
Regioselective Functionalization of Quinolines through C-H Activation: A Comprehensive Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
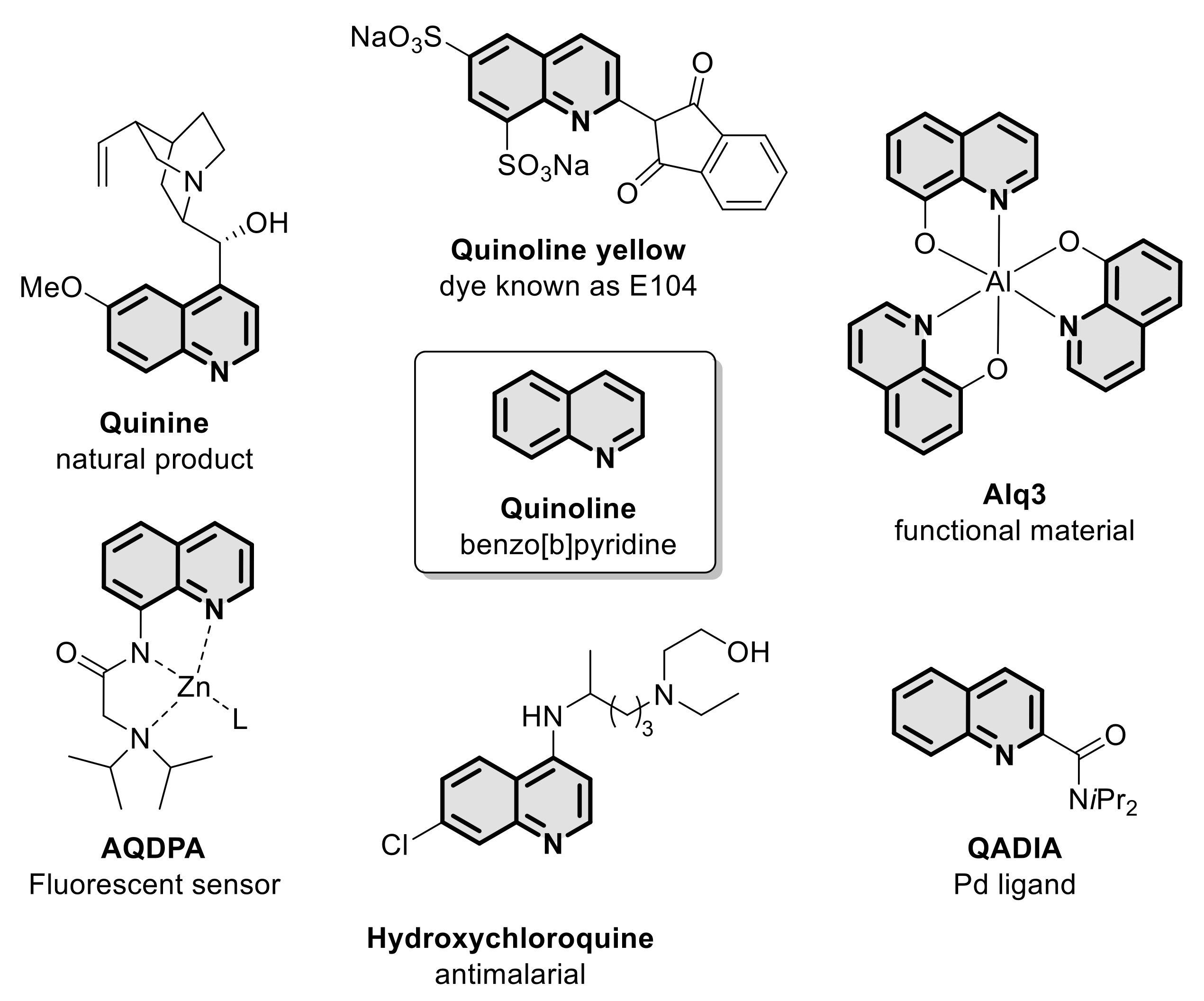
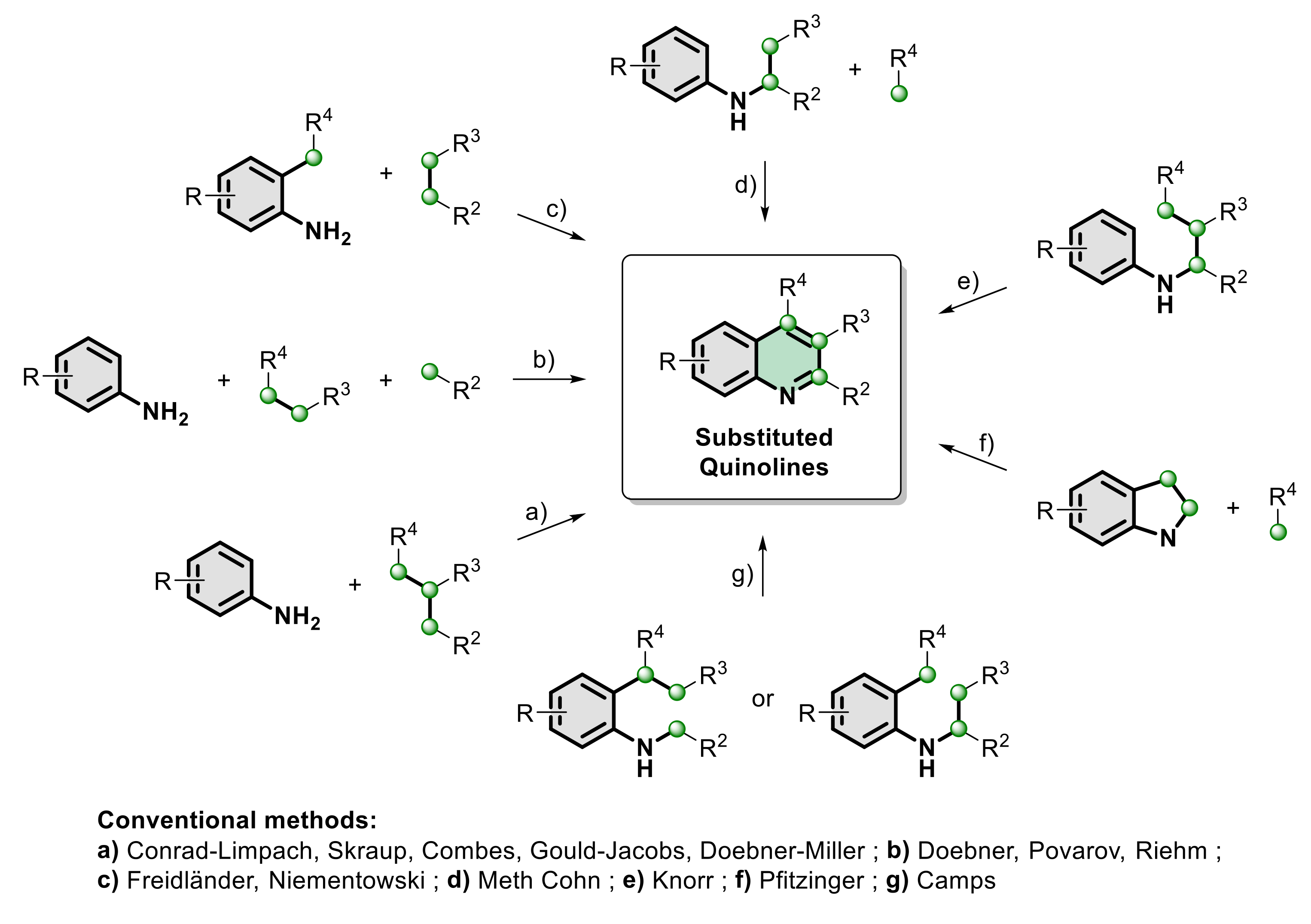
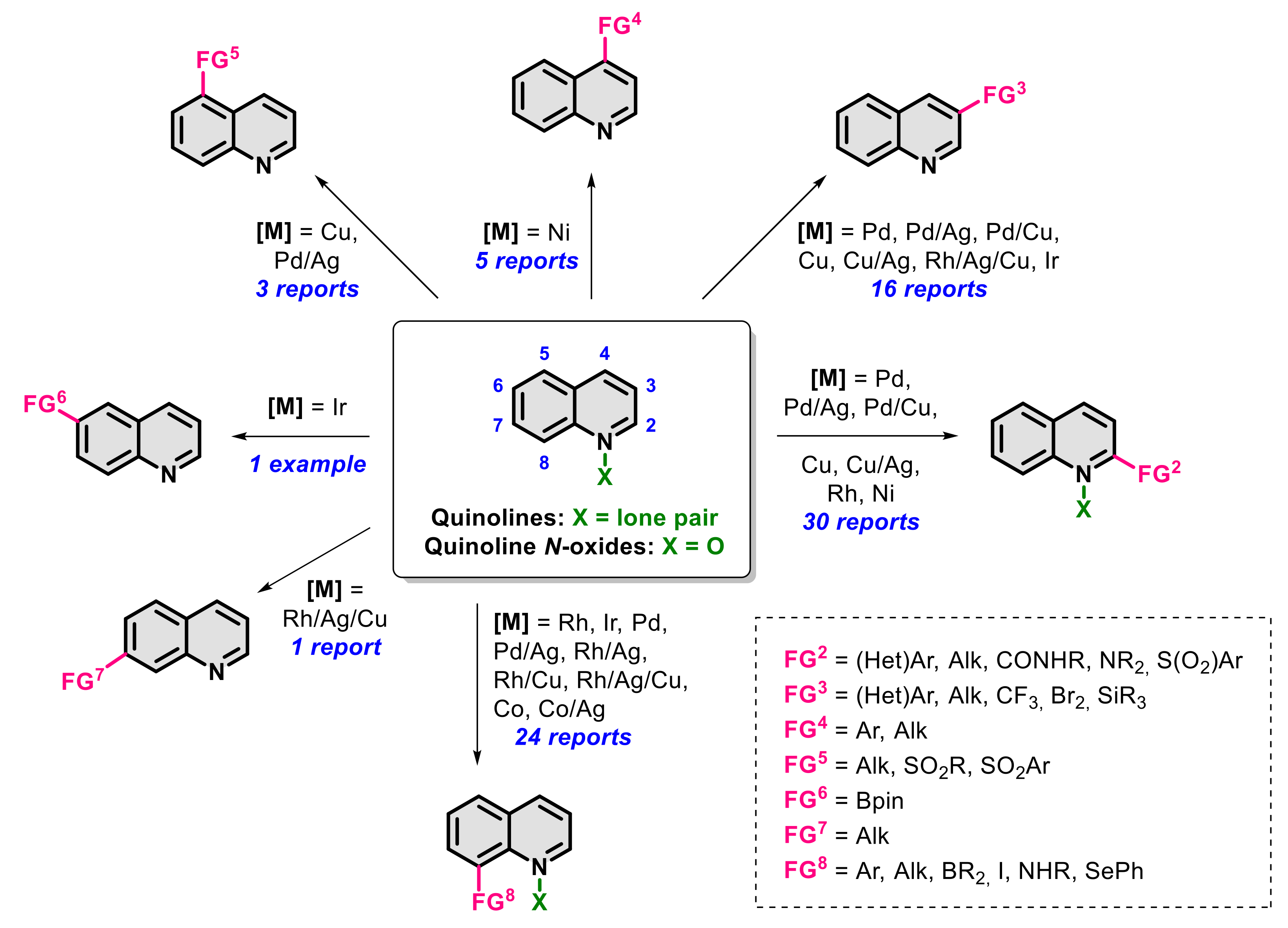
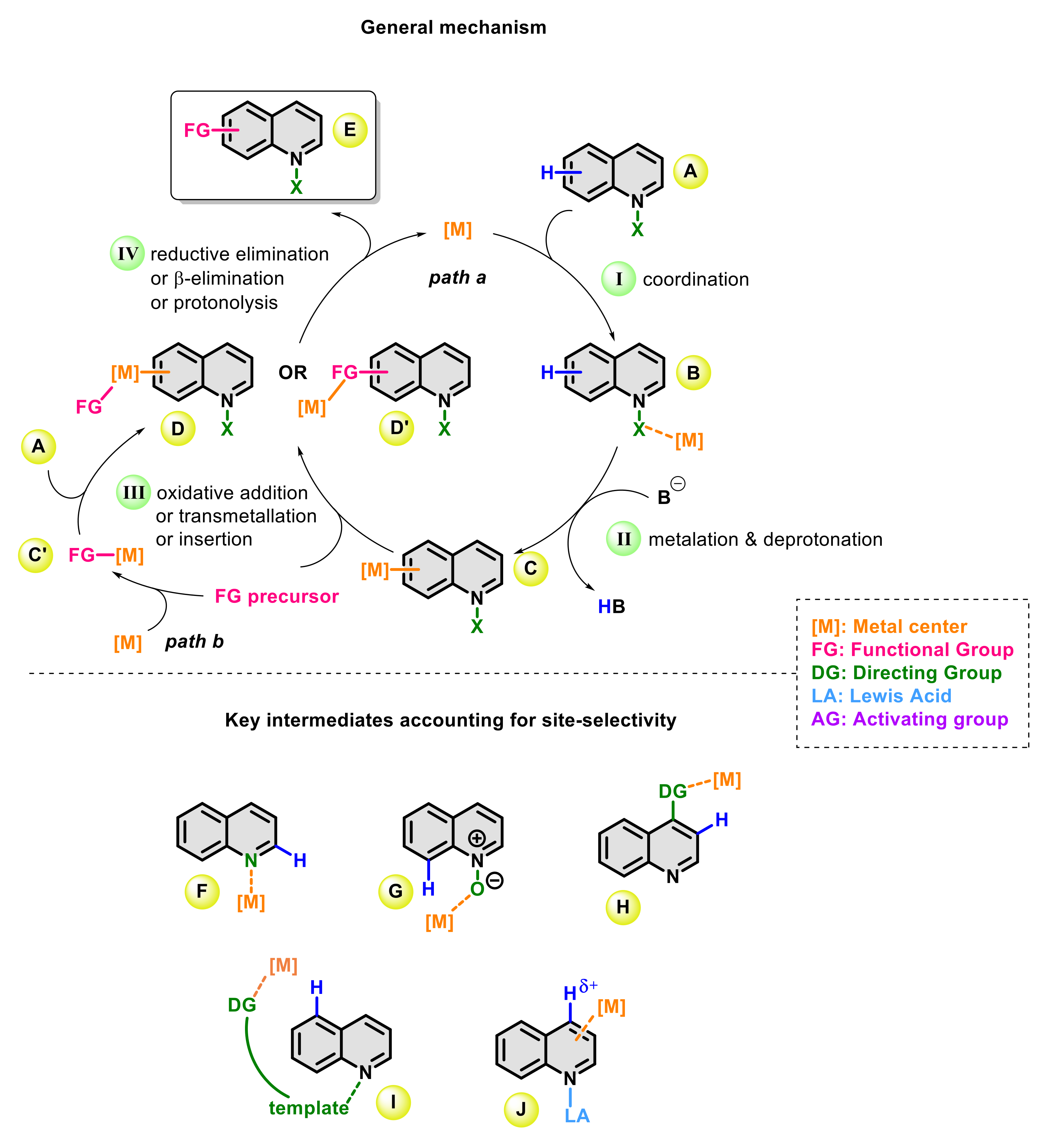
:1. Introduction
2. Functionalization in Position 2
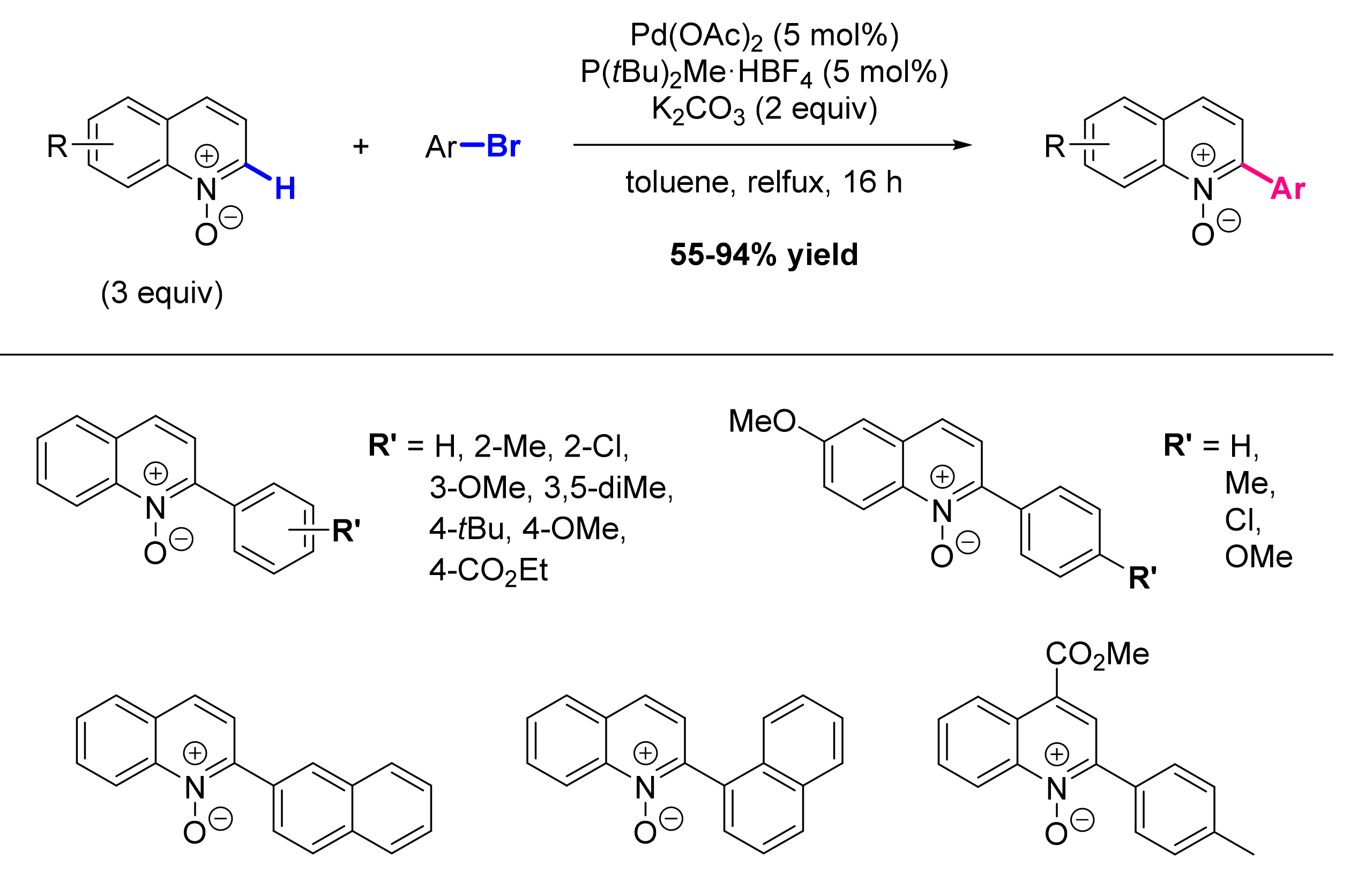
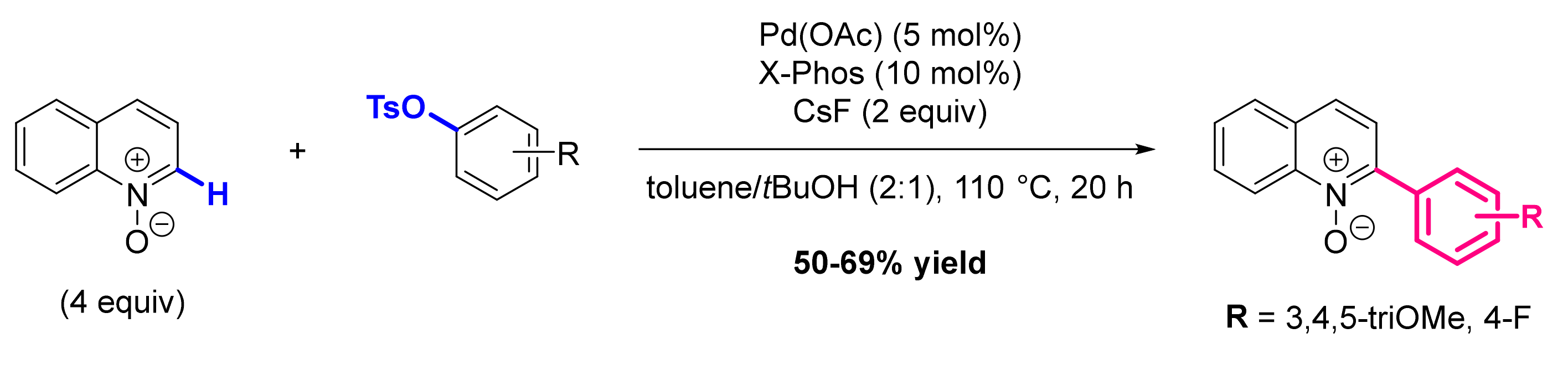
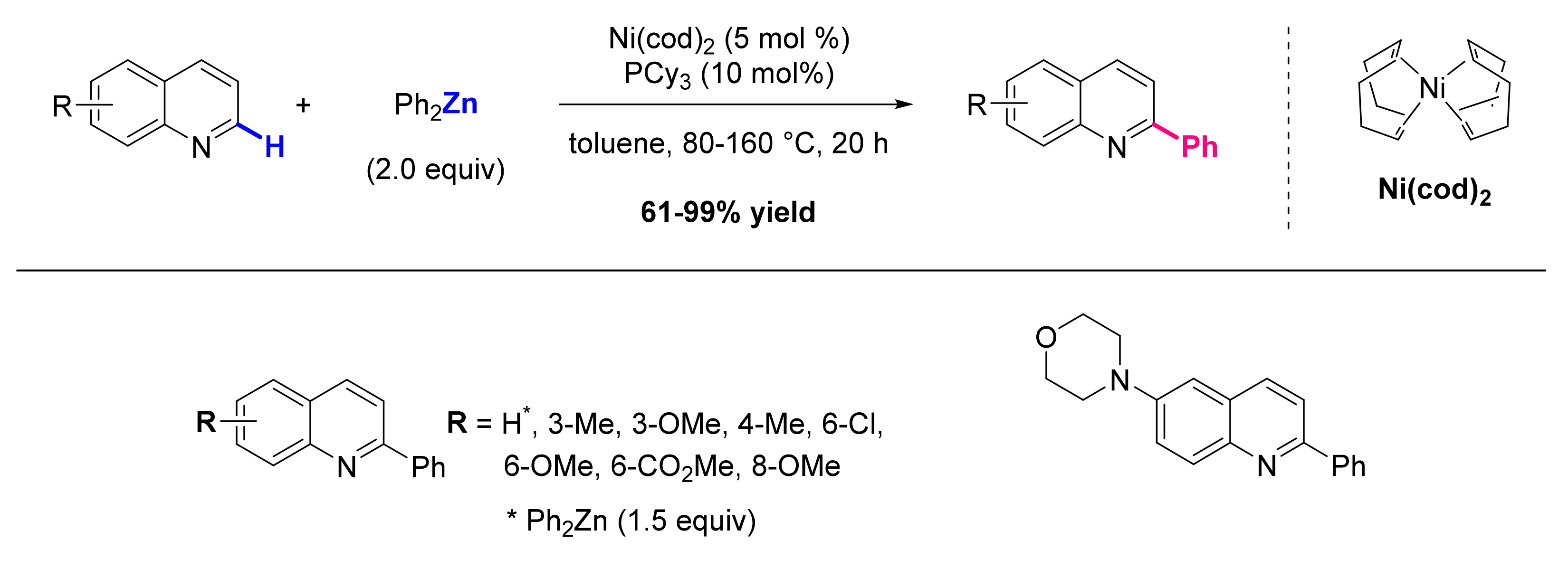
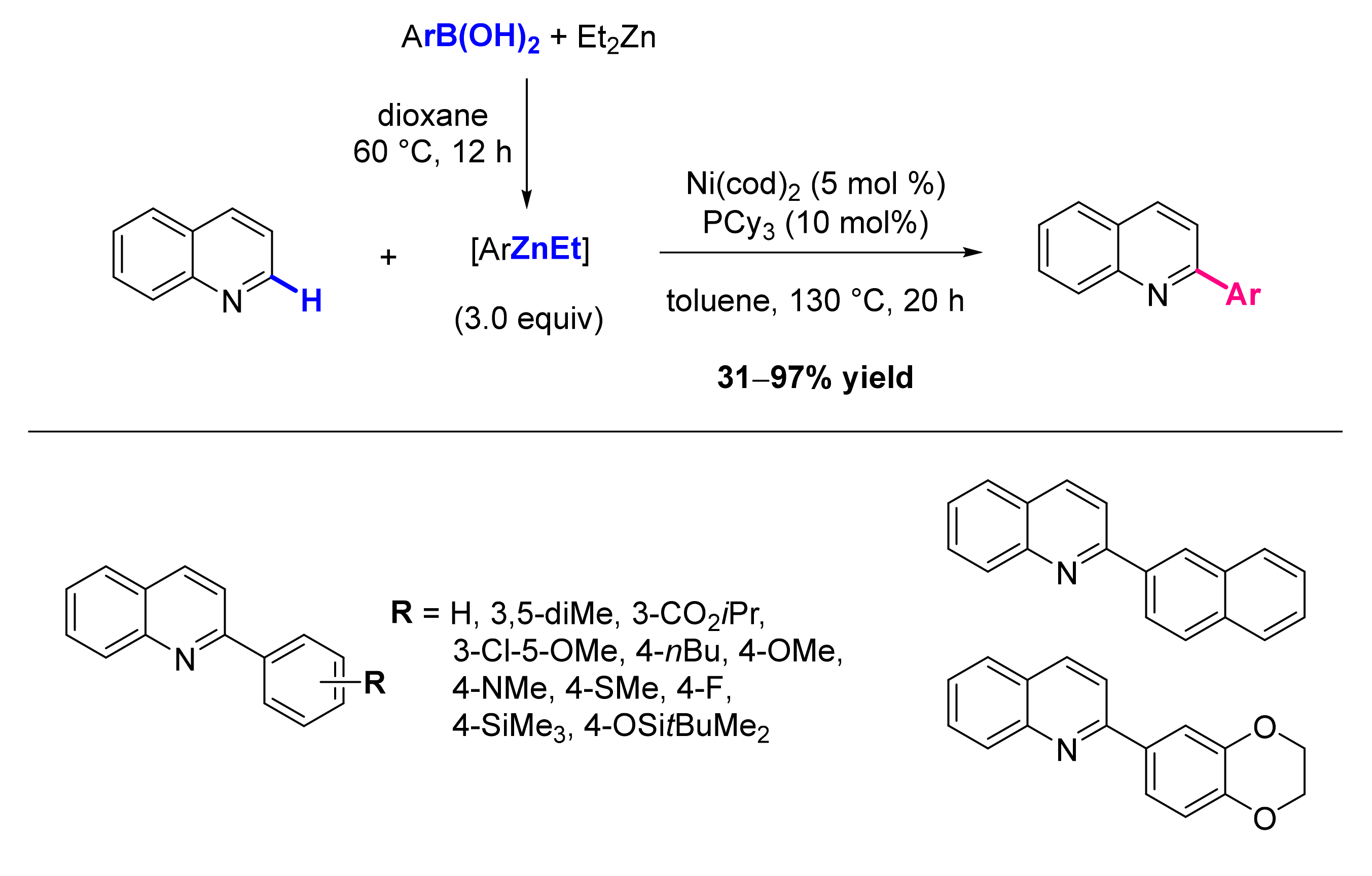
2.1. Formation of C-Ar Bonds
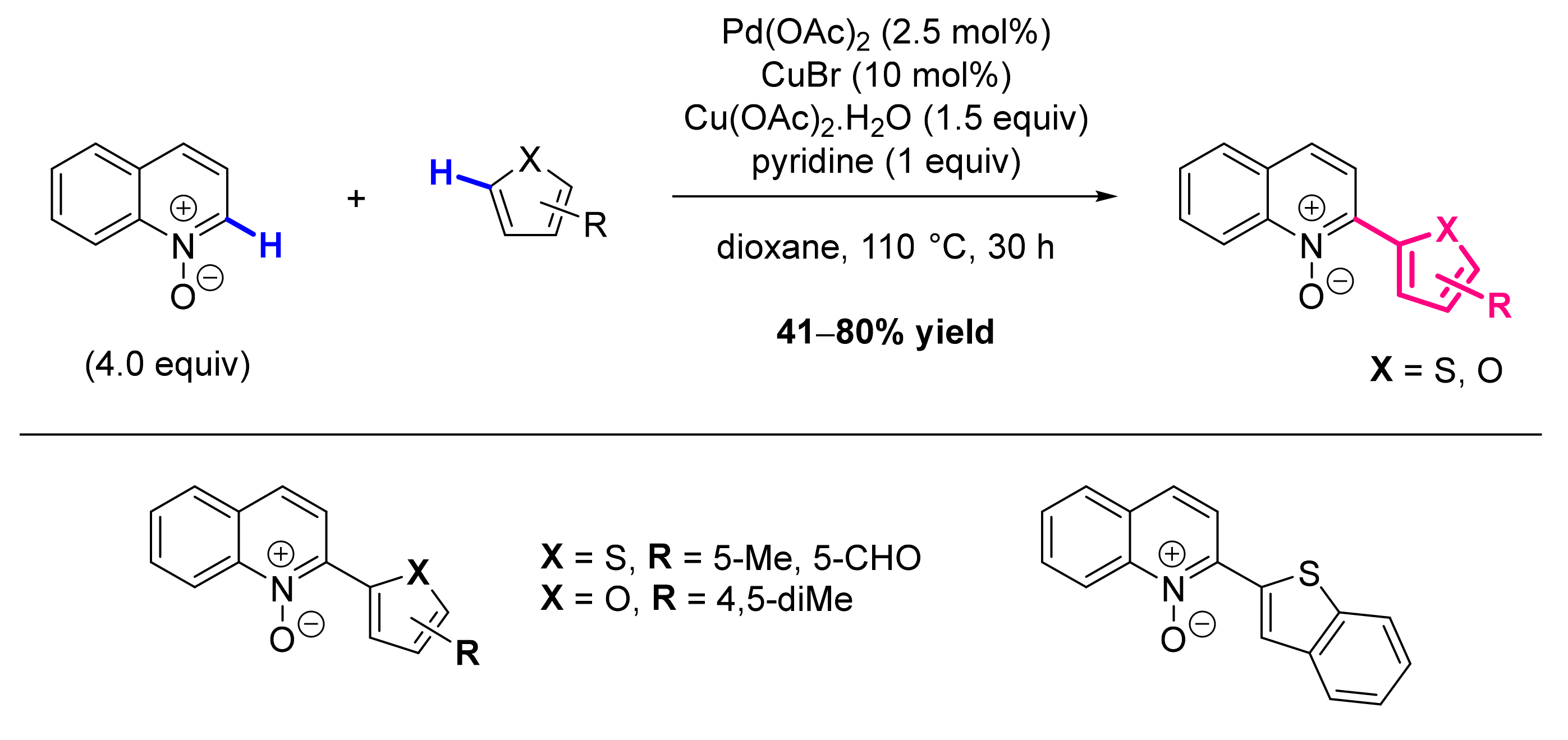
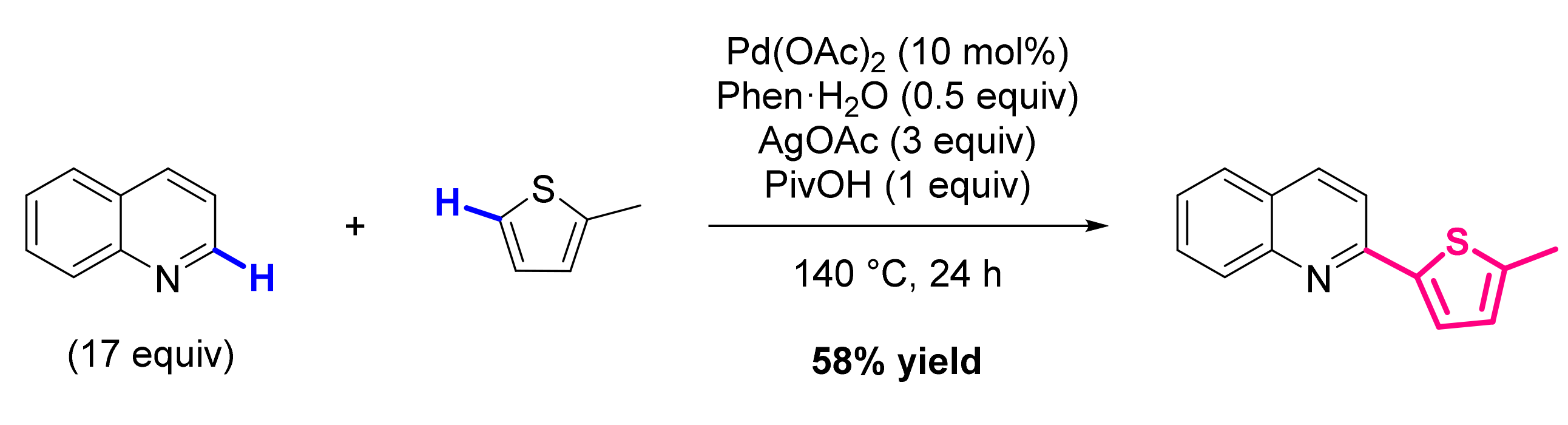
2.2. Formation of C-HetAr Bonds
2.3. Formation of C-C=C Bonds
2.4. Formation of C-Alkyl Bonds
2.5. Formation of C-C(O)NHR Bond
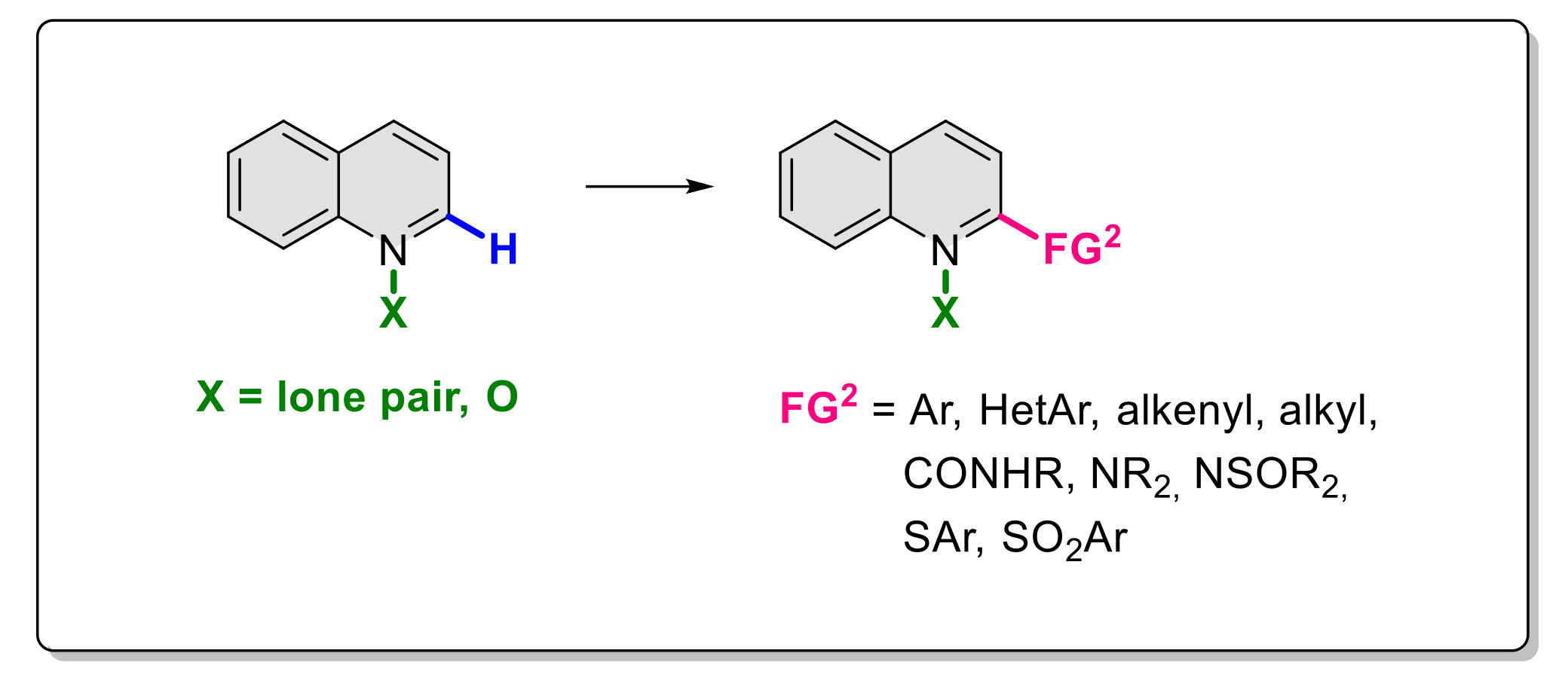
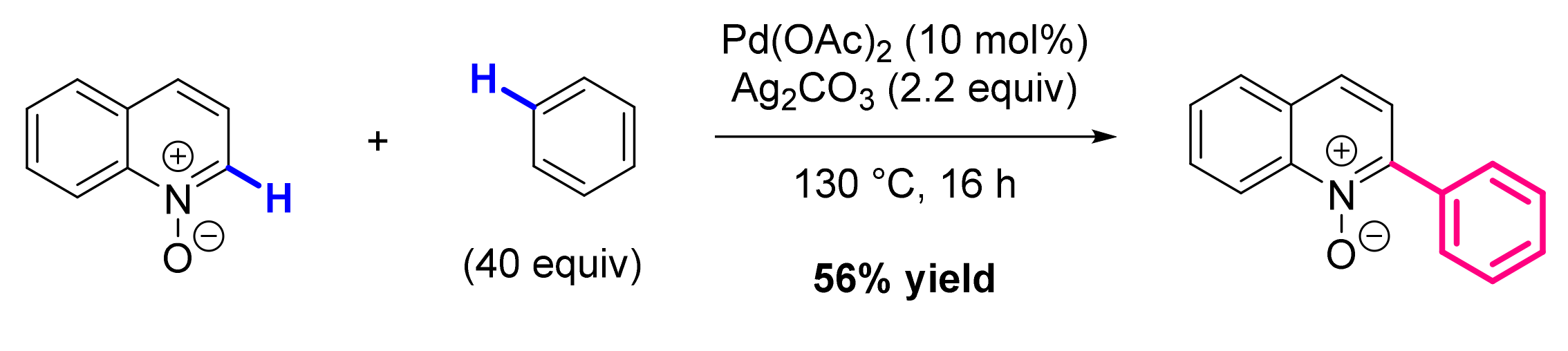
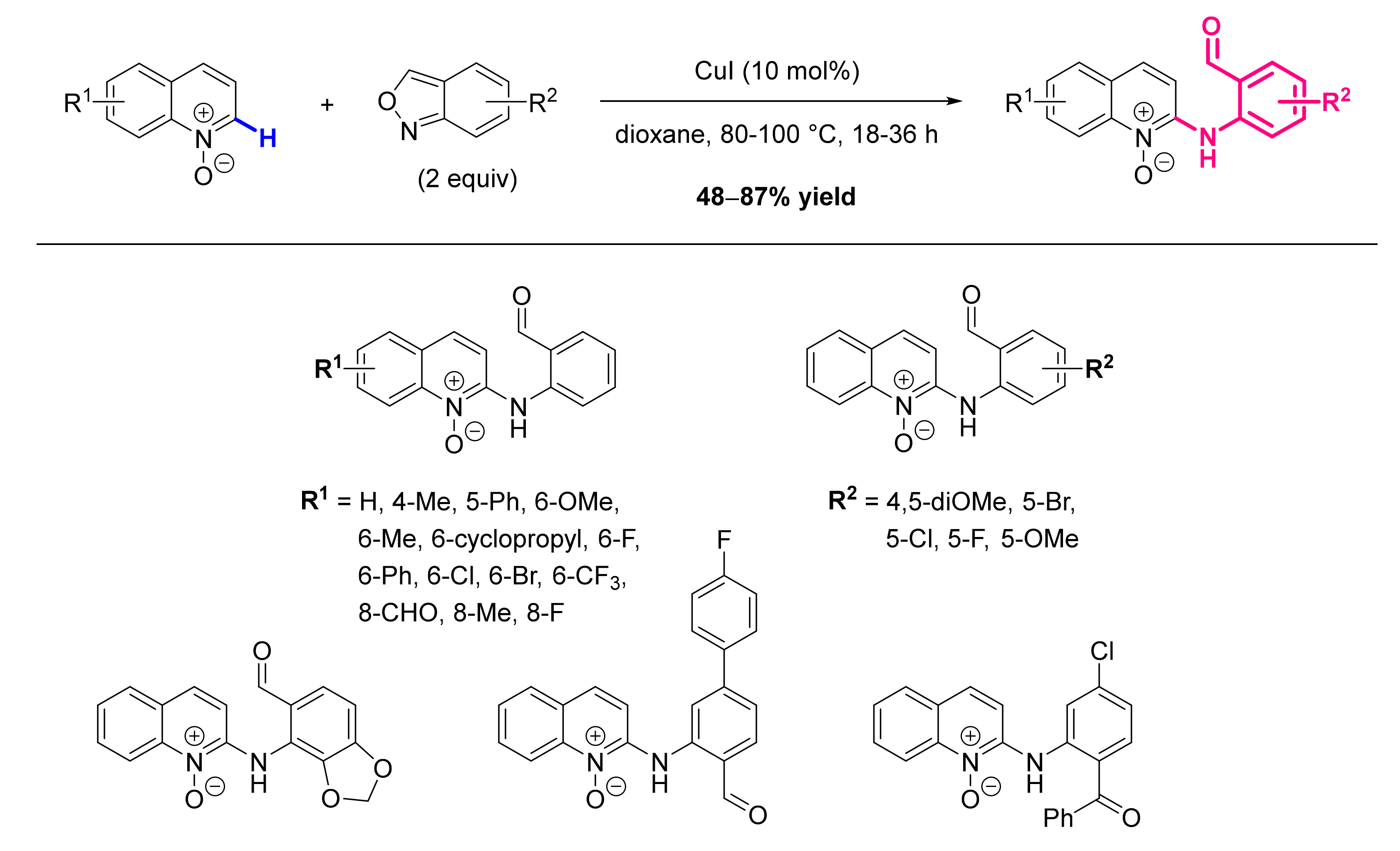
2.6. Formation of C-N Bond
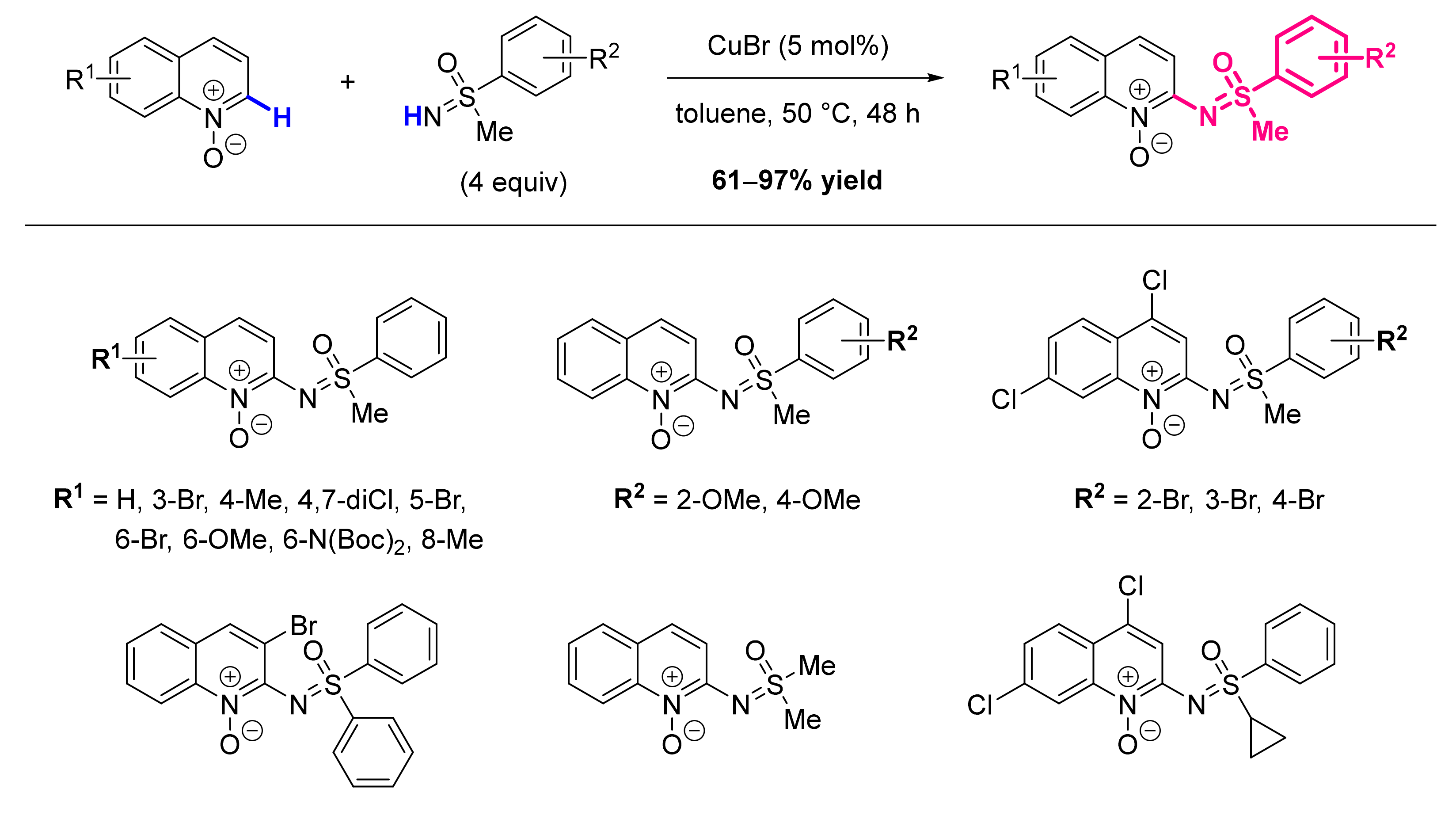
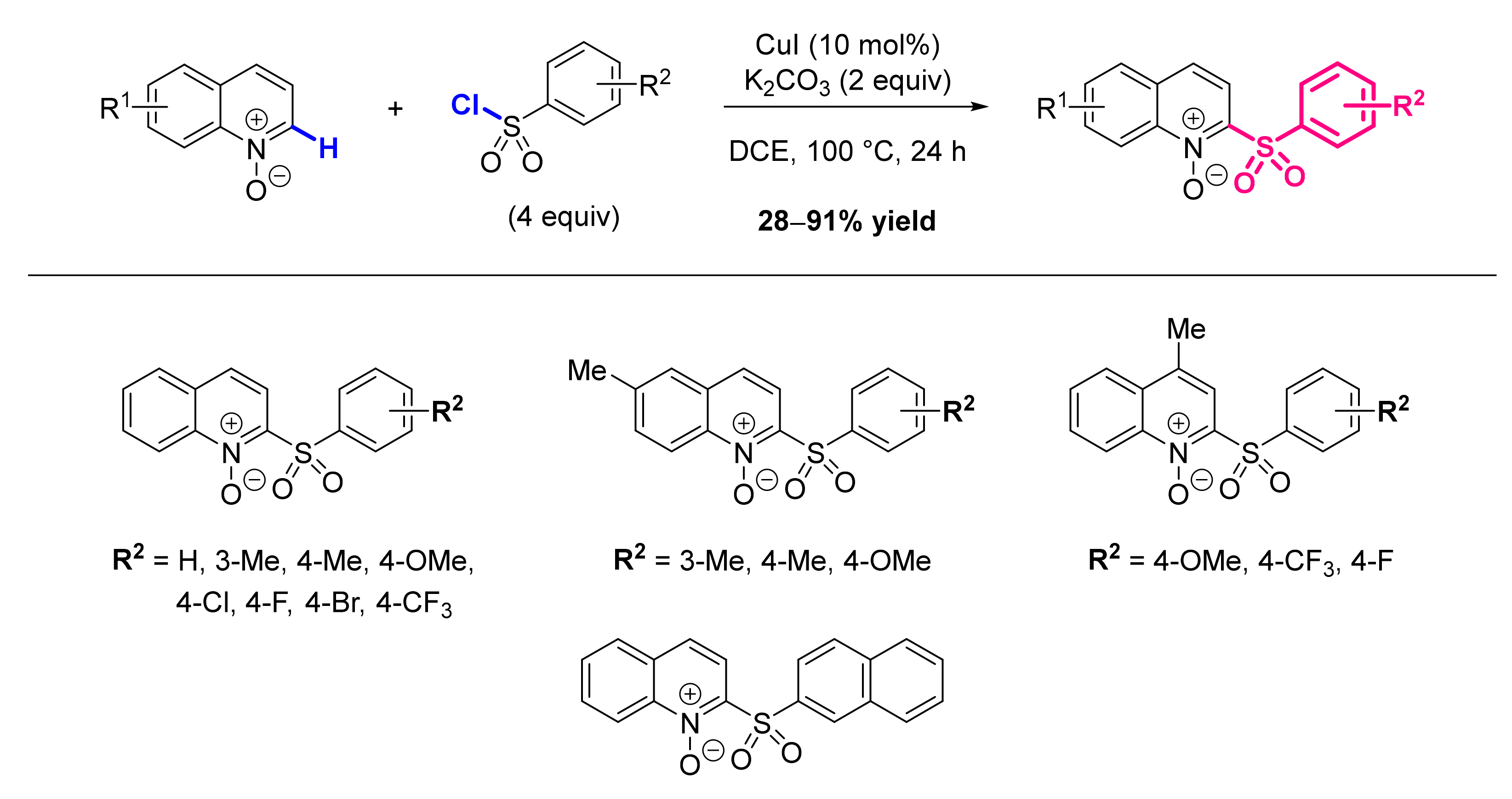
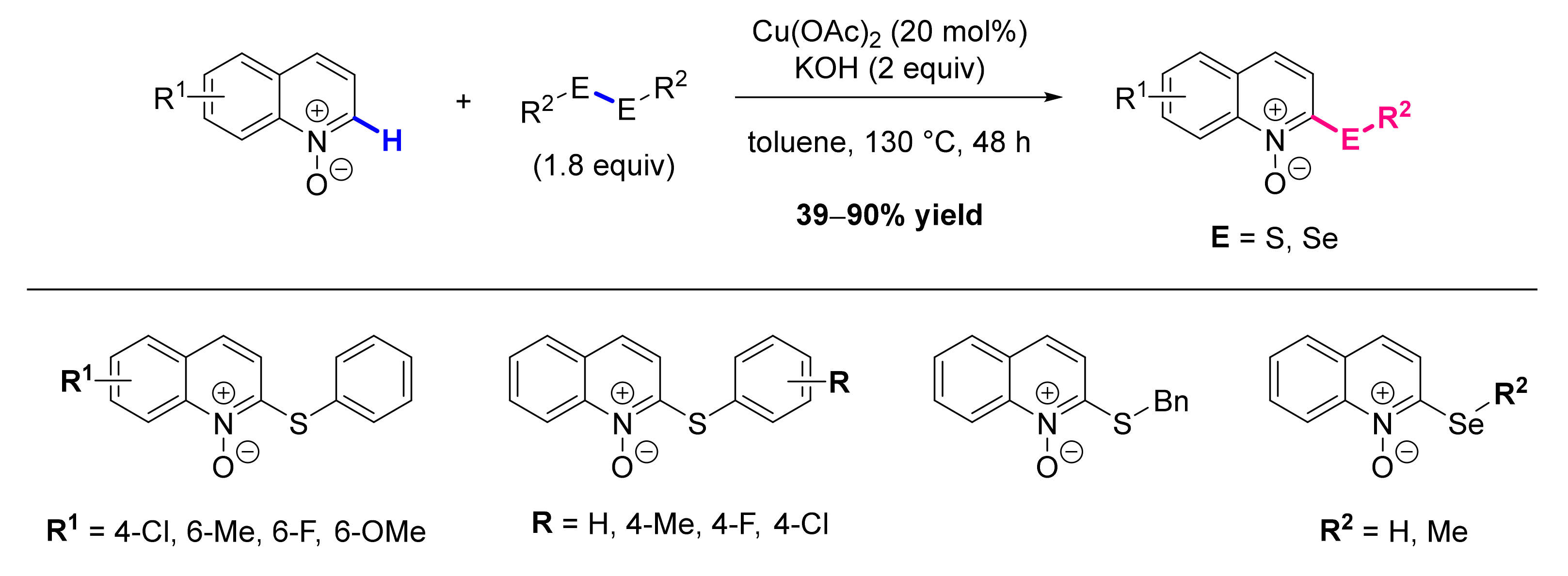
2.7. Formation of C-S Bond
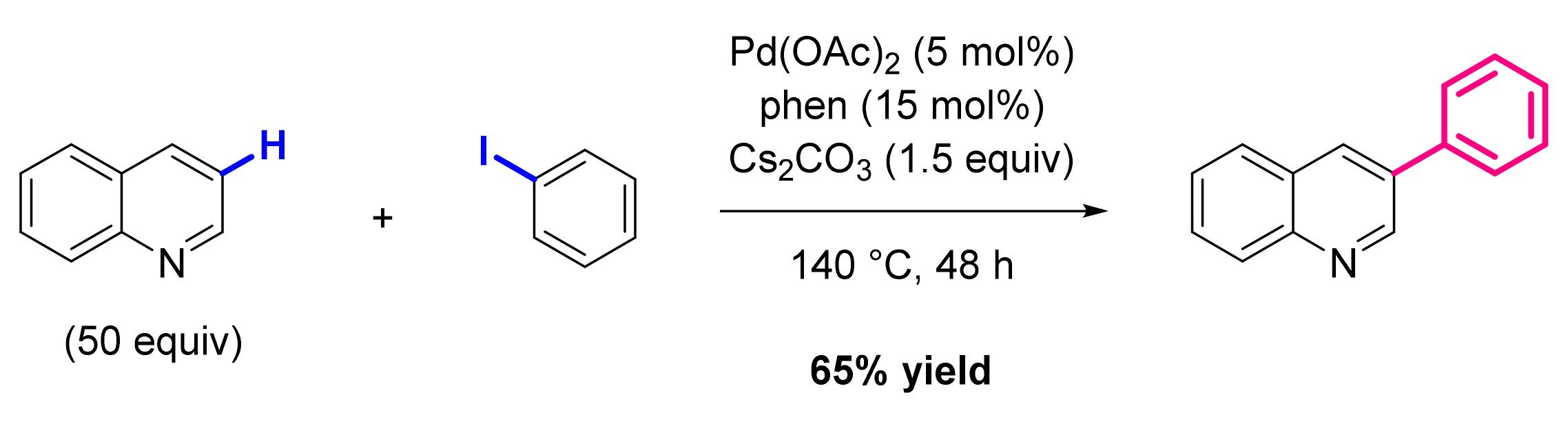
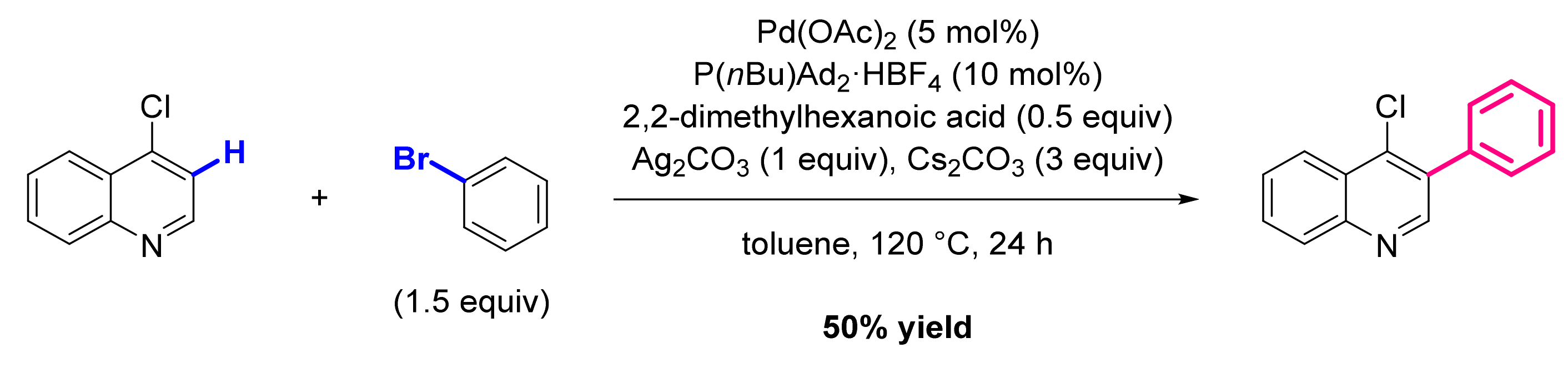
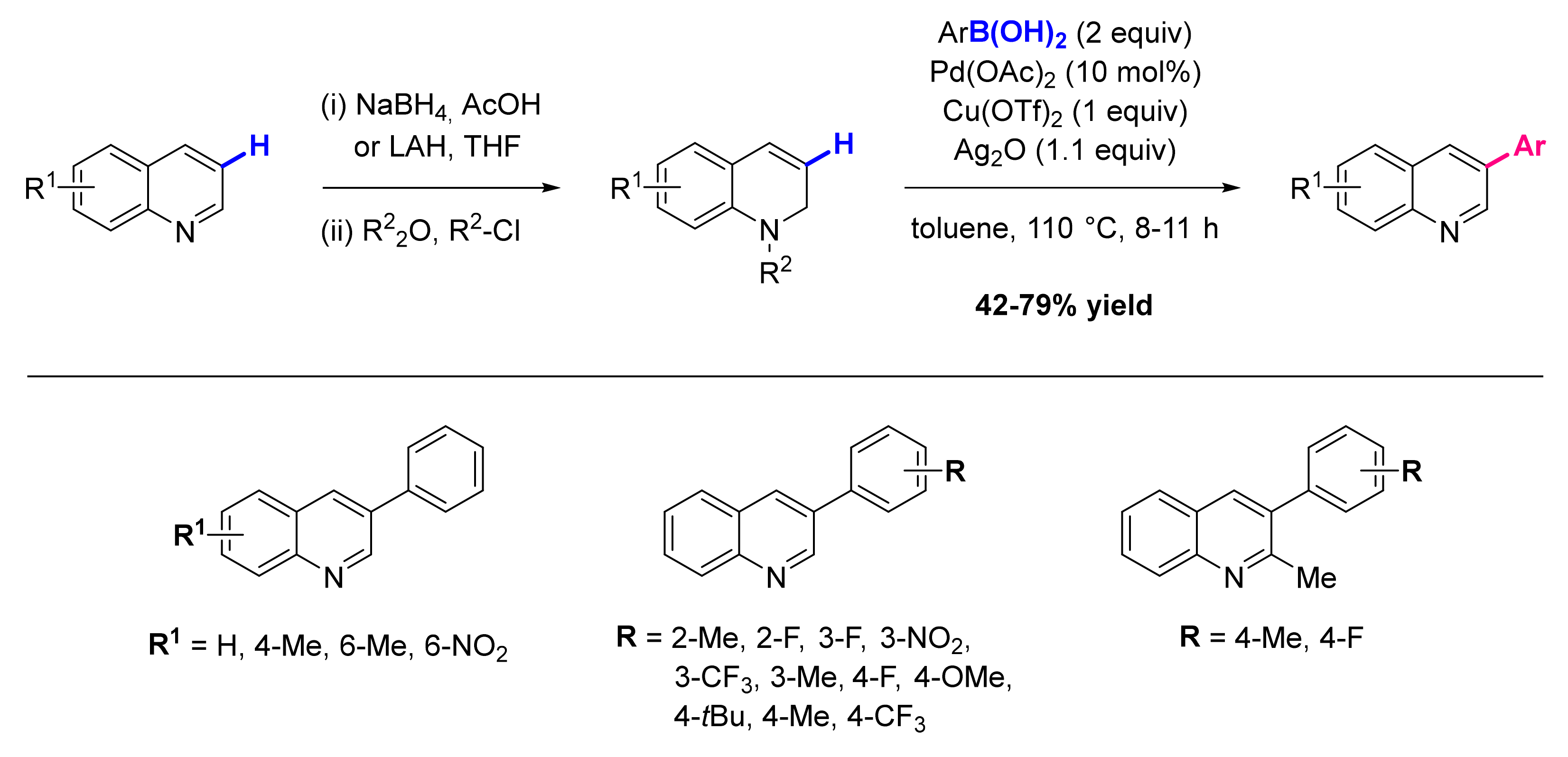
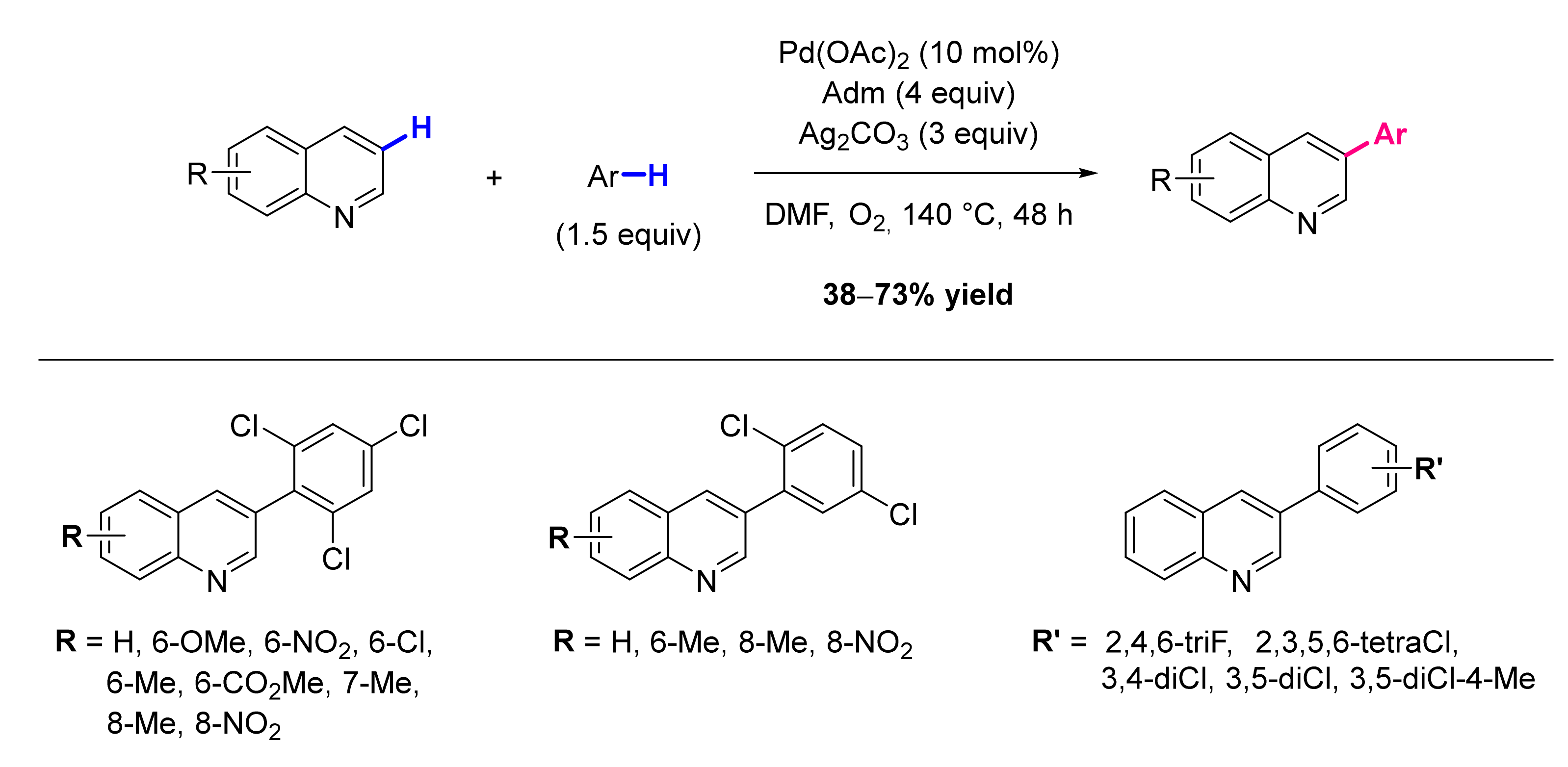
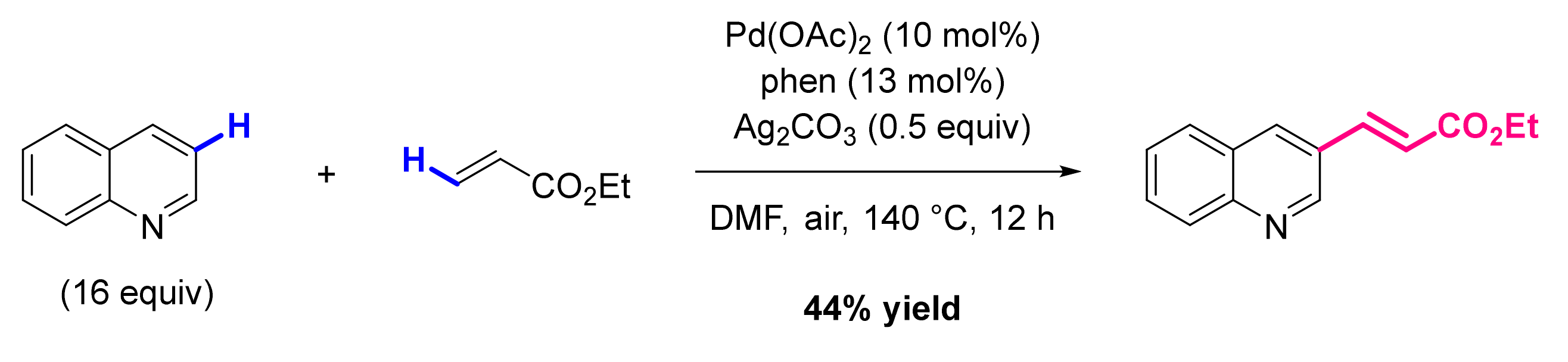
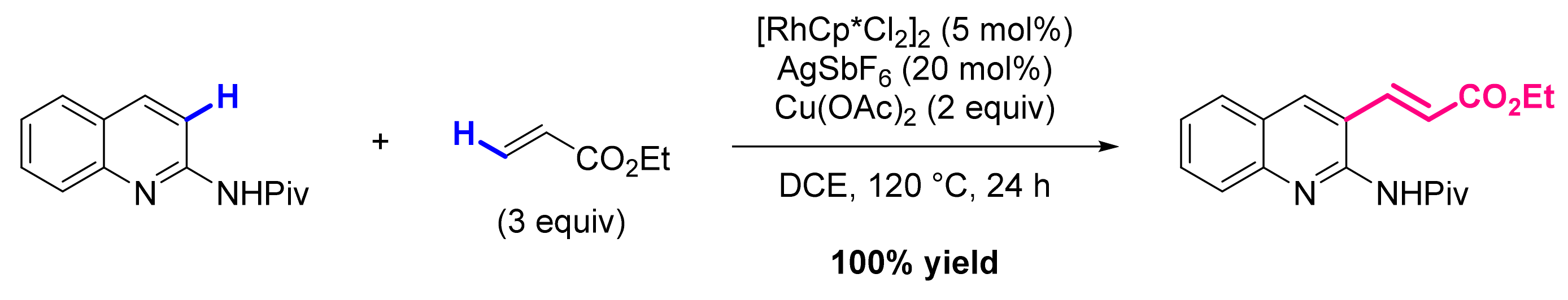
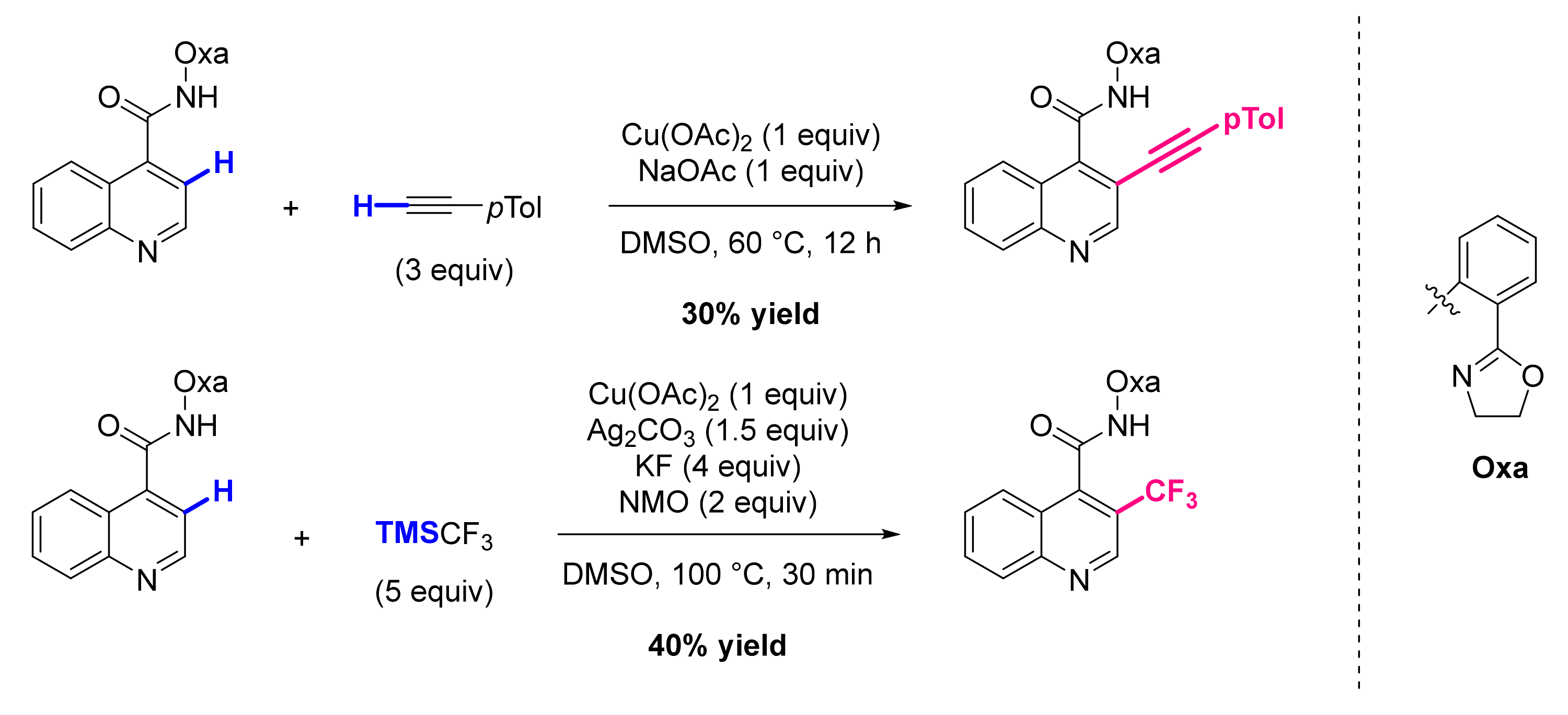
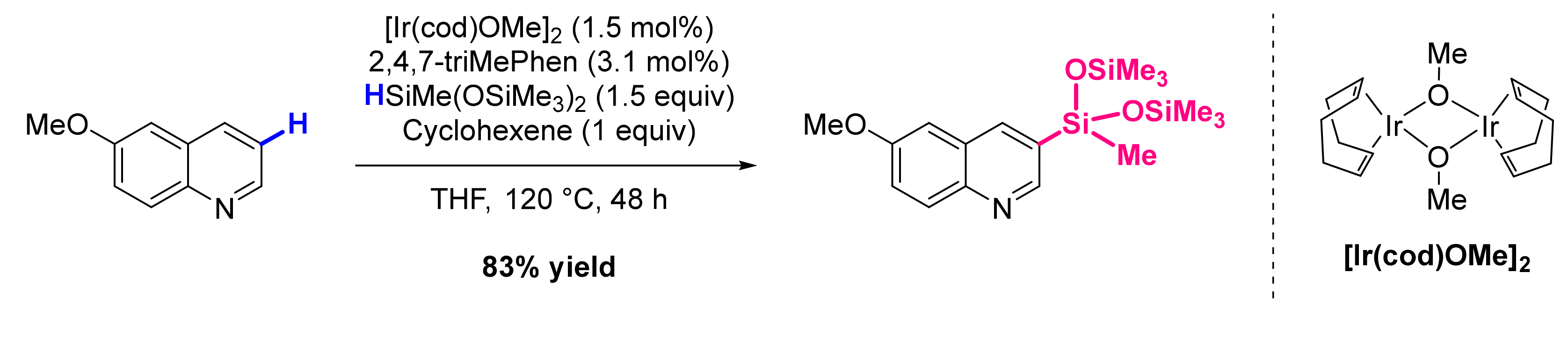
3. Functionalization in Position 3
3.1. Formation of C-Ar Bonds
3.2. Formation of C-Alkenyl Bonds
3.3. Formation of Other C-C Bonds
3.4. Formation of C-Het Bonds
4. Functionalization in Position 4
5. Functionalization in Position 5
6. Functionalization in Position 6
7. Functionalization in Position 7
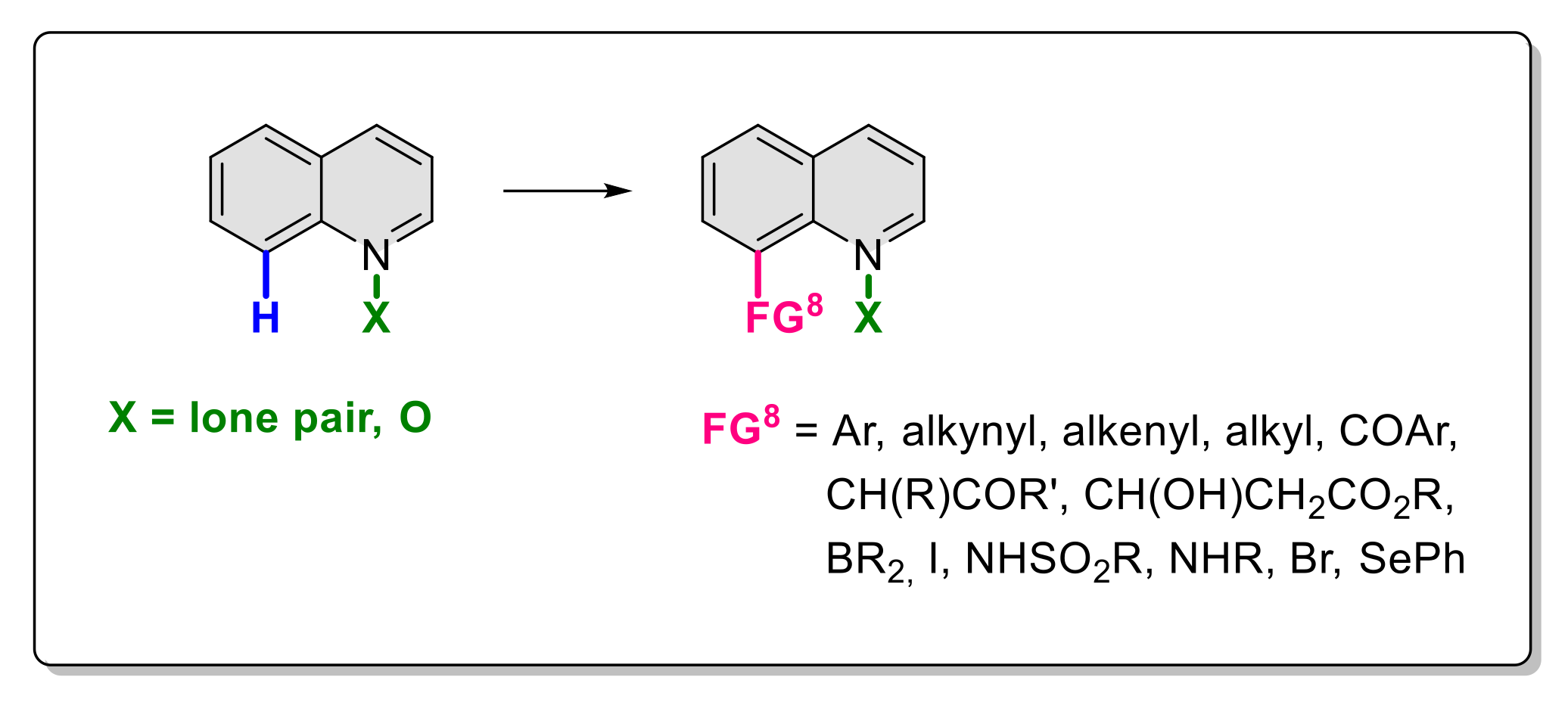
8. Functionalization in Position 8
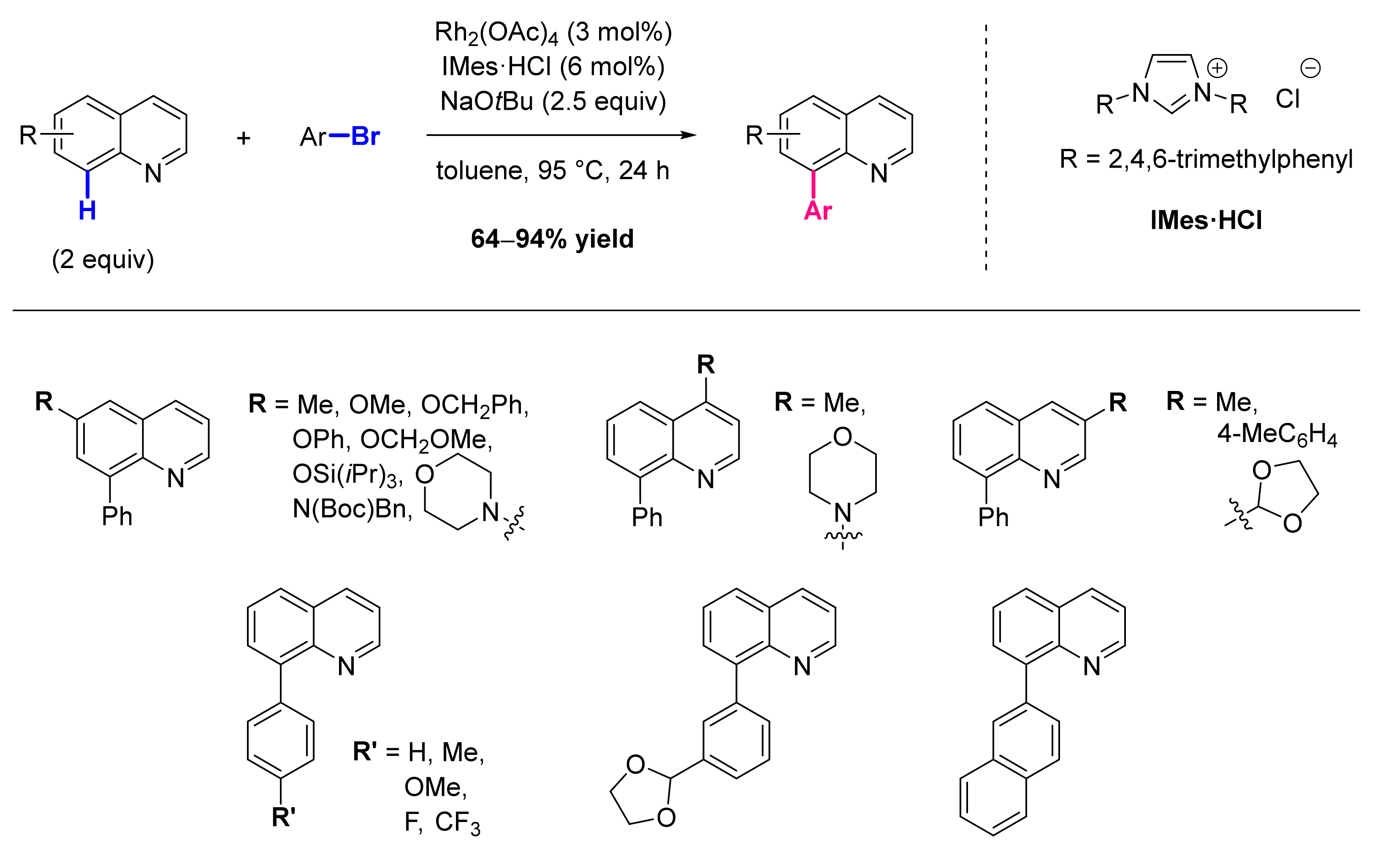
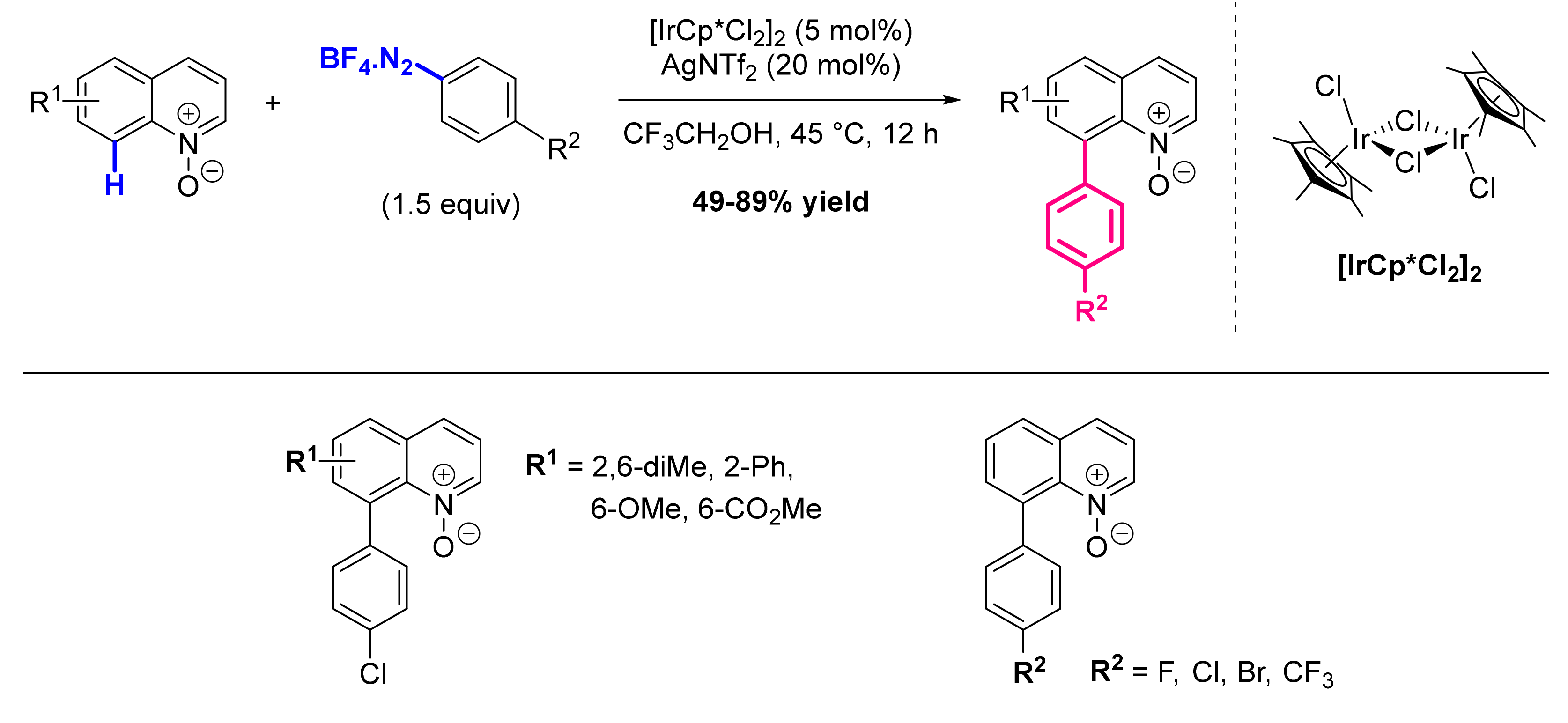
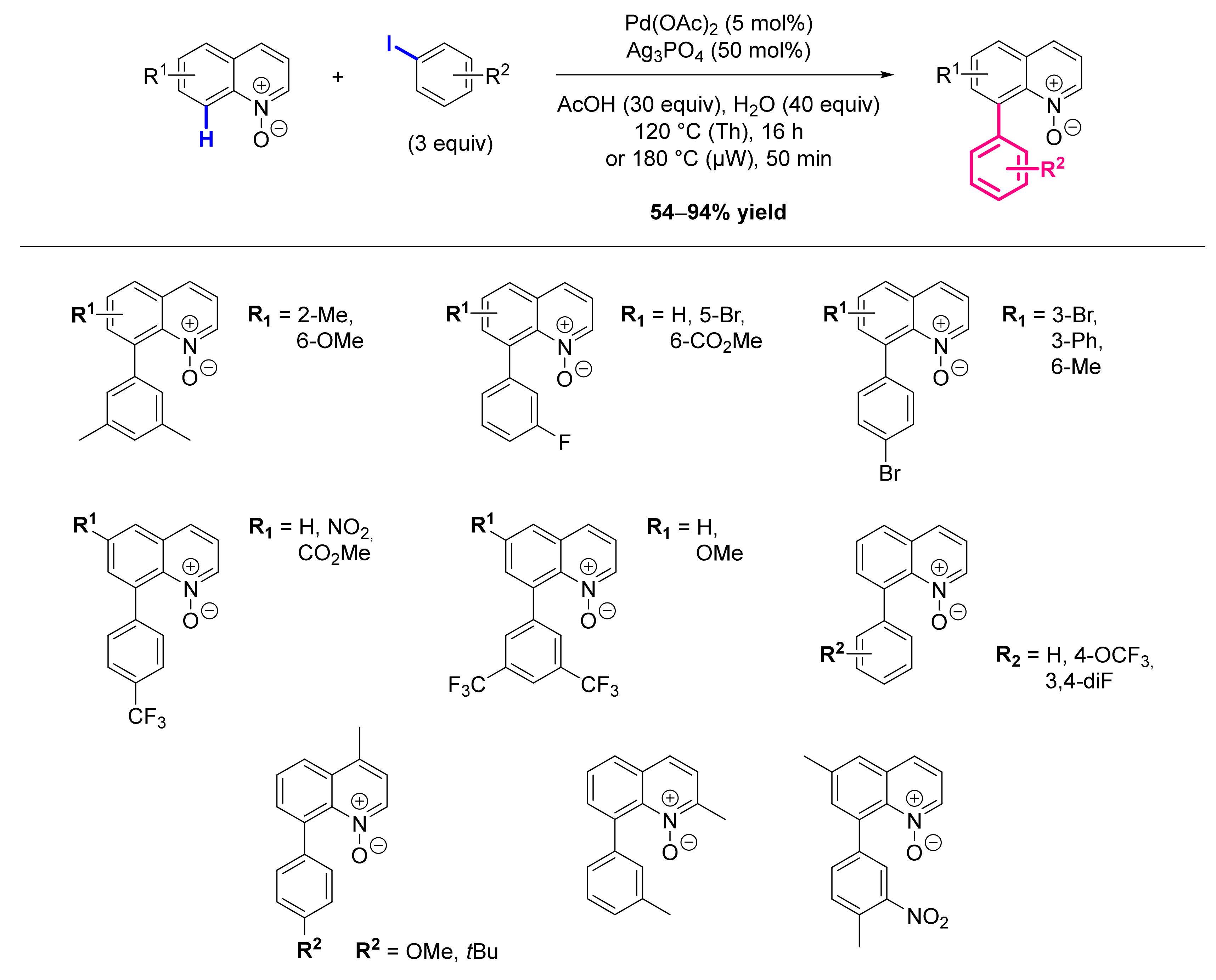
8.1. Formation of C-Ar Bonds
8.2. Formation of Other C-C Bonds
8.2.1. Without Incorporation of Oxygen (Path A)
8.2.2. With Incorporation of Oxygen (Path B)
8.3. Formation of C-heteroatom Bonds
9. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McAteer, C.H.; Balasubramanian, M.; Murugan, R. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 7, pp. 309–336. [Google Scholar]
- Jones, G. (Ed.) The Chemistry of Heterocyclic Compounds: Quinolines; Wiley: Chichester, UK, 2009; Volume 32, p. 1. ISBN 978-0-470-18853-8. [Google Scholar]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA; Wiley-VCH: Weinheim, Germany, 2003; Volume 6, pp. 316–336. [Google Scholar]
- Chung, P.-Y.; Bian, Z.-X.; Pun, H.-Y.; Chan, D.; Chan, A.S.-C.; Chui, C.-H.; Tang, J.C.-O.; Lam, K.-H. Recent Advances in Research of Natural and Synthetic Bioactive Quinolines. Future Med. Chem. 2015, 7, 947–967. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 1999, 16, 697–709. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2003, 20, 476–493. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2004, 21, 650–668. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2005, 22, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; MacMillan, J.B.; Gaudêncio, S.P.; Fenical, W.; La Clair, J.J. Ammosamides A and B Target Myosin. Angew. Chem. Int. Ed. 2009, 48, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.C.; MacMillan, J.B.; Gaudêncio, S.P.; Jensen, P.R.; Fenical, W. The Ammosamides: Structures of Cell Cycle Modulators from a Marine-Derived Streptomyces Species. Angew. Chem. Int. Ed. 2009, 48, 725–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Soni, K.; Sangani, C.; Yao, Y. An Overview of Privileged Scaffold: Quinolines and Isoquinolines in Medicinal Chemistry as Anticancer Agents. Curr. Top. Med. Chem. 2020, 20, 2599–2633. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Iannazzo, D.; Romeo, R.; Giofrè, S.V.; Legnani, L. Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Biologically Active Agents. Curr. Med. Chem. 2019, 26, 7166–7195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-Quinoline/Quinolone Hybrids as Potential Anti-Bacterial Agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar] [CrossRef]
