
The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study



Abstract
:1. Introduction
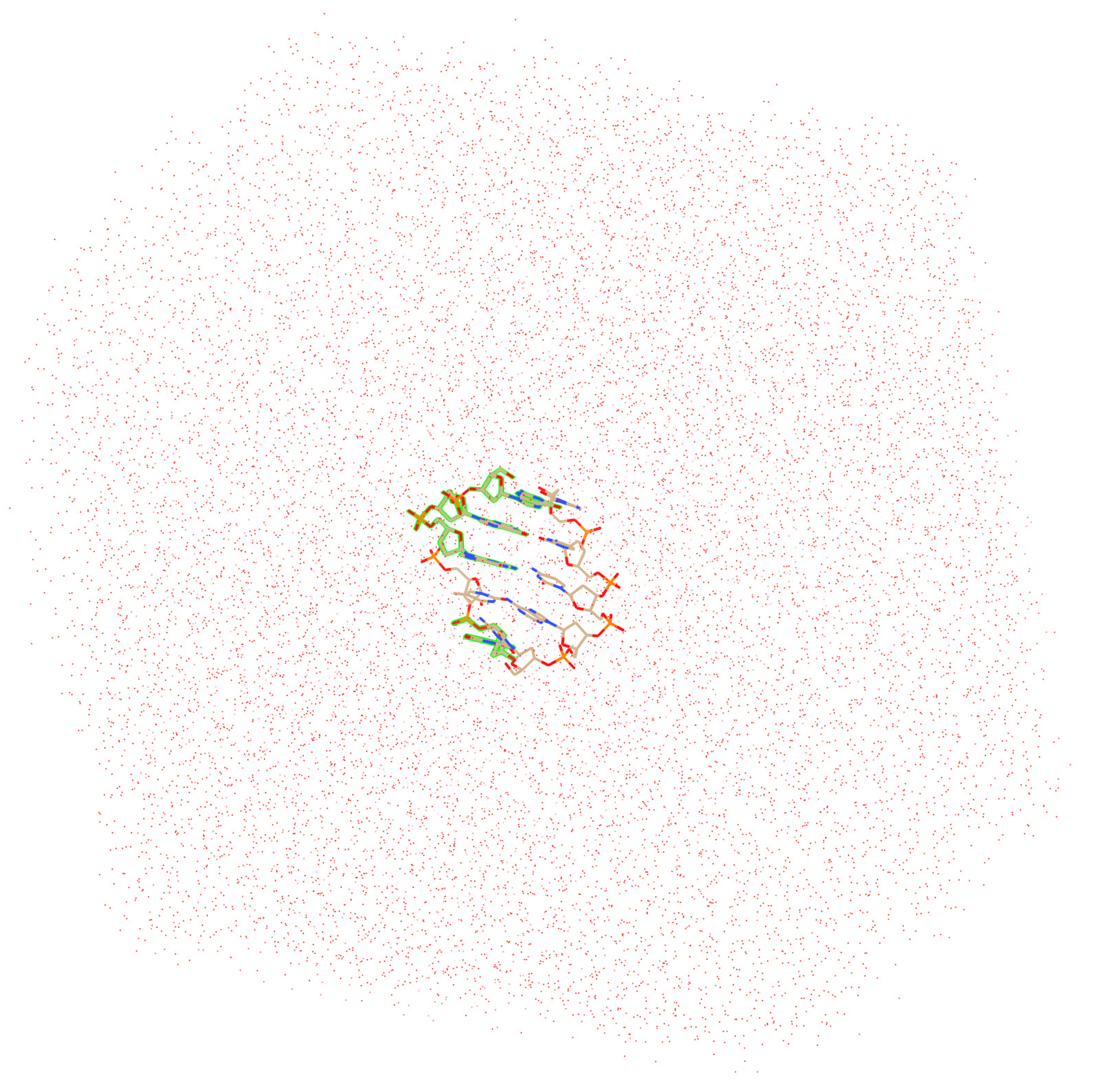
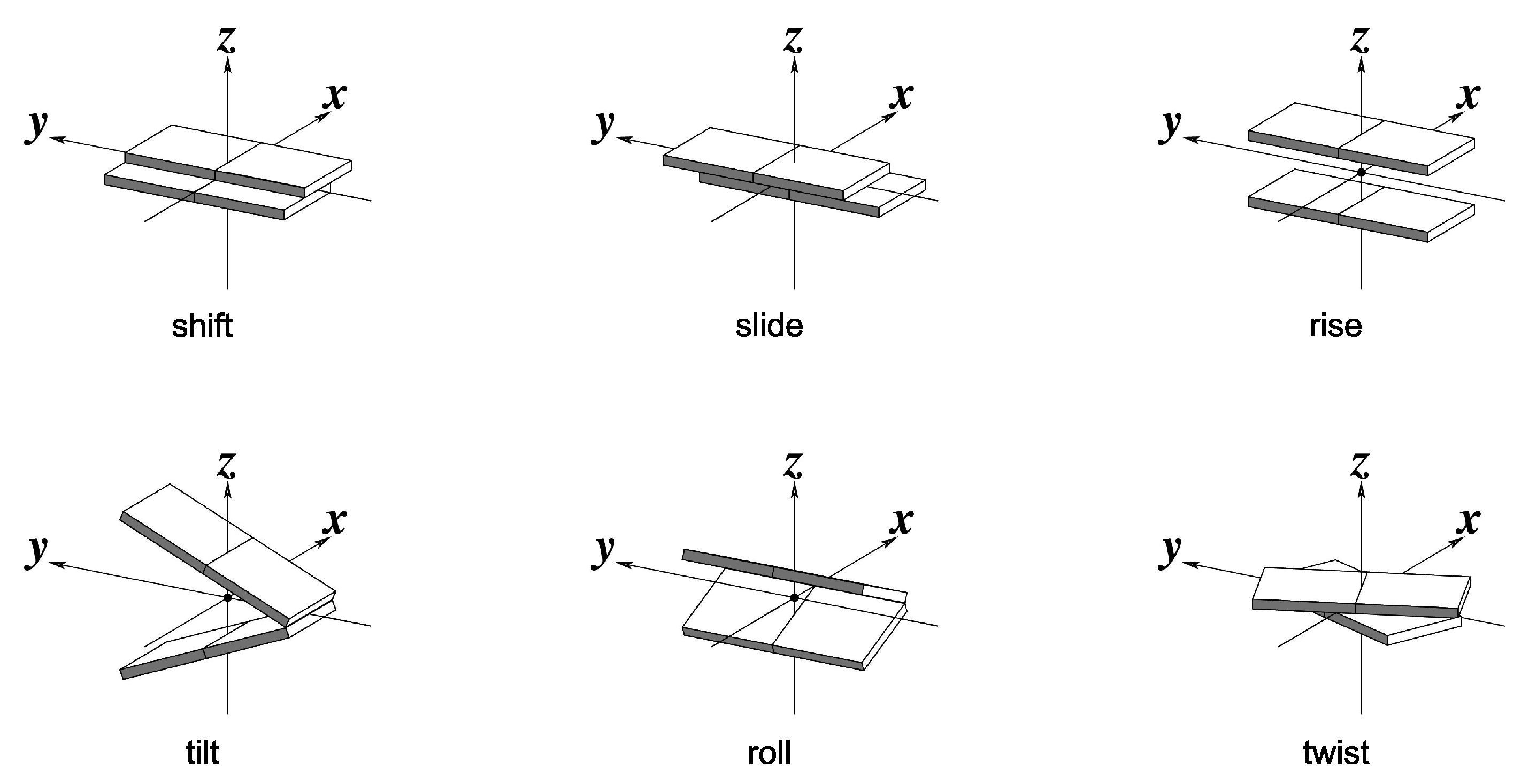
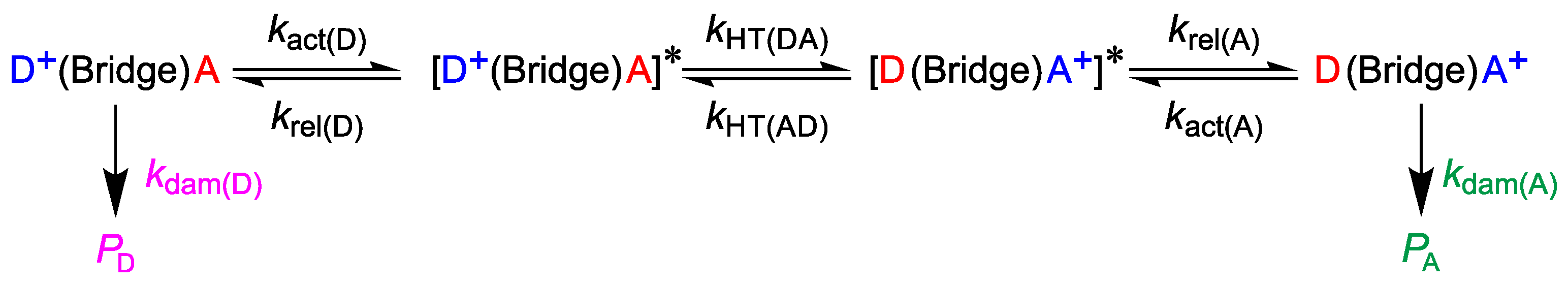
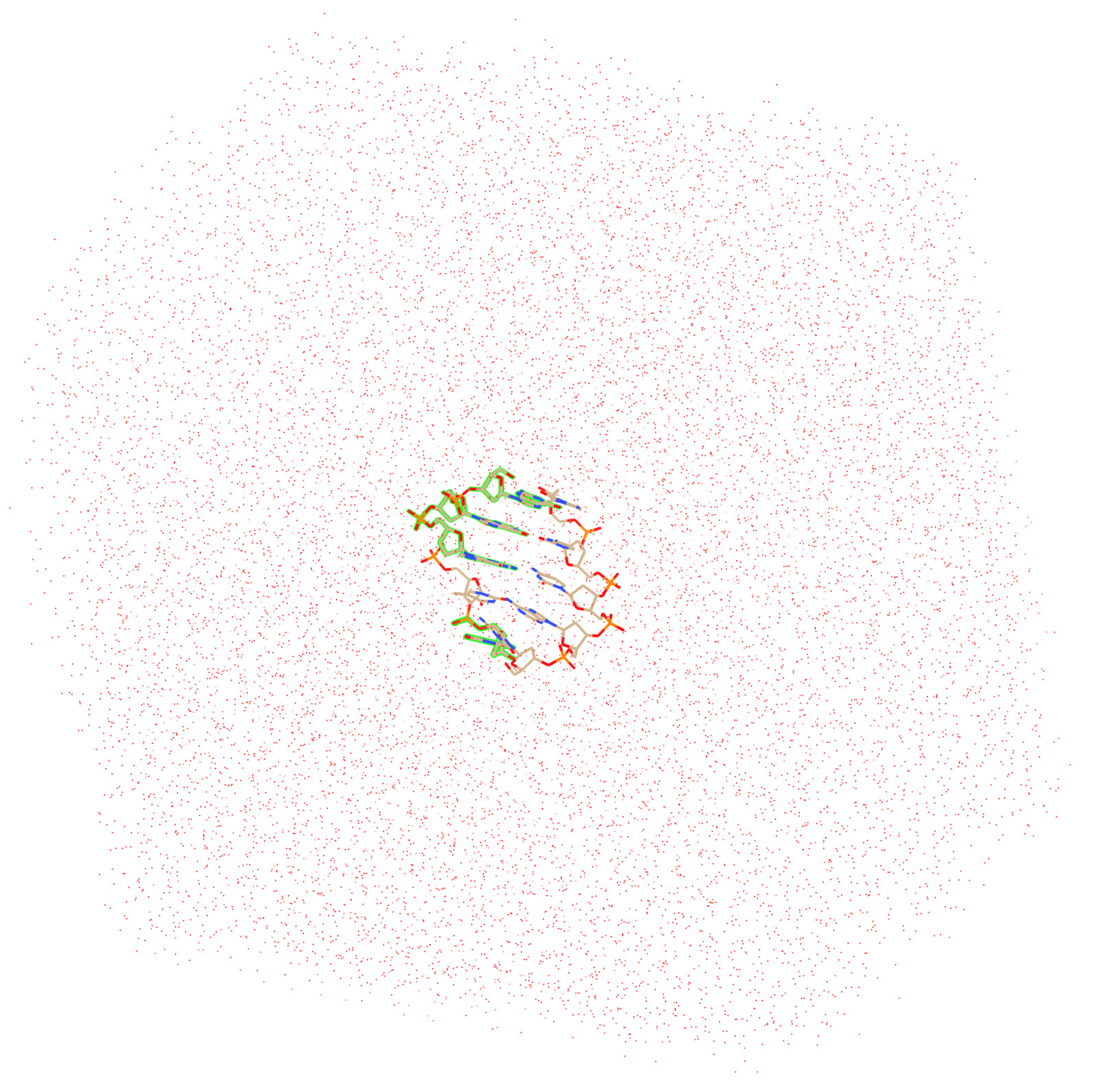
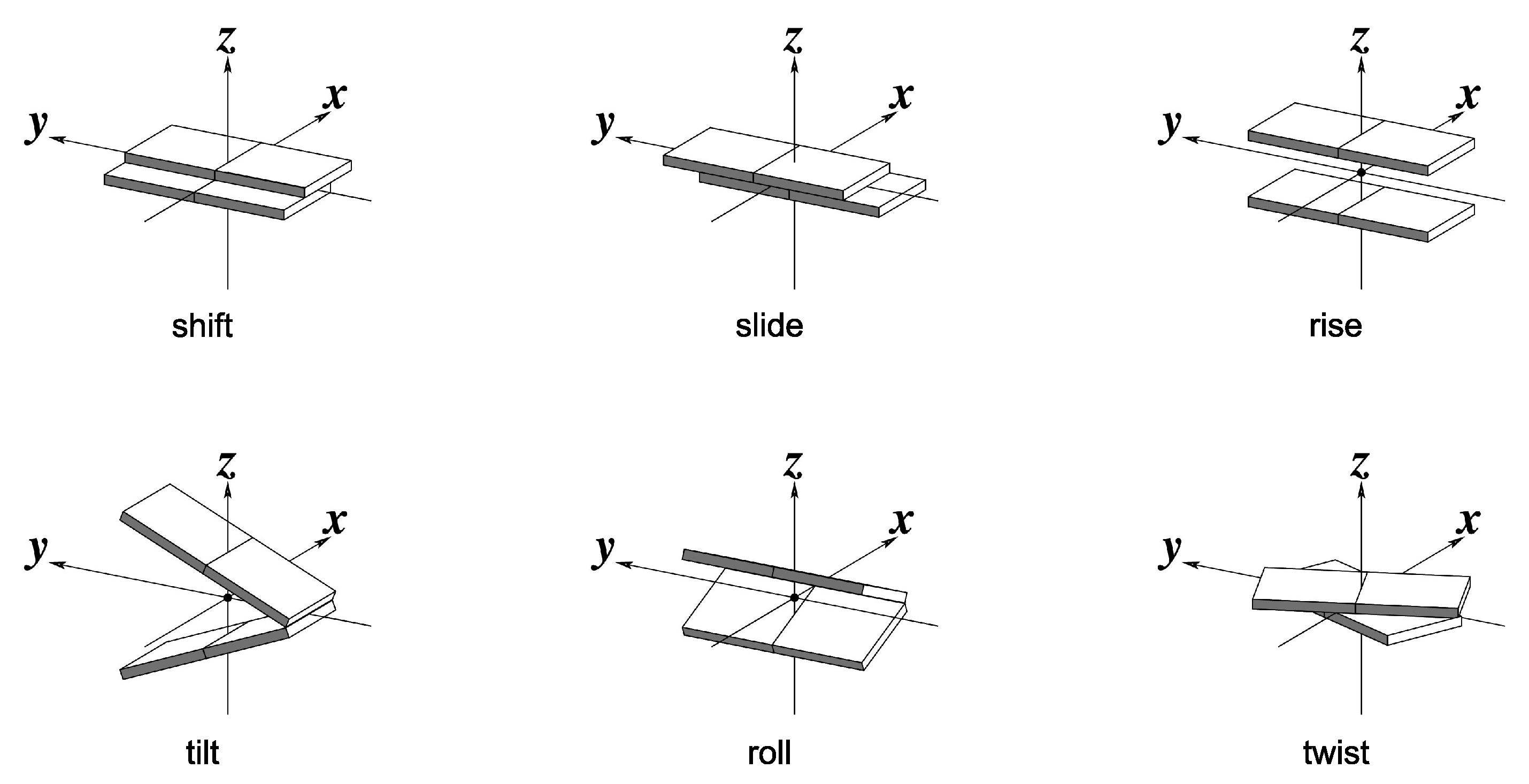
2. Computational Details
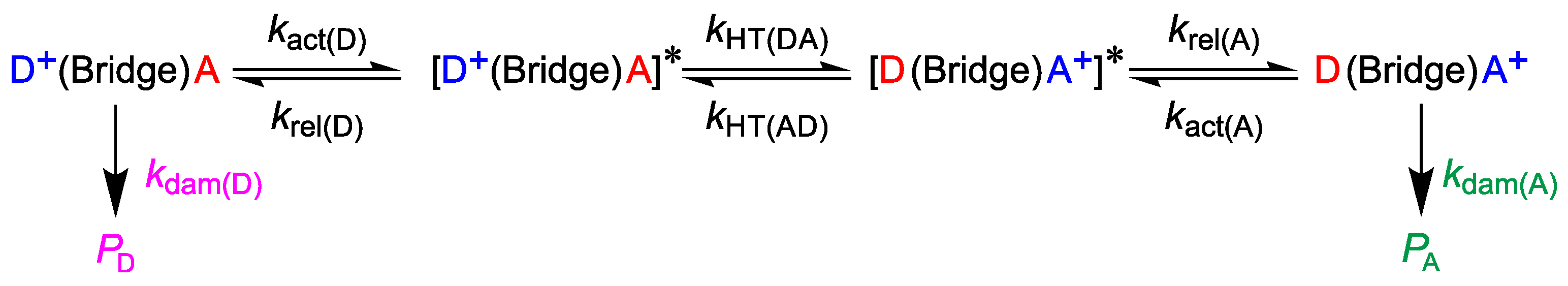
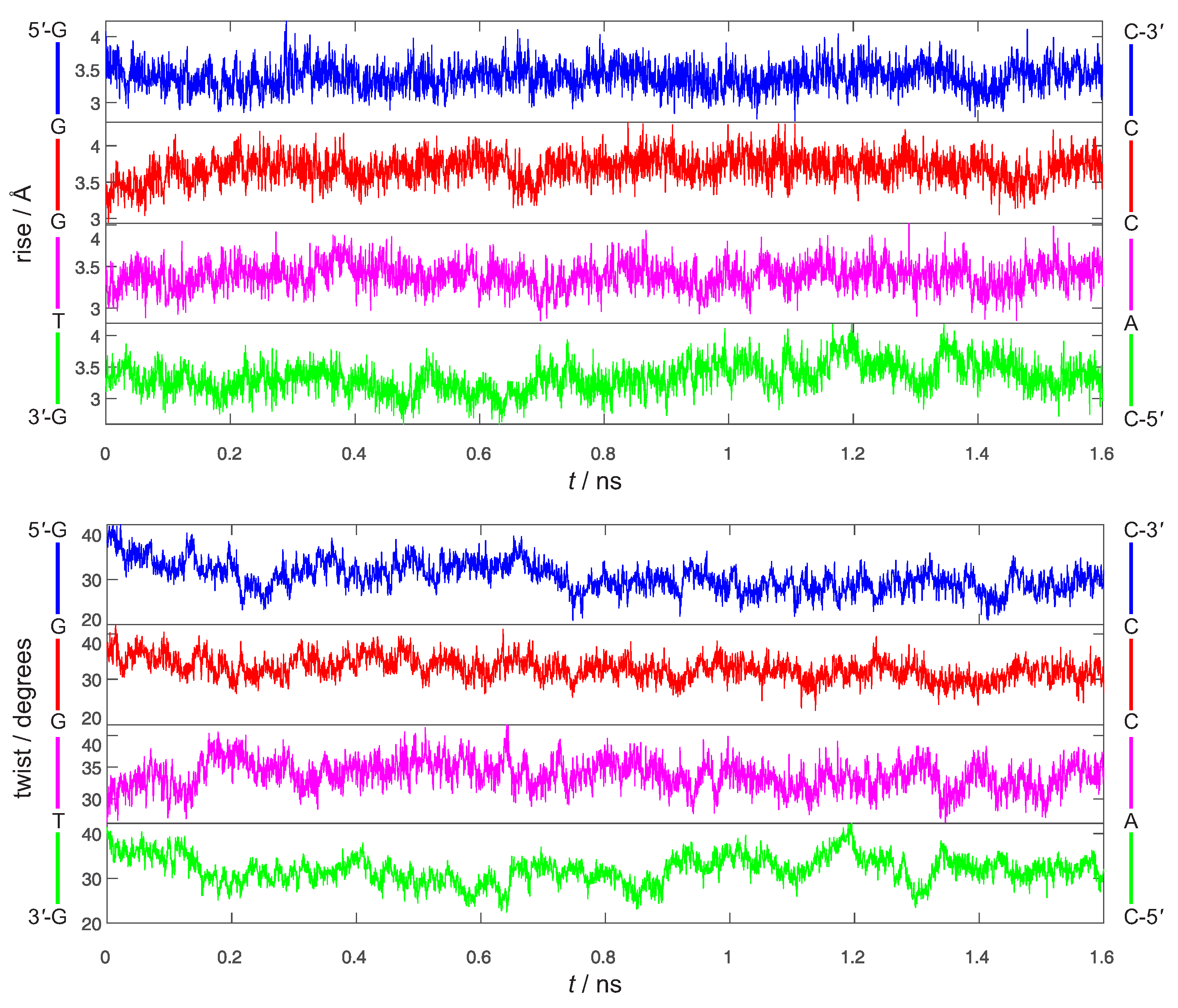
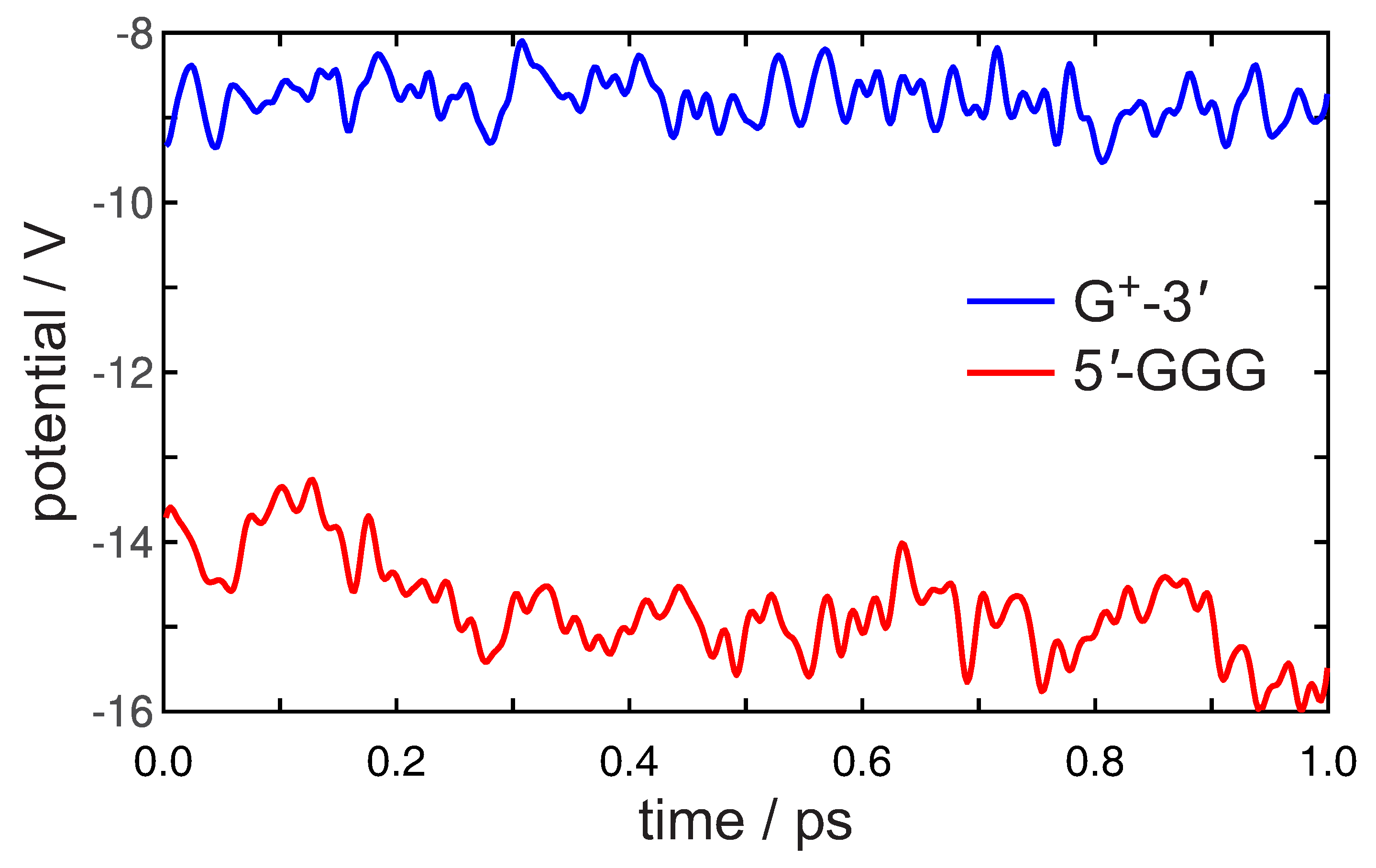
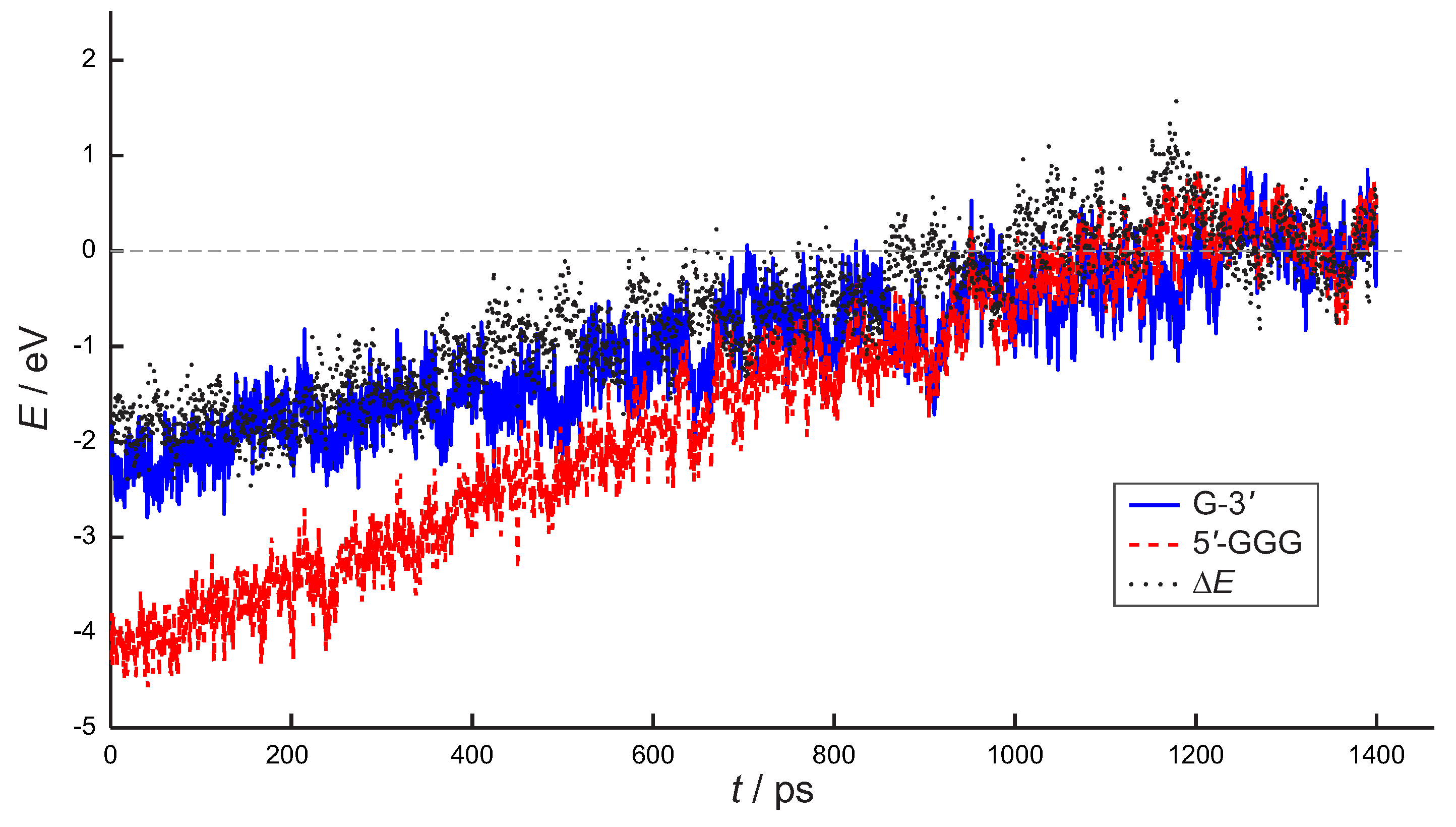
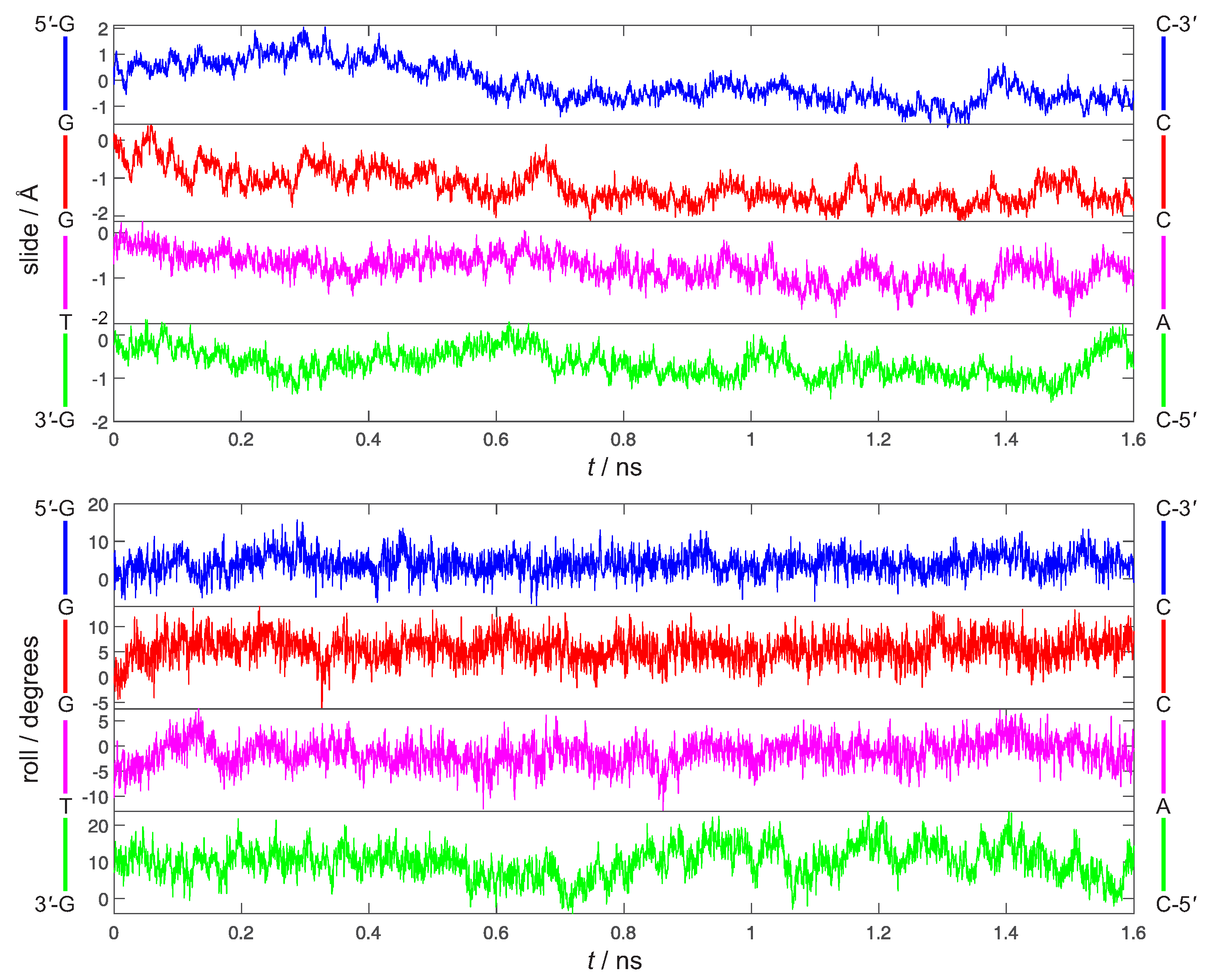
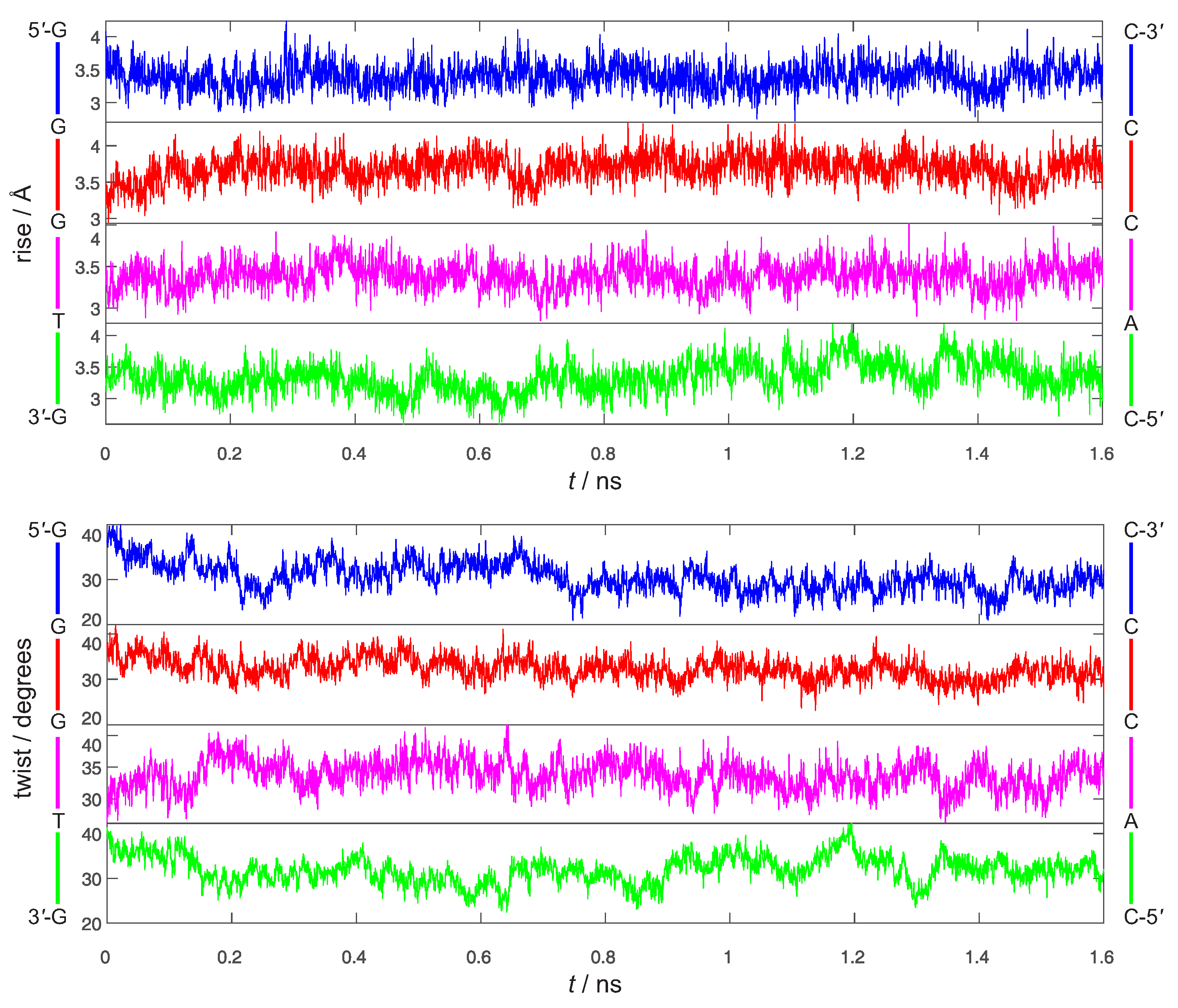
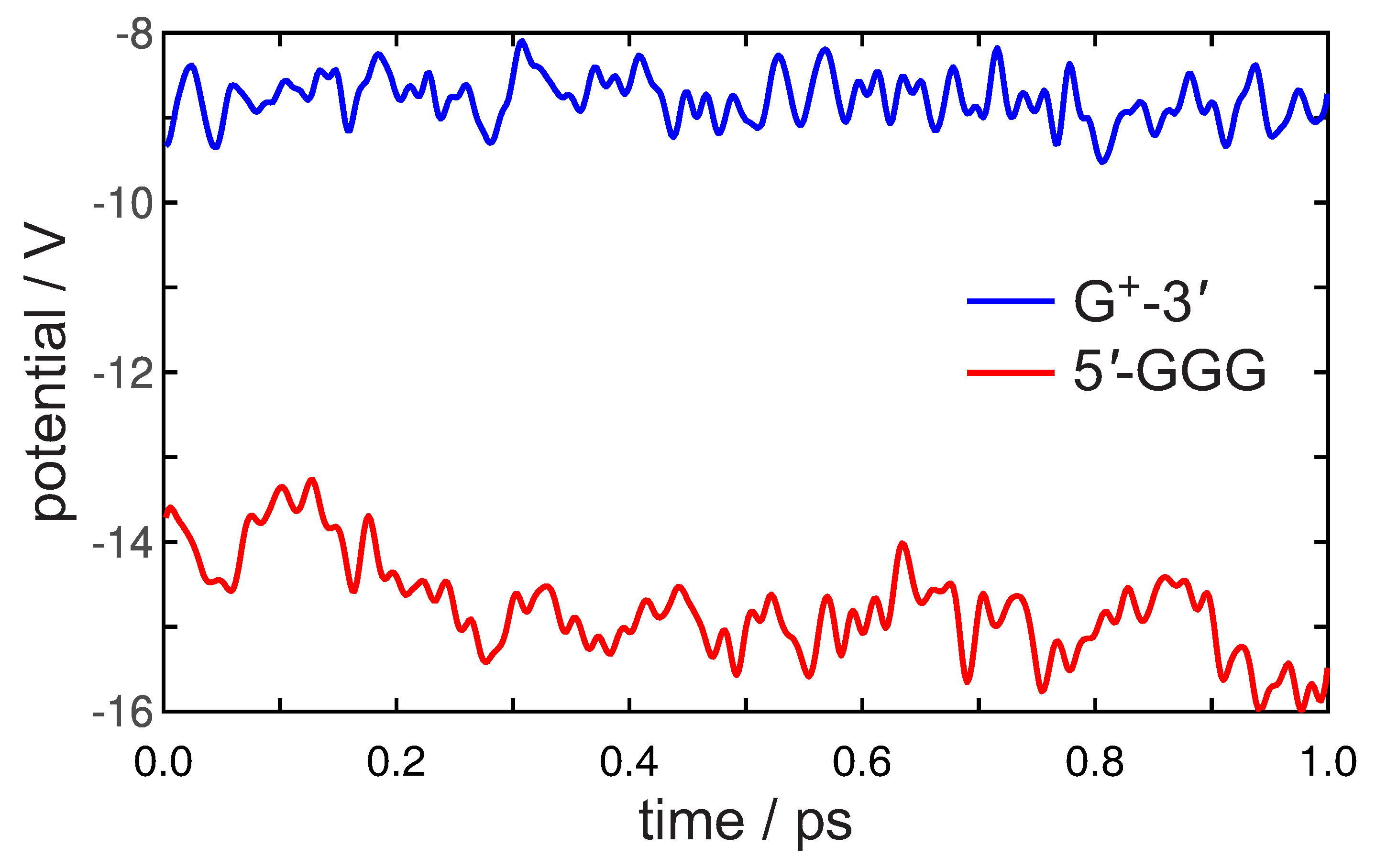
3. Preliminary Considerations
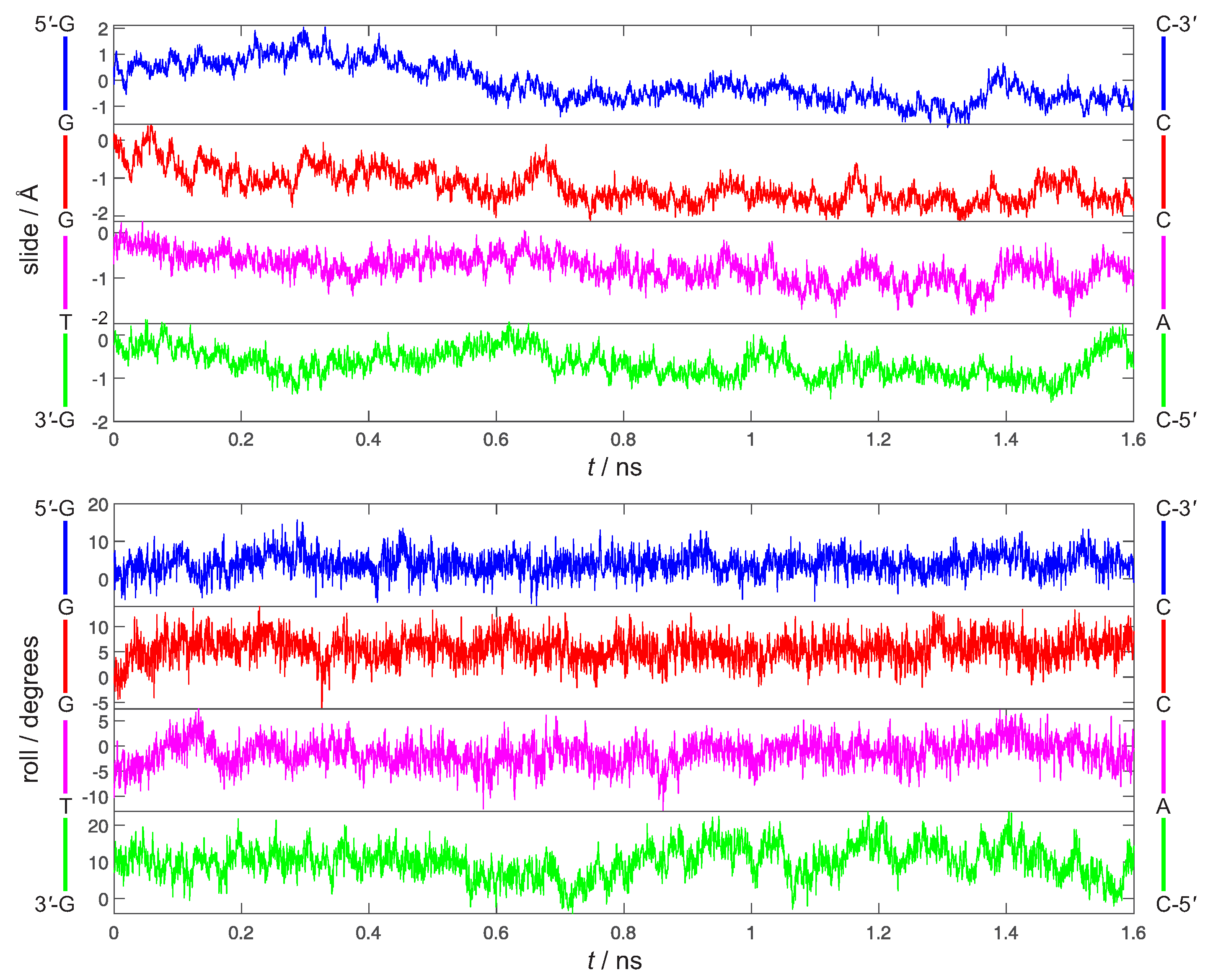
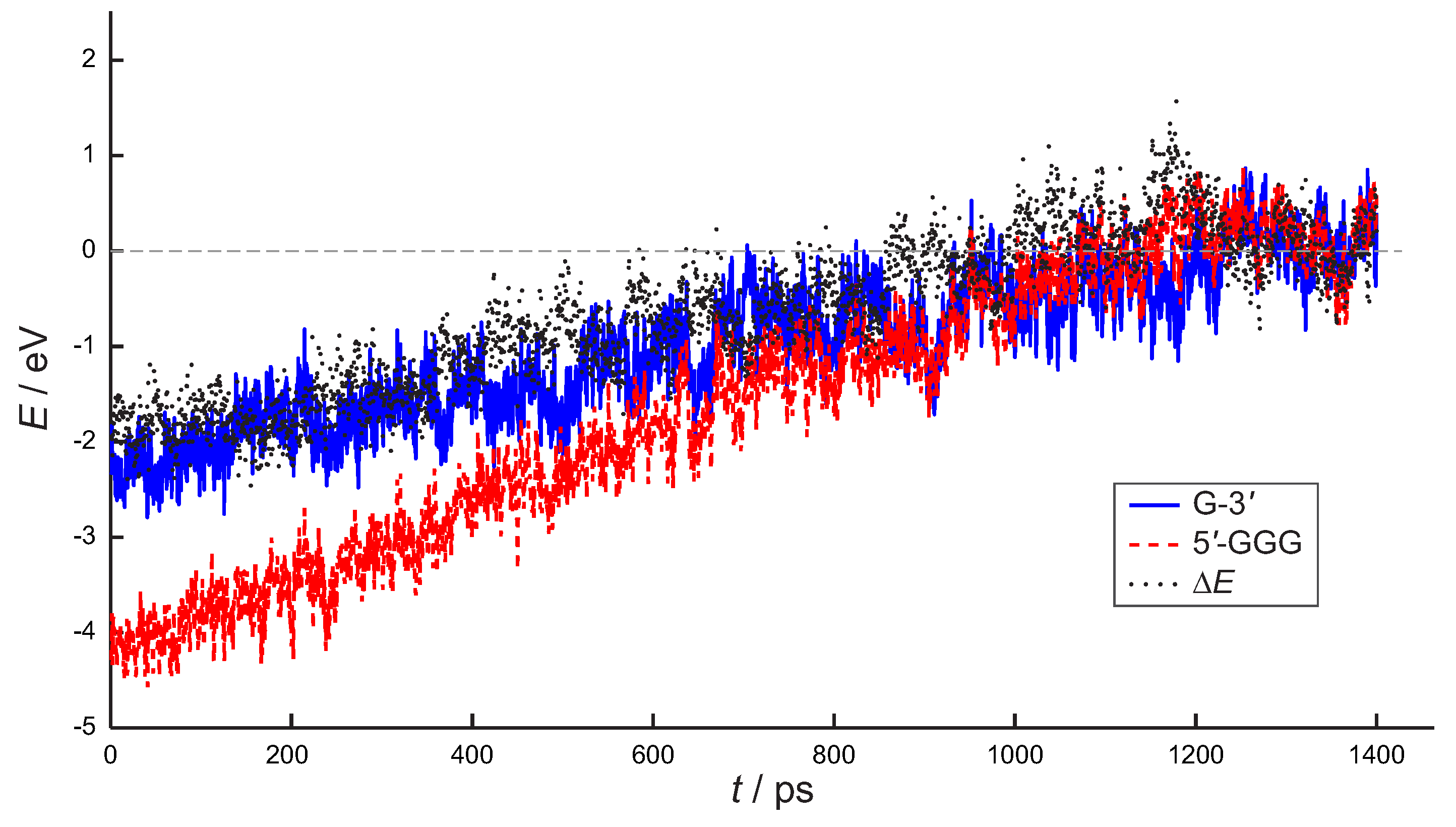
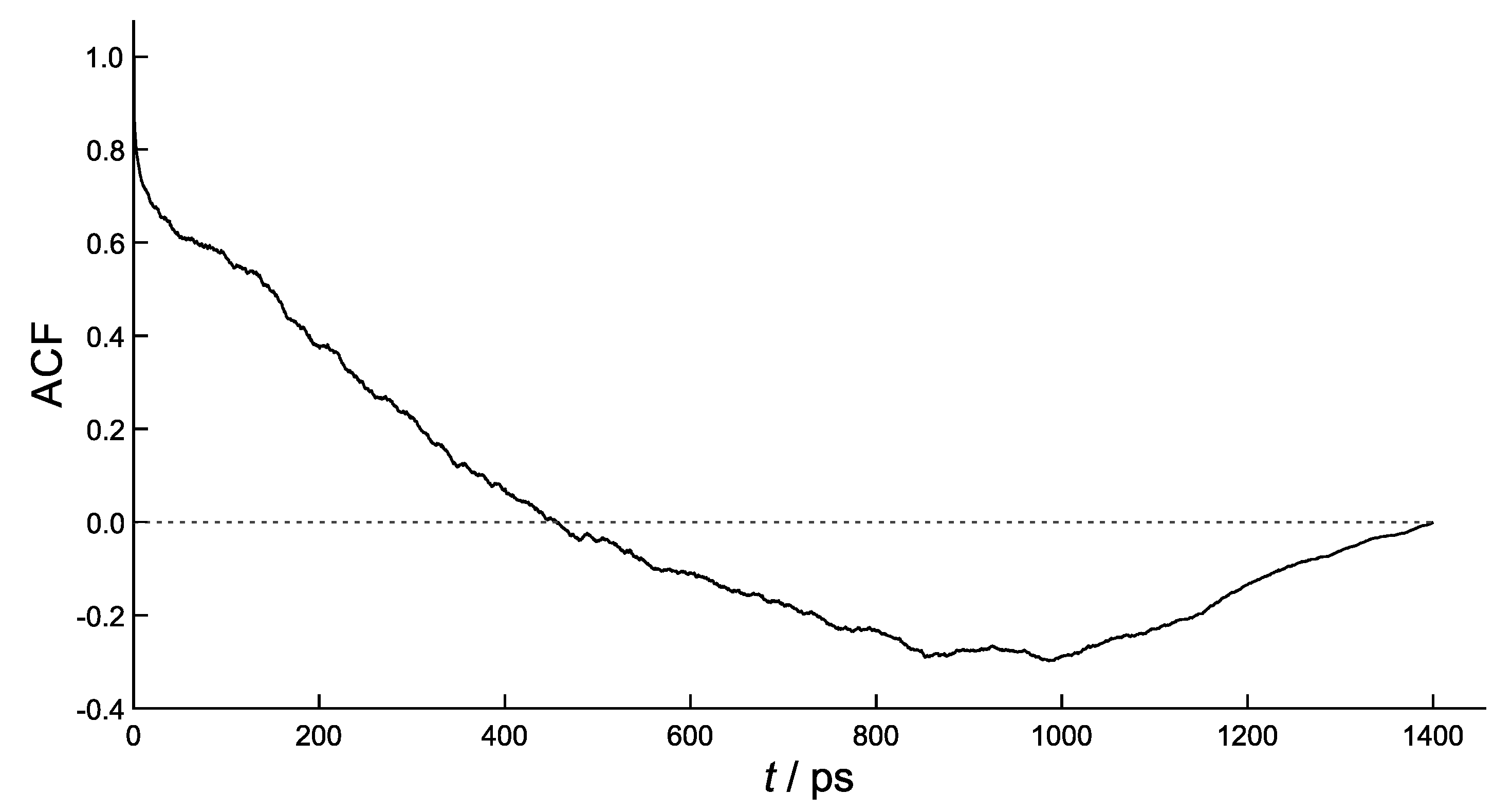
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to Nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Sevilla, M.D. Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem. Rev. 2010, 110, 7002–7023. [Google Scholar] [CrossRef] [Green Version]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef] [Green Version]
- Heller, A. Spiers Memorial Lecture. On the Hypothesis of Cathodic Protection of Genes. Faraday Discuss. 2000, 116, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rokhlenko, Y.; Cadet, J.; Geacintov, N.E.; Shafirovich, V. Mechanistic Aspects of Hydration of Guanine Radical Cations in DNA. J. Am. Chem. Soc. 2014, 136, 5956–5962. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Han, S.; Huang, W.; Yu, J. High Mobility Organic Field-Effect Transistor Based on Water-Soluble Deoxyribonucleic Acid via Spray Coating. Appl. Phys. Lett. 2015, 106, 043303. [Google Scholar] [CrossRef]
- Zhang, Y.; Zalar, P.; Kim, C.; Collins, S.; Bazan, G.C.; Nguyen, T.Q. DNA Interlayers Enhance Charge Injection in Organic Field-Effect Transistors. Adv. Mater. 2012, 24, 4255–4260. [Google Scholar] [CrossRef] [PubMed]
