4.1.2. Syntheses
Dicarboxylic Acid Anhydrides—General Procedure (1b,c), First, 19 mmol of a dicarboxylic acid was dissolved in 50 mL of acetic anhydride and refluxed for 4 h. The solvents were removed under reduced pressure, obtaining 18 mmol of a cyclic anhydride.
Pimelic Anhydride 1b, Starting from 3 g (19 mmol) of pimelic acid, 2.64 g (18 mmol, 95%) of pimelic anhydride 1b was obtained as a light-beige, low-melting solid. 1H NMR (500 MHz, DMSO-d6) δ: 2.50 (t, J = 7.3 Hz, 4H), 1.55 (qt, J = 7.4 Hz, 4H), 1.33 (qt, J = 6.9 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ: 170.08, 34.83, 27.85, 23.88.
Suberic Anhydride 1c, Starting from 10 g (57 mmol) of suberic acid, 9 g (56 mmol, 98%) of suberic anhydride 1c was obtained as a light-beige, low-melting solid. 1H NMR (500 MHz, DMSO-d6) δ: 2.49 (t, J = 7.1 Hz, 4H), 1.56 (m, 4H), 1.32 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ: 169.66, 34.47, 28.22, 24.03.
Mono-tert-butyl esters (3a–c)—General Procedure, First, 20 mmol of carboxylic acid anhydride, 2 mmol of DMAP, and 5.88 mmol of NHS were suspended in 75 mL of toluene. Subsequently, 40 mmol of tert-butanol and 6 mmol of TEA were added, and the resulting mixture was refluxed for 24 h in an oil bath. The mixture was cooled to room temperature; then, 75 mL of AcOEt was added. The resulting solution was washed with 1M HCl(aq) (3 × 70 mL) and with brine (3 × 70 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography or crystallization.
Mono-tert-butyl succinate 3a, Starting from 25.37 g (0.25 mol) of succinic anhydride 1a, 21.1 g (0.12 mol, 48%) of monoester 3a was obtained as a white solid with m.p. 42–44 °C (AcOEt/hexanes). 1H NMR (500 MHz, DMSO-d6) δ: 12.13 (brs, 1H), 2.38 (s, 4H), 1.39 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ: 173.67, 171.88, 80.08, 30.24, 29.10, 27.31.
Mono-tert-butyl pimelate 3b, Starting from 6 g (42.08 mmol) of pimelic anhydride 1b, 5.25 g (24.27 mmol, 58%) of monoester 3b was obtained as a light-yellow oil with Rf 0.67 (hexanes/AcOEt, 4/6, v/v). 1H NMR (500 MHz, DMSO-d6) δ: 11.98 (brs, 1H), 2.18 (m, 4H), 1.50 (m, 4H), 1.40 (s, 9H), 1.27 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ: 174.86, 172.79, 79.97, 35.08, 34.05, 28.42, 28.22, 24.80, 24.64.
Mono-tert-butyl suberate 3c, Starting from 9 g (58 mmol) of suberic anhydride 1c, 3.23 g (14.03 mmol, 24%) of monoester 3c was obtained as colorless oil with Rf 0.63 (hexanes/AcOEt/AcOH, 80/20/1, v/v/v). 1H NMR (500 MHz, DMSO-d6) δ: 11.97 (brs, 1H), 2.17 (m, 4H), 1.47 (m, 4H), 1.39 (s, 9H), 1.25 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ: 174.96, 172.60, 79.63, 35.14, 34.00, 28.64, 28.54, 28.20, 24.90, 24.78.
4,4,5,7-tetramethylchroman-2-one 6, To a mixture of 22.21 g (0.18 mol) of 3,5-dimethylphenol 5 dissolved in 40 mL of methanesulfonic acid, 23 g (0.20 mol) of methyl 3-methylbut-2-enoate 4 was added in one portion. The reaction mixture was stirred and heated at a 70 °C in an oil bath for 18 h. The mixture was cooled to room temperature, poured into 400 mL of cold water, and extracted with ethyl acetate (3 × 150 mL). The organic layer was washed with water (3 × 150 mL), saturated NaHCO3(aq) (3 × 150 mL), and water (2 × 150 mL), respectively. The acetate layer was dried over anhydrous MgSO4, the desiccant was removed, and the filtrate was concentrated under reduced pressure. The residue was recrystallized from diethyl ether obtaining 33.12 g (0.16 mol, 90%) of product 6 as colorless crystals, with m.p. 88–91 °C and Rf 0.35 (hexanes/AcOEt, 9/1, v/v). 1H NMR (500 MHz, DMSO-d6) δ: 6.78 (s, 1H), 6.72 (s, 1H), 2.65 (s, 2H), 2.42 (s, 3H), 2.21 (s, 3H), 1.34 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 168.35, 151.79, 137.28, 136.29, 129.46, 127.34, 115.75, 45.00, 35.06, 27.57, 23.00, 20.35. FTIR ν (cm−1): 3050, 2900, 1775, 1625, 1575.
2-(3-hydroxy-1,1-dimethylpropyl)-3,5-dimethylphenol 7, To a stirred suspension of 4 g (0.104 mol) of LiAlH4 in 100 mL of dry THF, 5.3 g (26 mmol) of lactone 6 was added in small portions. Once the addition was completed, the reaction mixture was stirred at room temperature for 3 h. The excess of unreacted LiAlH4 was decomposed by adding AcOEt, MeOH, and water, respectively. The slurry was filtered off under reduced pressure, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 100 mL of dichloromethane and dried over anhydrous MgSO4. After the removal of desiccant and concentration of filtrate under reduced pressure, the residue was recrystallized from dichloromethane. Obtained 3.47 g (17 mmol, 65%) of product 7 as colorless crystals, with m.p. 112–114 °C and Rf 0.27 (hexanes/AcOEt, 7/3, v/v). 1H NMR (500 MHz, DMSO-d6) δ: 8.96 (s, 1H), 6.43 (s, 1H), 6.28 (s, 1H), 4.11 (t, J = 4.9 Hz, 1H), 3.20 (m, 2H), 2.35 (s, 3H), 2.07 (s, 3H), 2.04 (t, J = 7.9 Hz, 2H), 1.44 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 157.42, 137.25, 135.16, 128.95, 125.79, 116.22, 59.59, 45.23, 40.00, 32.34, 25.98, 20.58.
2-{3-[(O-tert-butyldimethylsilyl)hydroxy]-1,1-dimethylpropyl}-3,5-dimethylphenol 8, The reaction was carried out under argon atmosphere. To an ice-cold stirred mixture of 250 mg (1.2 mmol) of phenol 7 and 244 mg (2 mmol) of DMAP, dissolved in 10 mL of dry dichloromethane, 217 mg (1.44 mmol) of TBDMSCl in 10 mL of dry dichloromethane was added dropwise. The mixture was stirred in an ice bath for 2 h and then for 3 h at room temperature. Subsequently, the mixture was washed with water (2 × 10 mL), 5% solution of NaHCO3(aq) (3 × 10 mL), and water (2 × 10 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was recrystallized from dichloromethane, obtaining 400 mg (1.2 mmol, 100%) of product 8 as white crystals, with m.p. 106–109 °C and Rf 0.52 (hexanes/AcOEt, 9/1, v/v). 1H NMR (500 MHz, DMSO-d6) δ: 8.99 (s, 1H), 6.43 (s, 1H), 6.28 (s, 1H), 3.39 (t, J = 7.4 Hz, 2H), 2.36 (s, 3H), 2.09 (m, 2H), 2.05 (s, 3H), 1.42 (s, 6H), 0.79 (s, 9H), −0.08 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 157.25, 137.27, 134.91, 128.29, 125.93, 115.85, 61.50, 45.50, 32.37, 26.44, 25.86, 20.68, 18.15, −4.71.
2-{3-[(O-tert-butyldimethylsilyl)hydroxy]-1,1-dimethylpropyl}-O′-acyloyl-3,5-dimethylphenol (9a–c)—General Procedure, A stirred mixture of 15.5 mmol of phenol, 23.5 mmol of carboxylic acid, and 2.05 mmol of DMAP in 100 mL of dry dichloromethane was cooled in an ice bath to 0 °C, and then, 30.5 mmol of DCC dissolved in 20 mL of dry dichloromethane was added dropwise. The mixture was stirred at room temperature for 48 h. The precipitate of DCU was filtered off, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 100 mL of chloroform and washed with saturated NaHCO3(aq) (2 × 50 mL), 5% NaHSO4(aq) (2 × 50 mL), and water (2 × 50 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt, 9/1, v/v as a mobile phase.
tert-Butyl-2-{3-[(O-tert-butyldimethylsilyl)hydroxy]-1,1-dimethylpropyl}-3,5-dimethylphenyl succinate 9a, Starting from 810 mg (4.65 mmol) of carboxylic acid 3a and 1 g (3.1 mmol) of phenol 8, 1.41 g (2.94 mmol, 95%) of ester 9a was obtained as a colorless oil. Rf 0.52 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C27H46O5Si 478.3115; found 479.3251 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 6.82 (s, 1H), 6.55 (s, 1H), 3.42 (t, J = 7.2 Hz, 2H), 2.75 (t, J = 6.7 Hz, 2H), 2.56 (t, J = 6.3 Hz, 2H), 2.48 (s, 3H), 2.17 (s, 2H), 1.96 (t, J = 7.2 Hz, 2H), 1.41 (s, 6H), 1.39 (s, 9H), 0.81 (s, 9H), −0.06 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 171.74, 171.44, 150.06, 138.24, 135.58, 134.00, 132.25, 123.37, 80.45, 60.83, 46.02, 39.17, 32.05, 30.07, 28.15, 26.23, 25.15, 20.01, 18.08, −4.87.
