.S.; Cukierman, S. Proton Transfer in Water Wires in Proteins: Modulation by Local Constraint and Polarity in Gramicidin A Channels. Biophys. J. 2007, 93, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezierska, A.; Panek, J.J. Theoretical study of intramolecular hydrogen bond in selected symmetric “proton sponges” on the basis of DFT and CPMD methods. J. Mol. Model. 2020, 26, 37. [Google Scholar] [CrossRef]
- Król-Starzomska, I.; Filarowski, A.; Rospenk, M.; Koll, A.; Melikova, S. Proton Transfer Equilibria in Schiff Bases with Steric Repulsion. J. Phys. Chem. A 2004, 108, 2131–2138. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. First-Principle Molecular Dynamics Study of Selected Schiff and Mannich Bases: Application of Two-Dimensional Potential of Mean Force to Systems with Strong Intramolecular Hydrogen Bonds. J. Chem. Theory Comput. 2008, 4, 375–384. [Google Scholar] [CrossRef]
- Jezierska-Mazzarello, A.; Panek, J.J.; Vuilleumier, R.; Koll, A.; Ciccotti, G. Direct observation of the substitution effects on the hydrogen bridge dynamics in selected Schiff bases—A comparative molecular dynamics study. J. Chem. Phys. 2011, 134, 034308. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J.; Koll, A. Spectroscopic Properties of a Strongly Anharmonic Mannich Base N-oxide. ChemPhysChem 2008, 9, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Panek, J.J.; Błaziak, K.; Jezierska, A. Hydrogen bonds in quinoline N-oxide derivatives: First-principle molecular dynamics and metadynamics ground state study. Struct. Chem. 2016, 27, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Panek, J.J.; Jezierska, A. N-oxide Derivatives: Car–Parrinello Molecular Dynamics and Electron Localization Function Study on Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2018, 122, 6605–6614. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J. “Zwitterionic Proton Sponge” Hydrogen Bonding Investigations on the Basis of Car–Parrinello Molecular Dynamics. J. Chem. Inf. Model. 2015, 55, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Pekin, G.; Ganzera, M.; Senol, S.; Bedir, E.; Korkmaz, K.; Stuppner, H. Determination of Naphthazarin Derivatives in Endemic Turkish Alkanna Species by Reversed Phase High Performance Liquid Chromatography. Planta Med. 2007, 73, 267–272. [Google Scholar] [CrossRef]
- Sánchez-Calvo, J.M.; Barbero, G.R.; Guerrero-Vásquez, G.; Durán, A.G.; Macías, M.; Rodríguez-Iglesias, M.A.; Molinillo, J.M.G.; Macías, F.A. Synthesis, antibacterial and antifungal activities of naphthoquinone derivatives: A structure–activity relationship study. Med. Chem. Res. 2016, 25, 1274–1285. [Google Scholar] [CrossRef]
- Masuda, K.; Funayama, S.; Komiyama, K.; Unezawa, I.; Ito, K. Constituents of Tritonia crocosmaeflora, I. Tricrozarin A, a Novel Antimicrobial Naphthazarin Derivative. J. Nat. Prod. 1987, 50, 418–421. [Google Scholar] [CrossRef]
- Rudnicka, M.; Ludynia, M.; Karcz, W. The Effect of Naphthazarin on the Growth, Electrogenicity, Oxidative Stress, and Microtubule Array in Z. mays Coleoptile Cells Treated With IAA. Front. Plant Sci. 2019, 9, 1940. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. The Chemistry and Biology of Alkannin, Shikonin, and Related Naphthazarin Natural Products. Angew. Chem. Int. Ed. 1999, 38, 270–301. [Google Scholar] [CrossRef]