- Zalar, P.; Kamkar, D.; Naik, R.; Ouchen, F.; Grote, J.G.; Bazan, G.C.; Nguyen, T.Q. DNA Electron Injection Interlayers for Polymer Light-Emitting Diodes. J. Am. Chem. Soc. 2011, 133, 11010–11013. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.F.; Venkatraman, V.; Grote, J.G.; Steckl, A.J. Exploring the Potential of Nucleic Acid Bases in Organic Light Emitting Diodes. Adv. Mater. 2015, 27, 7552–7562. [Google Scholar] [CrossRef]
- Liedl, T.; Sobey, T.L.; Simmel, F.C. DNA-Based Nanodevices. Nano Today 2007, 2, 36–41. [Google Scholar] [CrossRef]
- Liang, L.; Fu, Y.; Wang, D.; Wei, Y.; Kobayashi, N.; Minari, T. DNA as Functional Material in Organic-Based Electronics. Appl. Sci. 2018, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, Y.; Kitagawa, Y.; Takano, Y.; Yamaguchi, K.; Nakamura, T.; Saito, I. Experimental and Theoretical Studies on the Selectivity of GGG Triplets toward One-Electron Oxidation in B-Form DNA. J. Am. Chem. Soc. 1999, 121, 8712–8719. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; D’Ursi, A.M.; Fusco, S.; Masi, A.; Scrima, M.; Chatgilialoglu, C.; Peluso, A. Delocalized Hole Domains in Guanine-Rich DNA Oligonucleotides. J. Phys. Chem. B 2015, 119, 5462–5466. [Google Scholar] [CrossRef]
- Capobianco, A.; Landi, A.; Peluso, A. Modeling DNA Oxidation in Water. Phys. Chem. Chem. Phys. 2017, 19, 13571–13578. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Peluso, A. The Oxidization Potential of AA Steps in Single Strand DNA Oligomers. RSC Adv. 2014, 4, 47887–47893. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Peluso, A. Hole Delocalization over Adenine Tracts in Single Stranded DNA Oligonucleotides. Phys. Chem. Chem. Phys. 2015, 17, 4750–4756. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Celentano, M.; D’Ursi, A.M.; Scrima, M.; Peluso, A. Stacking Interactions between Adenines in Oxidized Oligonucleotides. J. Phys. Chem. B 2013, 117, 8947–8953. [Google Scholar] [CrossRef]