tert-Butyl-2-{3-[(O-tert-butyldimethylsilyl)hydroxy]-1,1-dimethylpropyl}-3,5-dimethylphenyl pimelate 9b, Starting from 2.67 g (12 mmol) of carboxylic acid 3b and 2.58 g (8 mmol) of phenol 8, 3.40 g (6.52 mmol, 82%) of ester 9b was obtained as a colorless oil. Rf 0.50 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C30H52O5Si 520.3584; found 521.3579 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 6.83 (s 1H), 6.58 (s, 1H), 3.42 (t, J = 7.4 Hz, 2H), 2.54 (m, 2H), 2.48 (s, 3H), 2.21 (m, 2H), 2.18 (s, 3H), 1.95 (t, J = 7.5 Hz, 2H), 1.63 (qt, J = 7.5 Hz, 2H), 1.54 (qt, J = 7.4 Hz, 2H), 1.40 (m, 17H), 0.81 (s, 9H), −0.06 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 172.57, 172.44, 150.09, 138.19, 135.77, 134.12, 132.18, 123.59, 79.86, 79.69, 60.63, 45.94, 39.15, 35.03, 34.49, 32.05, 28.31, 28.21, 26.23, 25.30, 24.76, 24.26, 20.12, 18.29.
tert-Butyl-2-{3-[(O-tert-butyldimethylsilyl)hydroxy]-1,1-dimethylpropyl}-3,5-dimethylphenyl suberate 9c, Starting from 3.09 g (13.40 mmol) of carboxylic acid 3c and 3.60 g (11.16 mmol) of phenol 8, 4.82 g (9.01 mmol, 81%) of ester 9c was obtained as a colorless oil. Rf 0.64 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C31H54O5Si 534.3741; found 535.3743 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 6.81 (s, 1H), 6.56 (s, 1H), 3.40 (t, J = 7.3 Hz, 2H), 2.52 (m, 2H), 2.46 (s, 3H), 2.17 (s, 3H), 1.93 (t, J = 7.3 Hz, 2H), 1.62 (qt, J = 7.2 Hz, 2H), 1.50 (qt, J = 7.5 Hz, 2H), 1.39 (m, 15H), 1.35–1.09 (m, 6H), 0.78 (s, 9H), −0.06 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 172.63, 172.50, 150.04, 138.15, 135.76, 134.07, 132.21, 123.64, 79.71, 60.55, 45.81, 39.15, 35.13, 34.45, 32.01, 28.52, 28.20, 27.80, 26.21, 25.80, 25.32, 24.79, 24.35, 20.13, 18.18, −5.02.
2-(3-hydroxy-1,1-dimethylpropyl}-O′-acyloyl-3,5-dimethylphenol (10a–c)—General Procedure, The silyl ether was dissolved in 200 mL of a mixture of THF/H2O/AcOH, 1/1/3, v/v/v and stirred at room temperature for 3 h. The solvent was evaporated under reduced pressure. The residue was dissolved in 100 mL of AcOEt and washed with water (30 mL), saturated NaHCO3(aq) (3 × 30 mL), and water (3 × 30 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt, 7/3, v/v as a mobile phase.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl succinate 10a, Starting from 204 mg (0.43 mmol) of ester 9a, 112 mg (0.31 mmol, 72%) of alcohol 10a was obtained as a colorless oil. Rf 0.41 (hexanes/AcOEt, 7/3, v/v). HRMS-ESI: m/z calcd. for C21H32O5 364.2251; found 365.2580 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 6.81 (s, 1H), 6.54 (s, 1H), 4.20 (t, J = 5 Hz, 1H), 3.20 (m, 2H), 2.76 (t, J = 6.6 Hz, 2H), 2.55 (t, J = 6.3 Hz, 2H), 2.48 (s, 3H), 2.15 (s, 3H), 1.90 (t, J = 7.7 Hz, 2H), 1.40 (s, 9H), 1.39 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 171.25, 170.92, 149.28, 137.73, 135.04, 133.79, 131.55, 122.79, 79.78, 58.06, 45.43, 38.41, 31.30, 29.68, 27.48, 24.58, 19.37.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl pimelate 10b, Starting from 3.40 g (6.52 mmol) of ester 9b, 1.40 g (3.44 mmol, 53%) of alcohol 10b was obtained as a colorless oil. Rf 0.46 (hexanes/AcOEt, 7/3, v/v). HRMS-ESI: m/z calcd. for C24H38O5 406.2790; found 407.2789 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 6.81 (s, 1H), 6.52 (s, 1H), 3.53 (t, J = 6.9 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 2.52 (s, 3H), 2.24 (t, J = 7.3 Hz, 2H), 2.22 (s, 3H), 2.04 (t, J = 7.4 Hz, 2H), 1.77 (qt, J = 7.5 Hz, 2H), 1.64 (qt, J = 7.7 Hz, 2H), 1.48 (s, 6H), 1.46–1.38 (m, 11H). 13C NMR (125 MHz, CDCl3) δ: 168.41, 168.27, 145.02, 133.71, 131.42, 129.05, 127.73, 118.52, 75.40, 55.76, 41.01, 34.37, 30.58, 30.09, 27.27, 23.86, 23.37, 20.58, 19.99, 19.60, 15.42.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl suberate 10c, Starting from 4.82 g (9.01 mmol) of ester 9c, 3.25 g (7.72 mmol, 86%) of alcohol 10c was obtained as a colorless oil. Rf 0.50 (hexanes/AcOEt, 7/3, v/v). HRMS-ESI: m/z calcd. for C25H40O5 420.2876; found 421.2875 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 6.84 (s, 1H), 6.54 (s, 1H), 3.56 (t, J = 7.3 Hz, 2H), 2.56 (m, 5H), 2.22 (m, 6H), 2.07 (t, J = 7.1 Hz, 2H), 1.78 (qt, J = 7.6 Hz, 2H), 1.63 (qt, J = 7.4 Hz, 2H), 1.49 (s, 6H), 1.45 (s, 9H), 1.41 (m, 4H). 13C NMR (125 MHz, CDCl3) δ: 174.14, 173.67, 155.42, 137.84, 135.95, 128.08, 126.93, 116.11, 80.39, 633.25, 40.81, 39.66, 35.51, 34.336, 31.64, 28.67, 28.61, 28.12, 25.60, 24.87, 24.733, 20.24.
2-(3-oxo-1,1-dimethylpropyl}-O-acyloyl-3,5-dimethylphenol—General Procedure (11a–c), The reaction was carried out under argon atmosphere. To a stirred solution of 8.55 mmol of alcohol dissolved in 100 mL of dry dichloromethane, 17.2 mmol of PCC was added in one portion. The reaction mixture was stirred at room temperature for 4 h and then was filtered through silica gel using a mixture of hexane/AcOEt, 7/3, v/v as eluent.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl succinate 11a, Starting from 429 mg (1.18 mmol) of alcohol 10a, 362 mg (1 mmol, 85%) of aldehyde 11a was obtained as a light-yellow oil. Rf 0.33 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C21H30O5 362.2091; found 363.2430 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 9.44 (t, J = 2.3 Hz, 1H), 6.85 (s, 1H), 6.58 (s, 1H), 2.88 (d, J = 2.1 Hz, 2H), 2.77 (t, J = 6.6 Hz, 2H), 2.55 (t, J = 6.6 Hz, 2H), 2.48 (s, 3H), 2.17 (s, 3H), 1.47 (s, 6H), 1.38 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ: 202.74, 171.99, 171.61, 149.60, 138.10, 136.12, 133.51, 132.17, 123.56, 80.59, 56.31, 37.96, 31.40, 30.10, 28.09, 25.19, 19.90.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl pimelate 11b, Starting from 1.40 g (3.44 mmol) of alcohol 10b, 1.35 g (3.34 mmol, 97%) of aldehyde 11b was obtained as a light-yellow oil. Rf 0.32 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C24H36O5 404.2563; found 405.2559 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 9.45 (t, J = 2.4 Hz, 1H), 6.86 (s, 1H), 6.61 (s, 1H), 2.81 (d, J = 2.2 Hz, 2H), 2.57 (t, J = 7.1 Hz, 2H), 2.50 (s, 3H), 2.21 (t, J = 7.1 Hz, 2H), 2.18 (s, 3H), 1.64 (qt, J = 7.3 Hz, 2H), 1.54 (qt, J = 7.6 Hz, 2H), 1.49 (s, 6H), 1.45–1.30 (m, 11H). 13C NMR (125 MHz, DMSO-d6) δ: 203.08, 172.94, 149.63, 138.21, 136.30, 133.61, 132.56, 123.63, 79.90, 56.66, 38.15, 35.03, 34.45, 31.55, 28.30, 28.23, 25.33, 24.74, 24.27, 20.14.
tert-Butyl-2-(3-hydroxy-1,1-dimethylpropyl}-3,5-dimethylphenyl suberate 11c, Starting from 3.25 g (7.72 mmol) of alcohol 10c, 2.92 g (6.98 mmol, 90%) of aldehyde 11c was obtained as a light-yellow oil. Rf 0.37 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C25H38O5 418.2719; found 419.2721 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 9.45 (t, J = 2.2 Hz, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 2.80 (d, J = 2.7 Hz, 2H), 2.57 (t, J = 6.6 Hz, 2H), 2.49 (s, 3H), 2.18 (m, 5H), 1.62 (qt, J = 7.2 Hz, 2H), 1.50 (qt, J = 7.5 Hz, 2H), 1.47 (s, 6H), 1.39 (s, 9H), 1.33 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ: 203.06, 172.72, 172.69, 149.66, 137.97, 136.42, 133.40, 132.34, 123.64, 79.86, 56.36, 38.09, 35.11, 34.46, 31.53, 28.51, 28.47, 28.20, 25.32, 24.86, 24.35, 20.14.