- Chokkar, N.; Kalra, S.; Chauhan, M.; Kumar, R. A Review on Quinoline Derived Scaffolds as Anti-HIV Agents. Mini Rev. Med. Chem. 2019, 19, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Kumari, L.; Salahuddin; Mazumder, A.; Pandey, D.; Yar, M.S.; Kumar, R.; Mazumder, R.; Sarafroz, M.; Ahsan, M.J.; Kumar, V.; et al. Synthesis and Biological Potentials of Quinoline Analogues: A Review of Literature. Mini Rev. Org. Chem. 2019, 16, 653–688. [Google Scholar] [CrossRef]
- Haeusler, I.L.; Chan, X.H.S.; Guérin, P.J.; White, N.J. The Arrhythmogenic Cardiotoxicity of the Quinoline and Structurally Related Antimalarial Drugs: A Systematic Review. BMC Med. 2018, 16, 200. [Google Scholar] [CrossRef]
- Nqoro, X.; Tobeka, N.; Aderibigbe, B.A. Quinoline-Based Hybrid Compounds with Antimalarial Activity. Molecules 2017, 22, 2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musiol, R. An Overview of Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.M.A. Therapeutic Significance of Quinolines: A Patent Review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 1201–1221. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Singh, S.; Kaur, G.; Mangla, V.; Gupta, M.K. Quinoline and Quinolones: Promising Scaffolds for Future Antimycobacterial Agents. J. Enzym. Inhib. Med. Chem. 2015, 30, 492–504. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, S.A. Quinoline: A Promising Antitubercular Target. Biomed. Pharmacother. 2014, 68, 1161–1175. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of Sp3 C-H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef] [PubMed]
- Bailie, D.S.; Clendenning, G.M.A.; McNamee, L.; Muldoon, M.J. Anionic N,O-Ligated Pd(II) Complexes: Highly Active Catalysts for Alcohol Oxidation. Chem. Commun. 2010, 46, 7238–7240. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-A.; Wang, Z.-X. Zinc and Aluminum Complexes Supported by Quinoline-Based N,N,N-Chelate Ligands: Synthesis, Characterization, and Catalysis in the Ring-Opening Polymerization of ε-Caprolactone and rac-Lactide. Organometallics 2011, 30, 4364–4373. [Google Scholar] [CrossRef]
- Corbet, M.; De Campo, F. 8-Aminoquinoline: A Powerful Directing Group in Metal-Catalyzed Direct Functionalization of C-H Bonds. Angew. Chem. Int. Ed. 2013, 52, 9896–9898. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, X.; Liu, F.; Tan, Y.; Jiang, Y. A simple and novel amide ligand based on quinoline derivative used for palladium-catalyzed Suzuki coupling reaction. J. Organomet. Chem. 2015, 794, 27–32. [Google Scholar] [CrossRef]
- Rit, R.K.; Yadav, M.R.; Ghosh, K.; Sahoo, A.K. Reusable directing groups [8-aminoquinoline, picolinamide, sulfoximine] in C(sp3)-H bond activation: Present and future. Tetrahedron 2015, 71, 4450–4459. [Google Scholar] [CrossRef]
- Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Quinine-Catalyzed Asymmetric Synthesis of 2,2′-Binaphthol-Type Biaryls under Mild Reaction Conditions. Angew. Chem. Int. Ed. Engl. 2016, 55, 6525–6529. [Google Scholar] [CrossRef] [Green Version]
- Khan, B.; Khan, A.A.; Kant, R.; Koley, D. Directing Group-Assisted Copper(II)-Catalyzed Ortho-Carbonylation to Benzamide Using 2,2′-Azobisisobutyronitrile (AIBN). Adv. Synth. Catal. 2016, 358, 3753–3758. [Google Scholar] [CrossRef]
- Chen, C.H.; Shi, J. Metal chelates as emitting materials for organic electroluminescence. Coord. Chem. Rev. 1998, 171, 161–174. [Google Scholar] [CrossRef]