- Capobianco, A.; Velardo, A.; Peluso, A. Single-Stranded DNA Oligonucleotides Retain Rise Coordinates Characteristic of Double Helices. J. Phys. Chem. B 2018, 122, 7978–7989. [Google Scholar] [CrossRef]
- Harris, M.A.; Mishra, A.K.; Young, R.M.; Brown, K.E.; Wasielewski, M.R.; Lewis, F.D. Direct Observation of the Hole Carriers in DNA Photoinduced Charge Transport. J. Am. Chem. Soc. 2016, 138, 5491–5494. [Google Scholar] [CrossRef] [PubMed]
- Schuster, G.B. Long-Range Charge Transfer in DNA: Transient Structural Distortions Control the Distance Dependence. Acc. Chem. Res. 2000, 33, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Caruso, T.; Carotenuto, M.; Vasca, E.; Peluso, A. Direct Experimental Observation of the Effect of the Base Pairing on the Oxidation Potential of Guanine. J. Am. Chem. Soc. 2005, 127, 15040–15041. [Google Scholar] [CrossRef] [PubMed]
- Caruso, T.; Capobianco, A.; Peluso, A. The Oxidation Potential of Adenosine and Adenosine-Thymidine Base-Pair in Chloroform Solution. J. Am. Chem. Soc. 2007, 129, 15347–15353. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528. [Google Scholar] [CrossRef]
- Giese, B.; Amaudrut, J.; Köhler, A.K.; Spormann, M.; Wessely, S. Direct Observation of Hole Transfer through DNA by Hopping between Adenine Bases and by Tunneling. Nature 2001, 412, 318–320. [Google Scholar] [CrossRef]
- Bixon, M.; Giese, B.; Wessely, S.; Langenbacher, T.; Michel-Beyerle, M.E.; Jortner, J. Long-Range Charge Hopping in DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11713–11716. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, R.; Capobianco, A.; Landi, A.; Peluso, A. Vibronic Couplings and Coherent Electron Transfer in Bridged Systems. Phys. Chem. Chem. Phys. 2015, 17, 30937–30945. [Google Scholar] [CrossRef] [Green Version]