3-[2-(O-acyloyl)hydroxyl-4,6-dimethyl]phenyl-3-methylbutanoic acid (12a–c)—General Procedure, To a stirred, ice-cold mixture of 2.48 mmol of aldehyde, 100 μL of 50% H2O2 and 80 mg of NaH2PO4 in 3 mL of MeCN/H2O, 3/1, v/v, 450 mg of NaClO2 in 2 mL of water was added dropwise. The reaction mixture was stirred at 0 °C for 1 h and then warmed to room temperature. The excess of oxidizing reagents was decomposed by a dropwise addition of saturated aqueous solution Na2S2O3. Subsequently, the mixture was acidified with 3M HCl to pH 2 and extracted with AcOEt (3 × 30 mL). The organic layer was washed with water (30 mL), brine (2 × 30 mL), and water (30 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The product was purified by liquid column chromatography using a mixture of solvents hexanes/AcOEt/AcOH.
3-{2-[O-(4-tert-butoxysuccinoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoic acid12a, Starting from 1.46 g (3.68 mmol) of aldehyde 11a, 1.09 g (2.88 mmol, 78%) of carboxylic acid 12a was obtained as a light-yellow oil. Rf 0.46 (hexanes/AcOEt/AcOH, 70/30/1, v/v/v). HRMS-ESI: m/z calcd. For C21H30O6 378.2042; found 379.2370 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.79 (s, 1H), 6.80 (s, 1H), 6.55 (s, 1H), 2.78 (t, J = 6.7 Hz, 2H), 2.71 (s, 2H), 2.58 (t, J = 6 Hz, 2H), 2.47 (s, 3H), 2.17 (s, 3H), 1.48 (s, 6H), 1.41 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ: 173.10, 171.82, 171.56, 149.74, 138.00, 135.70, 134.25, 132.00, 123.17, 80.51, 48.14, 38.45, 31.26, 30.21, 28.36, 25.26, 20.12.
3-{2-[O-(4-tert-butoxypimeoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoic acid12b, Starting from 1.35 g (3.34 mmol) of aldehyde 11b, 740 mg (1.76 mmol, 53%) of carboxylic acid 12b was obtained as a colorless oil. Rf 0.40 (hexanes/AcOEt/AcOH, 80/20/1, v/v/v). HRMS-ESI: m/z calcd. for C24H36O6 420.2512; found 421.2515 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.83 (brs, 1H), 6.81 (s, 1H), 6.57 (s, 1H), 2.70 (s, 2H), 2.56 (t, J = 7.5 Hz, 2H), 2.50 (s, 3H), 2.21 (t, J = 7 Hz, 2H), 2.17 (s, 3H), 1.64 (qt, J = 7.4 Hz, 2H), 1.54 (qt, J = 7.7 Hz, 2H), 1.47 (s, 6H), 1.43–1.30 (m, 11H). 13C NMR (125 MHz, DMSO-d6) δ: 179.37, 171.69, 167.95, 149.83, 138.21, 135.71, 134.46, 132.17, 123.43, 79.85, 79.63, 47.85, 38.69, 35.07, 34.49, 31.48, 28.31, 28.24, 25.28, 24.76, 24.29, 20.13.
3-{2-[O-(4-tert-butoxysuberoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoic acid 12c, Starting from 2.92 g (6.98 mmol) of aldehyde 11c, 2.41 g (5.55 mmol, 80%) of carboxylic acid 12c was obtained as a colorless oil. HRMS-ESI: m/z calcd. for C25H38O6 434.2668; found 435.2672 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.81 (s, 1H), 6.79 (s, 1H), 6.56 (s, 1H), 2.70 (s, 2H), 2.55 (t, J = 7.2 Hz, 2H), 2.49 (s, 3H), 2.19 (t, J = 7.3 Hz, 2H), 2.17 (s, 3H), 1.63 (qt, J = 7.4 Hz, 2H), 1.50 (qt, J = 7.7 Hz, 2H), 1.46 (s, 6H), 1.39 (s, 9H), 1.32 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ: 173.01, 172.73, 172.49, 149.86, 138.10, 135.64, 134.38, 132.09, 123.36, 79.83, 47.78, 38.68, 35.12, 34.50, 31.47, 28.54, 28.49, 28.24, 25.29, 24.87, 24.40, 20.15.
Benzyl 3-{2-[O-(4-tertbutoxyacyloyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoate (13a–c)—General Procedure, To a mixture of 2.64 mmol of carboxylic acid dissolved in 20 mL of dry DMF, 5.28 mmol of potassium bicarbonate was added, and the resulting suspension was allowed to stir at room temperature for 15 min. Subsequently, 4 mmol of benzyl bromide was added in one portion, and then, the reaction mixture was allowed to stir at 40 °C for 3 h. The mixture was cooled to room temperature, and 30 mL of 5% solution of NaHCO3(aq) was added. The resulting mixture was extracted with ethyl acetate (3 × 50 mL), and then, the organic layer was washed with 5% solution of NaHCO3(aq) (2 × 50 mL) and brine (2 × 50 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography using a mixture of solvents hexanes/AcOEt, 8/2, v/v as a mobile phase.
Benzyl 3-{2-[O-(4-tertbutoxysuccinoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoate 13a, Starting from 1 g (2.64 mmol) of carboxylic acid 12a, 1 g (2.13 mmol, 81%) of ester 13a was obtained as a light-yellow oil. Rf 0.37 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C28H36O6 468.2512; found 469.2840 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 7.13 (m, 3H), 7.23 (m, 2H), 6.80 (s, 1H), 6.62 (s, 1H), 5.02 (s, 2H), 2.93 (s, 2H), 2.81 (t, J = 6.6 Hz, 2H), 2.64 (t, J = 7 Hz, 2H), 2.52 (s, 3H), 2.25 (s, 3H), 1.59 (s, 6H), 1.49 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 171.61, 171.55, 171.31, 149.50, 139.97, 136.16, 136.02, 133.38, 132.41, 128.39, 128.24, 127.98, 123.00, 80.71, 65.92, 48.04, 38.96, 31.40, 30.15, 27.92, 25.42, 20.29.
Benzyl 3-{2-[O-(4-tertbutoxypimeoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoate 13b, Starting from 750 mg (1.78 mmol) of carboxylic acid 12b, 680 mg (1.33 mmol, 75%) of ester 13b was obtained as a colorless oil. Rf 0.44 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C31H42O6 510.2981; found 511.2983 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 7.32 (m, 3H), 7.21 (m, 2H), 6.80 (s, 1H), 6.58 (s, 1H), 5.02 (s, 2H), 2.53 (m, 5H), 2.26 (m, 5H), 1.76 (qt, J = 7.6 Hz, 2H), 1.35 (qt, J = 7.7 Hz, 2H), 1.58 (s, 6H), 1.53–1.39 (m, 11H). 13C NMR (125 MHz, CDCl3) δ: 172.99, 172.44, 171.56, 149.47, 138.00, 136.13, 136.01, 133.38, 132.36, 128.36, 128.24, 127.97, 123.04, 80.06, 66.06, 47.90, 39.05, 35.30, 34.88, 31.57, 28.60, 28.18, 25.28, 24.86, 24.39, 20.22.
Benzyl 3-{2-[O-(4-tertbutoxysuberoyl)]hydroxyl-4,6-dimethyl}phenyl-3-methylbutanoate 13c, Starting from 750 mg (1.78 mmol) of carboxylic acid 12c, 680 mg (1.33 mmol, 75%) of ester 13c was obtained as a colorless oil. Rf 0.44 (hexanes/AcOEt, 9/1, v/v). HRMS-ESI: m/z calcd. for C32H44O6 524.3138; found 525.3134 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 7.32 (m, 3H), 7.21 (m, 2H), 6.80 (s, 1H), 6.57 (s, 1H), 5.01 (s, 2H), 2.89 (s, 2H), 2.51 (m, 5H), 2.25 (s, 3H), 2.23 (t, J = 7.6 Hz, 2H), 1.73 (qt, J = 7.4 Hz, 2H), 1.61 (qt, J = 7.5 Hz, 2H), 1.57 (s, 6H), 1.47 (s, 9H), 1.38 (m, 4H). 13C NMR (125 MHz, CDCl3) δ: 173.18, 172.60, 171.60, 159.39, 149.50, 138.01, 136.14, 136.01, 133.33, 132.37, 128.37, 128.22, 127.99, 123.03, 80.01, 66.02, 47.78, 38.96, 35.49, 34.95, 31.52, 28.86, 28.76, 28.14, 25.33, 24.90, 24.51, 20.28.
Benzyl 3-[2-[O-acyloyl]hydroxyl-4,6-dimethyl]phenyl-3-methylbutanoate (14a–c)—General Procedure, First, 2.13 mmol of tert-butyl ester was dissolved in 20 mL of a mixture of DCM/TFA, 3/1, v/v and allowed to stir at room temperature for 1 h. The solvents were removed under reduced pressure, and the residue was purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt/AcOH, 70/30/1, v/v/v as a mobile phase.