- Jégou, G.; Jenekhe, S.A. Highly Fluorescent Poly(arylene ethynylene)s Containing Quinoline and 3-Alkylthiophene. Macromolecules 2001, 34, 7926–7928. [Google Scholar] [CrossRef]
- Tong, H.; Wang, L.; Jing, X.; Wang, F. “Turn-On” Conjugated Polymer Fluorescent Chemosensor for Fluoride Ion. Macromolecules 2003, 36, 2584–2586. [Google Scholar] [CrossRef]
- Kim, J.I.; Shin, I.-S.; Kim, H.; Lee, J.-K. Efficient Electrogenerated Chemiluminescence from Cyclometalated Iridium(III) Complexes. J. Am. Chem. Soc. 2005, 127, 1614–1615. [Google Scholar] [CrossRef]
- Hughes, G.; Bryce, M.R. Electron-Transporting Materials for Organic Electroluminescent and Electrophosphorescent Devices. J. Mater. Chem. 2005, 15, 94–107. [Google Scholar] [CrossRef]
- Tokoro, Y.; Nagai, A.; Kokado, K.; Chujo, Y. Synthesis of Organoboron Quinoline-8-Thiolate and Quinoline-8-Selenolate Complexes and Their Incorporation into the π-Conjugated Polymer Main-Chain. Macromolecules 2009, 42, 2988–2993. [Google Scholar] [CrossRef]
- Liu, H.; Wu, F.; Zhang, B.; Tan, C.; Chen, Y.; Hao, G.; Tan, Y.; Jiang, Y. A Simple Quinoline-Derived Fluorescent Sensor for the Selective and Sequential Detection of Copper(II) and Sulfide Ions and Its Application in Living-Cell Imaging. RSC Adv. 2016, 6, 77508–77514. [Google Scholar] [CrossRef]
- Czaplinska, B.; Spaczynska, E.; Musiol, R. Quinoline Fluorescent Probes for Zinc—From Diagnostic to Therapeutic Molecules in Treating Neurodegenerative Diseases. Med. Chem. 2018, 14, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, Y.; Dai, Q.; Liang, H.; Song, J.; Qu, J.; Wong, W.-Y. A simple amide fluorescent sensor based on quinoline for selective and sensitive recognition of zinc(II) ions and bioimaging in living cells. Dye. Pigm. 2018, 158, 312–318. [Google Scholar] [CrossRef]
- Manske, R.H. The Chemistry of Quinolines. Chem. Rev. 1942, 30, 113–144. [Google Scholar] [CrossRef]
- Weyesa, A.; Mulugeta, E. Recent Advances in the Synthesis of Biologically and Pharmaceutically Active Quinoline and Its Analogues: A Review. RSC Adv. 2020, 10, 20784–20793. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Mendez, L.Y.; Gomez, C.M. Recent Progress in the Synthesis of Quinolines. Curr. Org. Chem. 2005, 9, 141–161. [Google Scholar] [CrossRef]
- Madapa, S.; Tusi, Z.; Batra, S. Advances in the Syntheses of Quinoline and Quinoline-Annulated Ring Systems. Curr. Org.Chem. 2008, 12, 1116–1183. [Google Scholar] [CrossRef]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24476. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B.J. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- Bryson, T.A.; Gibson, J.M.; Stewart, J.J.; Voegtle, H.; Tiwari, A.; Dawson, J.H.; Marley, W.; Harmon, B. Synthesis of quinolines, pyridine ligands and biological probes in green media. Green Chem. 2003, 5, 177–180. [Google Scholar] [CrossRef]
- Huang, H.; Jiang, H.; Chen, K.; Liu, H. A Simple and Convenient Copper-Catalyzed Tandem Synthesis of Quinoline-2-carboxylates at Room Temperature. J. Org. Chem. 2009, 74, 5476–5480. [Google Scholar] [CrossRef]
- Kulkarni, A.; Török, B. Microwave-assisted multicomponent domino cyclization–aromatization: An efficient approach for the synthesis of substituted quinolines. Green Chem. 2010, 12, 875–878. [Google Scholar] [CrossRef]
- Mitamura, T.; Ogawa, A. Synthesis of 2,4-Diiodoquinolines via the Photochemical Cyclization of o-Alkynylaryl Isocyanides with Iodine. J. Org. Chem. 2011, 76, 1163–1166. [Google Scholar] [CrossRef]
- Li, X.; Mao, Z.; Wang, Y.; Chen, W.; Lin, X. Molecular iodine-catalyzed and air-mediated tandem synthesis of quinolines via three-component reaction of amines, aldehydes, and alkynes. Tetrahedron 2011, 67, 3858–3862. [Google Scholar] [CrossRef]