- Renger, T.; Marcus, R.A. Variable Range Hopping Electron Transfer Through Disordered Bridge States: Application to DNA. J. Phys. Chem. A 2003, 107, 8404–8419. [Google Scholar] [CrossRef]
- Bixon, M.; Jortner, J. Incoherent Charge Hopping and Conduction in DNA and Long Molecular Chains. Chem. Phys. 2005, 319, 273–282. [Google Scholar] [CrossRef]
- Grozema, F.C.; Tonzani, S.; Berlin, Y.A.; Schatz, G.C.; Siebbeles, L.D.A.; Ratner, M.A. Effect of Structural Dynamics on Charge Transfer in DNA Hairpins. J. Am. Chem. Soc. 2008, 130, 5157–5166. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Balaeff, A.; Skourtis, S.S.; Beratan, D.N. Biological Charge Transfer via Flickering Resonance. Proc. Natl. Acad. Sci. USA 2014, 111, 10049–10054. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.D.; Iv, M.; Peskin, U. Length-Independent Transport Rates in Biomolecules by Quantum Mechanical Unfurling. Chem. Sci. 2016, 7, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Landi, A.; Borrelli, R.; Capobianco, A.; Peluso, A. Transient and Enduring Electronic Resonances Drive Coherent Long Distance Charge Transport in Molecular Wires. J. Phys. Chem. Lett. 2019, 10, 1845–1851. [Google Scholar] [CrossRef]
- Landi, A.; Capobianco, A.; Peluso, A. Coherent Effects in Charge Transport in Molecular Wires: Toward a Unifying Picture of Long-Range Hole Transfer in DNA. J. Phys. Chem. Lett. 2020, 11, 7769–7775. [Google Scholar] [CrossRef]
- Jimenez, R.; Fleming, G.R.; Kumar, P.V.; Maroncelli, M. Femtosecond Solvation Dynamics of Water. Nature 1994, 369, 471–473. [Google Scholar] [CrossRef]