Benzyl 3-[2-[O-succinoyl]hydroxyl-4,6-dimethyl]phenyl-3-methylbutanoate 14a, Starting from 1 g (2.13 mmol) of ester 13a, 820 mg (2 mmol, 94%) of carboxylic acid 14a was obtained as a light-yellow oil. HRMS-ESI: m/z calcd. for C24H28O6 412.1892; found 413.1950 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 12.34 (brs, 1H), 7.31 (m, 3H), 7.18 (m, 2H), 6.79 (s, 1H), 6.57 (s, 1H), 4.95 (s, 2H), 2.90 (s, 2H), 2.74 (t, J = 6 Hz, 2H), 2.56 (t, J = 6.9 Hz, 2H), 2.47 (s, 3H), 2.18 (s, 3H), 1.47 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 173.90, 172.07, 171.42, 150.10, 138.06, 136.81, 135.77, 134.13, 132.30, 128.71, 128.25 123.30, 65.52, 47.73, 38.83, 31.37, 30.33, 28.89, 25.36, 20.19.
Benzyl 3-[2-[O-pimeoyl]hydroxyl-4,6-dimethyl]phenyl-3-methylbutanoate 14b, Starting from 680 mg (1.33 mmol) of ester 13b, 490 mg (1.08 mmol, 81%) of carboxylic acid 14b was obtained as a colorless oil. Rf 0.42 (hexanes/AcOEt/AcOH, 70/30/1, v/v/v). HRMS-ESI: m/z calcd. for C27H34O6 454.2355; found 455.2361 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 12.03 (brs, 1H), 7.31 (m, 3H), 7.18 (m, 2H), 6.80 (s, 1H), 6.58 (s, 1H), 4.95 (s, 2H), 2.86 (s, 2H), 2.52 (m, 2H), 2.46 (s, 3H), 2.21 (t, J = 7.5 Hz, 2H), 2.19 (s, 3H), 1.60 (qt, J = 7.8 Hz, 2H), 1.52–1.41 (m, 8H), 1.33 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ: 174.92, 172.48, 171.26, 149.83, 138.02, 136.46, 135.85, 133.85, 132.18, 128.73, 128.35, 123.53, 65.68, 47.70, 39.05, 34.40, 33.97, 31.48, 28.44, 25.33, 24.63, 24.26, 20.17.
Benzyl 3-[2-[O-suberoyl]hydroxyl-4,6-dimethyl]phenyl-3-methylbutanoate 14c, Starting from 2.36 g (4.50 mmol) of ester 13c, 1.97 g (4.20 mmol, 93%) of carboxylic acid 14c was obtained as a colorless oil. Rf 0.48 (hexanes/AcOEt/AcOH, 70/30/1, v/v/v). HRMS-ESI: m/z calcd. for C28H36O6 468.2512; found 469.2516 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.97 (brs, 1H), 7.30 (m, 3H), 7.18 (m, 2H), 6.78 (s, 1H), 6.57 (s, 1H), 4.94 (s, 2H), 2.84 (s, 2H), 2.50 (m, 2H), 2.45 (s, 3H), 2.18 (m, 5H), 1.58 (qt, J = 6.4 Hz, 2H), 1.47 (m, 8H), 1.29 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ: 174.91, 172.50, 171.27, 149.78, 138.05, 136.47, 135.88, 133.75, 132.14, 128.68, 128.29, 123.50, 65.59, 47.58, 38.98, 34.48, 34.02, 31.47, 28.62, 28.58, 25.31, 24.76, 24.39, 20.18.
N1,N7-bis(tert-butoxycarbonyl)spermidine 16, First, 1.23 g (8.47 mmol) of spermidine 15 was dissolved in 50 mL of dry THF and cooled in an ice bath to 0 °C. Then, 4.17 g (16.94 mmol) of Boc-ON, dissolved in 50 mL of dry THF, was added dropwise. The mixture was allowed to stir at 0 °C for 4 h, and then, solvents were evaporated under reduced pressure. The residue was dissolved in 50 mL of Et2O and washed with saturated NaOH(aq) until the yellow color disappeared. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The white crystalline residue was recrystallized from Et2O obtaining 1.88 g (5.44 mmol, 64%) of product 16 as white solid, with m.p. 83–85 °C. 1H NMR (500 MHz, CDCl3) δ: 5.22 (brs, 1H), 4.88 (brs, 1H), 3.21 (m, 2H), 3.14 (m, 2H), 2.67 (t, J = 6.6 Hz, 2H), 2.61 (t, J = 6.2 Hz, 2H), 1.65 (qt, J = 6.5 Hz, 2H), 1.53 (m, 4H), 1.45 (s, 18H). 13C NMR (125 MHz, CDCl3) δ: 156.09, 156.01, 78.94, 49.45, 47.75, 40.53, 39.29, 29.88, 28.44, 27.84, 27.41.
Boc2-spermidine-‘trimethyl lock’ benzyl ester building block (18a–c)—General Procedure, To the solution of 2.42 mmol of carboxylic acid 14a–c and 3.06 mmol of NHS in 20 mL of dry DCM, 4.34 mmol of DCC dissolved in 5 mL of dry DCM was added dropwise, and then, the mixture was allowed to stir at room temperature for 24 h. The precipitate of DCU was filtered off under reduced pressure, and the filtrate was diluted with 50 mL of DCM. Subsequently, the mixture was washed with water (2 × 30 mL), saturated solution of NaHCO3(aq) (2 × 30 mL), and water (2 × 30 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 50 mL of dry DCM, and 6.72 mmol of DIPEA and 3.2 mmol of Boc2-spermidine 16 were added. The mixture was allowed to stir at room temperature for 5 h; then, 20 mL of DCM was added, and the resulting solution was washed with brine (2 × 30 mL), 5% solution of NaHSO4(aq) (2 × 30 mL), and brine (2 × 30 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt, 4/6, v/v as a mobile phase.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-benzyloxycarbonyl-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,4-dioxo}butylspermidine 18a, Starting from 1 g (2.42 mmol) of carboxylic acid 14a and 1.1 g (3.2 mmol) of Boc2-spermidine 16, 1.1 g (1.4 mmol, 68%) of product 18a was obtained as a colorless oil with Rf 0.50 (hexanes/AcOEt, 4/6, v/v). HRMS-ESI: m/z calcd. for C41H61N3O9 739.4408; found 740.4537 [M + 1]+. 1H NMR (500 MHz, CD3OD) δ: 7.29 (m, 3H), 7.16 (m, 2H), 6.79 (s, 1H), 6.63 (m, 1H), 4.95 (s, 2H), 3.36 (m, 4H), 3.12–2.97 (m, 4H), 2.93 (s, 2H), 2.83 (m, 2H), 2.75 (m, 2H), 2.49 (s, 3H), 2.22 (s, 3H), 1.92–1.36 (m, 30H). 13C NMR (125 MHz, CD3OD) δ: 172.47, 172.42, 172.13, 171.88, 171.72, 157.11, 156.95, 149.70, 137.80, 136.08, 135.83, 133.14, 131.74, 127.97, 127.82, 127.57, 122.73, 78.09, 65.49, 45.49, 45.23, 43.08, 39.60, 39.35, 38.77, 37.39, 37.19, 33.40, 30.87, 29.91, 28.72, 27.38, 26.87, 25.48, 25.36, 24.66, 24.55, 24.16, 18.95.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-benzyloxycarbonyl-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,7-dioxo}heptylspermidine 18b, Starting from 490 mg (1.08 mmol) of carboxylic acid 14b and 374 mg (1.08 mmol) of Boc2-spermidine 16, 370 mg (0.47 mmol, 44%) of product 18b was obtained as a colorless oil with Rf 0.47 (hexanes/AcOEt, 4/6, v/v). HRMS-ESI: m/z calcd. for C44H67N3O9 781.4877; found 782.4851 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 7.30 (m, 3H), 7.19 (m, 2H), 6.78 (s, 1H), 6.57 (s, 1H), 5.00 (s, 2H), 3.46–3.02 (m, 8H), 2.89 (s, 2H), 2.54 (t, J = 7.2 Hz, 2H), 2.51 (s, 3H), 2.33 (t, J = 7.6 Hz, 2H), 2.24 (s, 3H), 1.83–1.38 (m, 36H). 13C NMR (125 MHz, CDCl3) δ: 173.14, 172.53, 172.32, 171.62, 156.08, 149.51, 138.02, 136.11, 136.01, 133.36, 132.33, 128.36, 128.21, 127.97, 123.05, 79.34, 78.77, 77.25, 65.98, 47.83, 47.40, 45.51, 42.38, 39.94, 39.06, 37.30, 34.86, 33.98, 32.83, 31.54, 29.92, 28.95, 28.50, 28.43, 27.97, 27.66, 26.15, 25.30, 25.11, 24.96, 24.48, 20.28, 14.21.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-benzyloxycarbonyl-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,8-dioxo}octylspermidine 18c, Starting from 1.97 g (4.20 mmol) of carboxylic acid 14c and 1.55 g (4.50 mmol) of Boc2-spermidine 16, 2.03 g (2.55 mmol, 61%) of product 18c was obtained as a colorless oil with Rf 0.45 (hexanes/AcOEt, 4/6, v/v). HRMS-ESI: m/z calcd. for C45H69N3O9 795.5034; found 796.5126 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 7.30 (m, 3H), 7.18 (m, 2H), 6.78 (s, 1H), 6.55 (s, 1H), 5.43 (m, 1H), 4.99 (s, 2H), 4.69 (m, 1H), 3.44–3.01 (m, 8H), 2.88 (s, 2H), 2.50 (m, 5H), 2.30 (m, 2H), 2.24 (s, 3H), 1.77–1.62 (m, 6H), 1.61–1.52 (m, 8H), 1.51–1.31 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 167.89, 166.80, 166.45, 151.25, 144.79, 133.27, 131.37, 131.23, 128.59, 127.58, 123.61, 123.45, 123.24, 118.30, 74.13, 61.07, 55.66, 42.98, 42.66, 40.65, 37.51, 35.07, 34.31, 32.36, 30.17, 28.16, 26.72, 24.43, 23.68, 22.91, 21.38, 20.55, 19.73, 16.31, 15.53.