- Reddy, T.R.; Reddy, L.S.; Reddy, G.R.; Yarbagi, K.; Lingappa, Y.; Rambabu, D.; Krishna, G.R.; Reddy, C.M.; Kumar, K.S.; Pal, M. Construction of a Quinoline Ring via a 3-Component Reaction in Water: Crystal Structure Analysis and H-Bonding Patterns of a 2-Aryl Quinoline. Green Chem. 2012, 14, 1870–1872. [Google Scholar] [CrossRef]
- Talwar, D.; Gonzalez-de-Castro, A.; Li, H.Y.; Xiao, J. Regioselective Acceptorless Dehydrogenative Coupling of N-Heterocycles toward Functionalized Quinolines, Phenanthrolines, and Indoles. Angew. Chem. Int. Ed. 2015, 54, 5223–5227. [Google Scholar] [CrossRef]
- Bharate, J.B.; Vishwakarma, R.A.; Bharate, S.B. Metal-Free Domino One-Pot Protocols for Quinoline Synthesis. RSC Adv. 2015, 5, 42020–42053. [Google Scholar] [CrossRef]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Synthesis of Quinolines: A Green Perspective. ACS Sustain. Chem. Eng. 2016, 4, 4064–4078. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, S.; Gao, Y.; Zhang, P.; Wu, Y.; Tang, G.; Zhao, Y. Tert-Butyl Hydroperoxide Mediated Cascade Synthesis of 3-Arylsulfonylquinolines. Org. Lett. 2016, 18, 1286–1289. [Google Scholar] [CrossRef]
- Sharma, R.; Kour, P.; Kumar, A. A Review on Transition-Metal Mediated Synthesis of Quinolines. J. Chem. Sci. 2018, 130, 73. [Google Scholar] [CrossRef] [Green Version]
- Xuan, D.D. Recent Progress in the Synthesis of Quinolines. Curr. Org. Chem. 2019, 16, 671–708. [Google Scholar] [CrossRef]
- Nainwal, L.M.; Tasneem, S.; Akhtar, W.; Verma, G.; Khan, M.F.; Parvez, S.; Shaquiquzzaman, M.; Akhter, M.; Alam, M.M. Green Recipes to Quinoline: A Review. Eur. J. Med. Chem. 2019, 164, 121–170. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira Pinto, L.S.; Alves Vasconcelos, T.R.; de Souza, M.V.N. Eco-Friendly Methodologies for the Synthesis of Quinoline Nucleus. Mini Rev. Org. Chem. 2019, 16, 602–608. [Google Scholar] [CrossRef]
- Mongin, F.; Quéguiner, G. Advances in the Directed Metallation of Azines and Diazines (Pyridines, Pyrimidines, Pyrazines, Pyridazines, Quinolines, Benzodiazines and Carbolines). Part 1: Metallation of Pyridines, Quinolines and Carbolines. Tetrahedron 2001, 57, 4059–4090. [Google Scholar] [CrossRef]
- Gros, P.; Fort, Y. NBuLi/Lithium Aminoalkoxide Aggregates: New and Promising Lithiating Agents for Pyridine Derivatives. Eur. J. Org. Chem. 2002, 2002, 3375–3383. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C.; Yus, M. Metalated Heterocycles and Their Applications in Synthetic Organic Chemistry. Chem. Rev. 2004, 104, 2667–2722. [Google Scholar] [CrossRef]
- Makosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen in Heterocyclic Chemistry. Chem. Rev. 2004, 104, 2631–2666. [Google Scholar] [CrossRef]
- Schlosser, M. The 2 × 3 Toolbox of Organometallic Methods for Regiochemically Exhaustive Functionalization. Angew. Chem. Int. Ed. Engl. 2005, 44, 376–393. [Google Scholar] [CrossRef]
- Haag, B.; Mosrin, M.; Ila, H.; Malakhov, V.; Knochel, P. Regio- and Chemoselective Metalation of Arenes and Heteroarenes Using Hindered Metal Amide Bases. Angew. Chem. Int. Ed. 2011, 50, 9794–9824. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Lesenyeho, L.G. Halogenated Quinolines as Substrates for the Palladium-Catalyzed Cross-Coupling Reactions to Afford Substituted Quinolines. J. Heterocycl. Chem. 2013, 50, 1–16. [Google Scholar] [CrossRef]
- Klatt, T.; Markiewicz, J.T.; Sämann, C.; Knochel, P. Strategies to Prepare and Use Functionalized Organometallic Reagents. J. Org. Chem. 2014, 79, 4253–4269. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and Exploiting C-H Bond Activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Godula, K.; Sames, D. C-H Bond Functionalization in Complex Organic Synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Bergman, R.G. C-H Activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-J.; Trost, B.M. Green Chemistry for Chemical Synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 13197–13202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, L.; O’Hara, F.; Gaunt, M.J. Recent Developments in Natural Product Synthesis Using Metal-Catalysed C-H Bond Functionalisation. Chem. Soc. Rev. 2011, 40, 1885–1898. [Google Scholar] [CrossRef]