- Fleming, G.R.; Cho, M. Chromophore-Solvent Dynamics. Annu. Rev. Phys. Chem. 1996, 47, 109–134. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Tagawa, S. Direct Observation of Guanine Radical Cation Deprotonation in Duplex DNA Using Pulse Radiolysis. J. Am. Chem. Soc. 2003, 125, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S. Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e− and OH Adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Celentano, M.; La Rocca, M.V.; Peluso, A. Proton Transfer in Oxidized Adenosine Self-Aggregates. J. Chem. Phys. 2013, 139, 145101-4. [Google Scholar] [CrossRef] [PubMed]
- Candeias, L.P.; Steenken, S. Structure and Acid-Base Properties of One-Electron-Oxidized Deoxyguanosine, Guanosine, and 1-Methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Caminal, C.; Guerra, M.; Mulazzani, Q.G. Tautomers of One-Electron-Oxidized Guanosine. Angew. Chem. Int. Ed. 2005, 44, 6030–6032. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Caminal, C.; Altieri, A.; Vougioukalakis, G.C.; Mulazzani, Q.G.; Gimisis, T.; Guerra, M. Tautomerism in the Guanyl Radical. J. Am. Chem. Soc. 2006, 128, 13796–13805. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Schuster, G.B. Emergent Functionality of Nucleobase Radical Cations in Duplex DNA: Prediction of Reactivity Using Qualitative Potential Energy Landscapes. J. Am. Chem. Soc. 2006, 128, 6070–6074. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A Versatile, Integrated Software System for the Analysis, Rebuilding and Visualization of Three-dimensional Nucleic-Acid Structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbovic, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E., III. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L. A modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecher, T.; Koslowski, T.; Case, D.A. Direct Simulation of Electron Transfer Reactions in DNA Radical Cations. J. Phys. Chem. B 2008, 112, 16935–16944. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.A.; Draper, D.E.; Pappu, R.V. Molecular Simulation Studies of Monovalent Counterion-Mediated Interactions in a Model RNA Kissing Loop. J. Mol. Biol. 2009, 390, 805–819. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.A.; Marucho, N.; Baker, N.A.; Pappu, R.V. Simulations of RNA Interactions with Monovalent Ions. In Methods in Enzymology; Academic Press: New York, NY, USA, 2009; Volume 469, pp. 411–432. [Google Scholar]
- Várnai, P.; Zakrzewska, K. DNA and Its Counterions: A Molecular Dynamics Study. Nucleic Acids Res. 2004, 32, 4269–4280. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Crowley, M.F.; Darden, T.A.; Cheatham, T.E., III; Deerfield, D.W., II. Adventures in Improving the Scaling and Accuracy of a Parallel Molecular Dynamics Program. J. Supercomput. 1997, 11, 255–278. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E., III. Convergence and Reproducibility in Molecular Dynamics Simulations of the DNA Duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowasser, R.; Hoffmann, R. What Do the Kohn-Sham Orbitals and Eigenvalues Mean? J. Am. Chem Soc. 1999, 121, 3414–3420. [Google Scholar] [CrossRef]
- Clemente, F.R.; Vreven, T.; Frisch, M.J. Getting the Most out of ONIOM: Guidelines and Pitfalls. In Quantum Biochemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; Chapter 2; pp. 61–83. [Google Scholar]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A Standard Reference Frame for the Description of Nucleic Acid Base-pair Geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.J.; Olson, W.K. 3DNA: A Software Package for the Analysis, Rebuilding and Visualization of Three-Dimensional Nucleic Acid Structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voityuk, A.A.; Jortner, J.; Bixon, M.; Rösch, N. Energetic of Hole Transfer in DNA. Chem. Phys. Lett. 2000, 324, 430–434. [Google Scholar] [CrossRef]