Boc2-spermidine-‘trimethyl lock’ carboxylic acid building block (19a–c)—General Procedure, First, 1.64 mmol of benzyl ester was dissolved in 30 mL of THF, and then, 500 mg of 10% Pd/C was added. The mixture was allowed to stir at room temperature under hydrogen (balloon) atmosphere for 4 h. Subsequently, the catalyst was filtered off under reduced pressure through a thin layer of celite. The filtrate was concentrated under reduced pressure, obtaining 1.41 mmol of carboxylic acid 19.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-carboxy-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,4-dioxo}butylspermidine 19a, Starting from 1.21 g (1.64 mmol) of ester 18a, 918 mg (1.41 mmol, 86%) of carboxylic acid 19a was obtained as a colorless oil, with Rf 0.73 (CHCl3/MeOH/H2O, 65/10/1, v/v/v). HRMS-ESI: m/z calcd. for C34H55N3O9 649.3938; found 650.3858 [M + 1]+. 1H NMR (500 MHz, CD3OD) δ: 6.82 (s, 1H), 6.61 (m, 1H), 3.38 (m, 4H), 3.09 (m, 2H), 3.01 (m, 2H), 2.87 (m, 2H), 2.84 (s, 2H), 2.80 (m, 2H), 2.55 (s, 3H), 2.20 (s, 3H), 1.90–1.28 (m, 30H). 13C NMR (125 MHz, CD3OD) δ: 174.05, 172.56, 172.51, 172.20, 171.80, 149.62, 137.85, 135.69, 133.66, 131.66, 122.66, 78.12, 47.35, 45.39, 45.17, 43.07, 39.55, 39.36, 38.39, 37.38, 37.23, 33.34, 30.64, 29.95, 27.56, 27.40, 26.84, 25.49, 25.33, 24.68, 24.55, 24.18, 18.91.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-carboxy-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,4-dioxo}heptylspermidine19b, Starting from 350 mg (0.448 mmol) of ester 18b, 298 mg (0.431 mmol, 96%) of carboxylic acid 19b was obtained as a light-yellow oil. HRMS-ESI: m/z calcd. for C37H61N3O9 691.4408; found 692.4414 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.70 (brs, 1H), 6.80 (s, 1H), 6.58 (s, 1H), 3.21 (m, 4H), 2.91 (m, 4H), 2.70 (s, 2H), 2.56 (t, J = 7.1 Hz, 2H), 2.50 (s, 3H), 2.27 (m, 2H), 2.17 (s, 3H), 1.78–1.20 (m, 36H). 13C NMR (125 MHz, DMSO-d6) δ: 173.04, 172.51, 172.04, 171.83, 156.20, 149.87, 138.09, 135.74, 134.31, 132.16, 123.41, 107.65, 79.74, 67.18, 47.87, 38.67, 34.52, 33.80, 32.38, 31.47, 29.36, 28.74, 28.30, 27.58, 27.25, 26.34, 25.29, 25.18, 24.89, 24.56, 24.09, 22.20, 20.17.
N1,N7-bis(tert-butoxycarbonyl)-N3-{4-[2-(2-carboxy-1,1-dimethylethyl)-3,5-dimethyl]phenoxy-1,4-dioxo}octylspermidine 19c, Starting from 2.03 g (2.55 mmol) of ester 18c, 1.95 g (2.50 mmol, 98%) of carboxylic acid 19c was obtained as a light-yellow oil with Rf 0.64 (CHCl3/MeOH/H2O, 65/10/1, v/v/v). HRMS-ESI: m/z calcd. for C38H63N3O9 705.4564; found 706.4572 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 11.84 (brs, 1H), 6.90–6.83 (m, 1H), 6.78–6.73 (m, 2H), 3.20 (m, 4H), 2.90 (m, 4H), 2.69 (s, 2H), 2.56 (t, J = 7.4 Hz, 2H), 2.49 (s, 3H), 2.24 (m, 2H), 2.17 (s, 3H), 1.62 (m, 3H), 3.72 (m, 10H), 3.45 (m, 26H). 13C NMR (125 MHz, DMSO-d6) δ: 173.08, 172.50, 172.08, 171.75, 156.03, 149.82, 138.11, 135.62, 134.27, 132.07, 123.44, 79.62, 47.86, 47.33, 44.97, 43.03, 38.70, 38.06, 37.87, 34.57, 32.45, 31.44, 28.88, 28.69, 28.34, 27.54, 26.31, 25.30, 24.49, 20.15.
Trisodium O,O′-bis(sulfate)deoxycholate 21a, To the solution of 1 g (2.55 mmol) of deoxycholic acid 20a in 10 mL of dry DMF, 7.32 g (46 mmol) of SO3/Py complex was added in one portion. The mixture was stirred at room temperature for 24 h. Subsequently, the mixture was alkalinized to pH 8 with a saturated solution of NaHCO3(aq). The solvents were evaporated under reduced pressure, the residue was suspended in 100 mL of MeOH, and then inorganic salts were filtered off. The filtrate was concentrated under reduced pressure, which was followed by treatment with MeCN. The formed precipitate was filtered off, obtaining 1.55 g (2.50 mmol, 98%) of product 21a as a light-beige solid with m.p. 202–204 °C and Rf 0.39 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 4.86 (s, 1H), 4.26 (m, 1H), 2.36–2.20 (m, 2H), 2.08 (m, 1H), 2.00–1.71 (m, 10H), 1.69–1.54 (m, 2H), 1.54–1.23 (m, 8H), 1.23–0.90 (m, 9H), 0.76 (s, 3H). 13C NMR (125 MHz, CD3OD) δ: 182.28, 81.21, 79.15, 45.99, 42.35, 36.06, 35.93, 35.60, 35.36, 35.14, 34.00, 33.62, 33.30, 32.56, 30.23, 28.95, 27.34, 27.05, 26.05, 24.68, 23.49, 22.30, 16.95, 11.61.
Tetrasodium O,O′,O″-tris(sulfate)cholate 21b, Compound 21b was prepared in a similar manner as 21a. Starting from 1 g (2.45 mmol) of cholic acid 20b and 10.5 g (66 mmol) of SO3/Py complex, 1.49 g (2 mmol, 82%) of product 21b was obtained as a light-beige solid. 1H NMR (500 MHz, CD3OD) δ: 4.68 (s, 1H), 4.46 (m, 1H), 4.15 (m, 1H), 2.52–2.21 (m, 5H), 2.18–1.62 (m, 12H), 1.50–1.22 (m, 5H), 1.17–0.90 (m, 8H), 0.78 (s, 3H). 13C NMR (125 MHz, CD3OD) δ: 182.48, 81.06, 79.52, 76.71, 45.90, 42.19, 42.05, 39.34, 36.19, 35.69, 35.39, 35.10, 34.14, 32.49, 30.30, 29.10, 27.59, 27.44, 27.27, 24.55, 22.52, 21.82, 17.01, 11.61.
Trisodium O,O′-bis(sulfate)deoxycholate active ester 22a, To the solution of 1.5 g (2.42 mmol) of deoxycholic acid derivative 21a in 30 mL of dry DMF, 355 μL (2.04 mmol) of DIPEA and 916 mg (3.03 mmol) of DEPBT were added, respectively. The mixture was stirred at room temperature for 2 h and then concentrated under reduced pressure. The oily residue was treated with MeCN and the formed precipitate was filtered off, obtaining 1.35 g (1.57 mmol, 65%) of crude active ester 22a as a light-yellow solid, which was used in the next step without further purification.
Tetrasodium O,O′,O″-tris(sulfate)cholate active ester 22b, Active ester 22b was prepared in a similar manner as 22a. Starting from 1.50 g (2.04 mmol) of cholic acid derivative 21b, 1.16 g (1.35 mmol, 66%) of active ester 22b was obtained as a light-yellow solid, which was used in the next step without further purification.
Cholic acid active ester 22c, First, 15 g (37 mmol) of cholic acid was dissolved in 250 mL of dry THF, which was followed by the addition of 4.26 g (37 mmol) of NHS. Subsequently, 9.16 g (44 mmol) of DCC dissolved in 30 mL of dry THF was added dropwise. The mixture was stirred at room temperature for 24 h; then, the precipitated DCU was filtered off under reduced pressure, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 300 mL of CHCl3 and washed with a saturated solution of NaHCO3(aq) (2 × 200 mL) and water (2 × 200 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure, obtaining 10.61 g (21 mmol, 57%) of crude active ester 22c, which was used in the next step without further purification.