- Song, G.; Kim, Y.; You, Y.; Cho, H.; Kim, S.; Sok, D.E.; Ahn, B. Naphthazarin derivatives (VI): Synthesis, inhibitory effect on DNA topoisomerase-I and antiproliferative activity of 2- or 6-(1-oxyiminoalkyl)- 5,8-dimethoxy-1,4-naphthoquinones. Arch. Pharm. 2000, 333, 87–92. [Google Scholar] [CrossRef]
- Park, E.M.; Lee, I.J.; Kim, S.H.; Song, G.Y.; Park, Y.M. Inhibitory effect of a naphthazarin derivative, S64, on heat shock factor (Hsf) activation and glutathione status following hypoxia. Cell Biol. Toxicol. 2003, 19, 273–284. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, Q.; Yang, C.; Zhu, Y.; Zhang, L.; Wang, L.; Liu, Z.; Zhang, G.; Zhang, C. Assembly Line and Post-PKS Modifications in the Biosynthesis of Marine Polyketide Natural Products. In Comprehensive Natural Products III; Elsevier: Amsterdam, The Netherlands, 2020; pp. 139–197. [Google Scholar] [CrossRef]
- Tchizhova, A.Y.; Anufriev, V.P.; Glazunov, V.P.; Denisenko, V.A. The chemistry of naphthazarin derivatives. Russ. Chem. Bull. 2000, 49, 466–471. [Google Scholar] [CrossRef]
- Herbstein, F.H.; Kapon, M.; Reisner, G.M.; Lehman, M.S.; Kress, R.B.; Wilson, R.B.; Shiau, W.i.; Duesler, E.N.; Paul, I.C.; Curtin, D.Y.; et al. Polymorphism of naphthazarin and its relation to solid-state proton transfer. Neutron and X-ray diffraction studies on naphthazarin C. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1985, 399, 295–319. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D. Energy of Intramolecular Hydrogen Bonding in ortho-Hydroxybenzaldehydes, Phenones and Quinones. Transfer of Aromaticity from ipso-Benzene Ring to the Enol System(s). Molecules 2017, 22, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezierska, A.; Kizior, B.; Szyja, B.M.; Panek, J.J. On the nature of inter- and intramolecular interactions involving benzo[h]quinoline and 10-hydroxybenzo[h]quinoline: Electronic ground state vs excited state study. J. Mol. Struct. 2021, 1234, 130126. [Google Scholar] [CrossRef]
- Jezierska, A.; Błaziak, K.; Klahm, S.; Lüchow, A.; Panek, J.J. Non-Covalent Forces in Naphthazarin—Cooperativity or Competition in the Light of Theoretical Approaches. Int. J. Mol. Sci. 2021, 22, 8033. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.Z.; Tayyari, S.F.; Tayyari, F.; Behforouz, M. Fourier transform infrared and Raman spectra, vibrational assignment and density functional theory calculations of naphthazarin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004, 60, 111–120. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Rodríguez, J.; Smith-Verdier, P.; Florencio, F.; García-Blanco, S. Crystal and molecular structure of 2, 3-dimethylnaph-thazarin: A charge-transfer complex. J. Mol. Struct. 1984, 112, 101–109. [Google Scholar] [CrossRef]
- Cannon, J.; Matsuki, Y.; Patrick, V.; White, A. Selective O-Demethylation of 5, 8-Dihydroxy-2, 3-Dimethoxy-6-Methylnaphtho-1, 4-Quinone: The Crystal Structures of 5, 8-Dihydroxy-2, 3-Dimethoxy-6-Methylnaphtho-1, 4-Quinone and 2, 5, 8-Triacetoxy-3-Methoxy-6-Methylnaphtho-1, 4-Quinone. Aust. J. Chem. 1987, 40, 1191. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Bader, R. Atoms in Molecules: A Quantum Theory; International Ser. of Monogr. on Chem.; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Kizior, B.; Panek, J.J.; Szyja, B.M.; Jezierska, A. Structure-Property Relationship in Selected Naphtho- and Anthra-Quinone Derivatives on the Basis of Density Functional Theory and Car–Parrinello Molecular Dynamics. Symmetry 2021, 13, 564. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Reviews in Computational Chemistry; John Wiley and Sons, Ltd.: Hoboken, NJ, USA, 2000; pp. 1–86. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Oliva-Enrich, J.M.; Yáñez, M.; Mó, O.; Montero-Campillo, M.M. The Importance of Strain (Preorganization) in Beryllium Bonds. Molecules 2020, 25, 5876. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [Green Version]