- White, M.C. Adding Aliphatic C-H Bond Oxidations to Synthesis. Science 2012, 335, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef] [PubMed]
- Seregin, I.V.; Gevorgyan, V. Direct Transition Metal-Catalyzed Functionalization of Heteroaromatic Compounds. Chem. Soc. Rev. 2007, 36, 1173–1193. [Google Scholar] [CrossRef] [PubMed]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- McGlacken, G.P.; Bateman, L.M. Recent advances in aryl–aryl bond formation by direct arylation. Chem. Soc. Rev. 2009, 38, 2447–2464. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C-H Bond Cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef]
- Satoh, T.; Miura, M. Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J. 2010, 16, 11212–11222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent Advances in the Transition Metal-Catalyzed Twofold Oxidative C-H Bond Activation Strategy for C-C and C-N Bond Formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.F. Borylation and Silylation of C-H Bonds: A Platform for Diverse C-H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Spring, D.R. Arene C-H Functionalisation Using a Removable/Modifiable or a Traceless Directing Group Strategy. Chem. Soc. Rev. 2014, 43, 6906–6919. [Google Scholar] [CrossRef]
- Yang, J. Transition Metal Catalyzed Meta-C-H Functionalization of Aromatic Compounds. Org. Biomol. Chem. 2015, 13, 1930–1941. [Google Scholar] [CrossRef]
- Mishra, N.K.; Park, J.; Oh, H.; Han, S.H.; Kim, I.S. Recent Advances in N-Heterocycles Synthesis through Catalytic C-H Functionalization of Azobenzenes. Tetrahedron 2018, 74, 6769–6794. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Catalytic Dehydrogenative Cross-Coupling DCC: Forming Carbon−Carbon Bonds by Oxidizing Two Carbon−Hydrogen Bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Daugulis, O. Palladium and Copper Catalysis in Regioselective, Intermolecular Coupling of C-H and C-Hal Bonds. Top. Curr. Chem. 2010, 292, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C-H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Pd-Catalyzed Oxidative Coupling with Organometallic Reagents via C-H Activation. Chem. Commun. 2010, 46, 677–685. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-Catalyzed C-H Bond Activation and Functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Song, G.; Wang, F.; Li, X. C-C, C-O and C-N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, N.; Hopkinson, M.N.; Wencel-Delord, J.; Glorius, F. Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chem. Int. Ed. 2012, 51, 10236–10254. [Google Scholar] [CrossRef] [PubMed]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(Sp2)-H and C(Sp3)-H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Kuhl, N.; Schröder, N.; Glorius, F. Formal SN-Type Reactions in Rhodium(III)-Catalyzed C-H Bond Activation. Adv. Synth. Catal. 2014, 356, 1443–1460. [Google Scholar] [CrossRef]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Rh(I)-Catalyzed Alkylation of Quinolines and Pyridines via C-H Bond Activation. J. Am. Chem. Soc. 2007, 129, 5332–5333. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Sawamura, M. Transition-Metal-Catalyzed Site-Selective C-H Functionalization of Quinolines beyond C2 Selectivity. ACS Catal. 2015, 5, 5031–5040. [Google Scholar] [CrossRef]
