
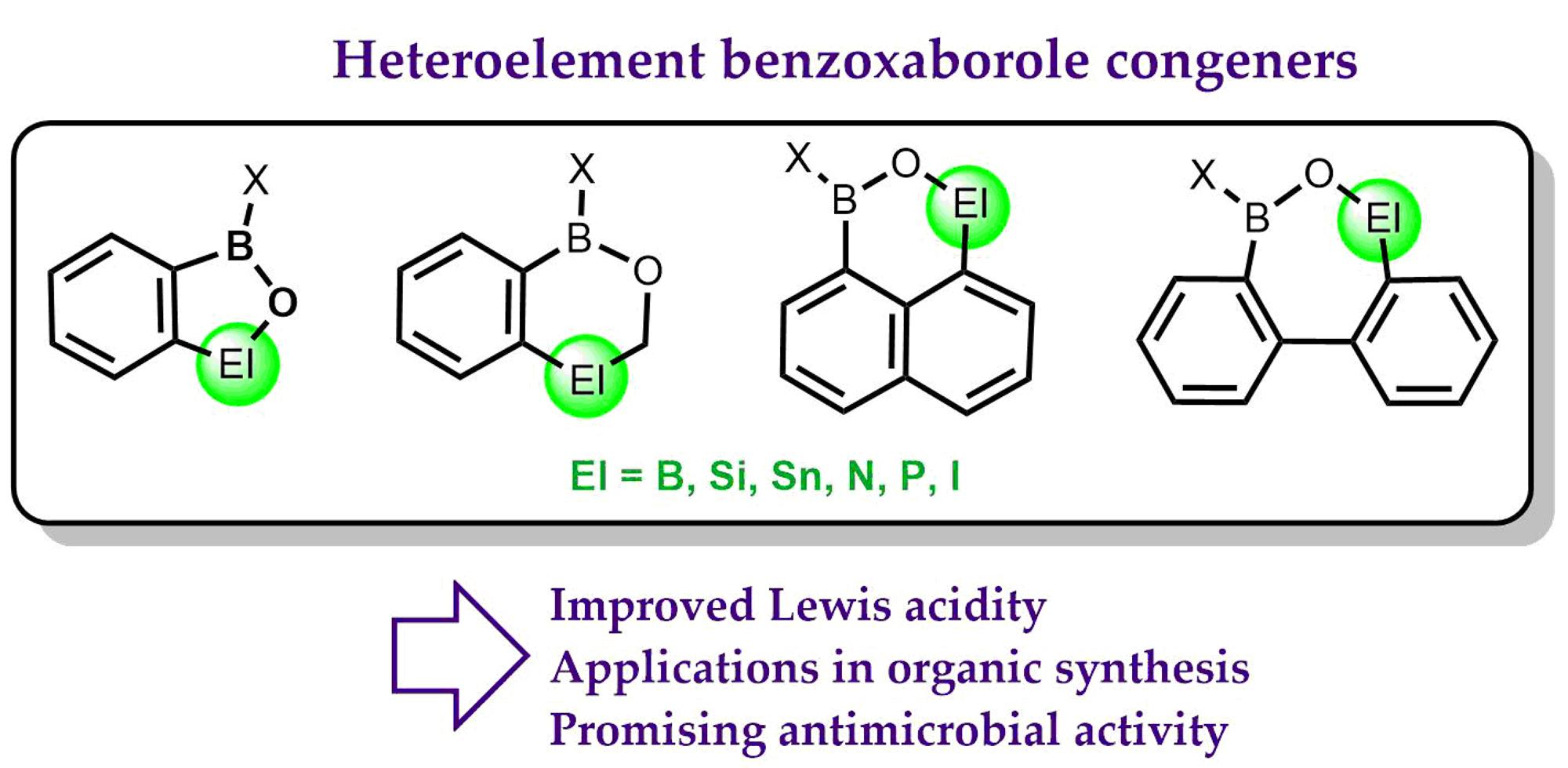
Heteroelement Analogues of Benzoxaborole and Related Ring Expanded Systems


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
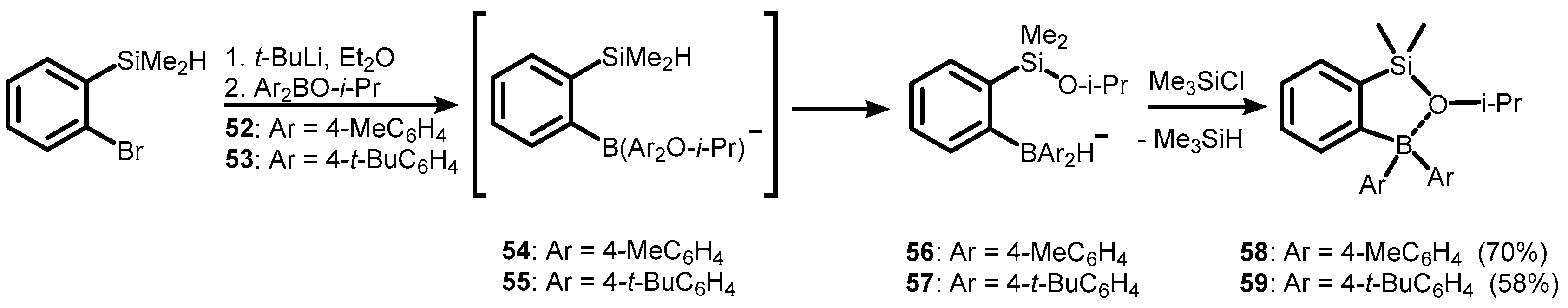{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
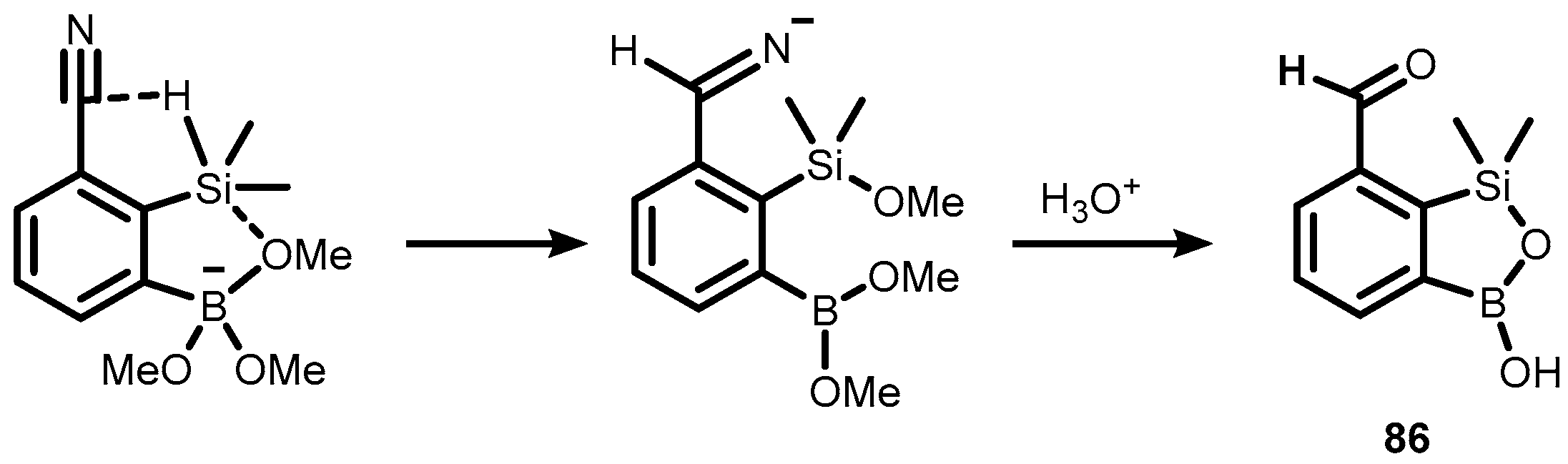{kind=link}
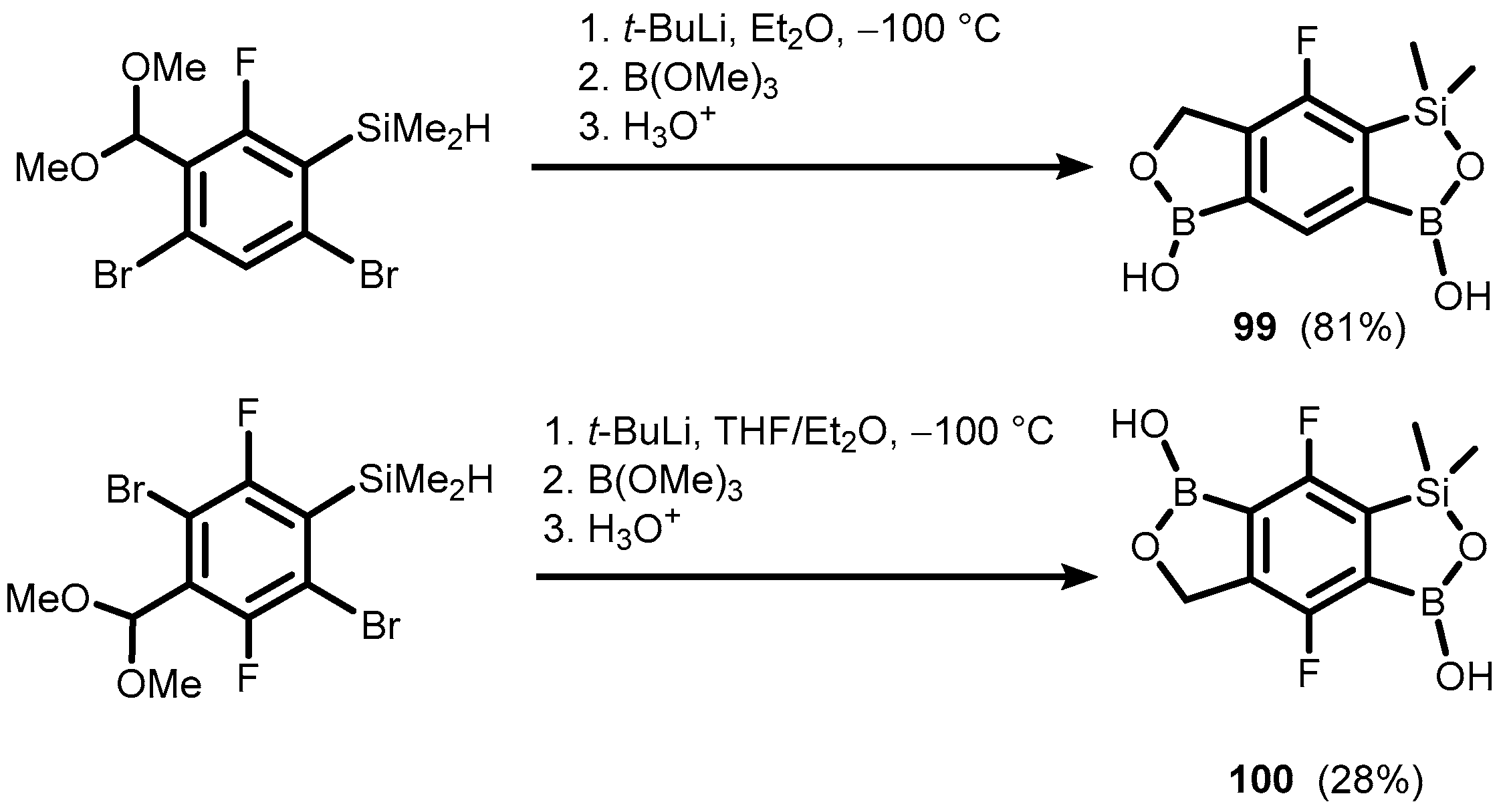{kind=link}
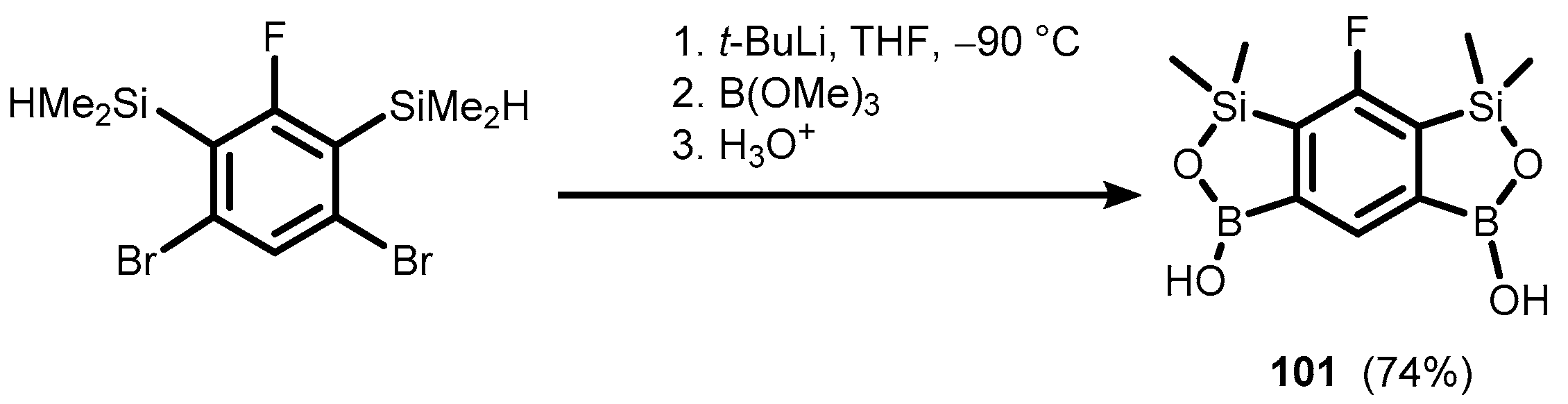{kind=link}
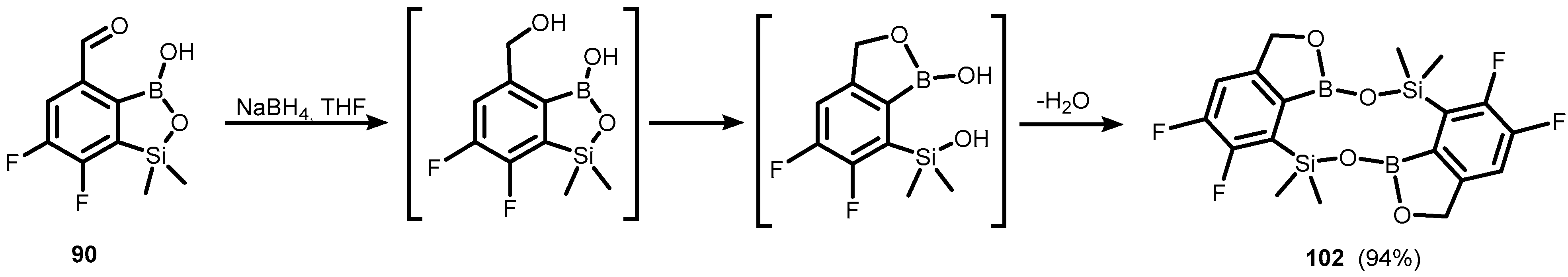{kind=link}
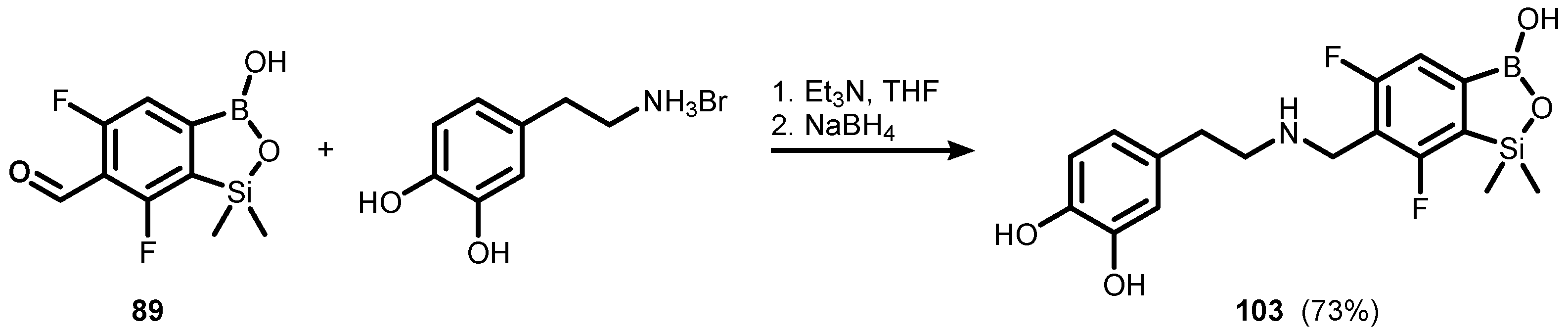{kind=link}
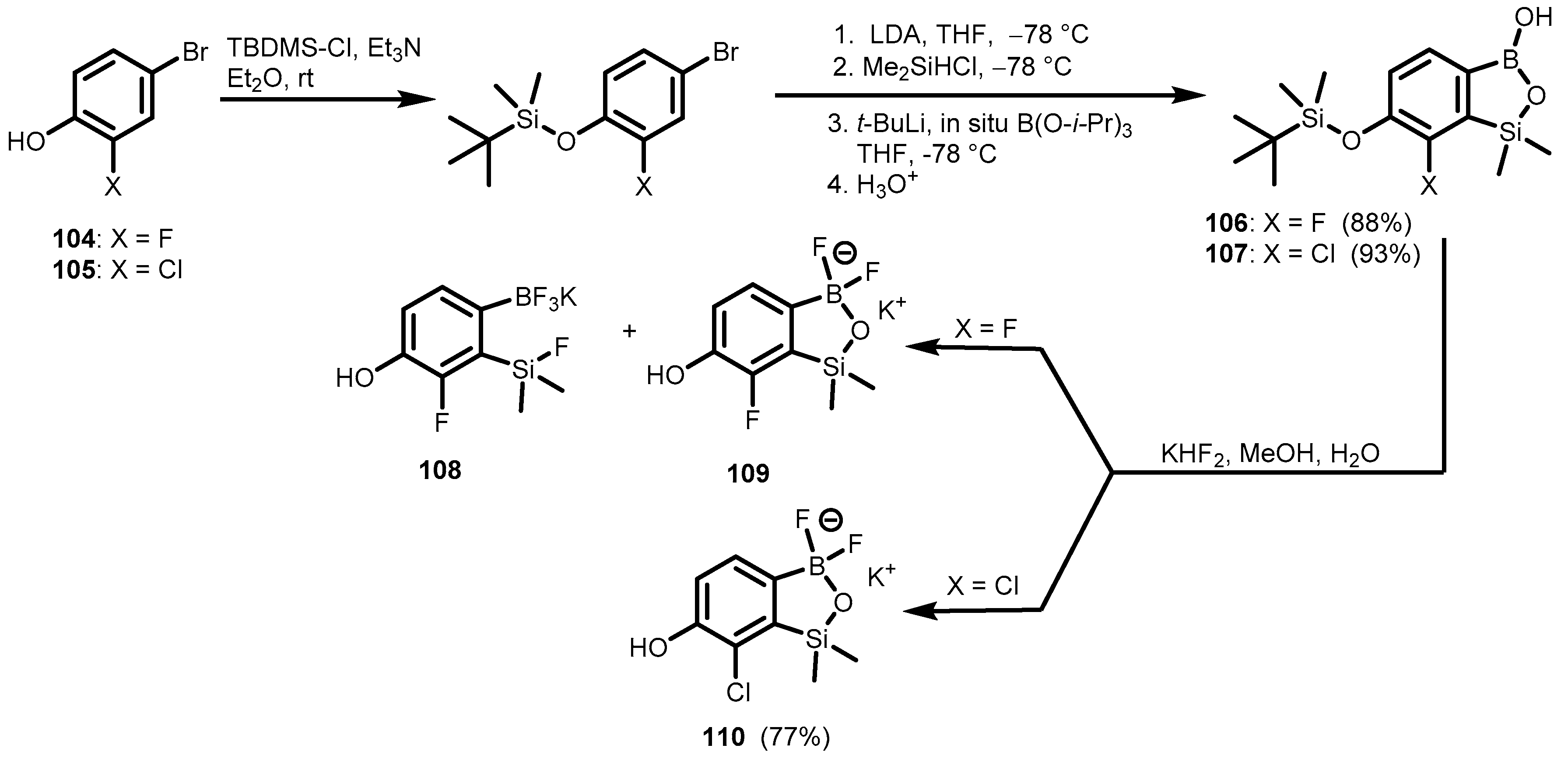{kind=link}
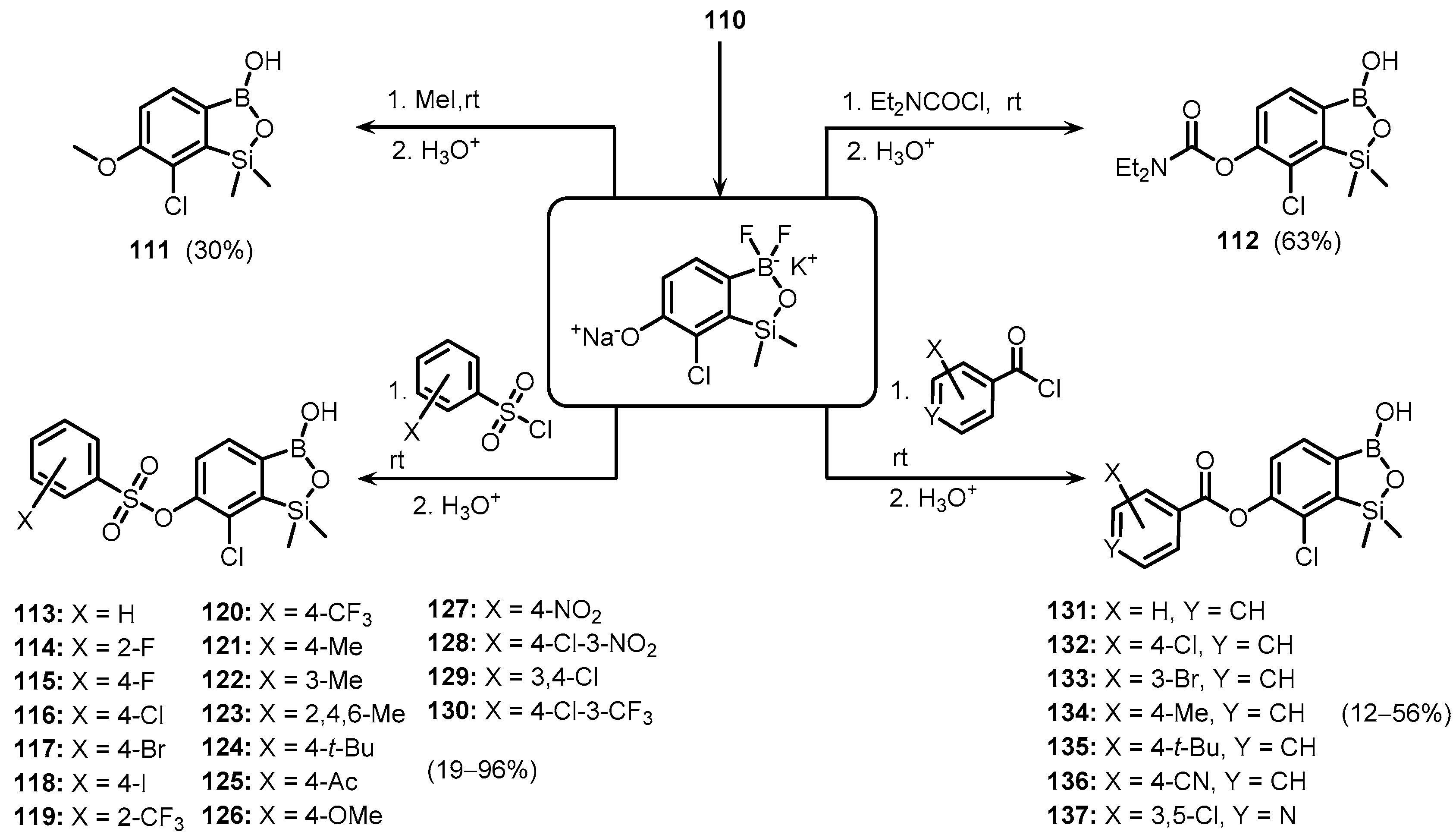{kind=link}
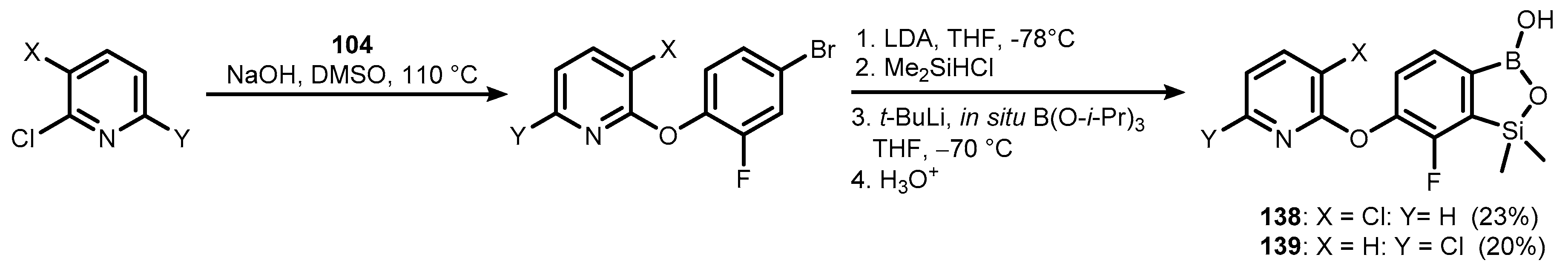{kind=link}
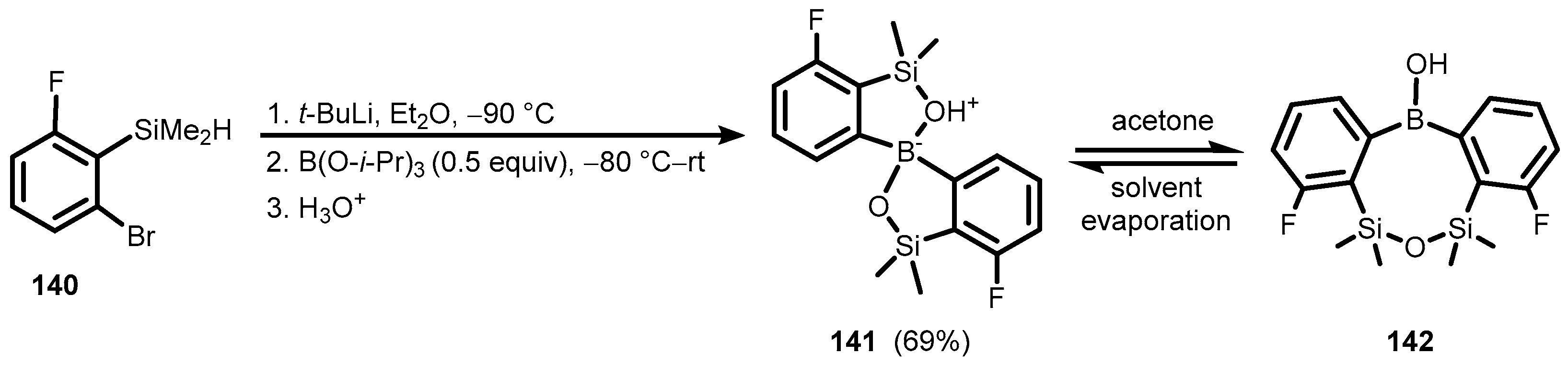{kind=link}
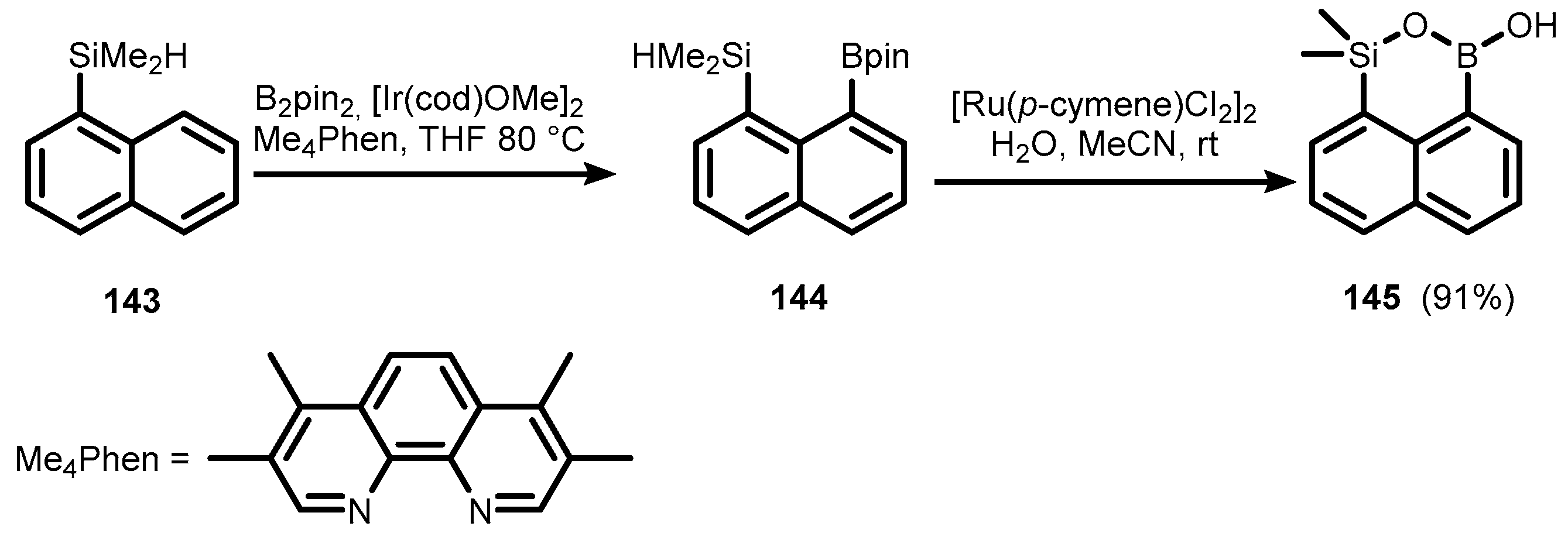{kind=link}
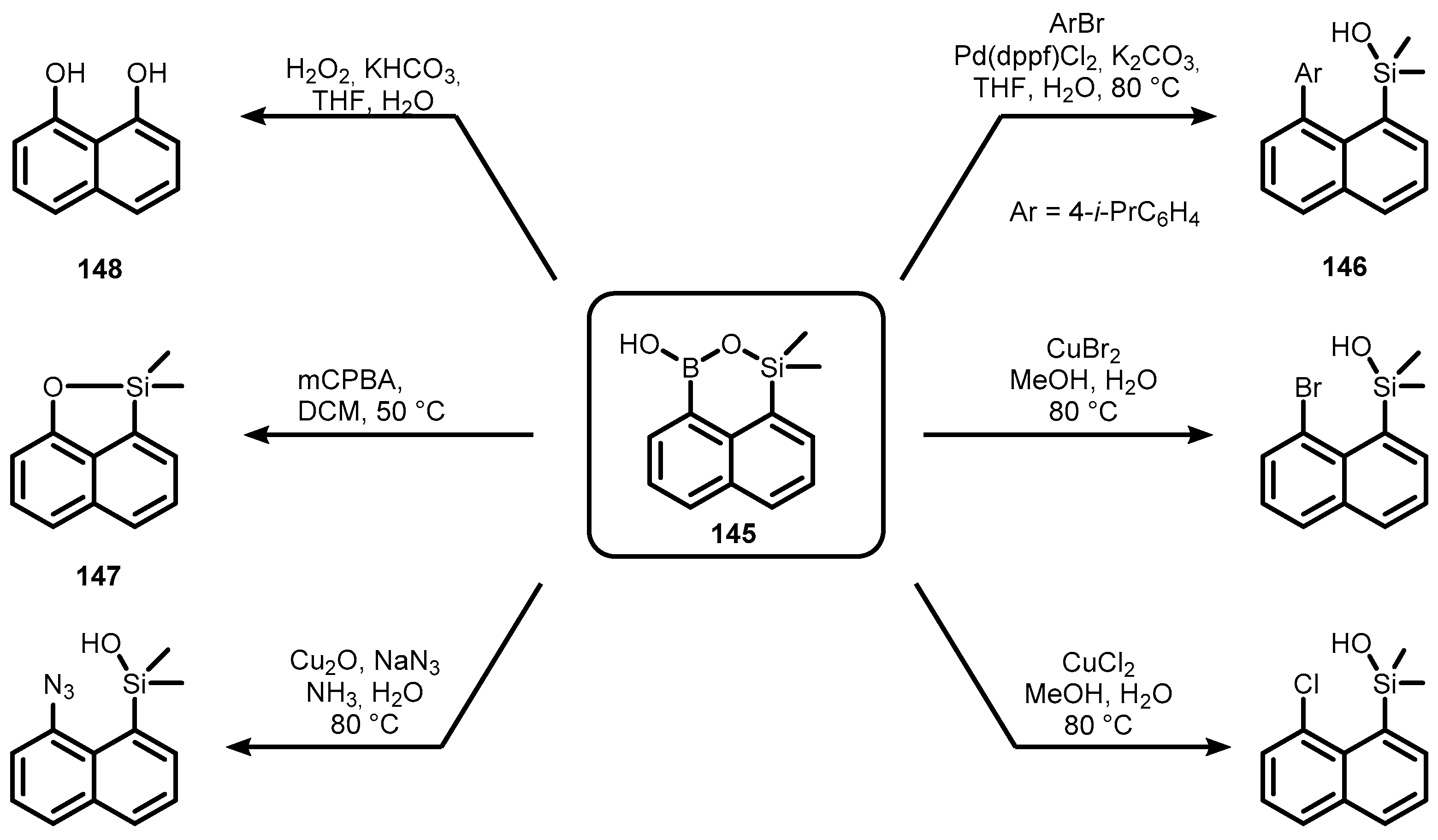{kind=link}
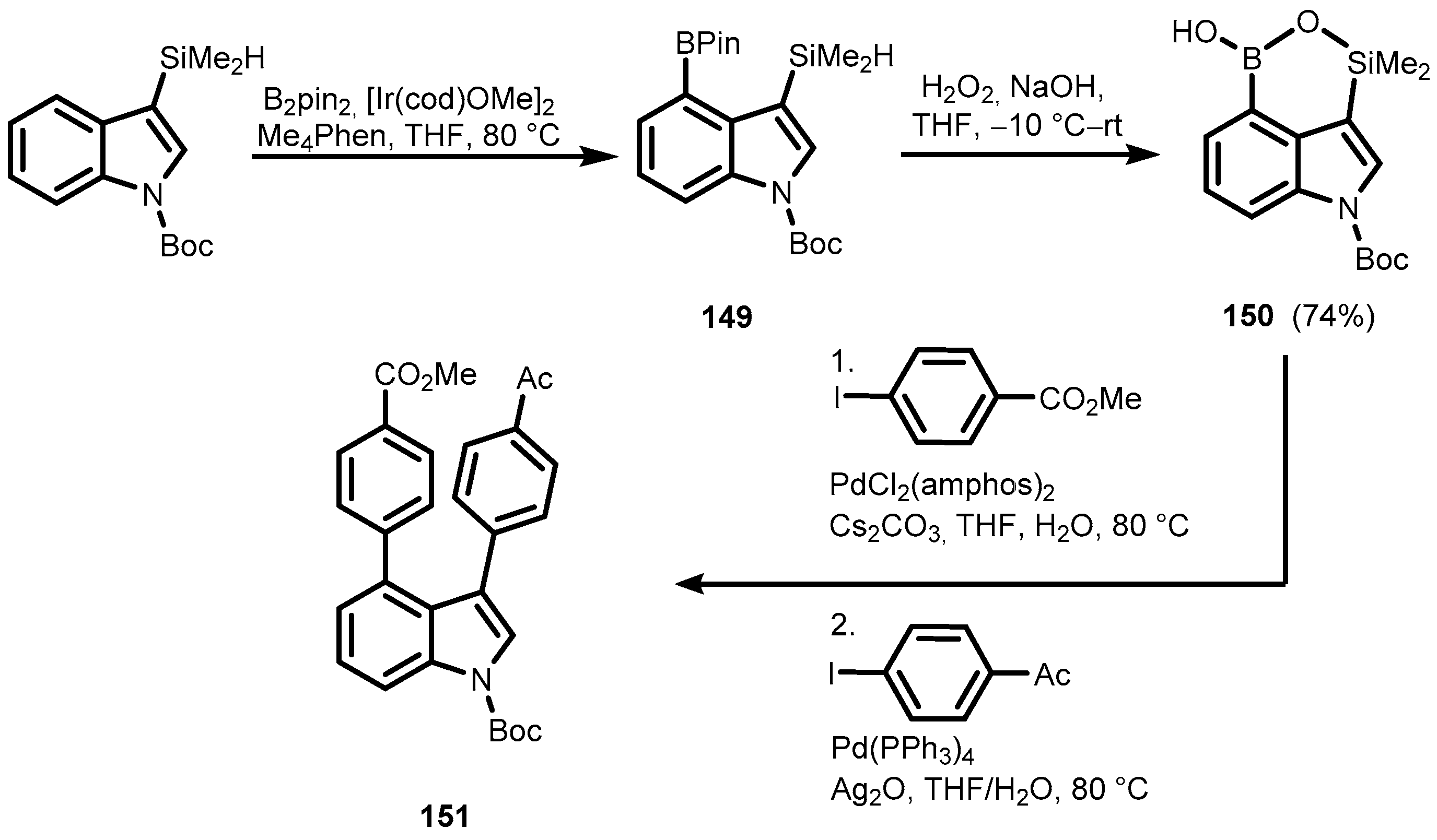{kind=link}
{kind=link}
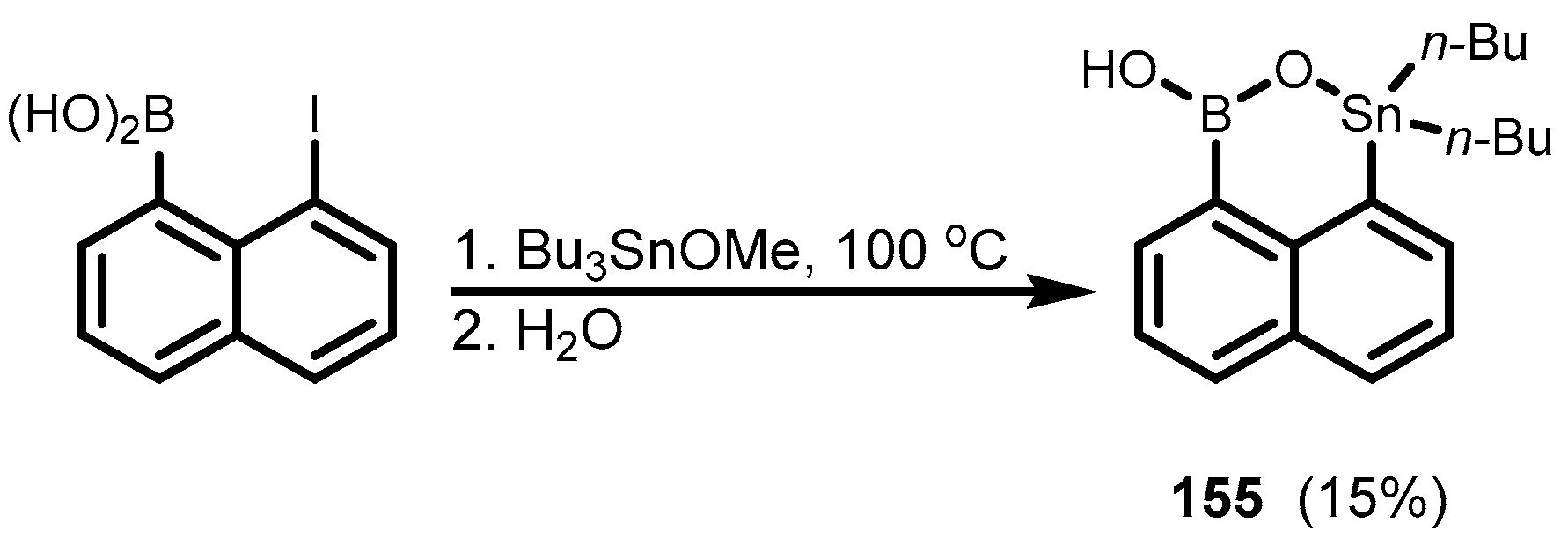{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
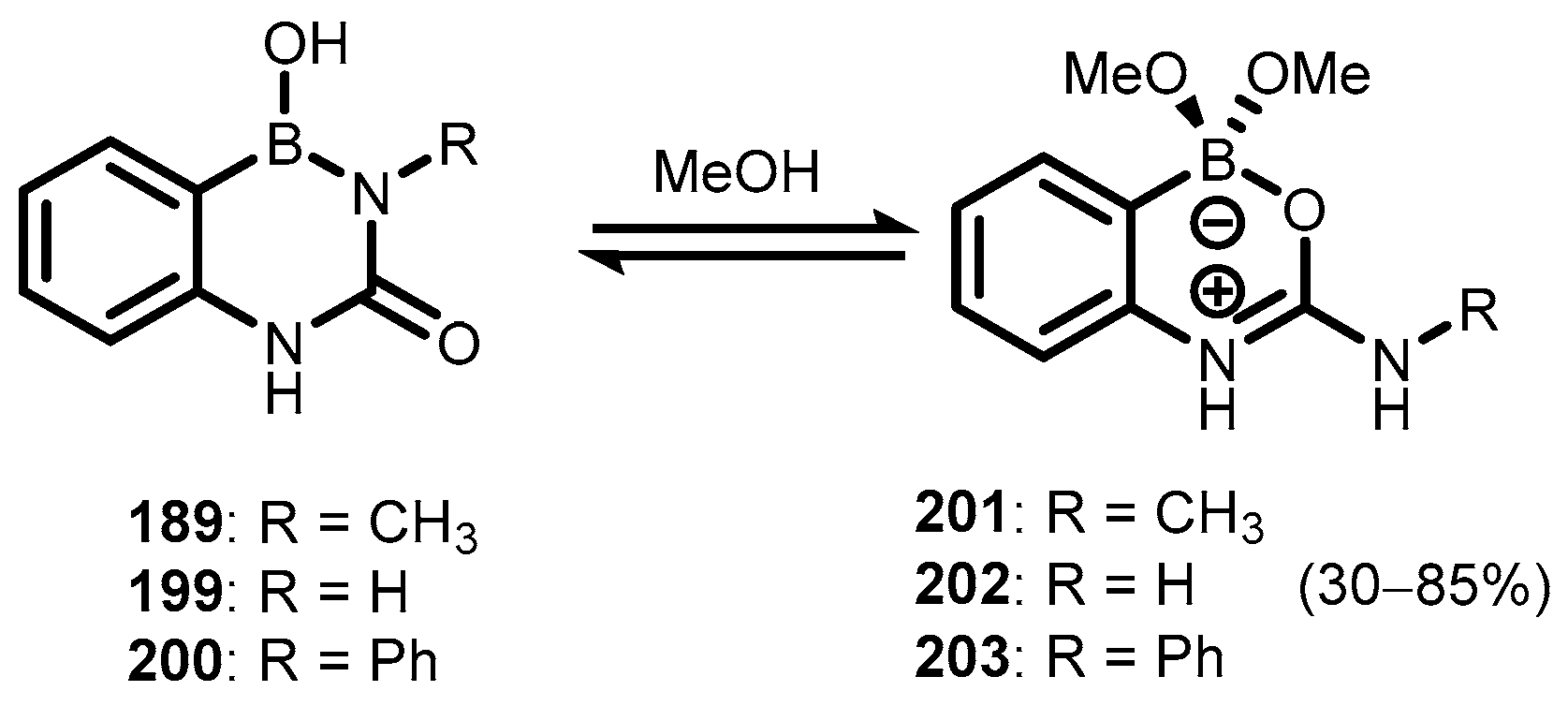{kind=link}
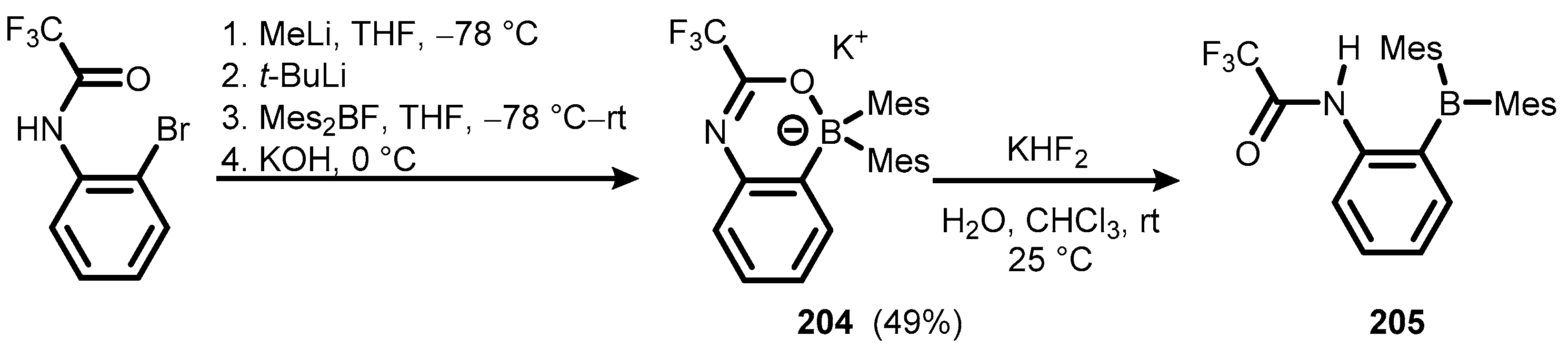{kind=link}
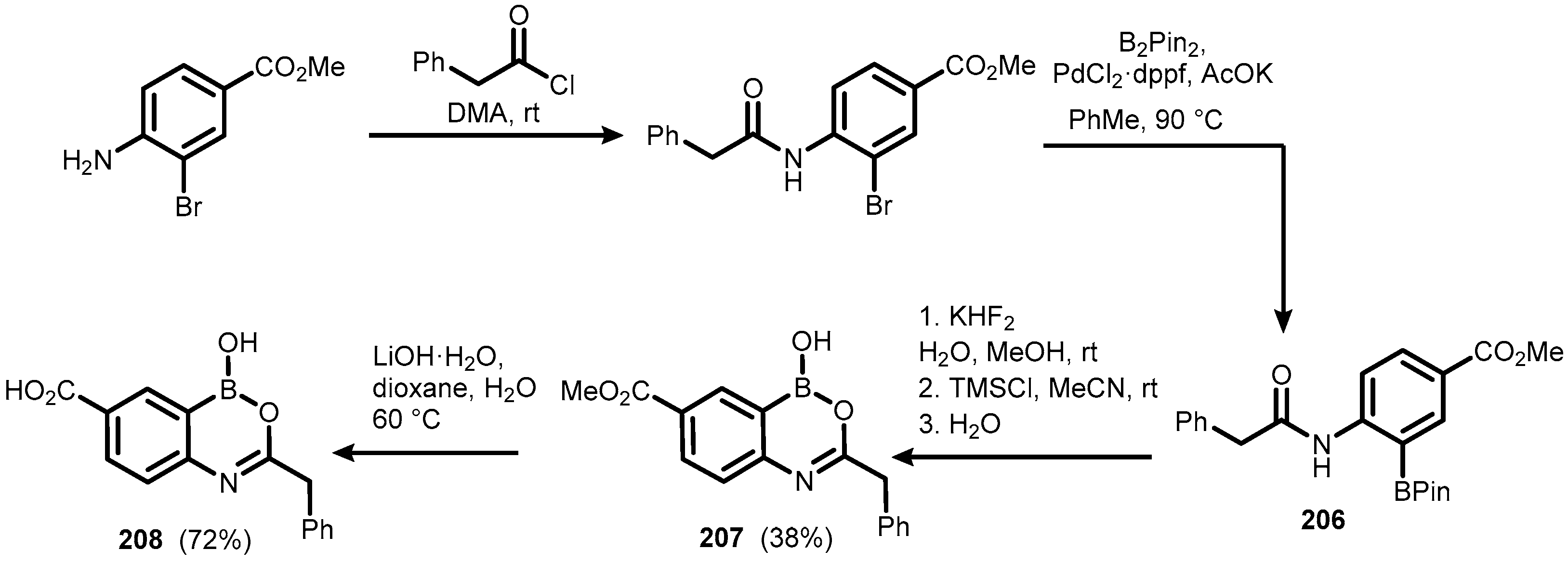{kind=link}
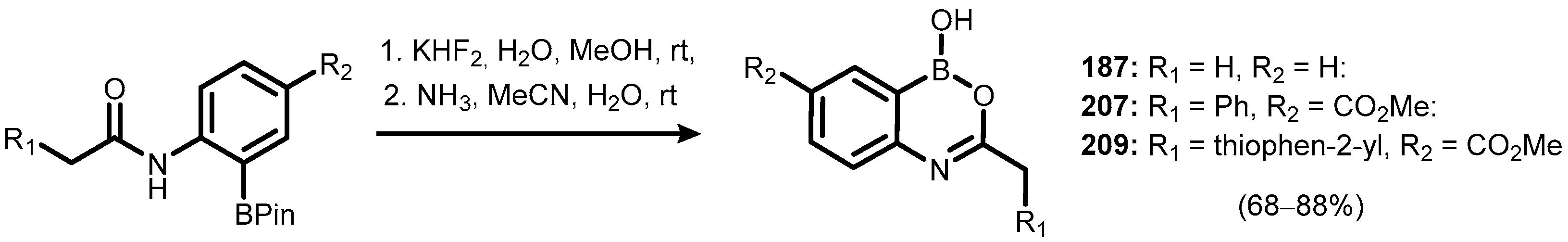{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
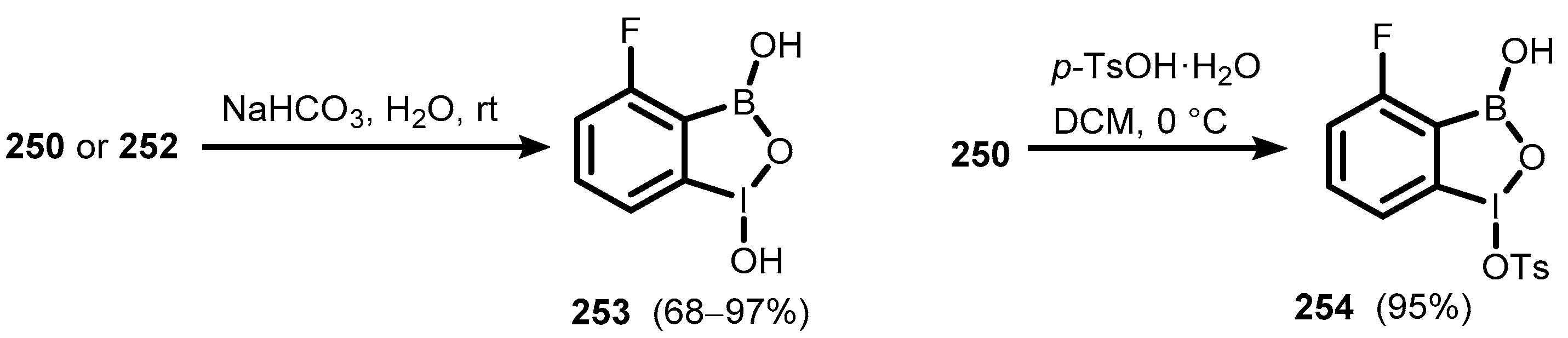{kind=link}
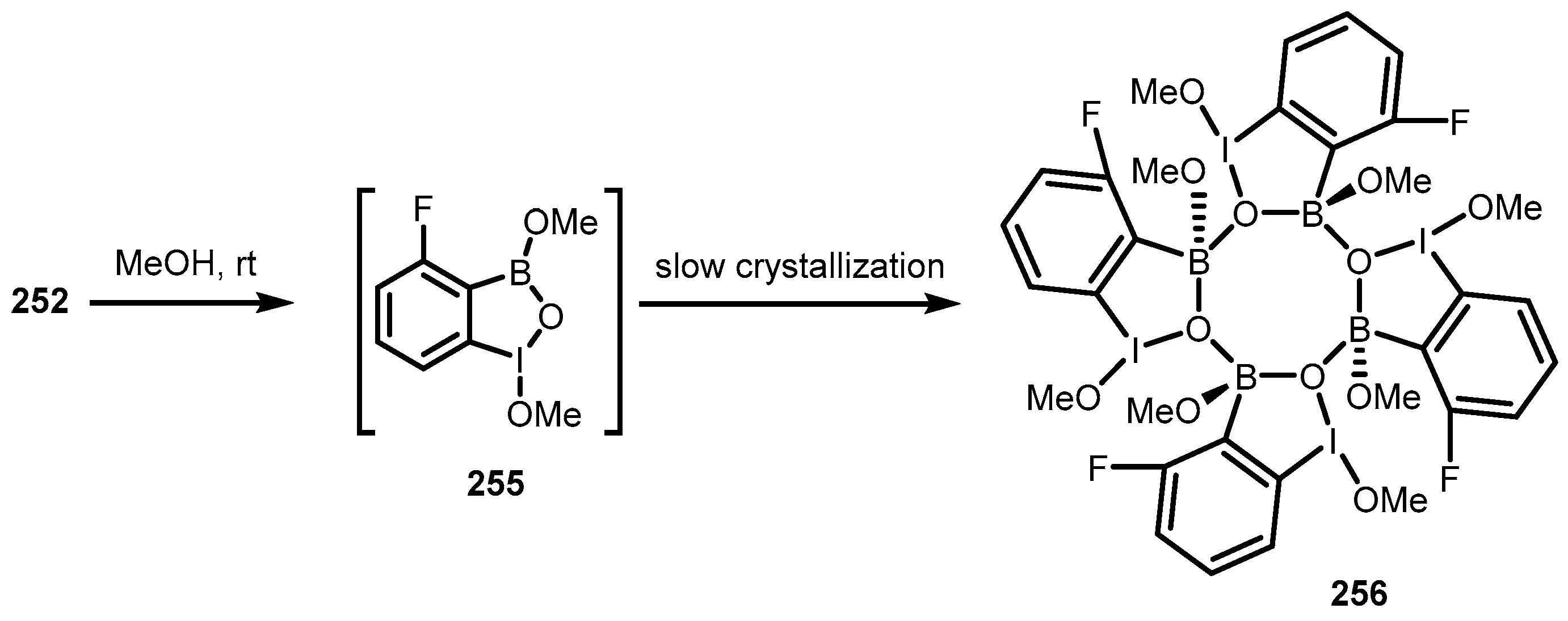{kind=link}
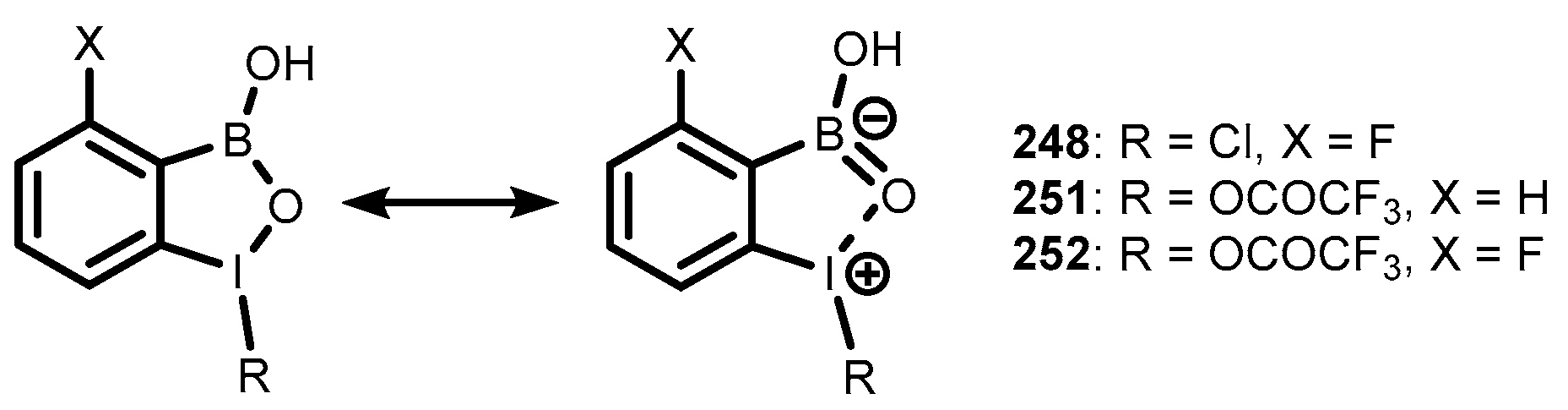{kind=link}
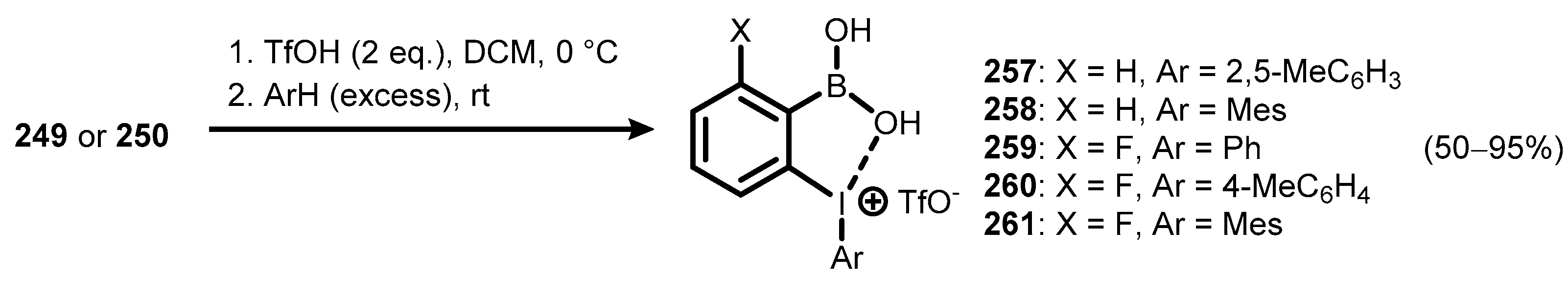{kind=link}
{kind=link}
{kind=link}
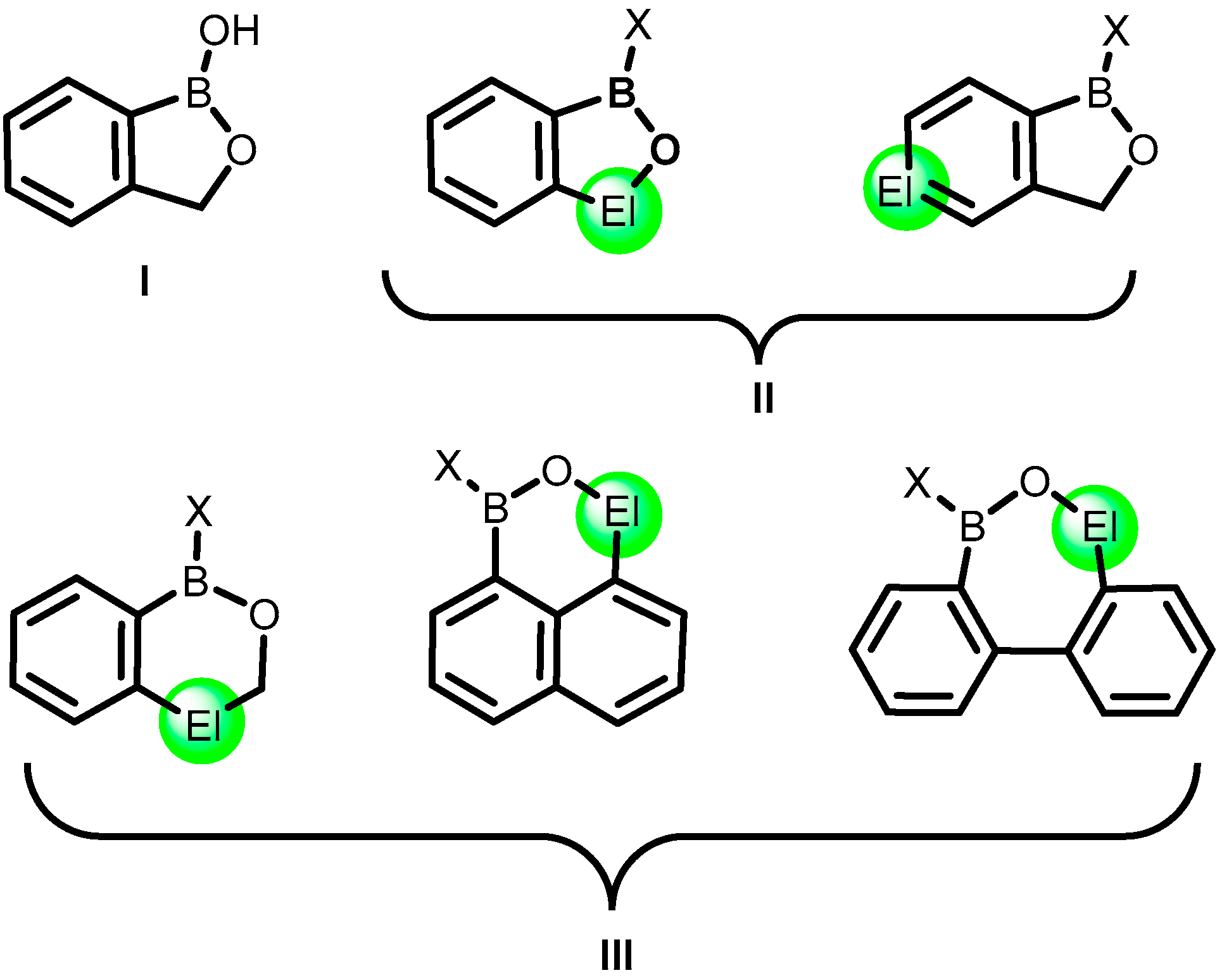
1. Introduction
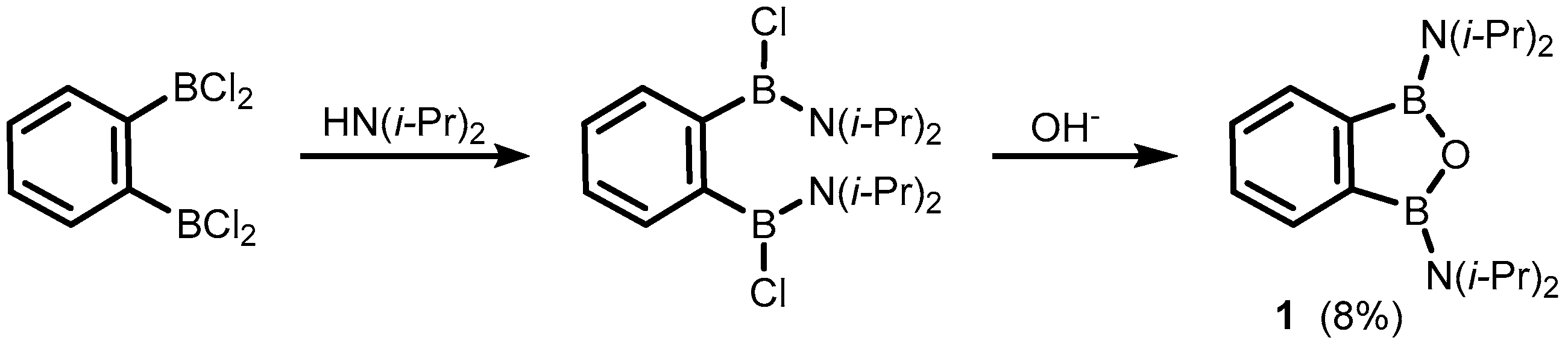
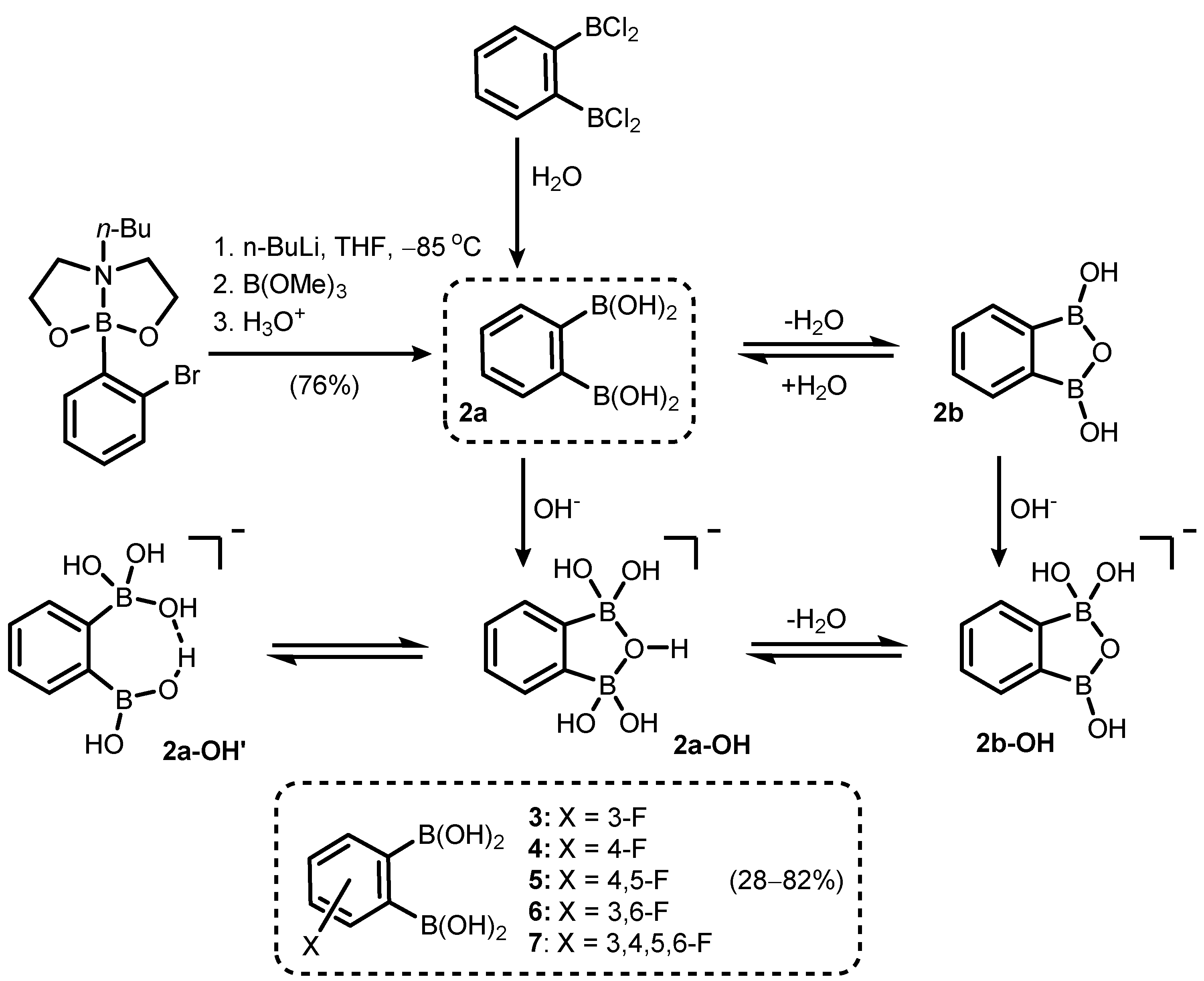
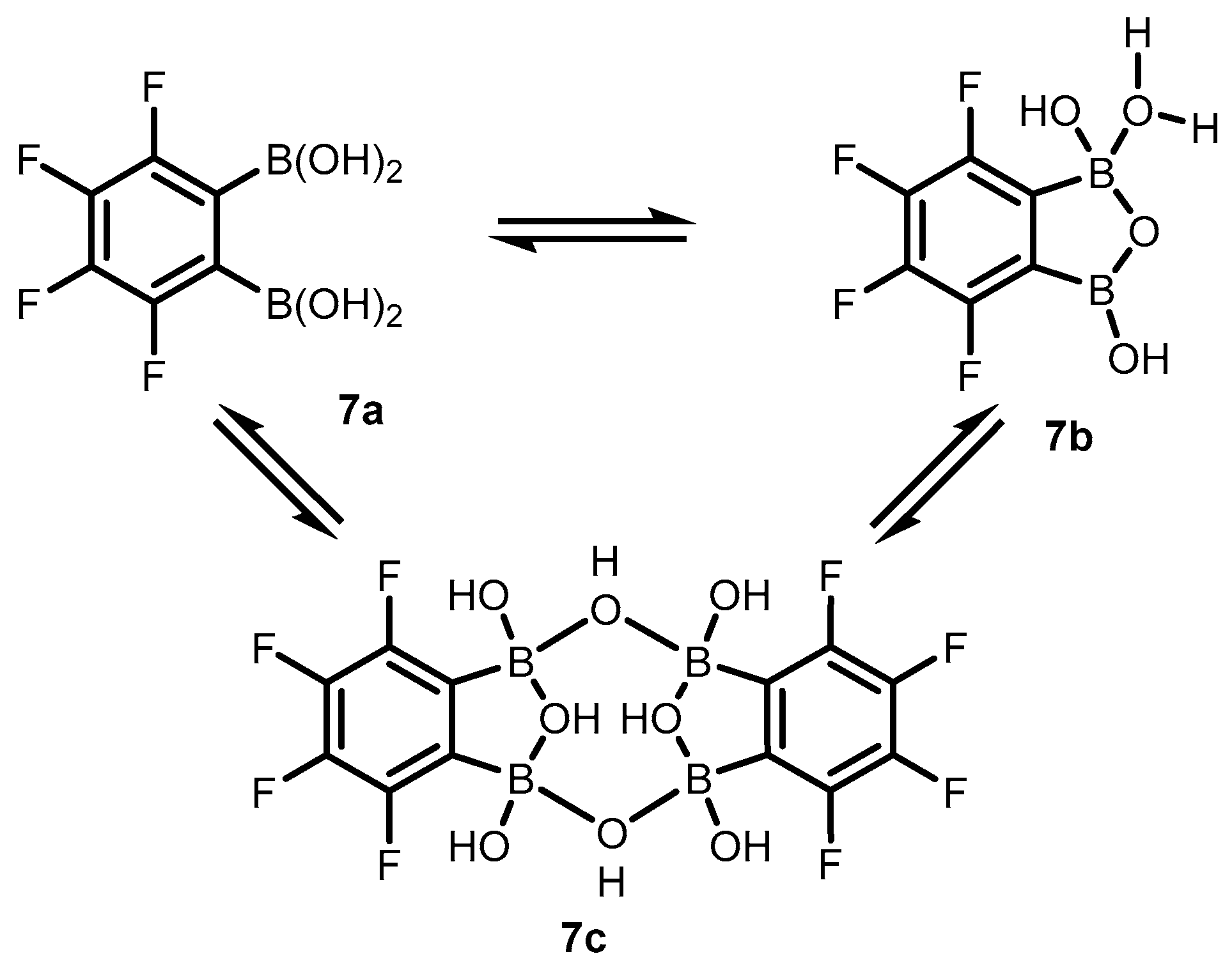
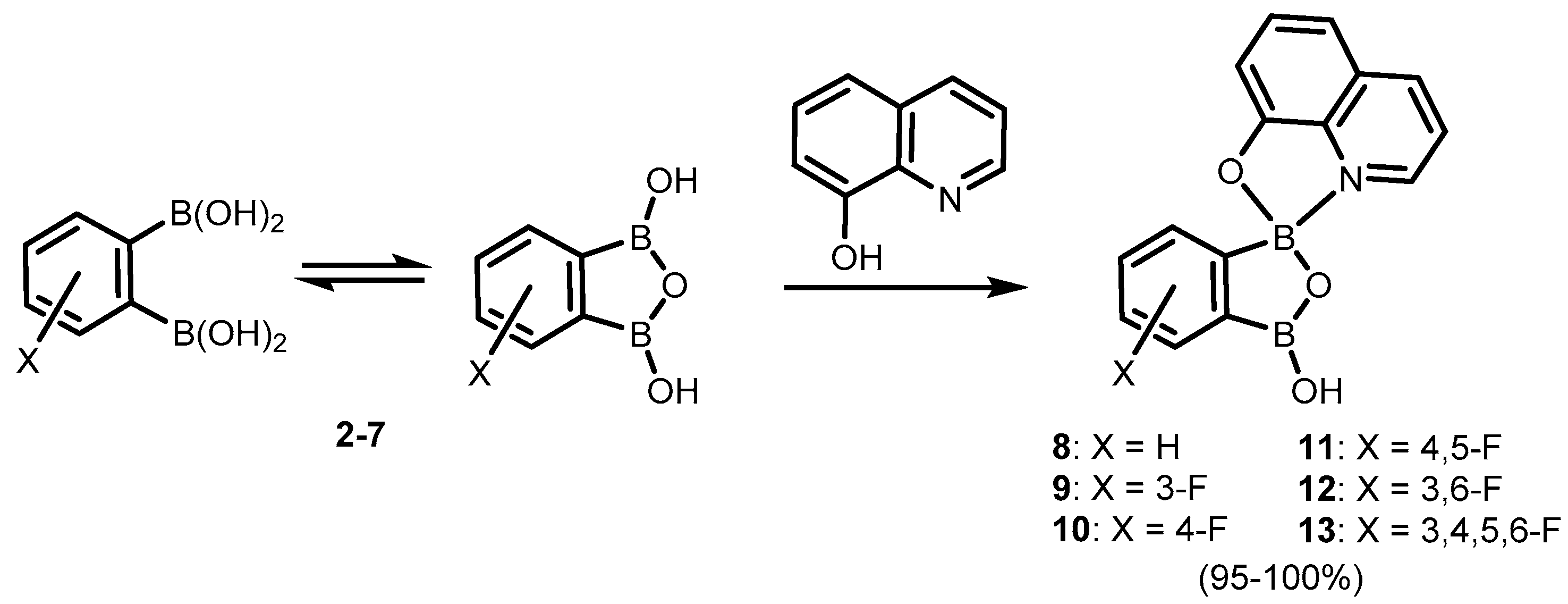
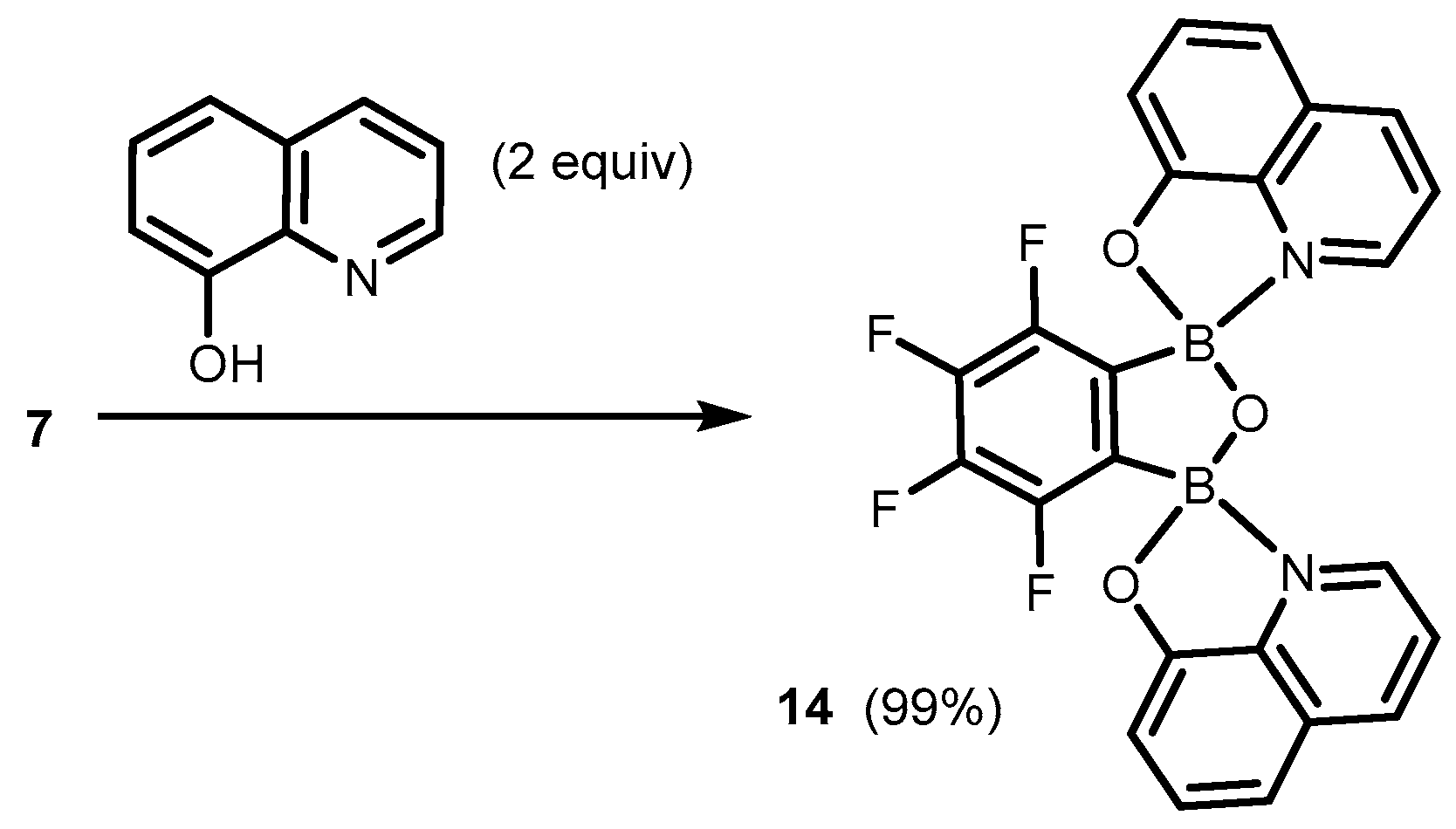
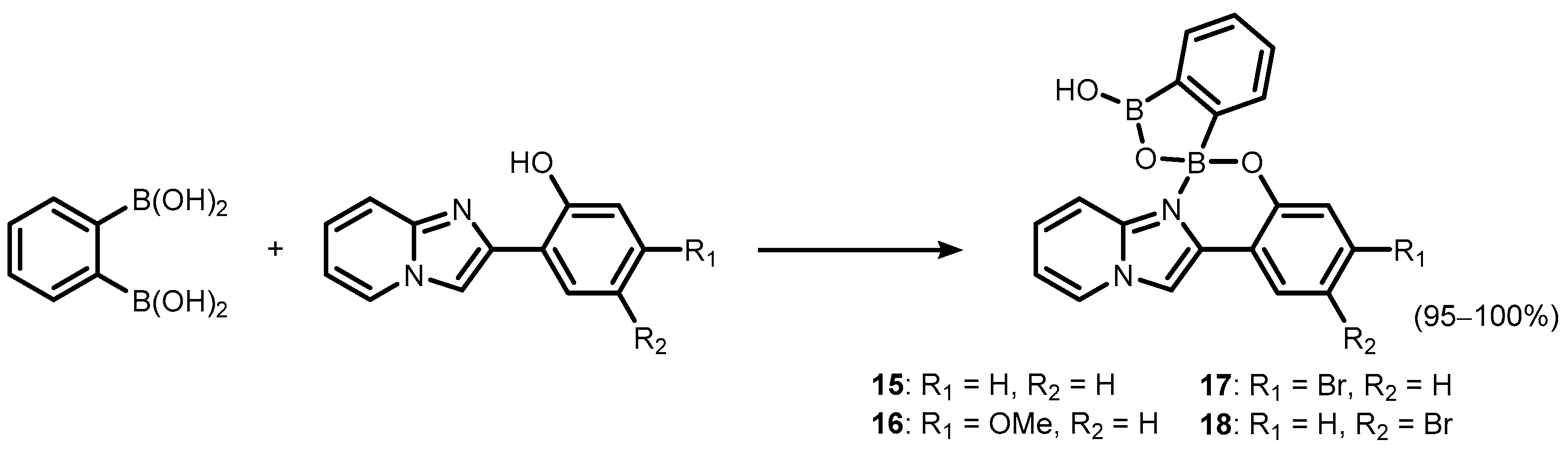
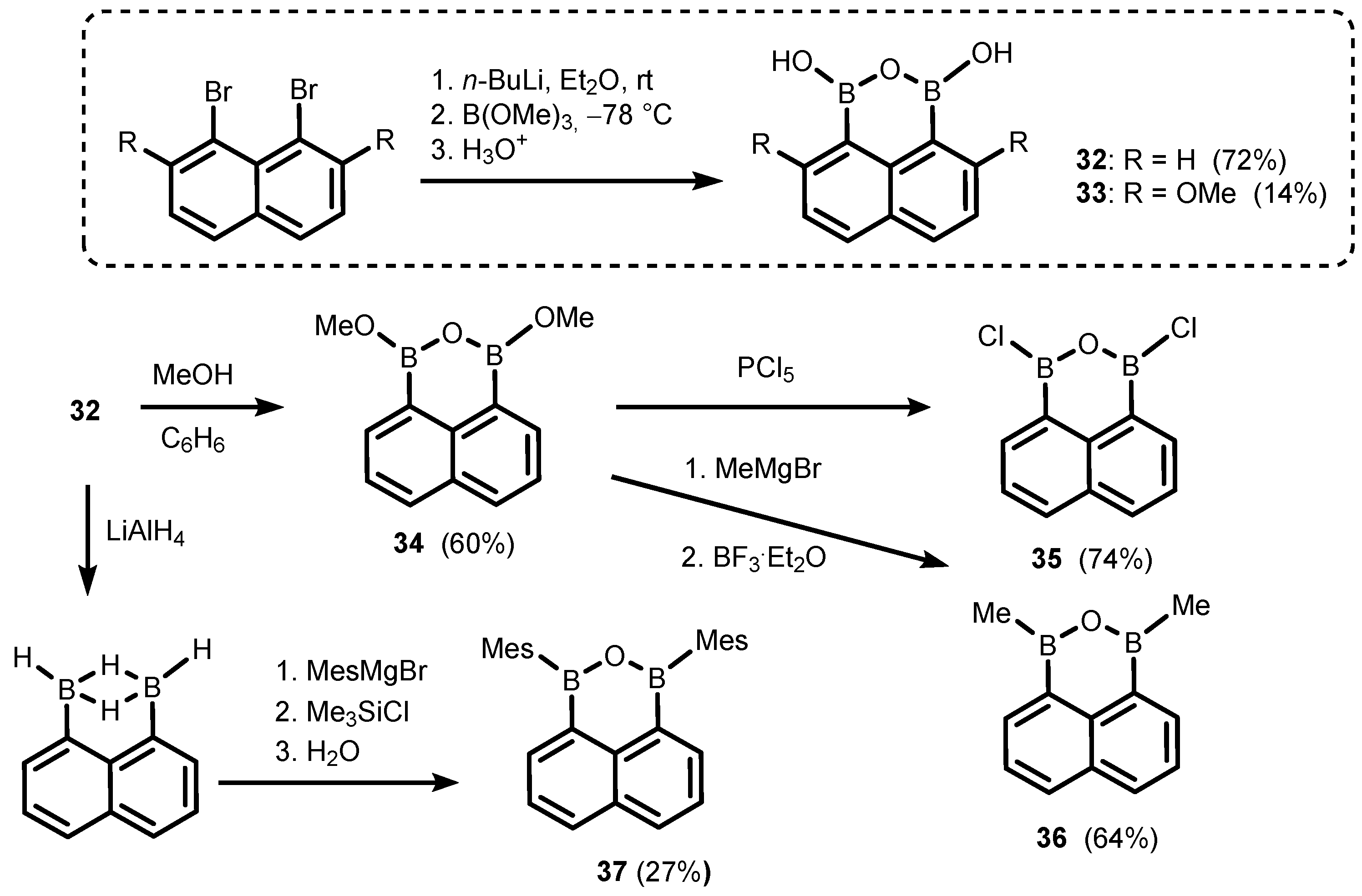
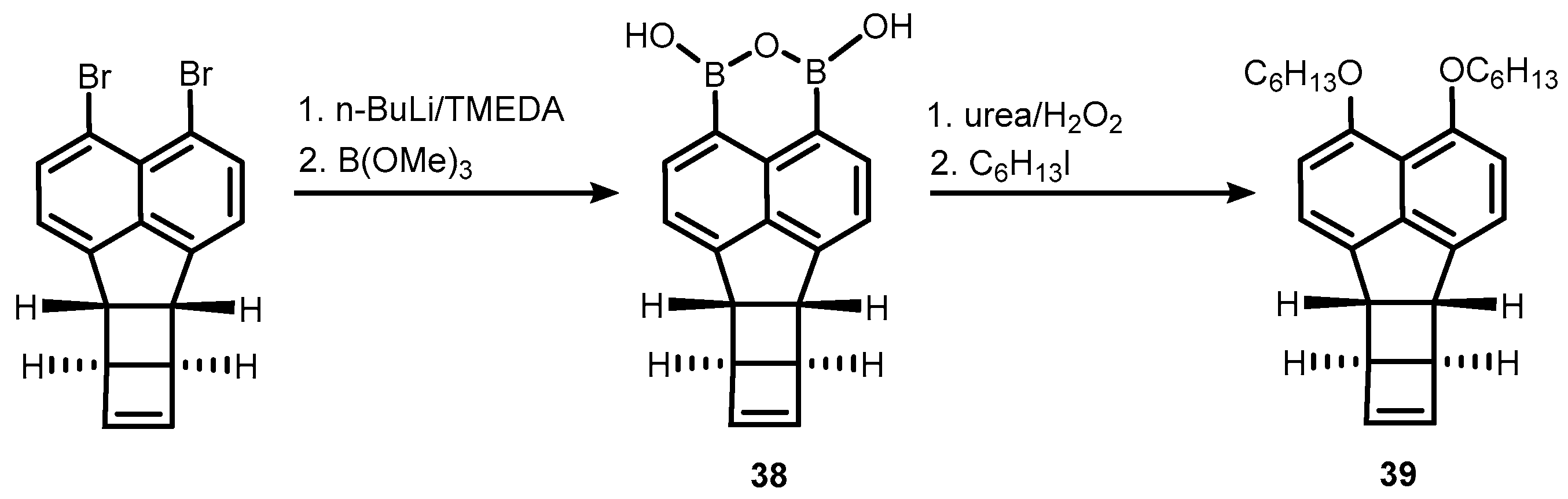
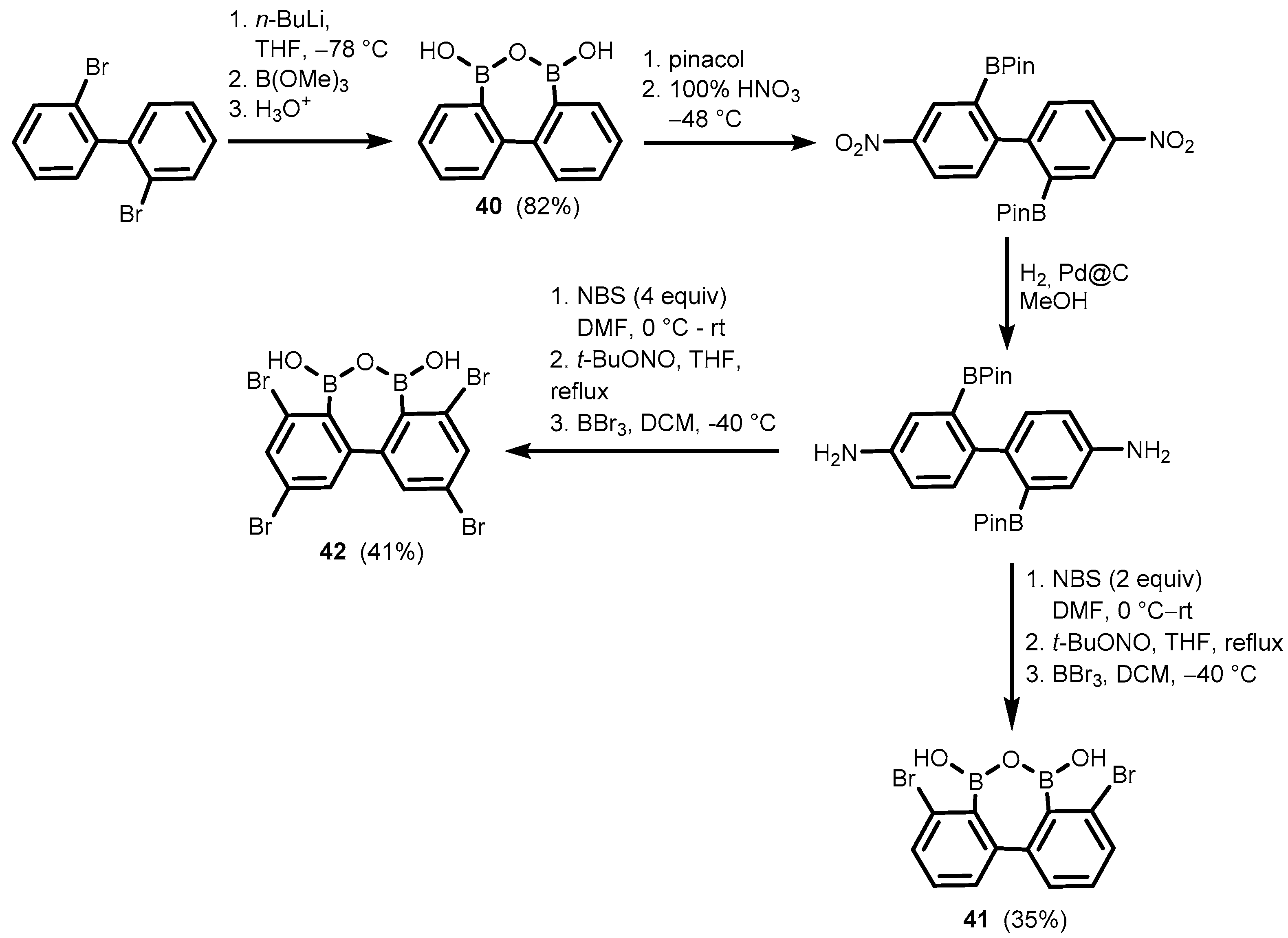
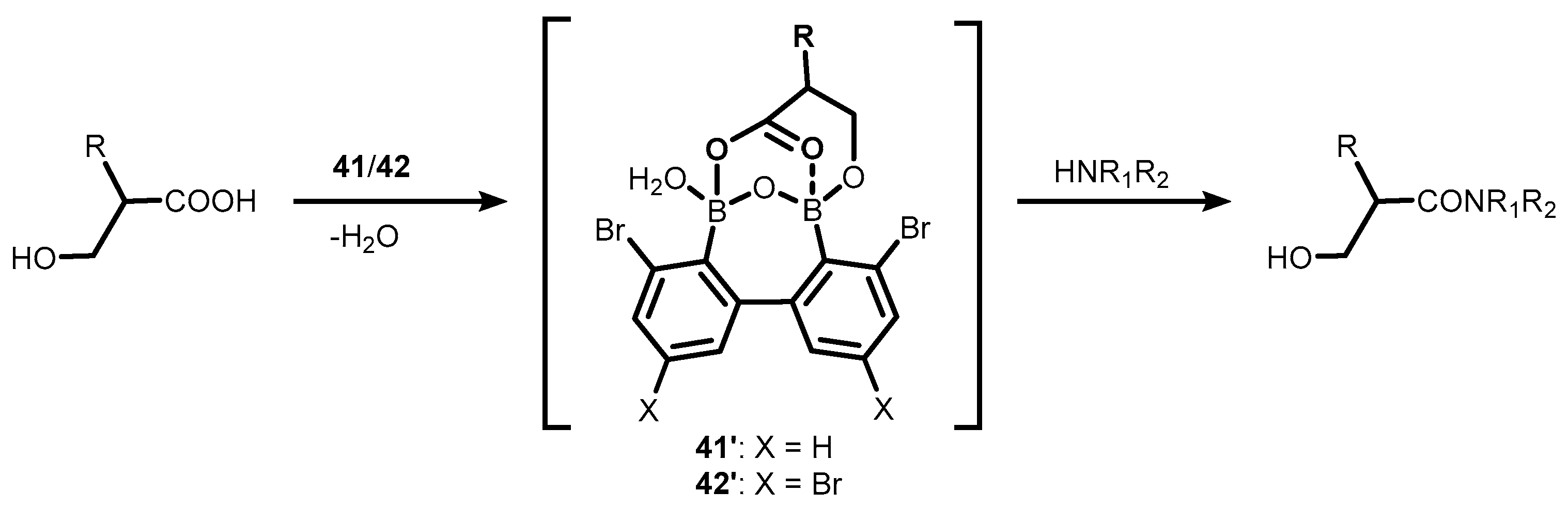
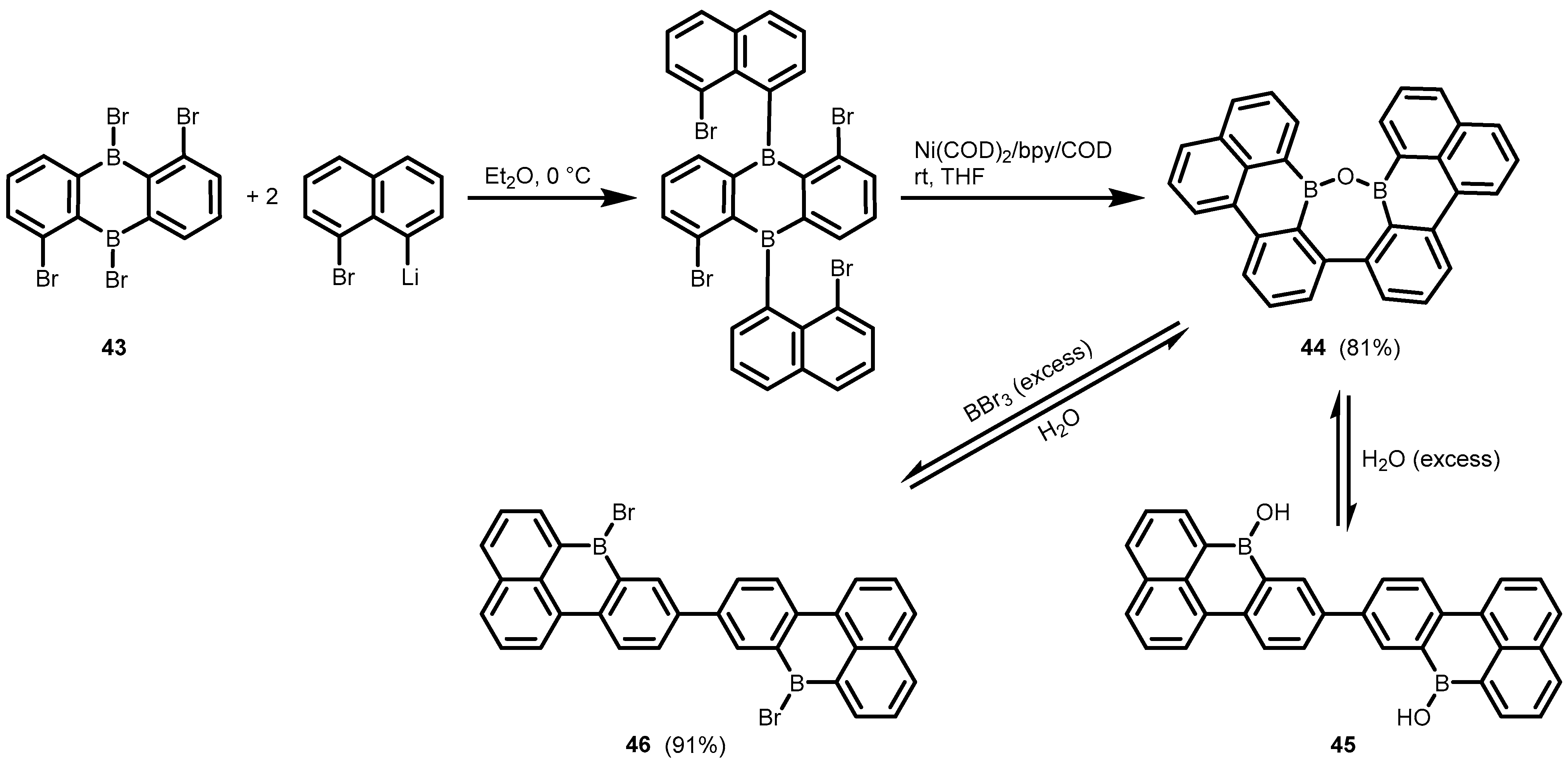
2. Benzoxadiboroles and Related Ring-Expanded Systems Comprising B-O-B Linkage
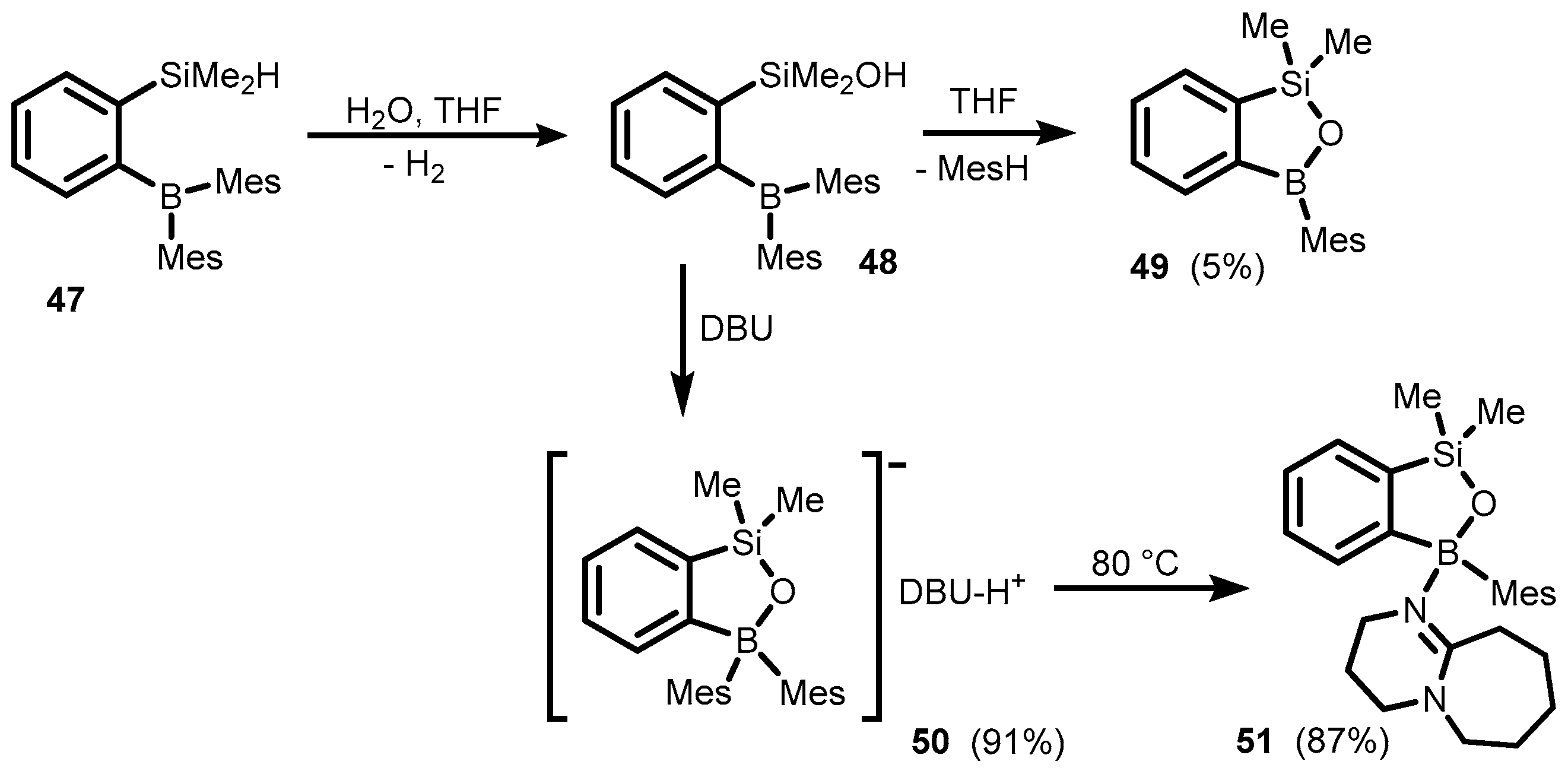
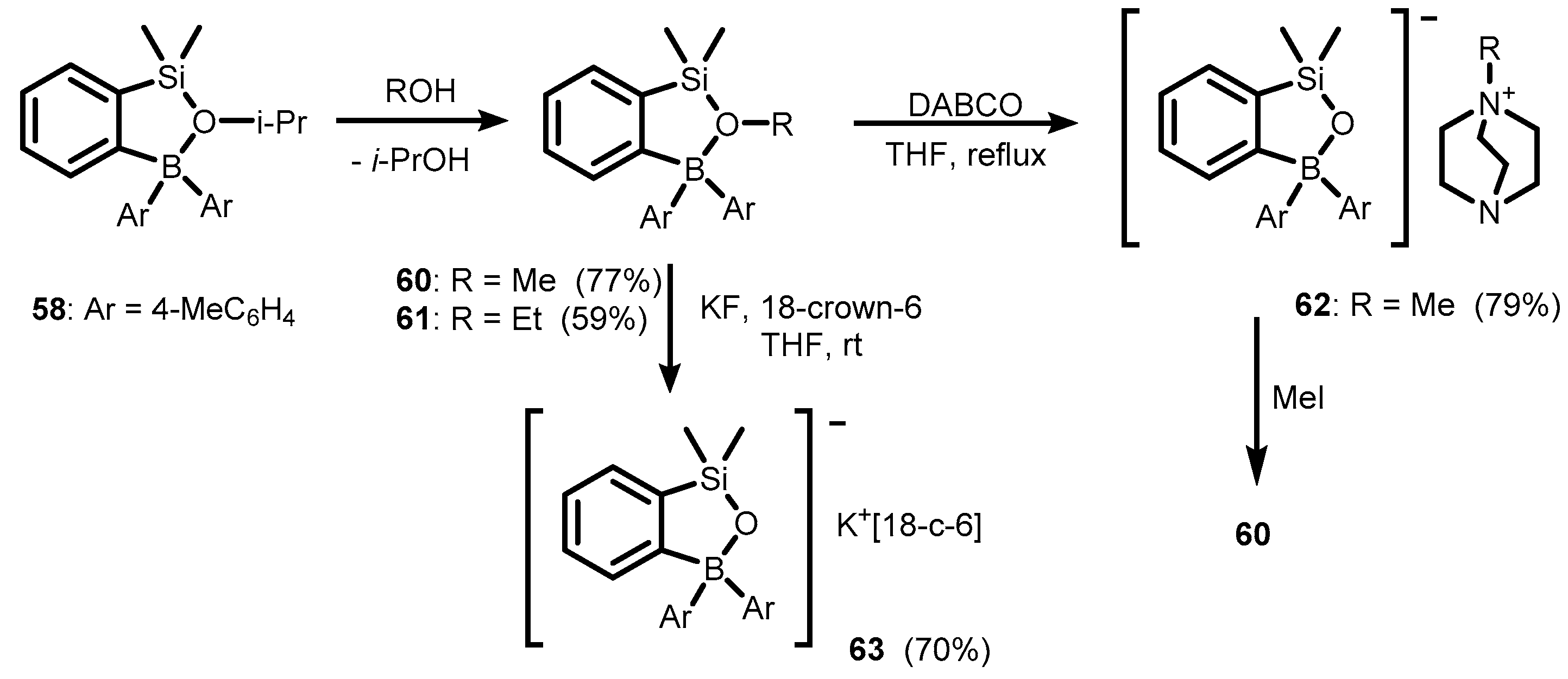
3. Benzosiloxaboroles and Related Ring-Expanded Systems Comprising B-O-Si Linkage
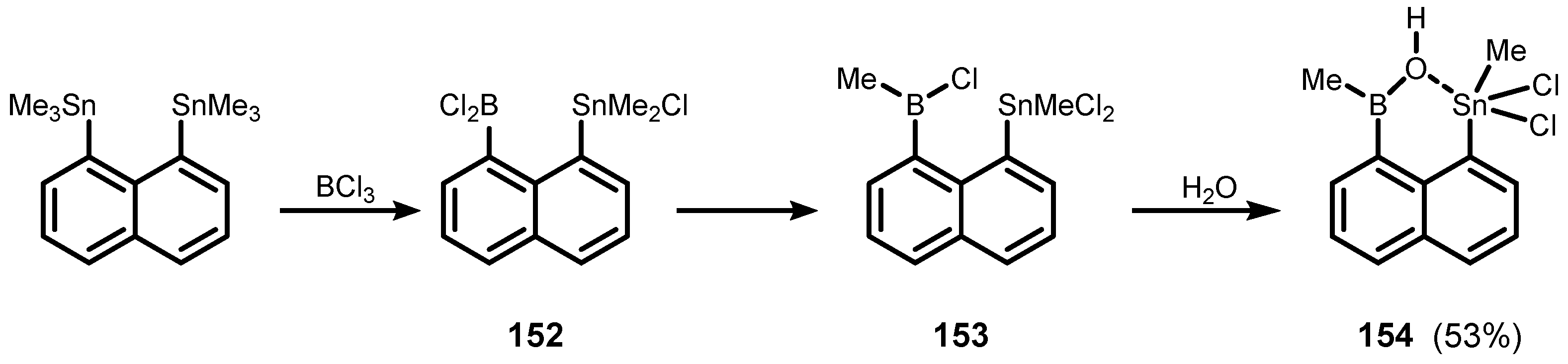
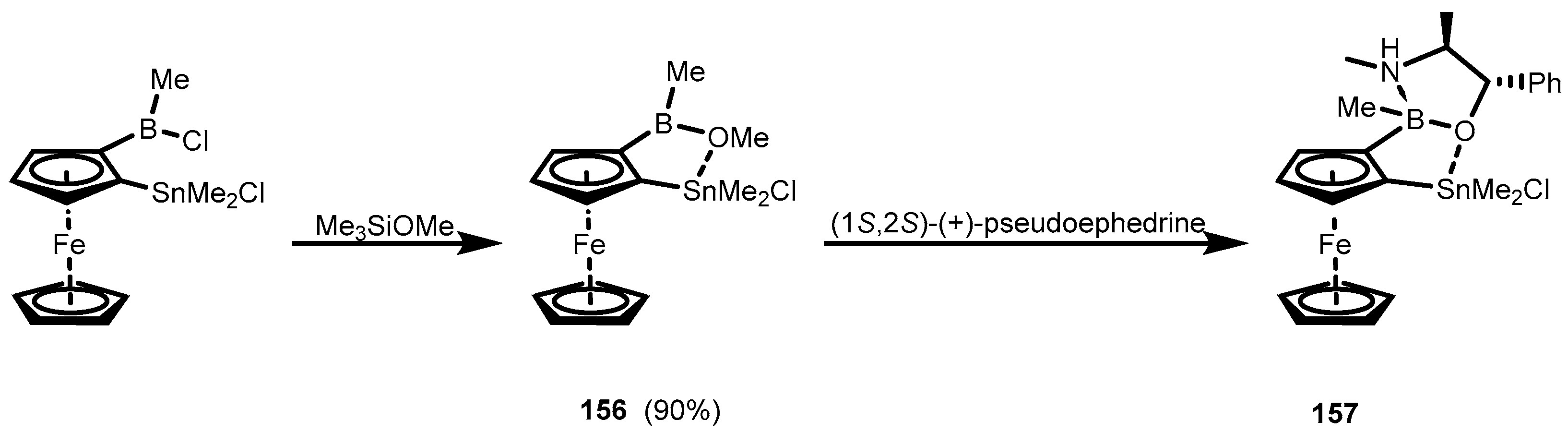
4. Benzoxaborole Congeners and Related Ring-Expanded Systems Comprising B-O-Sn Linkage
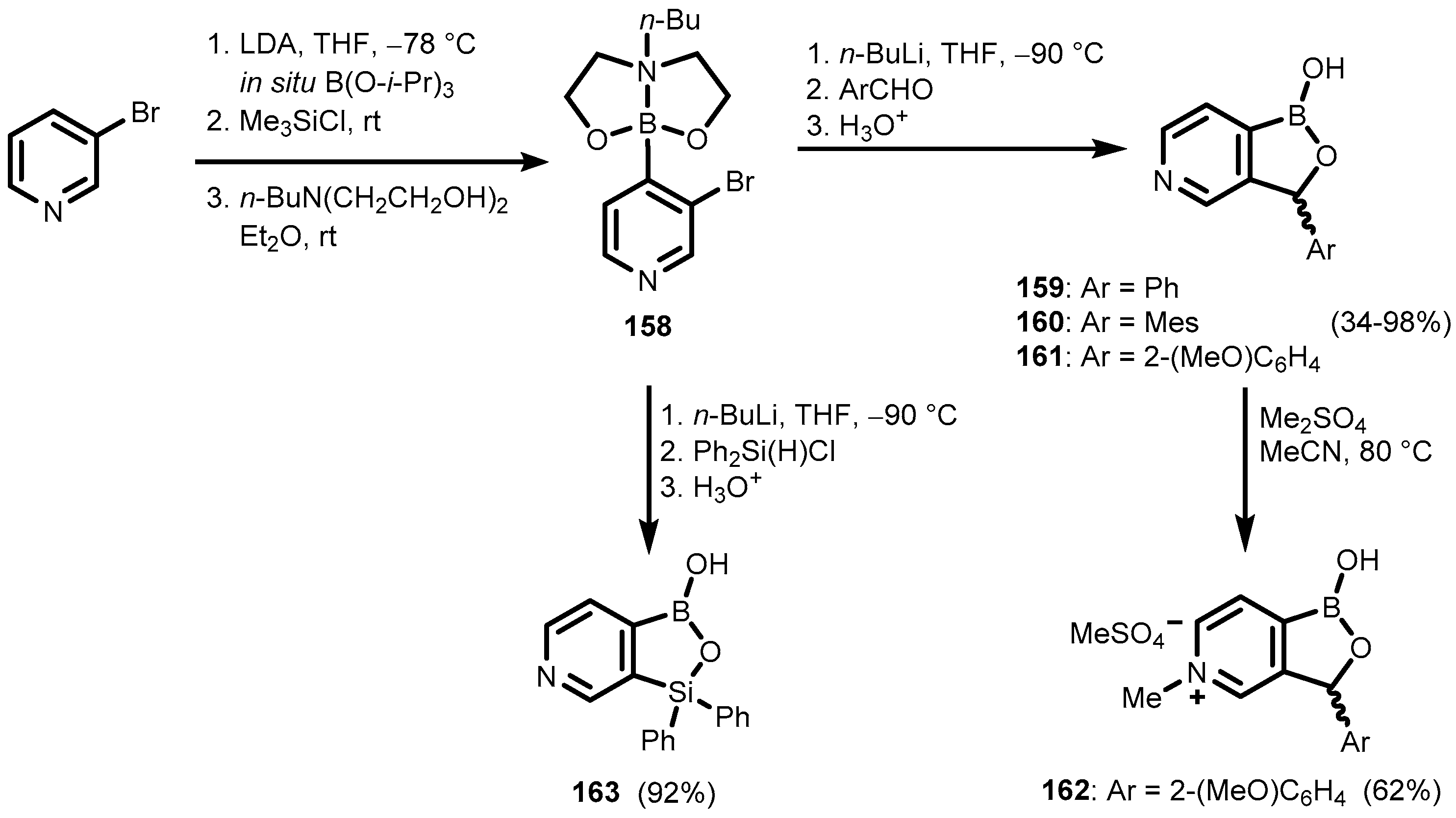
5. Benzoxaborole Aza Analogues and Related Ring-Expanded Systems
6. Benzophosphoxaboroles and Related Ring-Expanded Systems
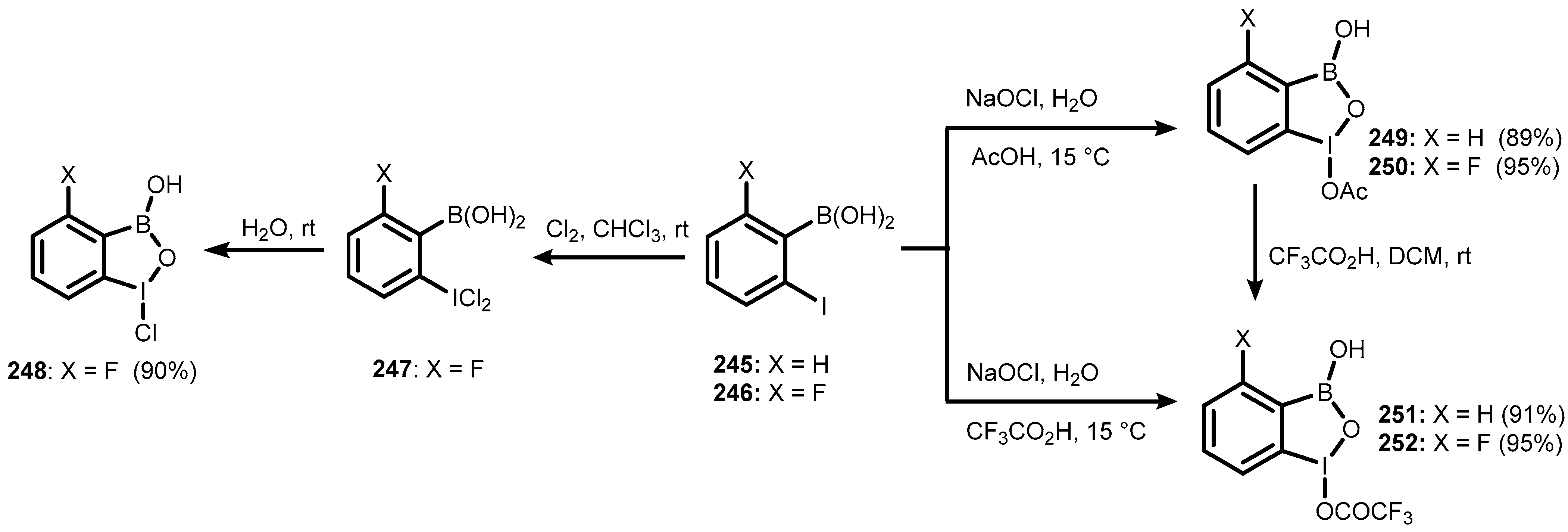
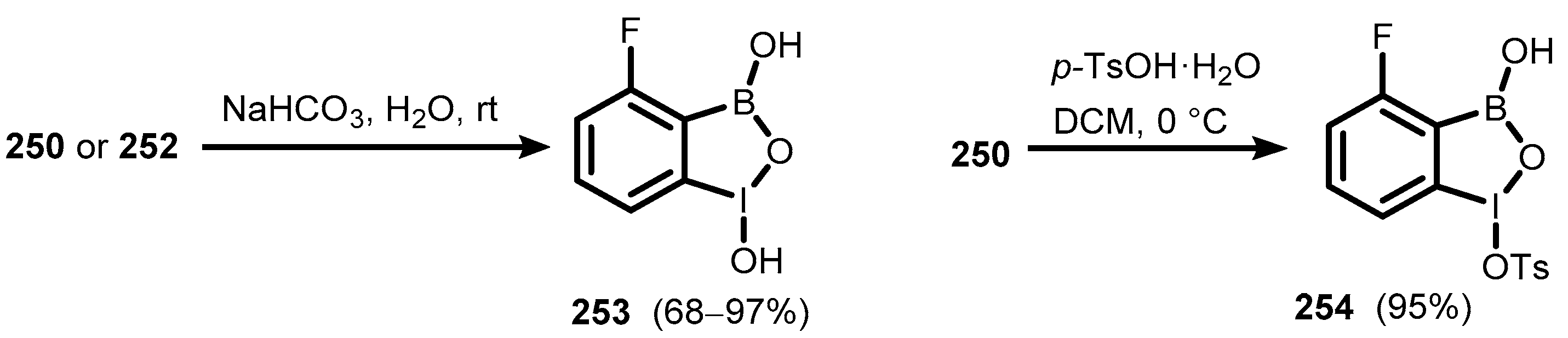
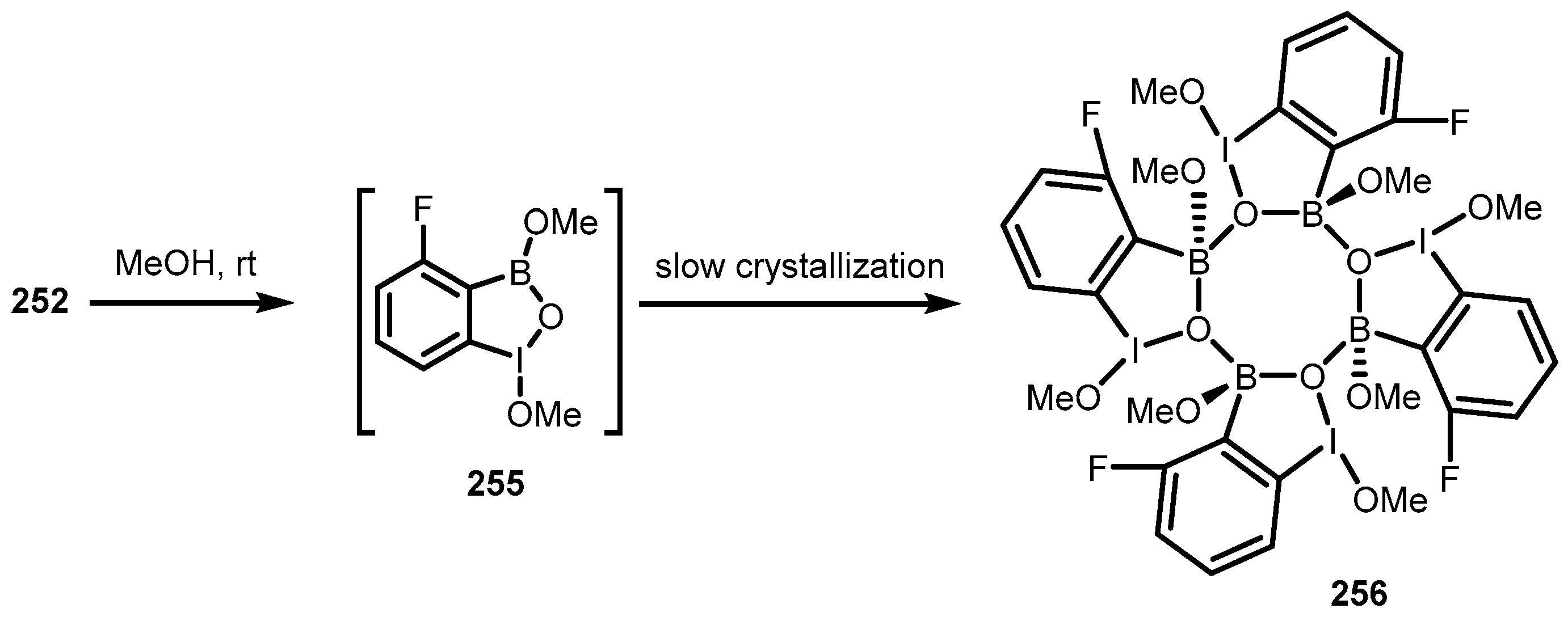
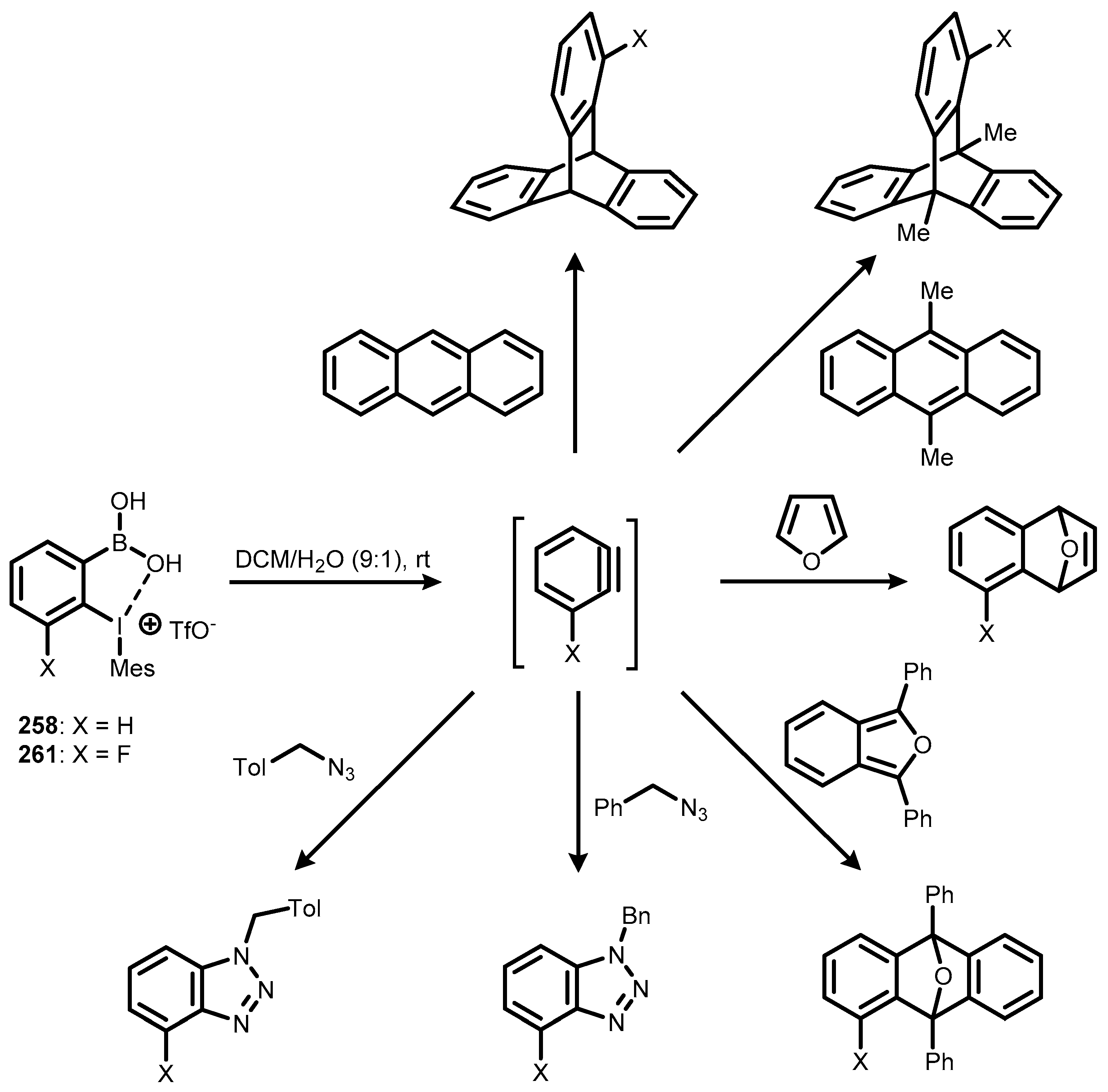
7. Benzoiodoxaboroles
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, C.T.; Tomsho, J.W.; Benkovic, S.J. The Unique Chemistry of Benzoxaboroles: Current and Emerging Applications in Biotechnology and Therapeutic Treatments. Bioorg. Med. Chem. 2014, 22, 4462–4473. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent Developments in the Chemistry and Biological Applications of Benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef]