- Troisi, A.; Orlandi, G. The Hole Transfer in DNA: Calculation of Electron Coupling between Close Bases. Chem. Phys. Lett. 2001, 344, 509–518. [Google Scholar] [CrossRef]
- Voityuk, A.A.; Jortner, J.; Bixon, M.; Rösch, N. Electronic Coupling between Watson-Crick Pairs for Hole Transfer and Transport in Desoxyribonucleic Acid. J. Chem. Phys. 2001, 114, 5614–5620. [Google Scholar] [CrossRef] [Green Version]
- Voityuk, A.A.; Siriwong, K.; Rösch, N. Environmental Fluctuations Facilitate Electron-Hole Transfer from Guanine to Adenine in DNA π Stacks. Angew. Chem. Int. Ed. 2004, 43, 624–627. [Google Scholar] [CrossRef]
- Denisov, V.P.; Carlström, G.; Venu, K.; Halle, B. Kinetics of DNA Hydration. J. Mol. Biol. 1997, 268, 118–136. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.T.; Leroy, J.L.; Guéron, M. Determination of the Residence Time of Water Molecules Hydrating B′-DNA and B-DNA, by One-Dimensional Zero-Enhancement Nuclear Overhauser Effect Spectroscopy. J. Mol. Biol. 1999, 286, 505–519. [Google Scholar] [CrossRef]
- Marcus, R.A. Solvent Dynamics-Modified RRKM Theory in Clusters. Chem. Phys. Lett. 1995, 244, 10–18. [Google Scholar] [CrossRef]
- El Hassan, M.A.; Calladine, C.R. Conformational Characteristics of DNA: Empirical Classifications and a Hypothesis for the Conformational Behaviour of Dinucleotide Steps. Philos. Trans. R. Soc. A 1997, 355, 43–100. [Google Scholar] [CrossRef]
- Heddi, B.; Oguey, C.; Lavelle, C.; Foloppe, N.; Hartmann, B. Intrinsic Flexibility of B-DNA: The Experimental TRX Scale. Nucleic Acids Res. 2010, 38, 1034–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Lankaš, F.; Jurečka, P. Base Pair Fraying in Molecular Dynamics Simulations of DNA and RNA. J. Chem. Theory Comput. 2014, 10, 3177–3189. [Google Scholar] [CrossRef]