- Stephens, D.E.; Larionov, O.V. Recent Advances in the C-H-Functionalization of the Distal Positions in Pyridines and Quinolines. Tetrahedron 2015, 71, 8683–8716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, B.; Dutta, H.S.; Koley, D. Remote C-H Functionalization of 8-Aminoquinolinamides. Asian J. Org. Chem. 2018, 7, 1270–1297. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Méndez, L.Y.V.; Galvis, C.E.P.; Villamizar, M.C.O. The Direct C-H Alkenylation of Quinoline N-Oxides as a Suitable Strategy for the Synthesis of Promising Antiparasitic Drugs. New J. Chem. 2020, 44, 12–19. [Google Scholar] [CrossRef]
- Dhiman, A.K.; Thakur, A.; Kumar, R.; Sharma, U. Rhodium-Catalyzed Selective C-H Bond Functionalization of Quinolines. Asian J. Org. Chem. 2020, 9, 1502–1518. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C-H Bond Functionalization Chemistry for the Expedient Construction of C-C Bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Farooq Zia, M.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C-H Functionalisation Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, Y.; Jie, X.; Zhao, H.; Li, G.; Su, W. Recent Advances in Directed C-H Functionalizations Using Monodentate Nitrogen-Based Directing Groups. Org. Chem. Front. 2014, 1, 843–895. [Google Scholar] [CrossRef]
- Frost, C.G.; Paterson, A.J. Directing Remote Meta-C-H Functionalization with Cleavable Auxiliaries. ACS Cent. Sci. 2015, 1, 418–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Thakur, K.; Kumar, R.; Kumar, I.; Sharma, U. Distant C-H Activation/Functionalization: A New Horizon of Selectivity Beyond Proximity. Catal. Rev. 2015, 57, 345–405. [Google Scholar] [CrossRef]
- Coe, B.J.; Glenwright, S.J. Trans-Effects in Octahedral Transition Metal Complexes. Coord. Chem. Rev. 2000, 203, 5–80. [Google Scholar] [CrossRef]
- Cho, S.H.; Hwang, S.J.; Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 2008, 130, 9254–9256. [Google Scholar] [CrossRef] [PubMed]
- Campeau, L.-C.; Stuart, D.R.; Leclerc, J.-P.; Bertrand-Laperle, M.; Villemure, E.; Sun, H.-Y.; Lasserre, S.; Guimond, N.; Lecavallier, M.; Fagnou, K. Palladium-Catalyzed Direct Arylation of Azine and Azole N-Oxides: Reaction Development, Scope and Applications in Synthesis. J. Am. Chem. Soc. 2009, 131, 3291–3306. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-Y.; Gorelsky, S.I.; Stuart, D.R.; Campeau, L.-C.; Fagnou, K. Mechanistic Analysis of Azine N-Oxide Direct Arylation: Evidence for a Critical Role of Acetate in the Pd(OAc)2 Precatalyst. J. Org. Chem. 2010, 75, 8180–8189. [Google Scholar] [CrossRef]
- Ackermann, L.; Fenner, S. Direct Arylations of Electron-Deficient (Hetero)Arenes with Aryl or Alkenyl Tosylates and Mesylates. Chem. Commun. 2010, 47, 430–432. [Google Scholar] [CrossRef]
- Ren, X.; Wen, P.; Shi, X.; Wang, Y.; Li, J.; Yang, S.; Yan, H.; Huang, G. Palladium-Catalyzed C-2 Selective Arylation of Quinolines. Org. Lett. 2013, 15, 5194–5197. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.M.; Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Rh(I)-Catalyzed Direct Arylation of Pyridines and Quinolines. J. Am. Chem. Soc. 2008, 130, 14926–14927. [Google Scholar] [CrossRef] [Green Version]
- Lehecq, A.; Rousée, K.; Schneider, C.; Levacher, V.; Hoarau, C.; Pannecoucke, X.; Bouillon, J.-P.; Couve-Bonnaire, S. Metal-Catalyzed Direct C-H Fluoroalkenylation of Pyridine N-Oxides and Related Derivatives. Eur. J. Org. Chem. 2017, 2017, 3049–3054. [Google Scholar] [CrossRef]