- Yang, F.; Zhu, M.; Zhang, J.; Zhou, H. Synthesis of Biologically Active Boron-Containing Compounds. Med. Chem. Commun. 2018, 9, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T.; Winum, J.-Y. Benzoxaborole Compounds for Therapeutic Uses: A Patent Review (2010–2018). Expert Opin. Ther. Pat. 2018, 28, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.E.; Boese, R.; Scheer, A. 1,2-Bis(diisopropylamino)-1,2-dihydro-1,2-benzodiboret-ein erstes thermisch stabiles 1,2-Dihydro-1,2-diboret. Chem. Ber. 1994, 127, 2349–2351. [Google Scholar] [CrossRef]
- Kaufmann, D. Borylierung von Arylsilanen, II Synthese und Reaktionen silylierter Dihalogenphenylborane. Chem. Ber. 1987, 120, 901–905. [Google Scholar] [CrossRef]
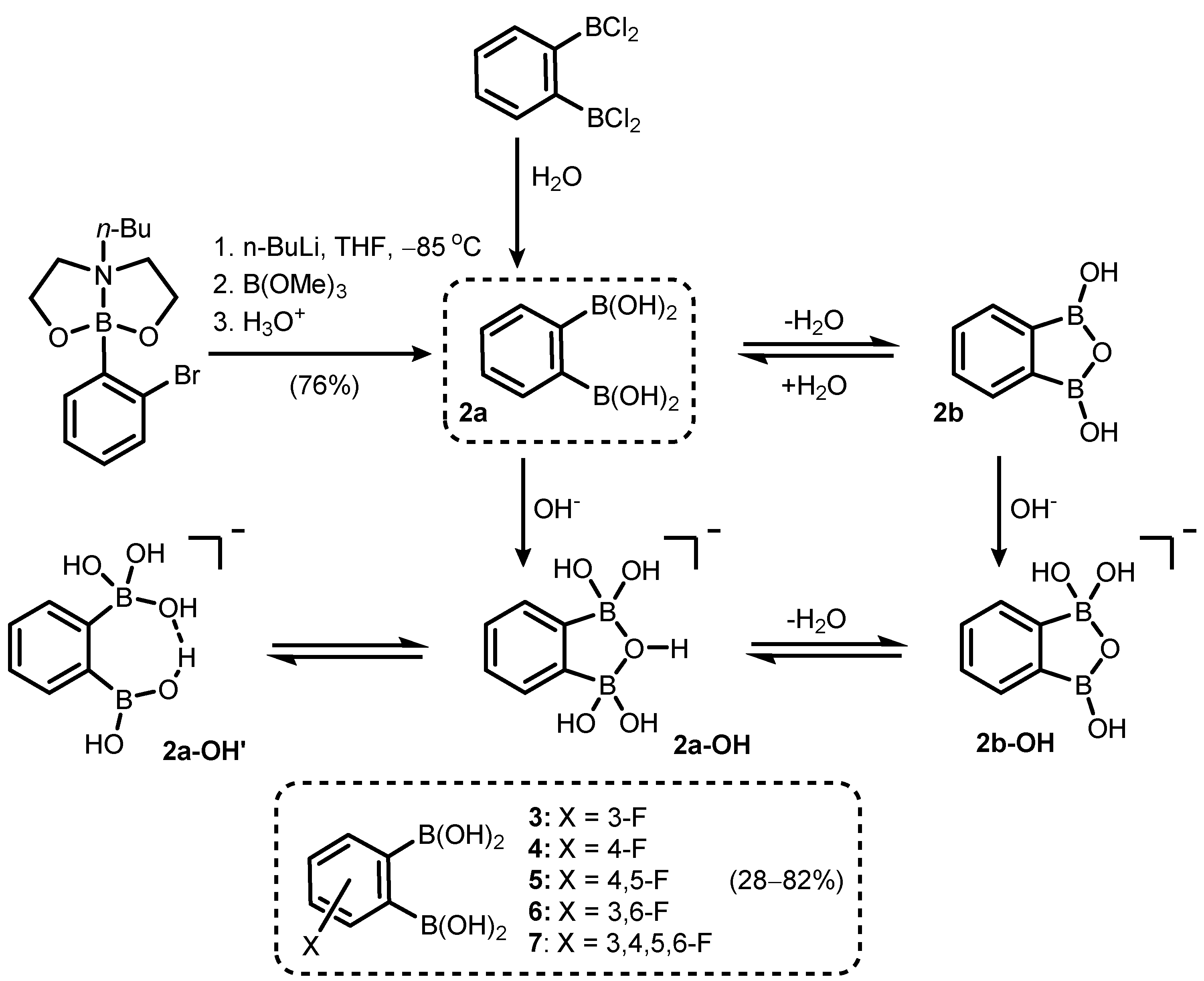
- Durka, K.; Jarzembska, K.N.; Kamiński, R.; Luliński, S.; Serwatowski, J.; Woźniak, K. Nanotubular Hydrogen-Bonded Organic Framework Architecture of 1,2-Phenylenediboronic Acid Hosting Ice Clusters. Cryst. Growth Des. 2013, 13, 4181–4185. [Google Scholar] [CrossRef]
- Durka, K.; Luliński, S.; Serwatowski, J.; Woźniak, K. Influence of Fluorination and Boronic Group Synergy on the Acidity and Structural Behavior of o-Phenylenediboronic Acids. Organometallics 2014, 33, 1608–1616. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Cyrański, M.; Durka, K.; Gozdalik, J.; Klimentowska, P.; Rusiecki, R.; Sporzyński, A.; Zarzeczańska, D. Structure and Properties of 1,3-Phenylenediboronic Acid: Combined Experimental and Theoretical Investigations. Crystals 2019, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Kubsik, M.; Nawara, K.; Witkowska, E.; Wiloch, M.; Luliński, S.; Waluk, J.; Głowacki, I.; et al. New Class of Easily-Synthesisable and Modifiable Organic Materials for Applications in Luminescent Devices. Dye. Pigm. 2017, 138, 267–277. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Kubsik, M. Engineering of Solvatomorphs of the Luminescent Complex of ortho -Phenylenediboronic Acid and 8-Hydroxyquinoline. Cryst. Growth Des. 2017, 17, 6836–6851. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Woźniak, K. Ground-State Charge-Density Distribution in a Crystal of the Luminescent Ortho-Phenylenediboronic Acid Complex with 8-Hydroxyquinoline. J. Phys. Chem. A 2018, 122, 4508–4520. [Google Scholar] [CrossRef]
- Kutniewska, S.E.; Jarzembska, K.N.; Kamiński, R.; Stasyuk, A.J.; Gryko, D.T.; Cyrański, M.K. Structural, Energetic and Spectroscopic Studies of New Luminescent Complexes Based on 2-(2′-Hydroxyphenyl)Imidazo[1,2-a]Pyridines and 1,2-Phenylenediboronic Acid. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2018, 74, 725–737. [Google Scholar] [CrossRef]
- Williams, V.C.; Piers, W.E.; Clegg, W.; Elsegood, M.R.J.; Collins, S.; Marder, T.B. New Bifunctional Perfluoroaryl Boranes. Synthesis and Reactivity of the ortho-Phenylene-Bridged Diboranes 1,2-[B(C6F5)2]2C6X4 (X = H, F). J. Am. Chem. Soc. 1999, 121, 3244–3245. [Google Scholar] [CrossRef]