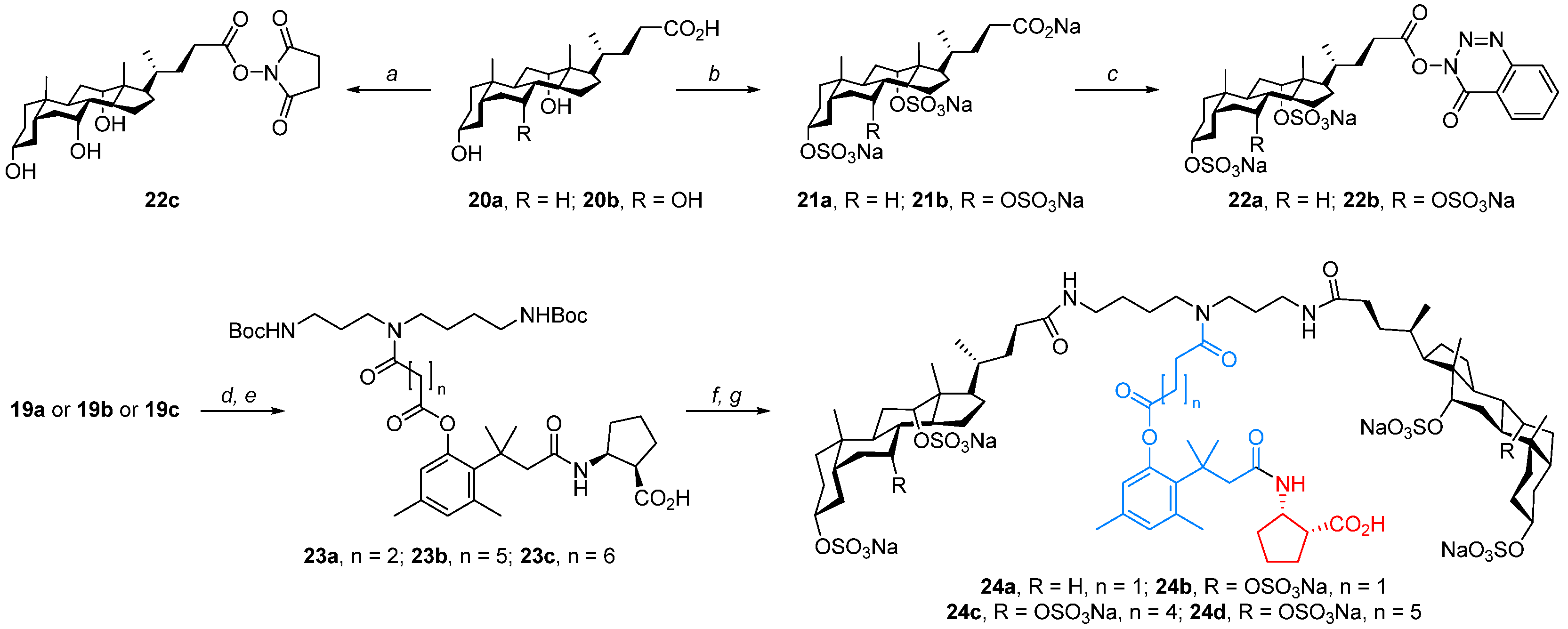
Molecular umbrella–‘trimethyl lock’–cargo conjugates (24a–d)—General Procedure, To a solution of 2.17 mmol of carboxylic acid 19a–c in 20 mL of dry DMF, 568 μL of DIPEA and 3.26 mmol of DEPBT were added, respectively. The mixture was stirred at room temperature for 2 h. Then, 100 mL of CHCl3 was added, and the resulting mixture was washed with 1 M solution of HCl(aq) (2 × 75 mL), brine (2 × 75 mL), saturated solution of NaHCO3(aq) (2 × 75 mL), and brine (2 × 75 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The crude active ester was roughly purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt, 4/6, v/v as a mobile phase. Then, 0.70 mmol of active ester was dissolved in 20 mL of dry DMF, and then 1.1 mL of DIPEA and 1.05 mmol of cispentacin were added, respectively. The mixture was stirred at room temperature for 2 h. Subsequently, 100 mL of CHCl3 was added, and the resulting solution was washed with 1 M solution of HCl(aq) (3 × 50 mL) and brine (3 × 50 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure, obtaining crude product 23a–c. Product 23a–c was dissolved in a mixture of DCM/TFA, 3/1, v/v and allowed to stir at room temperature for 1 h. Then, the solvents were evaporated under reduced pressure, obtaining the crude deprotection product which was used in the next step without further purification. To the solution of 0.30 mmol of deprotected spermidine derivative in dry DMF, 627 μL of DIPEA and 0.60 mmol of active ester 22a or 22b were added, respectively. The mixture was stirred at room temperature for 24 h; then, the solvents were evaporated under reduced pressure, and the residue was treated with MeCN. The precipitate was collected by filtration under reduced pressure and purified by liquid column chromatography.
Molecular umbrella–cispentacin conjugate 24a, Starting from 700 mg (1.1 mmol) of carboxylic acid 19a, 316 mg (0.18 mmol, 27%) of conjugate 24a was obtained as a white solid, with Rf 0.32 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 6.82 (s, 1H), 6.67 (s, 1H), 4.67 (s, 2H), 4.28 (m, 3H), 2.44 (m, 4H), 3.21 (m, 4H), 2.96 (m, 3H), 2.83 (m, 4H), 2.71 (m, 1H), 2.60–4.42 (m, 4H), 2.36–2.07 (m, 9H), 2.01–0.86 (m, 74H), 0.76 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.77, 175.53, 173.31, 172.39, 172.03, 163.52, 150.07, 138.06, 136.20, 133.59, 131.90, 122.96, 81.21, 79.37, 52.15, 48.51, 45.91, 45.36, 43.55, 42.31, 39.66, 38.50, 36.73, 36.52, 35.81, 35.15, 33.91, 33.63, 33.30, 32.64, 31.69, 31.26, 30.07, 27.54, 28.28, 27.16, 27.00, 26.67, 26.24, 26.07, 25.66, 24.77, 24.59, 23.51, 22.30, 21.46, 18.89, 16.83, 11.59.
Molecular umbrella–cispentacin conjugate 24b, Starting from 525 mg (0.69 mmol) of carboxylic acid 19a, 617 mg (0.31 mmol, 45%) of conjugate 24b was obtained as a white solid, with Rf 0.29 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 6.82 (s, 1H), 6.65 (s, 1H), 4.66 (s, 2H), 4.45 (m, 2H), 4.25 (m, 1H), 4.14 (m, 2H), 3.41 (m, 4H), 3.19 (m, 4H), 2.97–2.65 (m, 6H), 2.53 (m, 3H), 2.48–2.18 (m, 14H), 2.11 (m, 6H), 2.03–1.17 (m, 46H), 1.15–0.86 (m, 16H), 0.76 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 176.35, 175.69, 175.55, 175.41, 173.28, 172.15, 171.98, 171.87, 150.00, 138.02, 136.18, 133.57, 131.93, 122.98, 116.89, 80.77, 79.37, 76.43, 52.09, 48.48, 46.12, 45.84, 45.25, 43.42, 42.23, 41.89, 39.47, 39.16, 38.54, 38.44, 36.47, 35.67, 35.40, 35.10, 34.10, 32.81, 32.62, 31.72, 31.28, 31.12, 30.29, 30.06, 29.01, 28.18, 27.58, 27.42, 27.30, 27.16, 27.00, 26.29, 25.64, 24.59, 24.46, 22.49, 21.78, 21.48, 18.93, 16.88, 11.58.
Molecular umbrella–cispentacin conjugate 24c, Starting from 270 mg (0.39 mmol) of carboxylic acid 19b, 187 mg (0.09 mmol, 23%) of conjugate 24c was obtained as a white solid with Rf 0.25 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 6.84(s, 1H), 6.61 (s, 1H), 4.68 (s, 2H), 4.47 (s, 2H), 4.26 (m, 1H), 4.15 (m, 2H), 3.40 (m, 4H), 3.21 (m, 4H), 2.78 (m, 1H), 2.68 (m, 3H), 2.53 (s, 3H), 2.50–2.24 (m, 13H), 2.23 (s, 3H), 2.15–2.05 (m, 5H), 2.05–1.20 (m, 55H), 1.15–0.98 (m, 10H), 0.95 (s, 6H), 0.77 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.88, 175.58, 174.13, 173.89, 173.67, 172.15, 150.00, 138.20, 136.35, 133.43, 132.09, 123.14, 81.23, 79.82, 76.84, 52.21, 48.67, 48.50, 45.88, 42.29, 41.86, 39.61, 39.17, 38.45, 36.70, 35.74, 35.45, 35.03, 34.28, 34.15, 32.78, 32.44, 31.86, 31.28, 31.07, 30.36, 29.32, 28.57, 27.61, 27.44, 27.15, 26.90, 26.32, 25.94, 25.02, 24.60, 24.50, 24.16, 22.48, 21.73, 21.41, 18.92, 16.92, 16.87, 11.50.