- Berman, A.M.; Bergman, R.G.; Ellman, J.A. Rh(I)-Catalyzed Direct Arylation of Azines. J. Org. Chem. 2010, 75, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Wang, W.; Yang, F.; Lan, J.; Yang, L.; Gao, G.; You, J. Copper-Catalyzed Direct C Arylation of Heterocycles with Aryl Bromides: Discovery of Fluorescent Core Frameworks. Angew. Chem. Int. Ed. Engl. 2009, 48, 3296–3300. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, I.; Tobisu, M.; Chatani, N. Catalytic Arylation of a C-H Bond in Pyridine and Related Six-Membered N-Heteroarenes Using Organozinc Reagents. Chem. Asian J. 2012, 7, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Xi, P.; Yang, F.; Qin, S.; Zhao, D.; Lan, J.; Gao, G.; Hu, C.; You, J. Palladium(II)-Catalyzed Oxidative C−H/C-H Cross-Coupling of Heteroarenes. J. Am. Chem. Soc. 2010, 132, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Huang, Y.; Lan, J.; Song, F.; You, J. Pd-Catalyzed Oxidative C-H/C-H Cross-Coupling of Pyridines with Heteroarenes. Chem. Sci. 2013, 4, 2163–2167. [Google Scholar] [CrossRef]
- Gong, X.; Song, G.; Zhang, H.; Li, X. Palladium-Catalyzed Oxidative Cross-Coupling between Pyridine N-Oxides and Indoles. Org. Lett. 2011, 13, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- Liégault, B.; Fagnou, K. Palladium-Catalyzed Intramolecular Coupling of Arenes and Unactivated Alkanes in Air. Organometallics 2008, 27, 4841–4843. [Google Scholar] [CrossRef]
- Willis, N.J.; Smith, J.M. An Operationally Simple, Palladium Catalysed Dehydrogenative Cross-Coupling Reaction of Pyridine N-Oxides and Thiazoles “on Water”. RSC Adv. 2014, 4, 11059–11063. [Google Scholar] [CrossRef]
- Suresh, R.; Muthusubramanian, S.; Kumaran, R.S.; Manickam, G. C2-Arylation of Substituted Pyridine N-Oxides with Heteroaryl Carboxylic Acids by Palladium-Catalyzed Decarboxylative Coupling. Asian J. Org. Chem. 2014, 3, 604–608. [Google Scholar] [CrossRef]
- Nakao, Y.; Kanyiva, K.S.; Hiyama, T. A Strategy for C-H Activation of Pyridines: Direct C-2 Selective Alkenylation of Pyridines by Nickel/Lewis Acid Catalysis. J. Am. Chem. Soc. 2008, 130, 2448–2449. [Google Scholar] [CrossRef] [PubMed]
- Kanyiva, K.S.; Nakao, Y.; Hiyama, T. Nickel-catalyzed addition of pyridine-N-oxides across alkynes. Angew. Chem. Int. Ed. Engl. 2007, 46, 8872–8874. [Google Scholar] [CrossRef]
- Ogoshi, S.; Ikeda, H.; Kurosawa, H. Formation of an Aza-Nickelacycle by Reaction of an Imine and an Alkyne with Nickel(0): Oxidative Cyclization, Insertion, and Reductive Elimination. Angew. Chem. Int. Ed. Engl. 2007, 46, 4930–4932. [Google Scholar] [CrossRef]
- Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. Palladium-Catalyzed Alkenylation of Quinoline-N-Oxides via C-H Activation under External-Oxidant-Free Conditions. J. Am. Chem. Soc. 2009, 131, 13888–13889. [Google Scholar] [CrossRef]
- Guo, T.; Liu, Y.; Zhao, Y.-H.; Zhang, P.-K.; Han, S.-L.; Liu, H.-M. Palladium-Catalyzed External-Oxidant-Free Coupling Reactions between Isoquinoline/Quinoline N-Oxides with Olefins. Tetrahedron Lett. 2016, 57, 3920–3923. [Google Scholar] [CrossRef]
- Bellina, F.; Cauteruccio, S.; Rossi, R. Development and Application of Effective Protocols for the Synthesis of Arylheteroarenes and Biheteroaryls, Including Bioactive Derivatives, by Highly Regioselective Transition Metal-Catalyzed Direct Intermolecular Arylation Reactions of Five-Membered Heteroarenes with (Hetero)aryl Halides. Curr. Org. Chem. 2008, 12, 774–790. [Google Scholar] [CrossRef]
- Roudesly, F.; Veiros, L.F.; Oble, J.; Poli, G. Pd-Catalyzed Direct C-H Alkenylation and Allylation of Azine N-Oxides. Org. Lett. 2018, 20, 2346–2350. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Cho, S.H.; Chang, S. A Versatile Rhodium(I) Catalyst System for the Addition of Heteroarenes to Both Alkenes and Alkynes by a C-H Bond Activation. Angew. Chem. Int. Ed. 2012, 51, 3677–3681. [Google Scholar] [CrossRef]
- Wu, Z.; Pi, C.; Cui, X.; Bai, J.; Wu, Y. Direct C-2 Alkylation of Quinoline N-Oxides with Ethers via Palladium-Catalyzed Dehydrogenative Cross-Coupling Reaction. Adv. Synth. Catal. 2013, 355, 1971–1976. [Google Scholar] [CrossRef]