- Williams, V.C.; Irvine, G.J.; Piers, W.E.; Li, Z.; Collins, S.; Clegg, W.; Elsegood, M.R.J.; Marder, T.B. Novel Trityl Activators with New Weakly Coordinating Anions Derived from C6F4-1,2-[B(C6F5)2]2: Synthesis, Structures, and Olefin Polymerization Behavior. Organometallics 2000, 19, 1619–1621. [Google Scholar] [CrossRef]
- Lewis, S.P.; Taylor, N.J.; Piers, W.E.; Collins, S. Isobutene Polymerization Using a Chelating Diborane Co-Initiator. J. Am. Chem. Soc. 2003, 125, 14686–14687. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Lewis, S.P.; Collins, S.; Sciarone, T.J.J.; Henderson, L.D.; Chase, P.A.; Irvine, G.J.; Piers, W.E.; Elsegood, M.R.J.; Clegg, W. Formation of Chelated Counteranions Using Lewis Acidic Diboranes: Relevance to Isobutene Polymerization. Organometallics 2007, 26, 5667–5679. [Google Scholar] [CrossRef]
- Henderson, L.D.; Piers, W.E. Ion Pair Symmetrization in Metallocenium Cations Partnered with Diborane Derived Borate Counteranions. J. Organomet. Chem. 2007, 692, 4661–4668. [Google Scholar] [CrossRef]
- Henderson, L.D.; Piers, W.E.; Irvine, G.J.; McDonald, R. Anion Stability in Stannylium, Oxonium, and Silylium Salts of the Weakly Coordinating Anion [C6F4-1,2-{B(C6F5)2}2(μ-OCH3)]−. Organometallics 2002, 21, 340–345. [Google Scholar] [CrossRef]
- Lewis, S.P.; Chai, J.; Collins, S.; Sciarone, T.J.J.; Henderson, L.D.; Fan, C.; Parvez, M.; Piers, W.E. Isobutene Polymerization Using Chelating Diboranes: Polymerization in Aqueous Suspension and Hydrocarbon Solution. Organometallics 2009, 28, 249–263. [Google Scholar] [CrossRef]
- Sgro, M.J.; Dömer, J.; Stephan, D.W. Stoichiometric CO2 Reductions Using a Bis-Borane-Based Frustrated Lewis Pair. Chem. Commun. 2012, 48, 7253. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Smith, J.M.; Gilpin, J.; MacLean, D.B. Organoboron Compounds. XX. Chemistry of Some 1-Naphthaleneboronic Acids with Substituents in the 8-Position. J. Org. Chem. 1965, 30, 807–812. [Google Scholar] [CrossRef]
- Katz, H.E. Synthesis and Characterization of Novel 1H,3H-Naphth[1,8-cd][1,2,6]oxadiborins. J. Org. Chem. 1985, 50, 2575–2576. [Google Scholar] [CrossRef]
- Scholz, A.S.; Massoth, J.G.; Bursch, M.; Mewes, J.-M.; Hetzke, T.; Wolf, B.; Bolte, M.; Lerner, H.-W.; Grimme, S.; Wagner, M. BNB-Doped Phenalenyls: Modular Synthesis, Optoelectronic Properties, and One-Electron Reduction. J. Am. Chem. Soc. 2020, 142, 11072–11083. [Google Scholar] [CrossRef] [PubMed]
- Rzepa, H.S.; Arkhipenko, S.; Wan, E.; Sabatini, M.T.; Karaluka, V.; Whiting, A.; Sheppard, T.D. An Accessible Method for DFT Calculation of 11B NMR Shifts of Organoboron Compounds. J. Org. Chem. 2018, 83, 8020–8025. [Google Scholar] [CrossRef] [Green Version]
- Watkinson, M.; Whiting, A.; McAuliffe, C.A. Synthesis of a Bis-Manganese Water Splitting Complex. J. Chem. Soc. Chem. Commun. 1994, 2141. [Google Scholar] [CrossRef]
- Li, X.; Han, J.-W.; Zhang, Y.-X.; Wong, H.N.C. Palladium-Catalyzed Double Suzuki Reactions: Synthesis of Dibenzo[4,5:6,7]Cyclohepta[1,2,3-de]Naphthalenes. Asian J. Org. Chem. 2017, 6, 1876–1884. [Google Scholar] [CrossRef]