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The nature of the hydrated excess proton in water. Nature 1999, 397, 601–604. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Parrinello, M. Solvated excess protons in water: Quantum effects on the hydration structure. J. Phys. Condens. Matter 2000, 12, A153–A159. [Google Scholar] [CrossRef]
- Kong, L.; Wu, X.; Car, R. Roles of quantum nuclei and inhomogeneous screening in the X-ray absorption spectra of water and ice. Phys. Rev. B 2012, 86, 134203. [Google Scholar] [CrossRef] [Green Version]
- Stare, J.; Panek, J.; Eckert, J.; Grdadolnik, J.; Mavri, J.; Hadži, D. Proton Dynamics in the Strong Chelate Hydrogen Bond of Crystalline Picolinic Acid N-Oxide. A New Computational Approach and Infrared, Raman and INS Study. J. Phys. Chem. A 2008, 112, 1576–1586. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J.; Koll, A.; Mavri, J. Car-Parrinello simulation of an O–H stretching envelope and potential of mean force of an intramolecular hydrogen bonded system: Application to a Mannich base in solid state and in vacuum. J. Chem. Phys. 2007, 126, 205101. [Google Scholar] [CrossRef]
- Denisov, G.S.; Mavri, J.; Sobczyk, L. Potential energy shape for the proton motion in hydrogen bonds reflected in infrared and NMR spectra. In Hydrogen Bonding—New Insights, 1st ed.; Challenges and Advances in Computational Chemistry and Physics, 3; Grabowski, S.J., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 377–416. [Google Scholar]
- Stare, J.; Mavri, J. Numerical solving of the vibrational time-independent Schrödinger equation in one and two dimensions using the variational method. Comput. Phys. Commun. 2002, 143, 222–240. [Google Scholar] [CrossRef]
- CCDC Structural Database. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 7 May 2021).
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Čížek, J. On the Use of the Cluster Expansion and the Technique of Diagrams in Calculations of Correlation Effects in Atoms and Molecules. In Advances in Chemical Physics; John Wiley and Sons, Ltd.: London, UK, 1969; pp. 35–89. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- TURBOMOLE V6.5, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 1 June 2021).
- Keith, T.A. AIMAll; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Remigio, R.D.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockney, R.W. Potential Calculation and Some Applications. Methods Comput. Phys. 1970, 9, 135–211. [Google Scholar]
- Kittel, C. Introduction To Solid State Physics, 8th ed.; Wiley and Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- CPMD 3.17.1; Copyright IBM Corp. (1990–2004) Copyright MPI für Festkoerperforschung Stuttgart (1997–2001)
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot 4.4: An Interactive Plotting Program. 2010. Available online: http://gnuplot.sourceforge.net/ (accessed on 12 May 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Charge [e] | Compound 1 | Compound 2 | ||