- Sugiyama, H.; Saito, I. Theoretical Studies of GG-Specific Photocleavage of DNA via Electron Transfer: Significant Lowering of Ionization Potential and 5′-Localization of HOMO of Stacked GG Bases in B-form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Lewis, F.D.; Berlin, Y.A.; Ratner, M.A.; Siebbeles, L.D.A. Absolute Rates of Hole Transfer in DNA. J. Am. Chem. Soc. 2005, 127, 14894–14903. [Google Scholar] [CrossRef]
- Persson, R.A.X.; Pattni, V.; Singh, A.; Kast, S.M.; Heyden, M. Signatures of Solvation Thermodynamics in Spectra of Intermolecular Vibrations. J. Chem. Theory Comput. 2017, 13, 4467–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | ps | ps | exper. |
|---|---|---|---|
| 1 | 31 | 273 | 250 |
| 2 | 4.2 | 42 | 30 |
| 3 | 0.5 | 5.2 | 4.0 |
| 4 | 0.35 | 3.0 | 3.5 |
| 5 | 0.28 | 2.5 | 3.0 |
| 6 | 0.23 | 2.2 | 2.6 |
| 7 | 0.20 | 2.0 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landi, A.; Capobianco, A.; Peluso, A. The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study. Molecules 2021, 26, 5497. https://doi.org/10.3390/molecules26185497
Landi A, Capobianco A, Peluso A. The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study. Molecules. 2021; 26(18):5497. https://doi.org/10.3390/molecules26185497
Chicago/Turabian StyleLandi, Alessandro, Amedeo Capobianco, and Andrea Peluso. 2021. "The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study" Molecules 26, no. 18: 5497. https://doi.org/10.3390/molecules26185497
APA StyleLandi, A., Capobianco, A., & Peluso, A. (2021). The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study. Molecules, 26(18), 5497. https://doi.org/10.3390/molecules26185497