- Kusukawa, T.; Mura, R.; Ooe, M.; Sumida, R.; Nakagawa, A. Recognition of Carboxylic Acids and Phosphonic Acids Using 1,8-Diphenylnaphthalene-Based Diguanidine. Tetrahedron 2021, 77, 131770. [Google Scholar] [CrossRef]
- Yang, J.; Horst, M.; Werby, S.H.; Cegelski, L.; Burns, N.Z.; Xia, Y. Bicyclohexene-Peri-Naphthalenes: Scalable Synthesis, Diverse Functionalization, Efficient Polymerization, and Facile Mechanoactivation of Their Polymers. J. Am. Chem. Soc. 2020, 142, 14619–14626. [Google Scholar] [CrossRef]
- IJpeij, E.G.; Beijer, F.H.; Arts, H.J.; Newton, C.; de Vries, J.G.; Gruter, G.-J.M. A Suzuki Coupling Based Route to 2,2′-Bis(2-indenyl)biphenyl Derivatives. J. Org. Chem. 2002, 67, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Hübner, A.; Weber, M.; Bolte, M.; Lerner, H.-W.; Wagner, M. 9-H-9-Borafluorene Dimethyl Sulfide Adduct: A Product of a Unique Ring-Contraction Reaction and a Useful Hydroboration Reagent. Chem. Commun. 2011, 47, 11339–11341. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Hirata, M.; Koshizuka, M.; Ohse, N.; Kaito, R.; Makino, K. Diboronic Acid Anhydrides as Effective Catalysts for the Hydroxy-Directed Dehydrative Amidation of Carboxylic Acids. Org. Lett. 2019, 21, 4303–4308. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Takahashi, N.; Ohse, N.; Koshizuka, M.; Makino, K. Synthesis of Weinreb Amides Using Diboronic Acid Anhydride-Catalyzed Dehydrative Amidation of Carboxylic Acids. Chem. Commun. 2020, 56, 13145–13148. [Google Scholar] [CrossRef]
- Shimada, N.; Ohse, N.; Takahashi, N.; Urata, S.; Koshizuka, M.; Makino, K. Direct Synthesis of N-Protected Serine- and Threonine-Derived Weinreb Amides via Diboronic Acid Anhydride-Catalyzed Dehydrative Amidation: Application to the Concise Synthesis of Garner’s Aldehyde. Synlett 2021, 32, 1024–1028. [Google Scholar] [CrossRef]
- Koshizuka, M.; Makino, K.; Shimada, N. Diboronic Acid Anhydride-Catalyzed Direct Peptide Bond Formation Enabled by Hydroxy-Directed Dehydrative Condensation. Org. Lett. 2020, 22, 8658–8664. [Google Scholar] [CrossRef] [PubMed]
- Radtke, J.; Schickedanz, K.; Bamberg, M.; Menduti, L.; Schollmeyer, D.; Bolte, M.; Lerner, H.-W.; Wagner, M. Selective Access to Either a Doubly Boron-Doped Tetrabenzopentacene or an Oxadiborepin from the Same Precursor. Chem. Sci. 2019, 10, 9017–9027. [Google Scholar] [CrossRef]
- Kawachi, A.; Zaima, M.; Tani, A.; Yamamoto, Y. Dehydrogenative Condensation of (o-Borylphenyl)Hydrosilane with Alcohols and Amines. Chem. Lett. 2007, 36, 362–363. [Google Scholar] [CrossRef]
- Kawachi, A.; Zaima, M.; Yamamoto, Y. Intramolecular Reaction of Silanol and Triarylborane: Boron–Aryl Bond Cleavage and Formation of a Si–O–B Heterocyle. Organometallics 2008, 27, 4691–4696. [Google Scholar] [CrossRef]
- Shimizu, T.; Morisako, S.; Yamamoto, Y.; Kawachi, A. Intramolecular Activation of C–O Bond by an o-Boryl Group in o-(Alkoxysilyl)(Diarylboryl)Benzenes. ACS Omega 2020, 5, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Kawachi, A. Synthesis, Reactions, and Photophysical Properties of o-(Alkoxysilyl)(borafluorenyl)benzenes. J. Organomet. Chem. 2020, 912, 121179. [Google Scholar] [CrossRef]