Molecular umbrella–cispentacin conjugate 24d, Starting from 1.02 g (1.44 mmol) of carboxylic acid 19c, 824 mg (0.39 mmol, 27%) of conjugate 24d was obtained as a white solid with Rf 0.25 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 6.86 (s, 1H), 6.60 (s, 1H), 4.68 (s, 2H), 4.47 (s, 2H), 4.28 (m, 1H), 4.15 (m, 2H), 3.40 (m, 4H), 3.18 (m, 4H), 2.94–2.61 (m, 4H), 2.55 (s, 3H), 2.49–2.27 (m, 12H), 2.24 (s, 3H), 2.11 (m, 11H), 2.03–1.19 (m, 50H), 1.07 (m, 10H), 0.95 (m, 6H), 0.77 (m, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.93, 175.65, 174.17, 173.90, 164.92, 164.40, 151.30, 138.08, 135.99, 134.83, 133.28, 123.16, 81.19, 79.94, 76.81, 53.22, 48.54, 46.09, 45.80, 44.05, 42.15, 39.54, 39.33, 38.42, 36.58, 35.57, 35.46, 35.01, 34.32, 34.06, 32.61, 32.44, 31.86, 31.28, 31.10, 30.34, 30.04, 28.55, 27.41, 27.26, 26.28, 25.92, 25.14, 24.49, 24.13, 23.51, 23.71, 22.99, 21.93, 21.92, 21.46, 18.97, 16.51, 11.50.
Molecular umbrella–Lys(Mca) conjugate 26a, Starting from 292 mg (0.45 mmol) of carboxylic acid 19a, 210 mg (0.13 mmol, 29%) of conjugate 26a was obtained. The product was purified by liquid column chromatography using a mixtures of solvents CHCl3/MeOH/H2O, 65/10/1, v/v/v and CHCl3/MeOH/H2O, 7/4/1, v/v/v as mobile phases. HRMS-ESI: m/z calcd. for C90H135N5O18 1573.9802; found 1574.9853 [M + 1]+. 1H NMR (500 MHz, CD3OD) δ: 7.68 (d, J = 8.9 Hz, 1H), 6.96 (m, 1H), 6.91 (s, 1H), 6.81 (s, 1H), 6.63 (s, 1H), 6.26 (s, 1H), 4.17 (m, 1H), 3.94 (s, 2H), 3.88 (s, 3H), 3.79 (s, 2H), 3.74 (s, 2H), 3.36 (m, 6H), 3.24–3.08 (m, 6H), 2.91 (m, 2H), 2.81 (m, 3H), 2.55 (s, 4H), 2.27 (m, 6H), 2.18–1.19 (m, 57H), 1.18–0.85 (m, 18H), 0.86 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.37, 172.99, 171.95, 169.04, 163.16, 161.67, 155.34, 151.13, 150.04, 138.09, 136.01, 133.65, 131.91, 126.05, 122.81, 112.68, 100.47, 78.09, 72.51, 71.42, 67.55, 55.18, 54.73, 46.57, 46.02, 41.87, 41.46, 39.57, 39.16, 39.03, 38.83, 36.41, 35.56, 35.09, 34.51, 32.77, 31.92, 31.18, 30.00, 29.75, 29.31, 28.45, 28.21, 27.57, 27.39, 27.19, 26.47, 26.31, 22.57, 24.52, 22.83, 22.45, 21.85, 19.01, 16.37, 11.66.
Molecular umbrella–Lys(Mca) conjugate 26b, Starting from 221 mg (0.34 mmol) of carboxylic acid 19a, 219 mg (0.10 mmol, 29%) of conjugate 26b was obtained. The product was purified by liquid column chromatography using a mixture of solvents CHCl3/MeOH/H2O, 7/4/1, v/v/v as a mobile phase. 1H NMR (500 MHz, CD3OD) δ: 7.72 (d, J = 8.9 Hz, 1H), 6.98 (m, 1H), 6.91 (s, 1H), 6.79 (s, 1H), 6.65 (s, 1H), 6.29 (s, 1H), 4.66 (s, 2H), 4.44 (s, 2H), 4.13 (m, 3H), 3.90 (s, 3H), 3.77 (s, 2H), 3.40 (m, 4H), 3.19 (m, 6H), 2.90 (m, 2H), 2.82 (m, 3H), 2.56–1.18 (m, 67H), 1.18–0.87 (m, 18H), 0.74 (m, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.48, 173.21, 172.06, 169.00, 163.50, 163.19, 161.74, 155.17, 151.35, 149.91, 137.98, 136.08, 133.56, 131.87, 126.15, 122.85, 112.63, 112.39, 112.24, 100. 56, 81.01, 79.86, 76.43, 55.10, 53.75, 48.39, 46.03, 45.16, 43.45, 42.37, 42.30, 41.94, 39.51, 39.23, 38.73, 38.36, 36.44, 34.57, 34.93, 34.08, 32.78, 31.64, 31.16, 30.33, 30.22, 30.09, 28.96, 28.53, 28.19, 27.61, 27.46, 27.31, 27.18, 26.27, 25.66, 24.64, 24.47, 22.47, 21.77, 18.95, 16.85, 11.53.
Molecular umbrella–Nap–NH2conjugate 27, Starting from 580 mg (0.82 mmol) of carboxylic acid 19c, 900 mg (0.41 mmol, 50%) of fluorescent conjugate 27 was obtained as an orange solid with Rf 0.29 (CHCl3/MeOH/H2O, 7/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 8.52 (d, J = 7.5 Hz, 1H), 8.39 (dd, J = 8.2 Hz, 3.1 Hz, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.07 (t, J = 5.5 Hz, 1H), 7.98 (m, 1H), 7.76 (m, 1H), 7.64 (t, J = 7.9 Hz, 1H), 6.71 (d, J = 8.9 Hz, 1H), 6.61 (m, 1H), 6.38 (s, 1H), 4.66 (m, 2H), 4.62 (brs, 1H), 4.44 (m, 2H), 4.13 (m, 4H), 3.53 (m, 2H), 3.19 (m, 4H), 2.72 (m, 2H), 2.53 (m, 2H), 2.44 (m, 4H), 2.35 (m, 9H), 2.22 (m, 2H), 2.12 (m, 6H), 1.98 (m, 7H), 1.92–1.30 (m, 47H), 1.23 (m, 7H), 1.14–0.96 (m, 13H), 0.92 (m, 6H), 0.73 (m, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.93, 175.65, 174.17, 173.90, 164.92, 164.40, 151.30, 149.75, 138.08, 135.99, 134.83, 133.28, 131.93, 131.07, 129.63, 128.56, 124.44, 122.89, 121.68,120.27, 107.92, 103.66, 81.19, 79.94, 76.81, 60.97, 48.54, 46.09, 45.80, 44.05, 42.15, 41.74, 39.54, 39.33, 39.15, 38.42, 37.75, 36.58, 35.57, 35.46, 35.01, 34.32, 34.06, 32.61, 31.77, 31.10, 30.34, 30.04, 28.55, 27.41, 27.26, 26.28, 25.92, 25.14, 24.49, 24.13, 22.51, 21.71, 19.99, 18.93, 16.92, 15.46, 12.97, 11.51.
{N,N′-bis[4-(tert-butoxycarbonyl)amino]butyl-N,N′-bis[3-(tert-butylcarbonyl)amino]propyl}-3,3′-dithiodipropanoic amide 29, To a suspension of 1.76 g (4.34 mmol) of diester 28 in 50 mL of THF, 2 mL (8.68 mmol) of DIPEA and 3 g (8.68 mmol) of protected spermidine 16 was added, respectively. The mixture was stirred at room temperature for 24 h, and then, the solvents were evaporated under reduced pressure. The residue was dissolved in 75 mL of CHCl3 and washed with a saturated solution of NaHCO3(aq) (2 × 50 mL), brine (2 × 50 mL), 5% solution of NaHSO4(aq) (2 × 50 mL), and brine (2 × 50 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography using a mixture of solvents CHCl3/MeOH/H2O, 65/10/11, v/v/v as a mobile phase, obtaining 2.89 g (3.34 mmol, 77%) of product 29 as a colorless oil with Rf 0.70 (CHCl3/MeOH/H2O, 65/10/11, v/v/v). HRMS-ESI: m/z calcd. for C40H76N6O10S2 864.5064; found 865.5112 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 3.52–3.22 (m, 8H), 3.20–3.01 (m, 8H), 2.96 (m, 4H), 2.74 (m, 4H), 1.83–1.31 (m, 48H). 13C NMR (125 MHz, CDCl3) δ: 171.16, 170.43, 156.09, 79.21, 78.88, 49.02, 47.48, 45.66, 45.49, 42.73, 40.11, 39.89, 38.01, 37.29, 34.03, 33.88, 32.89, 37.78, 32.63, 32.52, 29.99, 28.47, 28.43, 27.94, 27.67, 27.49, 26.16, 25.66, 24.94.
N1,N7-bis(tert-butoxycarbonyl)-N3-(3-thiopropanoyl)spermidine 30, To a solution of 2.4 g (2.78 mmol) of spermidine derivative 29 in 50 mL of MeOH, 1.20 g (4.18 mmol) of TCEP solution in 10 mL of water, adjusted to pH 7 with NaHCO3, was added. The mixture was stirred at room temperature for 1 h, and then, MeOH was evaporated under reduced pressure. The aqueous residue was extracted with CHCl3 (3 × 25 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography using a mixture of solvents hexanes/AcOEt, 3/7, v/v as a mobile phase obtaining 1.72 g (4 mmol, 72%) of thiol 30 as a colorless oil, with Rf 0.55 (hexanes/AcOEt, 3/7, v/v). HRMS-ESI: m/z calcd. for C20H39N3O5S 433.2610; found 434.2623 [M + 1]+. 1H NMR (500 MHz, CDCl3) δ: 6.95–6.63 (m, 2H), 3.21 (m, 4H), 2.91 (m, 4H), 2.63 (m, 4H), 2.29 (m, 1H), 1.77–1.22 (m, 24 H). 13C NMR (125 MHz, CDCl3) δ: 170.48, 170.23, 156.10, 79.69, 77.89, 60.28, 47.25, 45.20, 45.00, 43.27, 38.13, 36.78, 29.28, 28.73, 28.26, 27.58, 27.23, 26.30, 25.20, 20.39.