|---|---|---|---|---|
| Molecular Form | PT Form | Molecular Form | PT Form | |
| Hydrogen Bridge 1 | ||||
| O8 | −1.134 | −1.094 | −1.134 | −1.097 |
| 0.641 | 0.643 | 0.641 | 0.642 | |
| O1 | −1.099 | −1.139 | −1.081 | −1.119 |
| C8 | 0.597 | 0.874 | 0.597 | 0.874 |
| C9 | −0.027 | −0.030 | −0.029 | −0.027 |
| C1 | 0.869 | 0.587 | 0.902 | 0.618 |
| Hydrogen Bridge 2 | ||||
| O5 | −1.134 | −1.094 | −1.139 | −1.104 |
| 0.641 | 0.642 | 0.642 | 0.645 | |
| O4 | −1.098 | −1.137 | −1.102 | −1.136 |
| C5 | 0.596 | 0.875 | 0.582 | 0.857 |
| C10 | −0.028 | −0.030 | −0.027 | −0.029 |
| C4 | 0.868 | 0.585 | 0.873 | 0.601 |
| Compound 1 | Compound 2 | |||
|---|---|---|---|---|
| BCP | ||||
| Molecular Form | ||||
| O8- | 0.339 | −2.536 | 0.340 | −2.540 |
| -O1 | 0.051 | 0.138 | 0.050 | 0.136 |
| O5- | 0.339 | −2.533 | 0.337 | −2.511 |
| -O4 | 0.051 | 0.137 | 0.053 | 0.140 |
| Proton-Transferred Form (PT) | ||||
| O8- | 0.054 | 0.139 | 0.053 | 0.137 |
| -O1 | 0.335 | −2.487 | 0.335 | −2.488 |
| O5- | 0.052 | 0.137 | 0.057 | 0.141 |
| -O4 | 0.336 | −2.505 | 0.330 | −2.441 |
| Compound | Dimer | Elst | Exch | Ind | Disp | SAPT0 | SAPT2 |
|---|---|---|---|---|---|---|---|
| 1 | d1 | −3.617 | 4.754 | −0.676 | −3.739 | −3.342 | −3.278 |
| 1 | d2 | −0.710 | 1.716 | −0.181 | −2.104 | −1.328 | −1.279 |
| 1 | d3 | −3.210 | 7.190 | −0.884 | −11.991 | −9.233 | −8.894 |
| 2 | d1 | −4.426 | 3.857 | −0.760 | −4.178 | −6.305 | −5.507 |
| 2 | d2 | −0.750 | 1.598 | −0.349 | −2.084 | −1.601 | −1.585 |
| 2 | d3 | −6.162 | 15.081 | −2.063 | −21.160 | −14.795 | −14.304 |
| 2 | d4 | −5.099 | 3.273 | −1.069 | −2.838 | −6.849 | −5.733 |
| Compound | Dimer | Elst | Exch | Ind | Disp | SAPT0 | SAPT2 |
|---|---|---|---|---|---|---|---|
| 1 | d1 | −6.116 | 7.711 | −1.112 | −4.766 | −5.410 | −4.283 |
| 1 | d3 | −9.129 | 20.285 | −2.485 | −21.867 | −13.681 | −13.196 |
| 2 | d1 | −5.830 | 8.537 | −1.187 | −5.958 | −5.443 | −4.438 |
| 2 | d3 | −14.669 | 29.178 | −3.754 | −29.914 | −19.906 | −19.158 |
| 2 | d4 | −7.830 | 8.427 | −2.254 | −4.258 | −7.290 | −5.916 |
| O8-H...O1 | O5-H...O4 | ||
|---|---|---|---|
| 1, gas phase | |||
| O8 donor | O1 acceptor | O5 donor | O4 acceptor |
| 89.7% | 10.3% | 90.2% | 9.8% |
| 1, solid state | |||
| O8 donor | O1 acceptor | O5 donor | O4 acceptor |
| 89.5% | 10.5% | 89.1% | 10.9% |
| 2, gas phase | |||
| O8 donor | O1 acceptor | O5 donor | O4 acceptor |
| 91.6% | 8.4% | 91.8% | 8.2% |
| 2, solid state | |||
| O8 donor | O1 acceptor | O5 donor | O4 acceptor |
| 41.6% | 58.4% | 53.1% | 46.9% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kułacz, K.; Pocheć, M.; Jezierska, A.; Panek, J.J. Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces. Molecules 2021, 26, 5642. https://doi.org/10.3390/molecules26185642
Kułacz K, Pocheć M, Jezierska A, Panek JJ. Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces. Molecules. 2021; 26(18):5642. https://doi.org/10.3390/molecules26185642
Chicago/Turabian StyleKułacz, Karol, Michał Pocheć, Aneta Jezierska, and Jarosław J. Panek. 2021. "Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces" Molecules 26, no. 18: 5642. https://doi.org/10.3390/molecules26185642
APA StyleKułacz, K., Pocheć, M., Jezierska, A., & Panek, J. J. (2021). Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces. Molecules, 26(18), 5642. https://doi.org/10.3390/molecules26185642