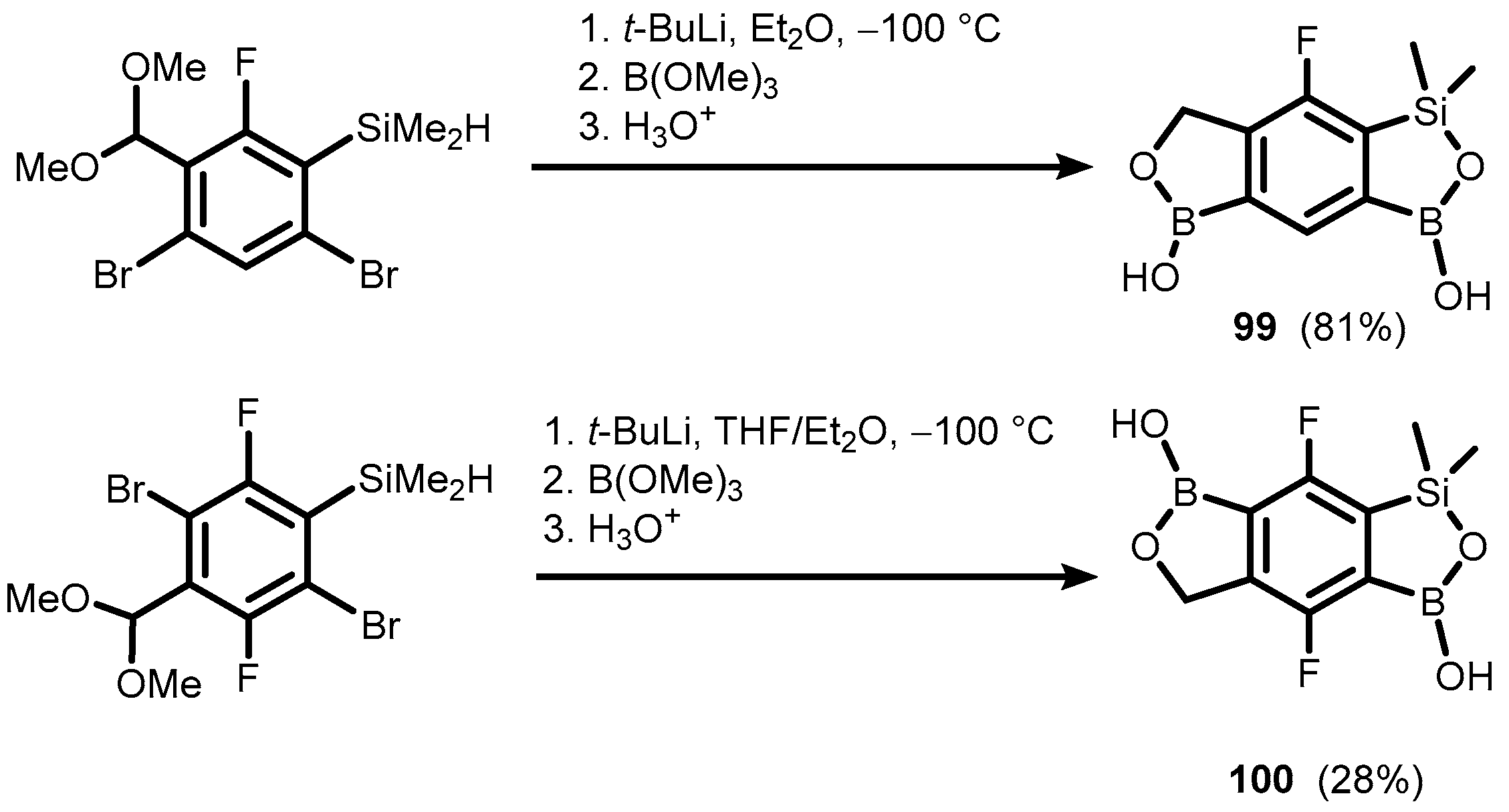
- Brzozowska, A.; Ćwik, P.; Durka, K.; Kliś, T.; Laudy, A.E.; Luliński, S.; Serwatowski, J.; Tyski, S.; Urban, M.; Wróblewski, W. Benzosiloxaboroles: Silicon Benzoxaborole Congeners with Improved Lewis Acidity, High Diol Affinity, and Potent Bioactivity. Organometallics 2015, 34, 2924–2932. [Google Scholar] [CrossRef]
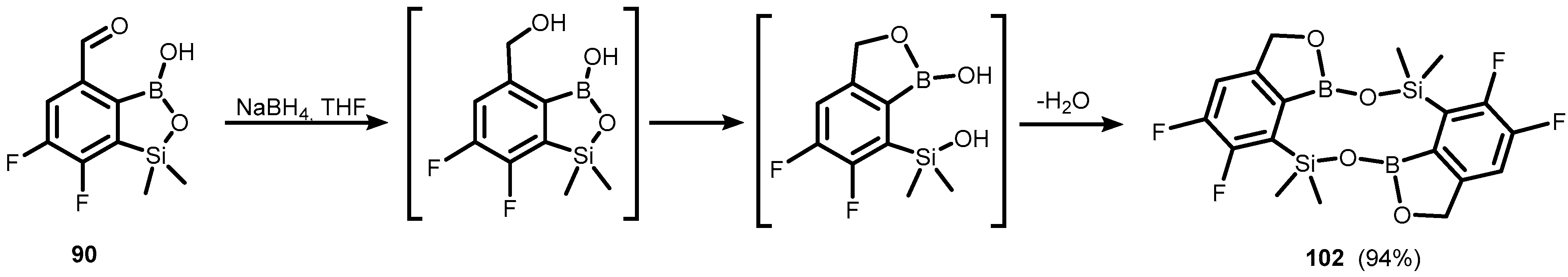
- Czub, M.; Durka, K.; Luliński, S.; Łosiewicz, J.; Serwatowski, J.; Urban, M.; Woźniak, K. Synthesis and Transformations of Functionalized Benzosiloxaboroles: Synthesis and Transformations of Functionalized Benzosiloxaboroles. Eur. J. Org. Chem. 2017, 818–826. [Google Scholar] [CrossRef]
- Durka, K.; Laudy, A.E.; Charzewski, Ł.; Urban, M.; Stępień, K.; Tyski, S.; Krzyśko, K.A.; Luliński, S. Antimicrobial and KPC/AmpC Inhibitory Activity of Functionalized Benzosiloxaboroles. Eur. J. Med. Chem. 2019, 171, 11–24. [Google Scholar] [CrossRef] [PubMed]
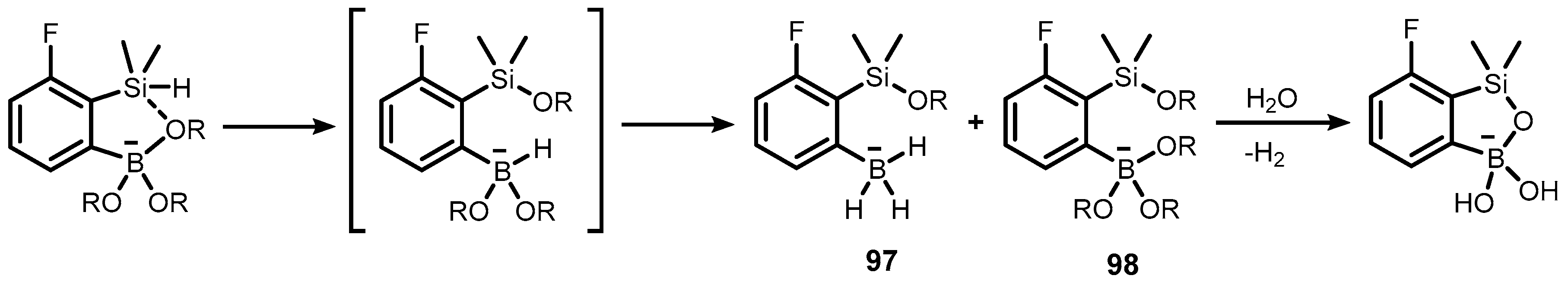
- Durka, K.; Urban, M.; Czub, M.; Dąbrowski, M.; Tomaszewski, P.; Luliński, S. An Intramolecular Ortho-Assisted Activation of the Silicon–Hydrogen Bond in Arylsilanes: An Experimental and Theoretical Study. Dalton Trans. 2018, 47, 3705–3716. [Google Scholar] [CrossRef] [PubMed]
- Pacholak, P.; Krajewska, J.; Wińska, P.; Dunikowska, J.; Gogowska, U.; Mierzejewska, J.; Durka, K.; Woźniak, K.; Laudy, A.E.; Luliński, S. Development of Structurally Extended Benzosiloxaboroles–Synthesis and in Vitro Biological Evaluation. RSC Adv. 2021, 11, 25104–25121. [Google Scholar] [CrossRef]
- Ćwik, P.; Ciosek-Skibińska, P.; Zabadaj, M.; Luliński, S.; Durka, K.; Wróblewski, W. Differential Sensing of Saccharides Based on an Array of Fluorinated Benzosiloxaborole Receptors. Sensors 2020, 20, 3540. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Hartwig, J.F. Iridium-Catalyzed, Silyl-Directed, Peri-Borylation of C–H Bonds in Fused Polycyclic Arenes and Heteroarenes. Angew. Chem. Int. Ed. 2018, 57, 10163–10167. [Google Scholar] [CrossRef]
- Sumida, Y.; Harada, R.; Sumida, T.; Hashizume, D.; Hosoya, T. Hydrosilyl Group-Directed Iridium-Catalyzed Peri-Selective C–H Borylation of Ring-Fused (Hetero)Arenes. Chem. Lett. 2018, 47, 1251–1254. [Google Scholar] [CrossRef]
- Schulte, M.; Gabbaï, F.P. Synthesis of heteronuclear bifunctional Lewis acids by transmetalation of 1,8-bis(trimethylstannyl)naphthalene with BCl3. Can. J. Chem. 2002, 80, 1308–1312. [Google Scholar] [CrossRef]
- Breitwieser, K.; Chen, P. Crystal Structure of a 1,1-Dibutyl-1H,3H-naphtho[1,8-Cd][1,2,6]oxastannaborinin-3-ol. Acta Crystallogr. E Cryst. Commun. 2021, 77, 180–183. [Google Scholar] [CrossRef]
- Boshra, R.; Venkatasubbaiah, K.; Doshi, A.; Jäkle, F. Resolution of Planar-Chiral Ferrocenylborane Lewis Acids: The Impact of Steric Effects on the Stereoselective Binding of Ephedrine Derivatives. Organometallics 2009, 28, 4141–4149. [Google Scholar] [CrossRef]