N1,N7-bis(tert-butoxycarbonyl)-N3-[3-(o-hydroxymethylphenyl)dithio]propanoyl]spermidine 32, First, 1.56 g (3.60 mmol) of thiol 30 was dissolved in 20 mL of dry MeOH, and then, 1 mL (7.42 mmol) of TEA and 995 mg (4.32 mmol) of activated disulfide 31 were added, respectively. The mixture was stirred at room temperature for 2 h, and then, the solvents were evaporated under reduced pressure. The residue was dissolved in 50 mL of CHCl3 and washed with a saturated solution of NaHCO3(aq) (2 × 50 mL), water (2 × 50 mL), 5% solution of NaHSO4(aq) (2 × 50 mL), and water (2 × 50 mL), respectively. The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by liquid column chromatography, using a mixture of solvents hexanes/AcOEt, 3/7, v/v as a mobile phase, obtaining 610 mg (1.07 mmol, 30%) of disulfide 32 as a colorless oil, with Rf 0.50 (hexanes/AcOEt, 3/7, v/v). HRMS-ESI: m/z calcd. for C27H45N3O6S2 571.2750; found 572.2698 [M + 1]+. 1H NMR (500 MHz, DMSO-d6) δ: 7.69 (m, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.30 (m, 2H), 6.93–6.66 (m, 2H), 5.31 (t, J = 5.4 Hz, 1H), 4.59 (d, J = 5.4 Hz, 2H), 3.24–2.99 (m, 6H), 2.92–2.83 (m, 4H), 2.65 (m, 2H), 1.59–1.42 (m, 3H), 1.33–1.20 (m, 3H), 1.37 (s, 18H). 13C NMR (125 MHz, DMSO-d6) δ: 170.96, 170.31, 156.08, 140.30, 135.26, 128.34, 127.53, 78.96, 70.75, 62.74, 60.52, 47.36, 45.42, 42.78, 39.98, 33.91, 32.20, 28.47, 27.55, 26.46, 26.12, 24.97, 21.11, 14.16.
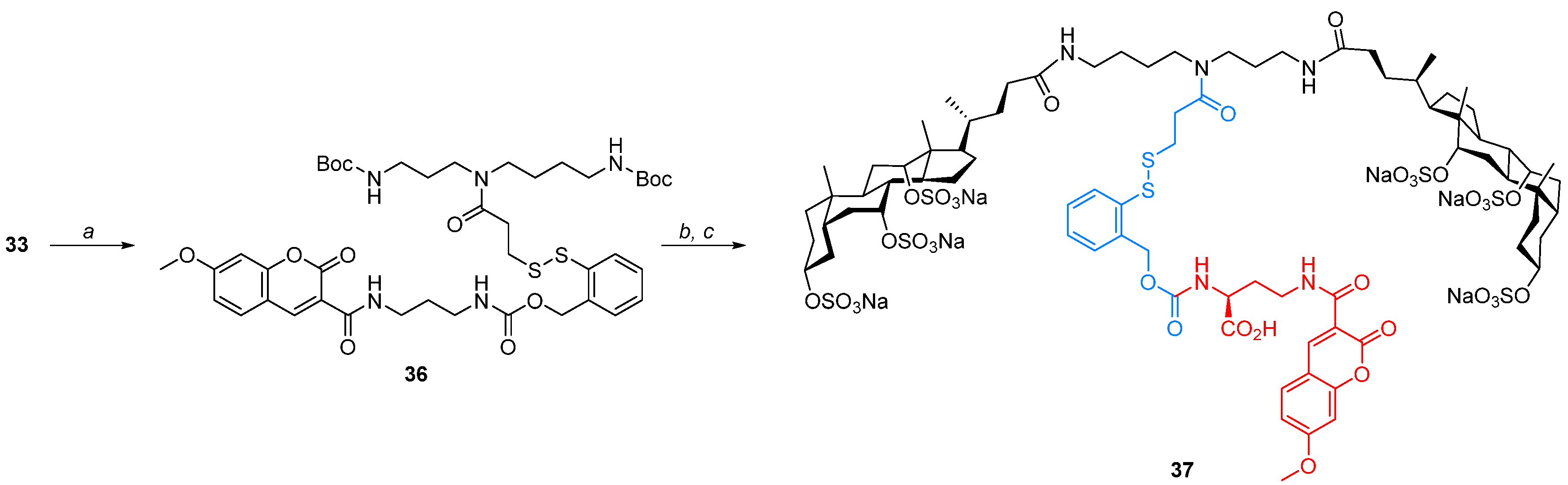
Molecular umbrella–o-dithobenzoylcarbamoyl linker–cargo conjugates (35 and 37)—General Procedure, To a solution of 1.05 mmol of disulfide 32 in 10 mL of MeCN, 1.05 mmol of TEA and 1.05 mmol of DSC were added, respectively. The mixture was stirred at room temperature for 2 h, and then, solvents were evaporated under reduced pressure. The residue was dissolved in 50 mL of AcOEt and washed with water (2 × 20 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 10 mL of MeCN and then transferred to the mixture of 1.58 mmol of a cargo molecule and 2.10 mmol of TEA dissolved in 10 mL of water. The mixture was stirred at room temperature for 2 h, and then, MeCN was evaporated under reduced pressure. The remaining aqueous residue was acidified with 1M HCl(aq) to pH 2, and the resulting solution was extracted with AcOEt (4 × 20 mL). The organic layer was dried over anhydrous MgSO4, the desiccant was filtered off, and the filtrate was concentrated under reduced pressure. The residue was roughly purified by liquid column chromatography, using a mixture of solvents CHCl3/MeOH/H2O, 65/10/1, v/v/v as a mobile phase, obtaining 0.62 mmol of a crude product as an oil. The resulting oil was dissolved in 8 mL of DCM/TFA, 3/1, v/v mixture and was allowed to stir at room temperature for 1 h. Subsequently, the solvents were evaporated, and the residue was dissolved in 20 mL of dry DMF. Then, 220 μ of DIPEA and 1.24 mmol of 22b were added, respectively. The mixture was stirred at room temperature for 5 h, and then, most of the solvents were evaporated under reduced pressure, and MeCN was added to the residue. The precipitate was collected and purified by liquid column chromatography using a mixture of solvents CHCl3/MeOH/H2O, 5/4/1, v/v/v as a mobile phase.
Molecular umbrella–o-dithobenzoylcarbamoyl linker–cispentacin conjugate 35, Starting from 600 mg (1.05 mmol) of disulfide 32, 230 mg (0.12 mmol, 11%) of conjugate 35 was obtained as a beige solid, with Rf 0.30 (CHCl3/MeOH/H2O, 5/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 8.00 (m, 2H), 7.80 (m, 1H), 7.52–7.37 (m, 2H), 7.32 (m, J = 6.1 Hz, 1H), 5.41–5.17 (m, 2H), 4.66 (s, 2H), 4.46 (s, 2H), 4.16 (m, 3H), 3.35 (m, 2H), 3.18 (m, 6H), 3.01 (m, 2H), 2.89 (m, 1H), 2.75 (t, 2H), 2.49–2.22 (m, 10H), 2.10 (m, 6H), 2.04–1.20 (m, 42H), 1.17–0.98 (m, 10H), 0.95 (s, 6H), 0.76 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 175.74, 164.79, 129.80, 128.70, 127.30, 125.61, 109.81, 81.06, 79.81, 76.60, 63.79, 54.28, 48.47, 46.06, 45.96, 45.38, 42.22, 42.04, 39.31, 35.68, 35.47, 35.11, 34.43, 34.11, 32.71, 31.99, 31.80, 31.62, 30.30, 29.40, 28.94, 28.29, 27.61, 27.43, 27.35, 27.15, 26.37, 25.80, 24.60, 22.52, 21.79, 16.89, 11.56.
Molecular umbrella–o-dithobenzoylcarbamoyl linker–Lys(Mca) conjugate 37, Starting from 630 mg (1.10 mmol) of disulfide 32, 230 mg (0.11 mmol, 10%) of conjugate 37 was obtained as a light-yellow solid, with Rf 0.50 (CHCl3/MeOH/H2O, 5/4/1, v/v/v). 1H NMR (500 MHz, CD3OD) δ: 8.82 (s, 1H), 7.78 (m, 2H), 7.42 (d, J = 7.5 Hz, 1H), 7.38 (t, J = 7.3 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.04 (m, 1H), 6.98 (m, 1H), 5.24 (m, 2H), 4.65 (m, 2H), 4.46 (m, 2H), 4.16 (m, 2H), 3.96 (m, 3H), 3.51 (m, 2H), 3.37 (m, 2H), 3.30–3.12 (m, 8H), 3.01 (m, 2H), 2.78 (m, 2H), 2.47–2.20 (m, 10H), 2.20–1.18 (m, 42H), 1.14–0.84 (m, 16H), 0.74 (m, 6H).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}