- Steciuk, I.; Durka, K.; Gontarczyk, K.; Dąbrowski, M.; Luliński, S.; Woźniak, K. Nitrogen–Boron Coordination versus OH⋯N Hydrogen Bonding in Pyridoxaboroles–Aza Analogues of Benzoxaboroles. Dalton Trans. 2015, 44, 16534–16546. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.J.; Lee, W.; Johnson, A.R.; Delatorre, K.J.; Chen, J.; Eigenbrot, C.; Heidmann, J.; Kakiuchi-Kiyota, S.; Katewa, A.; Kiefer, J.R.; et al. Stereochemical Differences in Fluorocyclopropyl Amides Enable Tuning of Btk Inhibition and Off-Target Activity. ACS Med. Chem. Lett. 2020, 11, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.R.; Reedy, A.J.; Lennarz, W.J. Synthesis of Aromatic Boronic Acids. Aldehydo Boronic Acids and a Boronic Acid Analog of Tyrosine. J. Am. Chem. Soc. 1958, 80, 835–838. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Dougherty, R.C. Boron-Containing Analogs of Isoquinoline. J. Am. Chem. Soc. 1962, 84, 2648–2649. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Dougherty, R.C. New Heteroaromatic Compounds. XX. Derivatives of 4,3-Borazaroisoquinoline. J. Am. Chem. Soc. 1964, 86, 433–436. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Jones, R. New Heteroaromatic Compounds. XXV. Studies of Salt Formation in Boron Oxyacids by Boron-11 Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1967, 89, 2408–2410. [Google Scholar] [CrossRef]
- Davis, F.A.; Dewar, M.J.S.; Jones, R. New Heteroaromatic Compounds. XXVII. Boron-11 Chemical Shifts of Some Heteroaromatic Boron Compounds. J. Am. Chem. Soc. 1968, 90, 706–708. [Google Scholar] [CrossRef]
- Dougherty, R.C. Mass Spectra of Heteroaromatic Boron Compounds. Tetrahedron 1968, 24, 6755–6772. [Google Scholar] [CrossRef]
- Dunn, H.E.; Catlin, J.C.; Snyder, H.R. Arylboronic Acids. Imino Derivatives from o-Formylbenzeneboronic Acid. J. Org. Chem. 1968, 33, 4483–4486. [Google Scholar] [CrossRef]
- Nanninga, D.; Kliegel, W. Nitrone von 2-formylphenylboronsäureestern. J. Organomet. Chem. 1983, 247, 247–252. [Google Scholar] [CrossRef]
- Groziak, M.P.; Chen, L.; Yi, L.; Robinson, P.D. Planar Boron Heterocycles with Nucleic Acid-Like Hydrogen-Bonding Motifs. J. Am. Chem. Soc. 1997, 119, 7817–7826. [Google Scholar] [CrossRef]
- Sarina, E.A.; Olmstead, M.M.; Nguyen, D.N.; Groziak, M.P. 1-Hydroxy-1H-benzo[d][1,2,6]oxazaborinin-4(3H)-one. Acta Crystallogr C Cryst. Struct. Commun. 2013, 69, 183–185. [Google Scholar] [CrossRef]
- Tickell, D.A.; Mahon, M.F.; Bull, S.D.; James, T.D. A Simple Protocol for NMR Analysis of the Enantiomeric Purity of Chiral Hydroxylamines. Org. Lett. 2013, 15, 860–863. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Lunde, C.S.; Freund, Y.R.; Hernandez, V.; Li, X.; Xia, Y.; Carter, D.S.; Berry, P.W.; Halladay, J.; Rock, F.; et al. Boron-Pleuromutilins as Anti-Wolbachia Agents with Potential for Treatment of Onchocerciasis and Lymphatic Filariasis. J. Med. Chem. 2019, 62, 2521–2540. [Google Scholar] [CrossRef] [Green Version]
- Soloway, A.H. Synthesis of Aromatic Diboronic Acids. J. Am. Chem. Soc. 1960, 82, 2442–2444. [Google Scholar] [CrossRef]
- Groziak, M.P. Boron Heterocycles as Platforms for Building New Bioactive Agents. In Progress in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2000; Volume 12, pp. 1–21. ISBN 978-0-08-043882-5. [Google Scholar]
- Groziak, M.P.; Ganguly, A.D.; Robinson, P.D. Boron Heterocycles Bearing a Peripheral Resemblance to Naturally-Occurring Purines: Design, Syntheses, Structures, and Properties. J. Am. Chem. Soc. 1994, 116, 7597–7605. [Google Scholar] [CrossRef]
- Hughes, M.P.; Shang, M.; Smith, B.D. High Affinity Carboxylate Binding Using Neutral Urea-Based Receptors with Internal Lewis Acid Coordination. J. Org. Chem. 1996, 61, 4510–4511. [Google Scholar] [CrossRef]
- Hughes, M.P.; Smith, B.D. Enhanced Carboxylate Binding Using Urea and Amide-Based Receptors with Internal Lewis Acid Coordination: A Cooperative Polarization Effect. J. Org. Chem. 1997, 62, 4492–4499. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.-C.; Soloway, A.H.; Beeson, J.C.; Ji, W.; Barnum, B.A.; Rong, F.-G.; Tjarks, W.; Jordan, G.T.; Liu, J.; Shore, S.G. Boron-Containing Heterocycles: Syntheses, Structures, and Properties of Benzoborauracils and a Benzoborauracil Nucleoside. J. Org. Chem. 1999, 64, 9566–9574. [Google Scholar] [CrossRef]
- Hudnall, T.W.; Melaïmi, M.; Gabbaï, F.P. Hybrid Lewis Acid/Hydrogen-Bond Donor Receptor for Fluoride. Org. Lett. 2006, 8, 2747–2749. [Google Scholar] [CrossRef]
- Inglis, S.R.; Zervosen, A.; Woon, E.C.Y.; Gerards, T.; Teller, N.; Fischer, D.S.; Luxen, A.; Schofield, C.J. Synthesis and Evaluation of 3-(Dihydroxyboryl)Benzoic Acids as d,d-Carboxypeptidase R39 Inhibitors. J. Med. Chem. 2009, 52, 6097–6106. [Google Scholar] [CrossRef] [PubMed]
- Inglis, S.R.; Woon, E.C.Y.; Thompson, A.L.; Schofield, C.J. Observations on the Deprotection of Pinanediol and Pinacol Boronate Esters via Fluorinated Intermediates. J. Org. Chem. 2010, 75, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Hiscock, J.R.; Wells, N.J.; Ede, J.A.; Gale, P.A.; Sambrook, M.R. Biasing Hydrogen Bond Donating Host Systems towards Chemical Warfare Agent Recognition. Org. Biomol. Chem. 2016, 14, 9560–9567. [Google Scholar] [CrossRef] [Green Version]
- Manankandayalage, C.; Unruh, D.K.; Krempner, C. Small Molecule Activation with Intramolecular “Inverse” Frustrated Lewis Pairs. Chem. Eur. J. 2021, 27, 6263–6273. [Google Scholar] [CrossRef] [PubMed]
- Porcel, S.; Bouhadir, G.; Saffon, N.; Maron, L.; Bourissou, D. Reaction of Singlet Dioxygen with Phosphine-Borane Derivatives: From Transient Phosphine Peroxides to Crystalline Peroxoboronates. Angew. Chem. Int. Ed. 2010, 49, 6186–6189. [Google Scholar] [CrossRef] [PubMed]
- Breunig, J.M.; Lehmann, F.; Bolte, M.; Lerner, H.-W.; Wagner, M. Synthesis and Reactivity of o-Phosphane Oxide Substituted Aryl(Hydro)Borates and Aryl(Hydro)Boranes. Organometallics 2014, 33, 3163–3172. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.H.; Wang, H. Synthesis of an Oxygen-Linked Germinal Frustrated Lewis Pair and Its Application in Small Molecule Activation. RSC Adv. 2018, 8, 26271–26276. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-F.; Kang, Y.; Ko, S.-B.; Rao, Y.; Sauriol, F.; Wang, S. Highly Congested Donor–Acceptor P–B Compound: Synthesis and Properties of a BMes2-and a PPh2-functionalized 1,8-naphthalene. Organometallics 2013, 32, 3063–3068. [Google Scholar] [CrossRef]
- Furan, S.; Hupf, E.; Lork, E.; Beckmann, J. Synthesis and Structure of 5-Diphenylphosphino- Acenaphth-6-yl Boronic Acid, Related Dialkyl Esters and Boroxine Rings. Z. Anorg. Allg. Chem. 2021, 647, 507–512. [Google Scholar] [CrossRef]
- Moebs-Sanchez, S.; Bouhadir, G.; Saffon, N.; Maron, L.; Bourissou, D. Tracking Reactive Intermediates in Phosphine-Promoted Reactions with Ambiphilic Phosphino-Boranes. Chem. Commun. 2008, 3435. [Google Scholar] [CrossRef] [PubMed]
- Declercq, R.; Bouhadir, G.; Bourissou, D.; Légaré, M.-A.; Courtemanche, M.-A.; Nahi, K.S.; Bouchard, N.; Fontaine, F.-G.; Maron, L. Hydroboration of Carbon Dioxide Using Ambiphilic Phosphine–Borane Catalysts: On the Role of the Formaldehyde Adduct. ACS Catal. 2015, 5, 2513–2520. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, V.O.; Volodin, A.D.; Korlyukov, A.A.; Dilman, A.D. Trapping of Difluorocarbene by Frustrated Lewis Pairs. Angew. Chem. Int. Ed. 2020, 59, 12428–12431. [Google Scholar] [CrossRef] [PubMed]
- Krachko, T.; Nicolas, E.; Ehlers, A.W.; Nieger, M.; Slootweg, J.C. Ring-Opening of Epoxides Mediated by Frustrated Lewis Pairs. Chem. Eur. J. 2018, 24, 12669–12677. [Google Scholar] [CrossRef] [PubMed]
- Nemykin, V.N.; Maskaev, A.V.; Geraskina, M.R.; Yusubov, M.S.; Zhdankin, V.V. Preparation and X-Ray Crystal Study of Benziodoxaborole Derivatives: New Hypervalent Iodine Heterocycles. Inorg. Chem. 2011, 50, 11263–11272. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Fuchs, J.M.; Middleton, K.R.; Maskaev, A.V.; Rohde, G.T.; Saito, A.; Postnikov, P.S.; Yusubov, M.S.; Nemykin, V.N.; Zhdankin, V.V. Pseudocyclic Arylbenziodoxaboroles: Efficient Benzyne Precursors Triggered by Water at Room Temperature. Chem. Eur. J. 2017, 23, 16738–16742. [Google Scholar] [CrossRef]






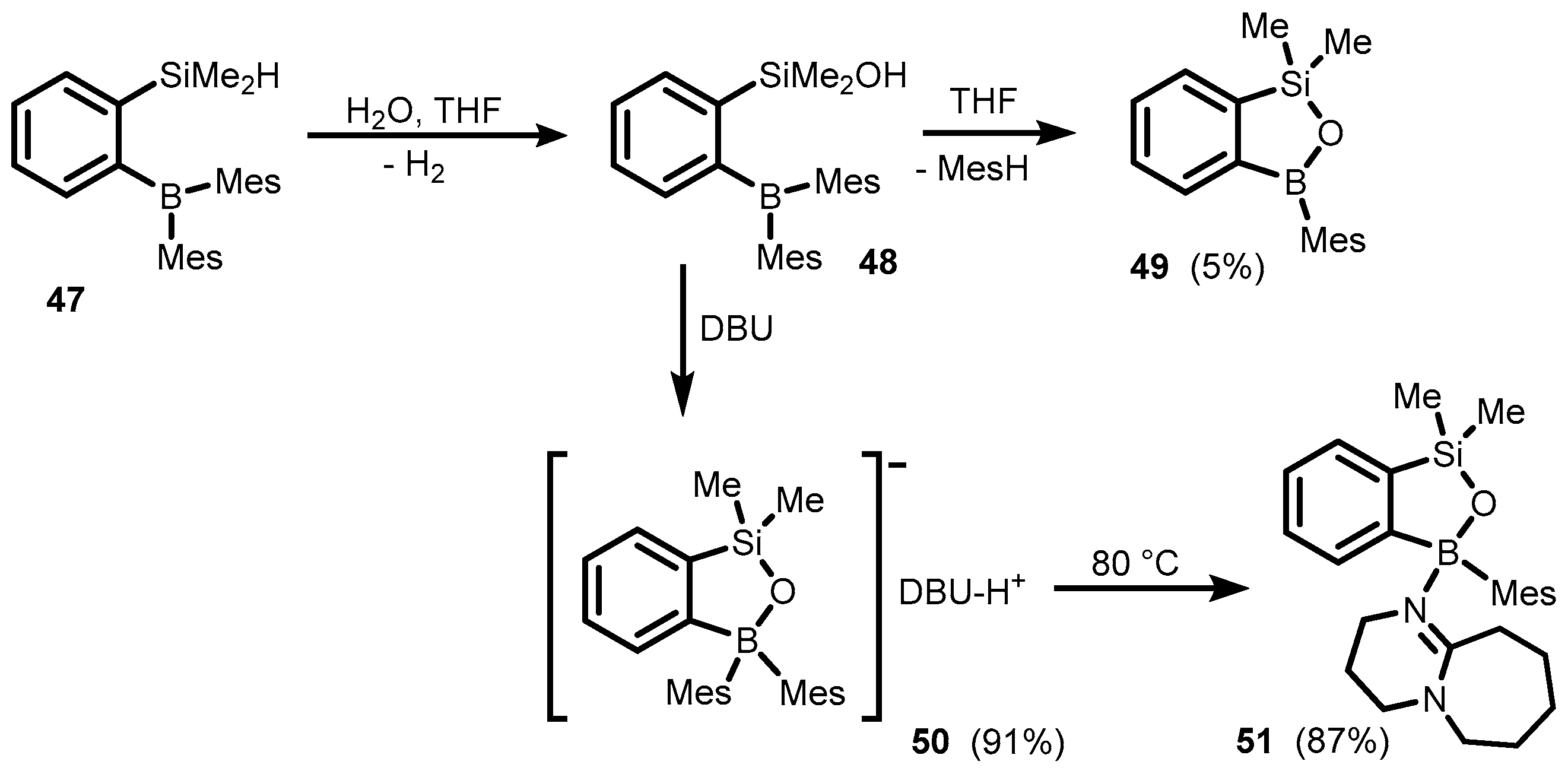
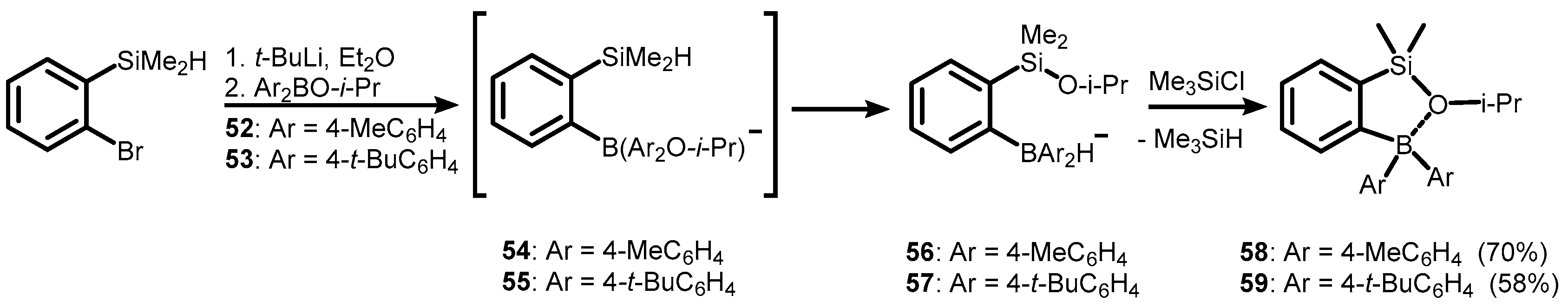

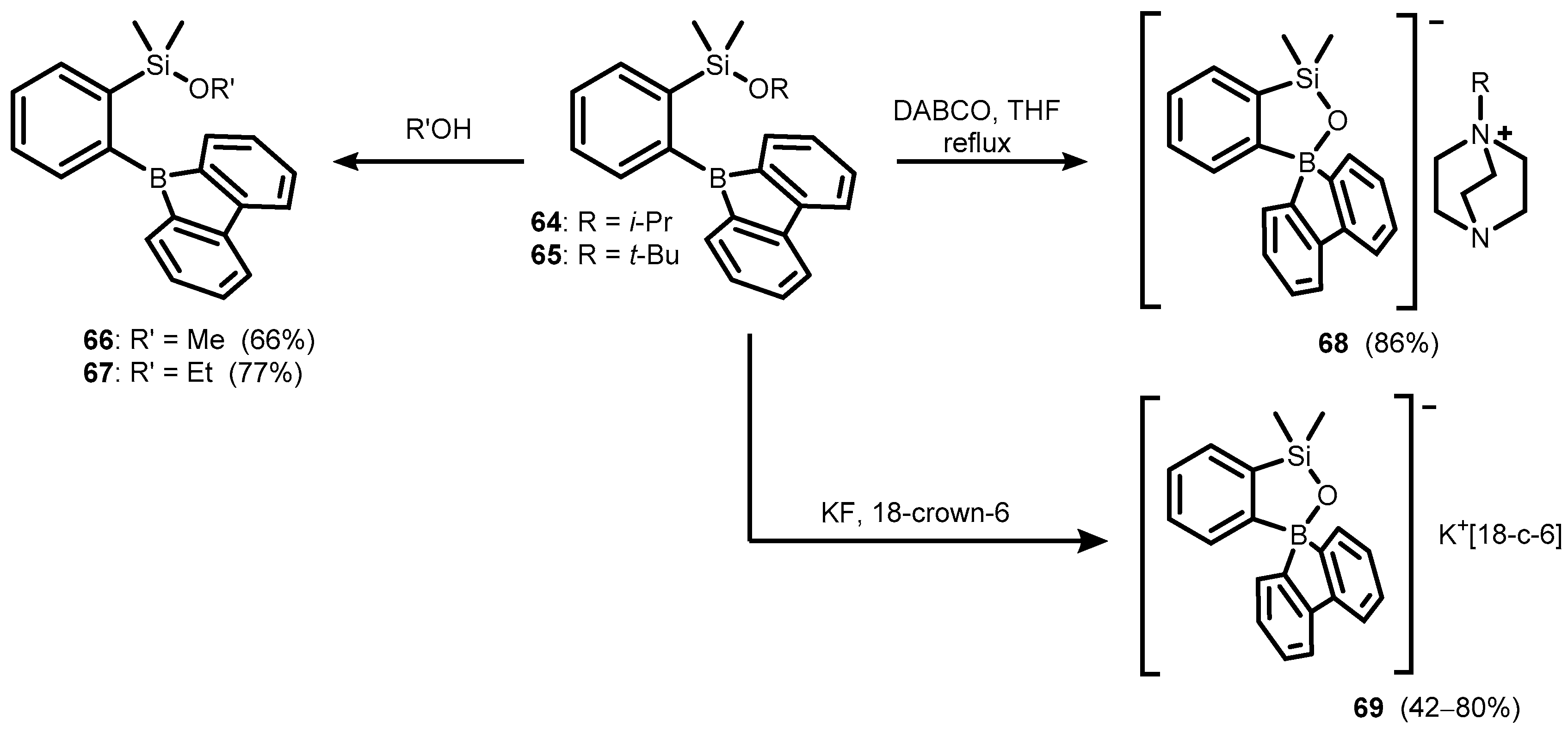

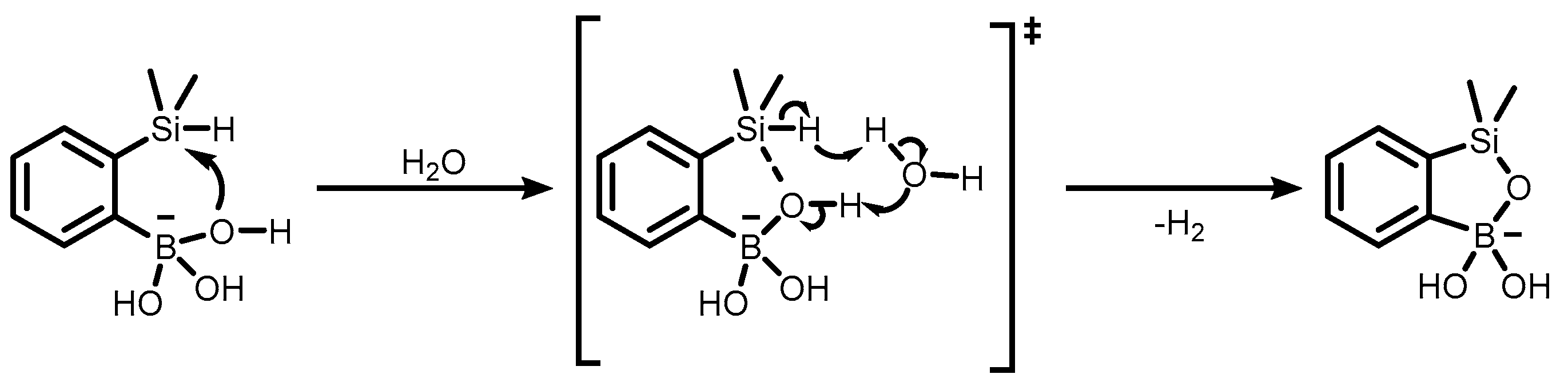

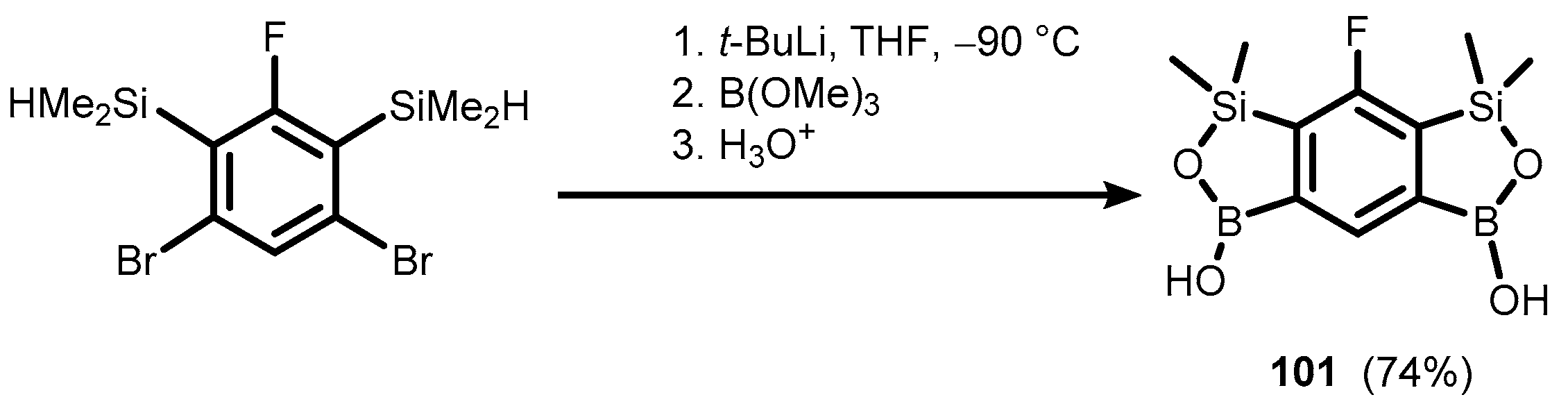
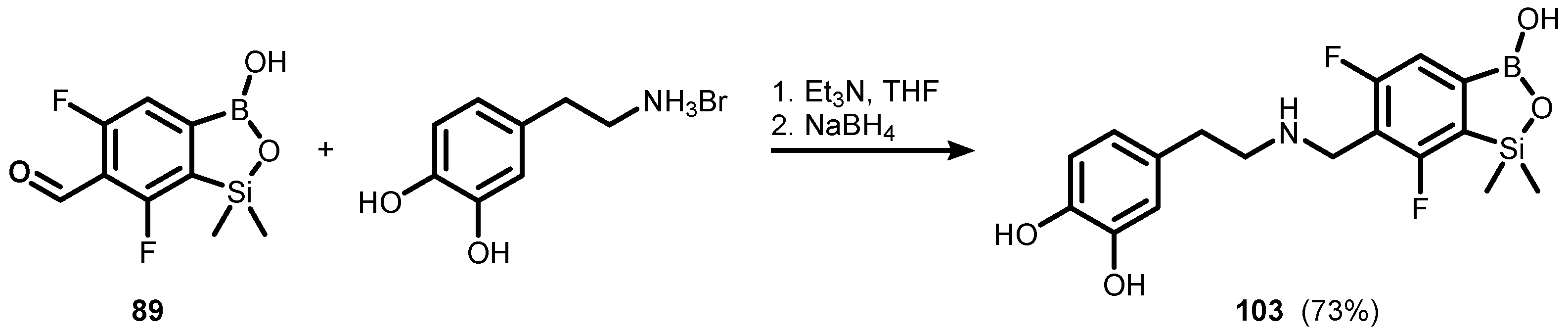
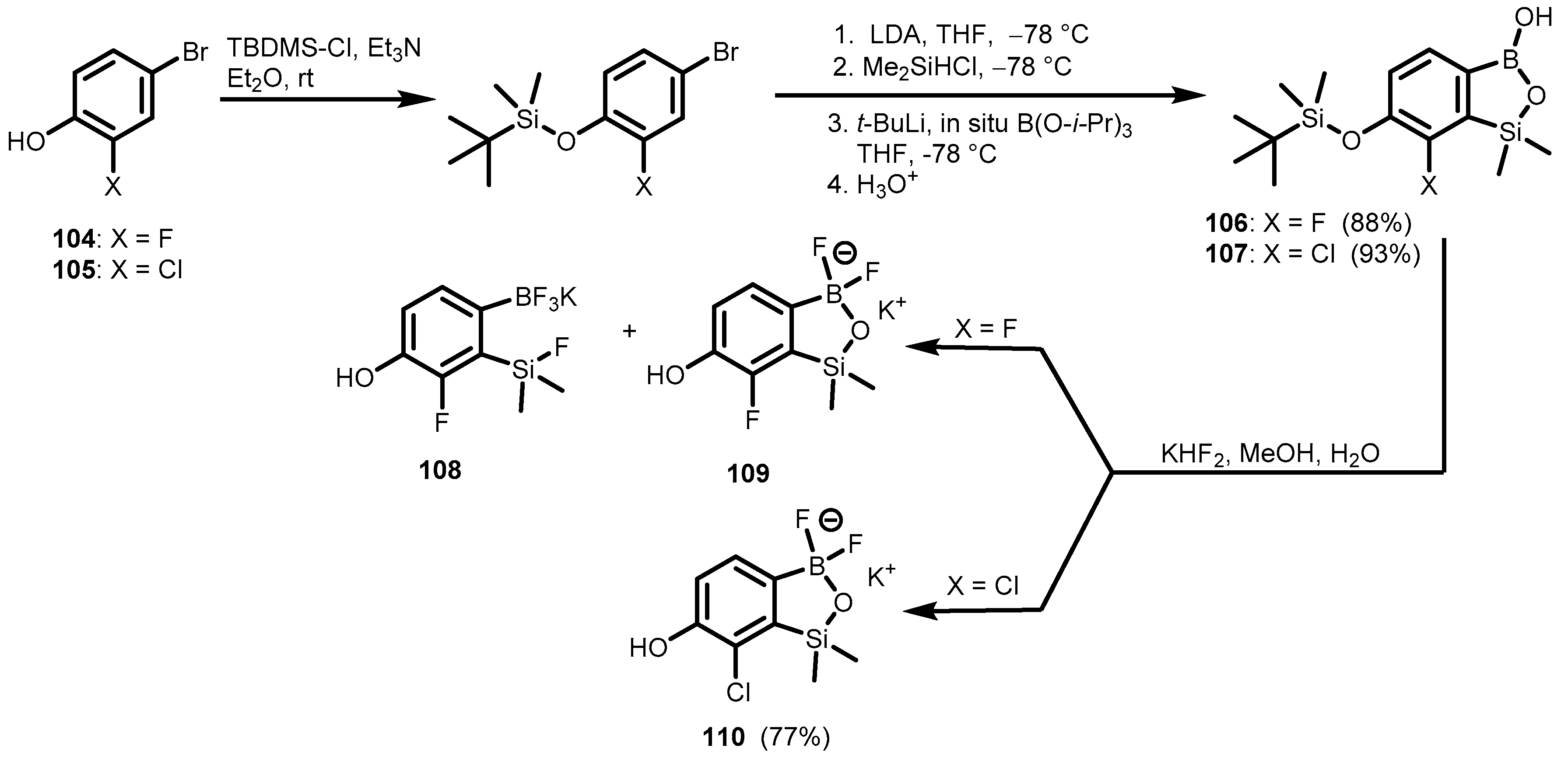
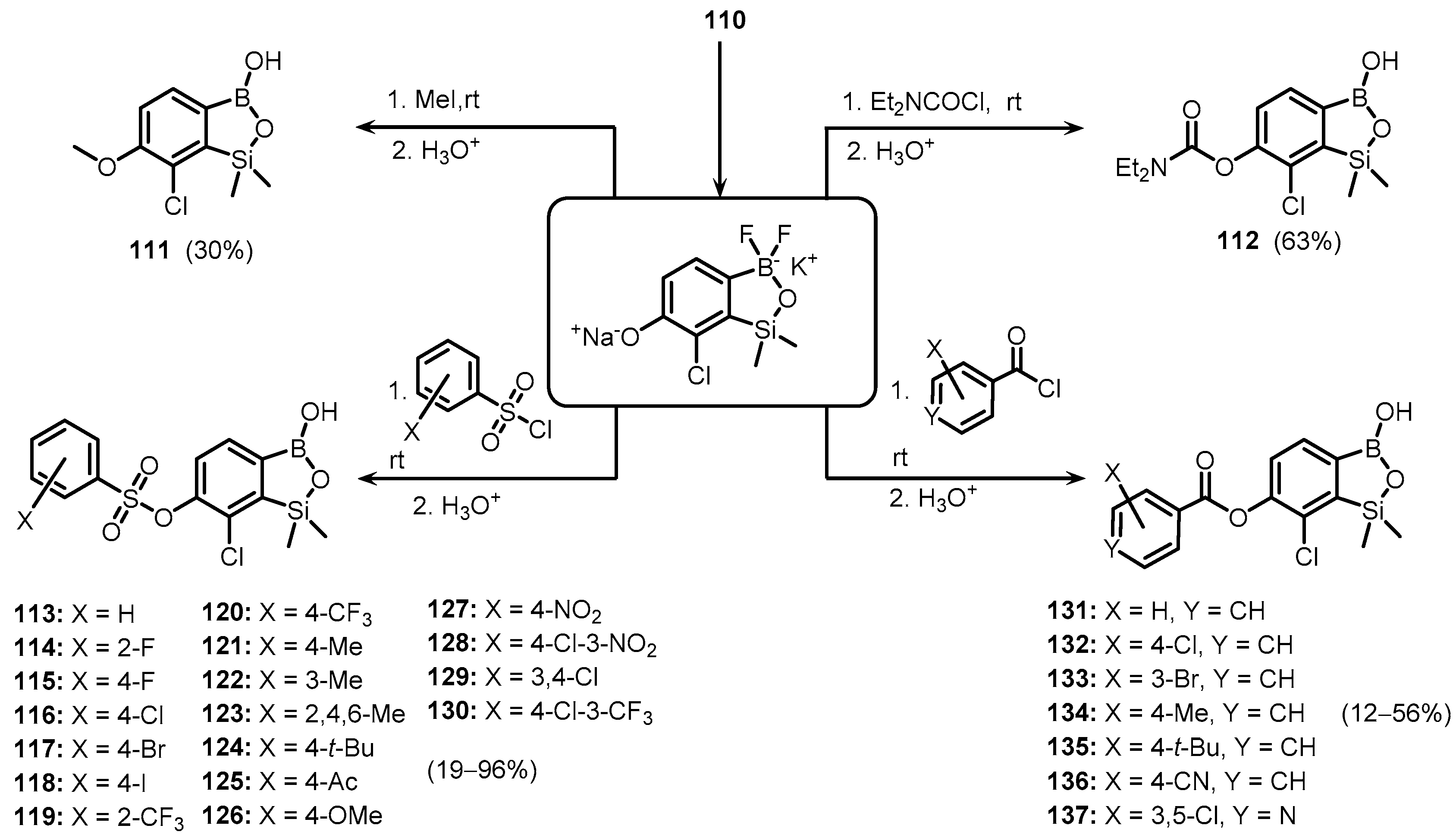
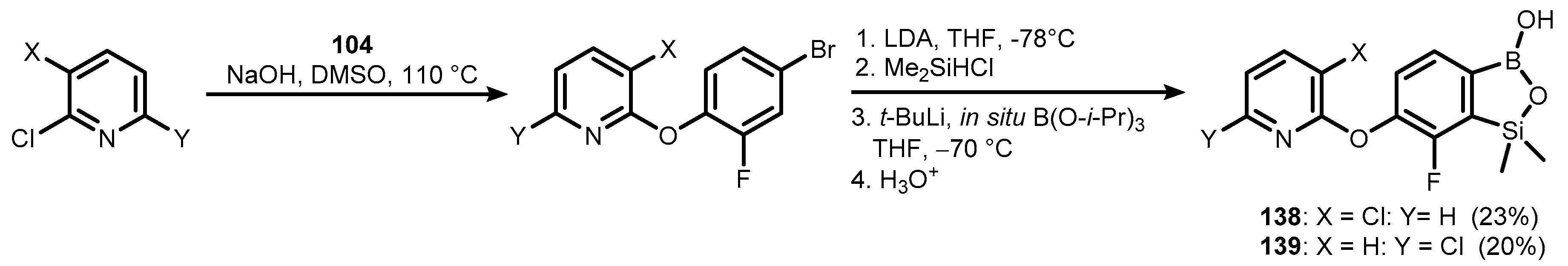













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowicki, K.; Pacholak, P.; Luliński, S. Heteroelement Analogues of Benzoxaborole and Related Ring Expanded Systems. Molecules 2021, 26, 5464. https://doi.org/10.3390/molecules26185464
Nowicki K, Pacholak P, Luliński S. Heteroelement Analogues of Benzoxaborole and Related Ring Expanded Systems. Molecules. 2021; 26(18):5464. https://doi.org/10.3390/molecules26185464
Chicago/Turabian StyleNowicki, Krzysztof, Piotr Pacholak, and Sergiusz Luliński. 2021. "Heteroelement Analogues of Benzoxaborole and Related Ring Expanded Systems" Molecules 26, no. 18: 5464. https://doi.org/10.3390/molecules26185464
APA StyleNowicki, K., Pacholak, P., & Luliński, S. (2021). Heteroelement Analogues of Benzoxaborole and Related Ring Expanded Systems. Molecules, 26(18), 5464. https://doi.org/10.3390/molecules26185464