- Jha, A.K.; Jain, N. The Microwave-Assisted Ortho-Alkylation of Azine N-Oxides with N-Tosylhydrazones Catalyzed by Copper(I) Iodide. Chem. Commun. 2016, 52, 1831–1834. [Google Scholar] [CrossRef]
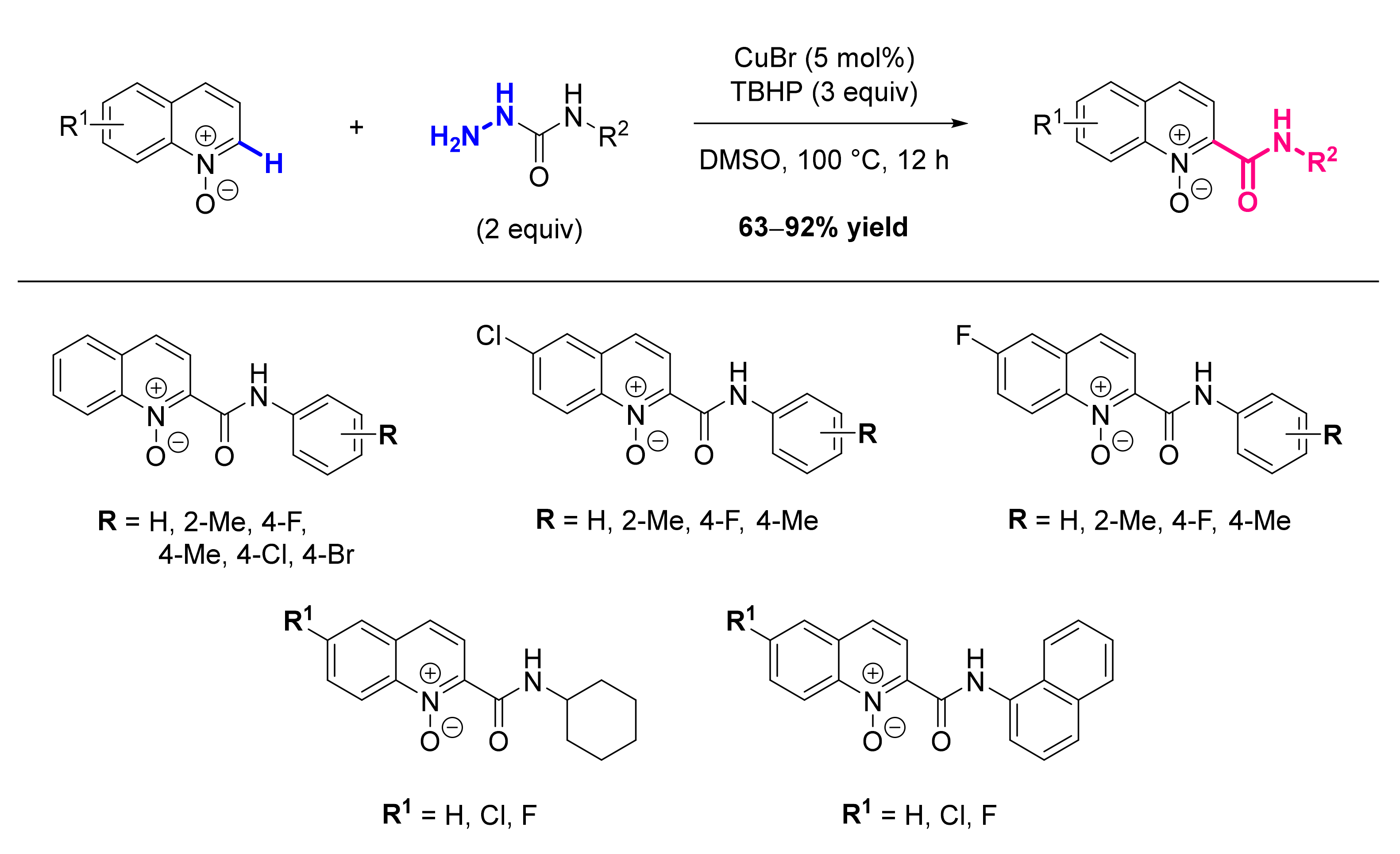
- Li, G.-H.; Dong, D.-Q.; Yang, Y.; Yu, X.-Y.; Wang, Z.-L. Direct Carbamoylation of Quinoline N-Oxides with Hydrazinecarboxamides via C-H Bond Activation Catalyzed by Copper Catalyst. Adv. Synth. Catal. 2019, 361, 832–835. [Google Scholar] [CrossRef]
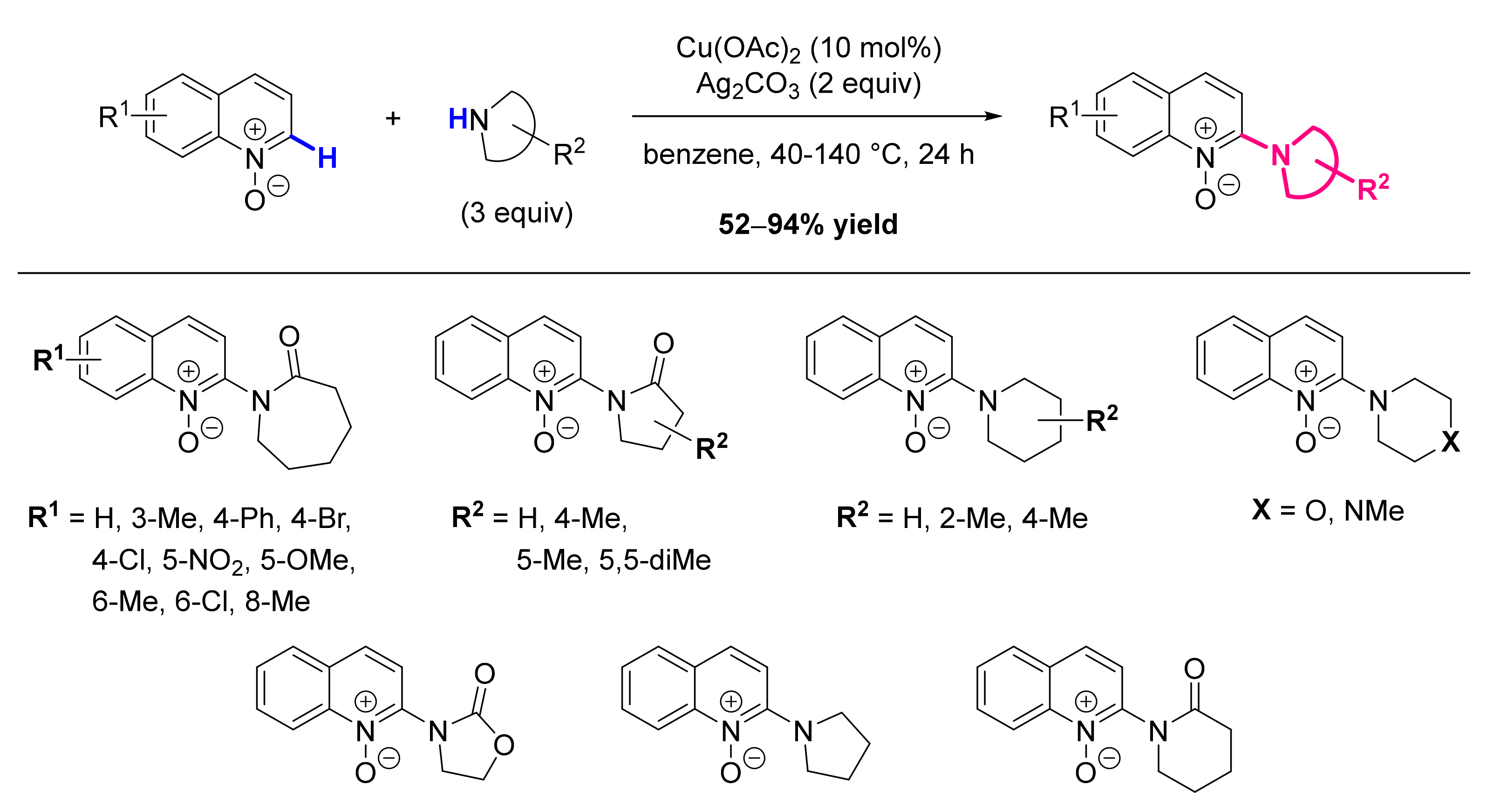
- Li, G.; Jia, C.; Sun, K. Copper-Catalyzed Intermolecular Dehydrogenative Amidation/Amination of Quinoline N-Oxides with Lactams/Cyclamines. Org. Lett. 2013, 15, 5198–5201. [Google Scholar] [CrossRef]
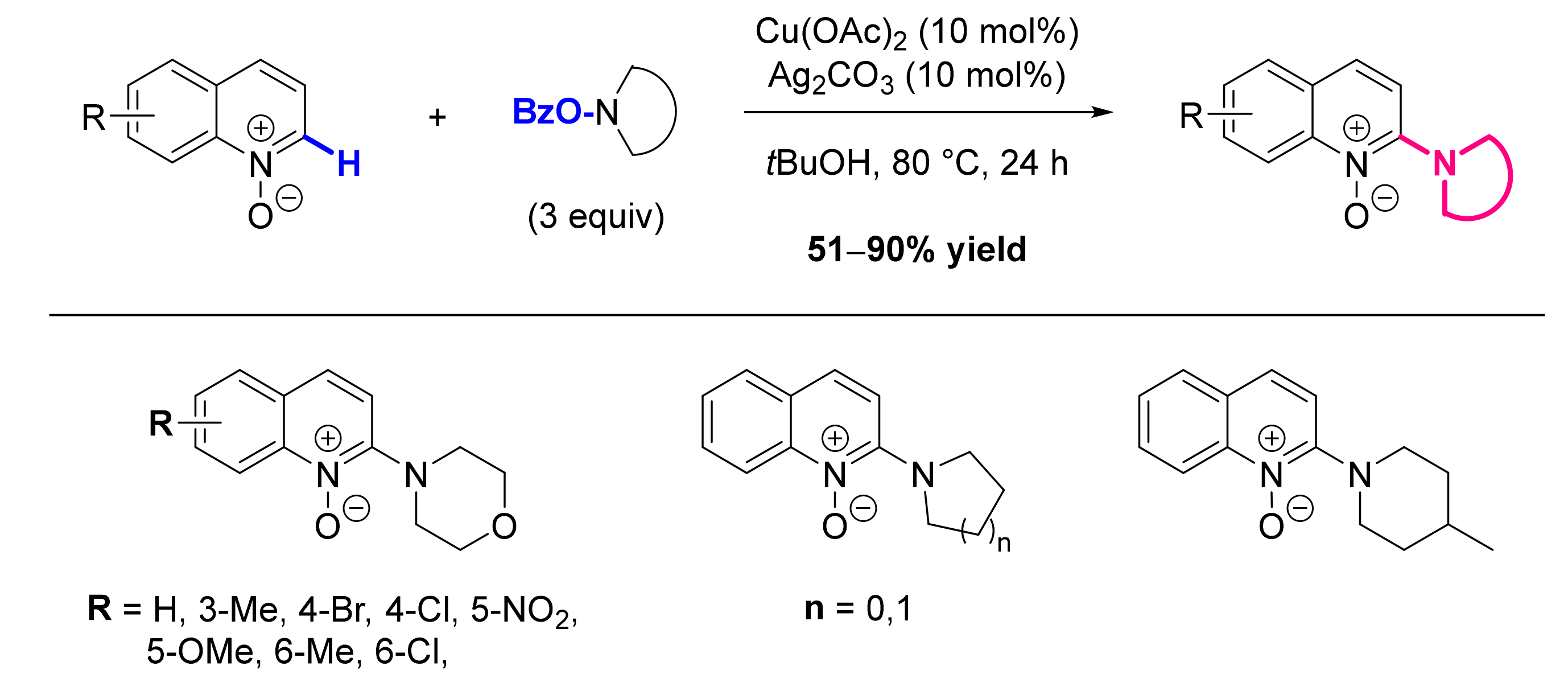
- Li, G.; Jia, C.; Sun, K.; Lv, Y.; Zhao, F.; Zhou, K.; Wu, H. Copper(II)-Catalyzed Electrophilic Amination of Quinoline N-Oxides with O-Benzoyl Hydroxylamines. Org. Biomol. Chem. 2015, 13, 3207–3210. [Google Scholar] [CrossRef]
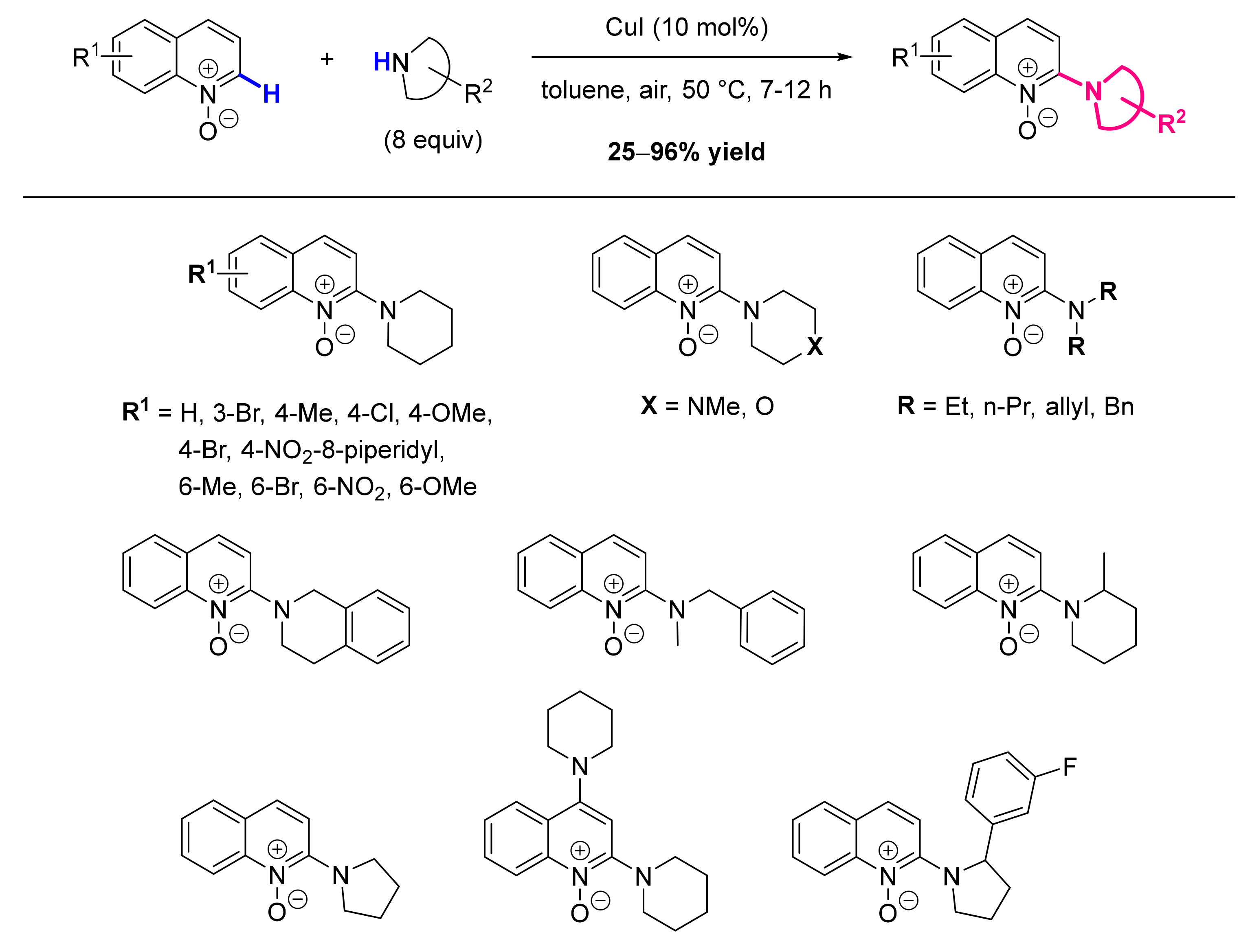
- Zhu, C.; Yi, M.; Wei, D.; Chen, X.; Wu, Y.; Cui, X. Copper-Catalyzed Direct Amination of Quinoline N-Oxides via C-H Bond Activation under Mild Conditions. Org. Lett. 2014, 16, 1840–1843. [Google Scholar] [CrossRef]
- Yu, H.; Dannenberg, C.A.; Li, Z.; Bolm, C. Copper-Catalyzed Direct Sulfoximination of Heteroaromatic N-Oxides by Dual C−H/N−H Dehydrogenative Cross-Coupling. Chem. Asian J. 2016, 11, 54–57. [Google Scholar] [CrossRef]
- Biswas, A.; Karmakar, U.; Nandi, S.; Samanta, R. Copper-Catalyzed Direct, Regioselective Arylamination of N-Oxides: Studies to Access Conjugated π-Systems. J. Org. Chem. 2017, 82, 8933–8942. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Song, H.; Cui, X.; Pi, C.; Du, W.; Wu, Y. Sulfonylation of Quinoline N-Oxides with Aryl Sulfonyl Chlorides via Copper-Catalyzed C-H Bonds Activation. Org. Lett. 2013, 15, 1270–1273. [Google Scholar] [CrossRef]
- Lai, M.; Zhai, K.; Cheng, C.; Wu, Z.; Zhao, M. Direct Thiolation of Aza-Heteroaromatic N-Oxides with Disulfides via Copper-Catalyzed Regioselective C-H Bond Activation. Org. Chem. Front. 2018, 5, 2986–2991. [Google Scholar] [CrossRef]
- Li, G.-H.; Dong, D.-Q.; Deng, Q.; Yan, S.-Q.; Wang, Z.-L. Copper-Catalyzed Deoxygenative C2-Sulfonylation of Quinoline N-Oxides with DABSO and Phenyldiazonium Tetrafluoroborates for the Synthesis of 2-Sulfonylquinolines via a Radical Reaction. Synthesis 2019, 51, 3313–3319. [Google Scholar] [CrossRef] [Green Version]
- Jonckers, T.H.M.; Maes, B.U.W.; Lemière, G.L.F.; Rombouts, G.; Pieters, L.; Haemers, A.; Dommisse, R.A. Synthesis of Isocryptolepine via a Pd-Catalyzed ‘Amination-Arylation’ Approach. Synlett 2003, 2003, 615–618. [Google Scholar] [CrossRef]
- Wasa, M.; Worrell, B.T.; Yu, J.-Q. Pd0/PR3-Catalyzed Arylation of Nicotinic and Isonicotinic Acid Derivatives. Angew. Chem. Int. Ed. 2010, 49, 1275–1277. [Google Scholar] [CrossRef] [Green Version]
- Netherton, M.R.; Fu, G.C. Air-Stable Trialkylphosphonium Salts: Simple, Practical, and Versatile Replacements for Air-Sensitive Trialkylphosphines. Applications in Stoichiometric and Catalytic Processes. Org. Lett. 2001, 3, 4295–4298. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Gao, G.-L.; Edmunds, A.J.F.; Worthington, P.A.; Morris, J.A.; Yu, J.-Q. Ligand-Promoted C3-Selective Arylation of Pyridines with Pd Catalysts: Gram-Scale Synthesis of (±)-Preclamol. J. Am. Chem. Soc. 2011, 133, 19090–19093. [Google Scholar] [CrossRef]
- Ye, M.; Gao, G.-L.; Yu, J.-Q. Ligand-Promoted C-3 Selective C-H Olefination of Pyridines with Pd Catalysts. J. Am. Chem. Soc. 2011, 133, 6964–6967. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Joo, J.M.; Rakshit, S.; Sames, D. C-H Arylation of Pyridines: High Regioselectivity as a Consequence of the Electronic Character of C-H Bonds and Heteroarene Ring. J. Am. Chem. Soc. 2011, 133, 16338–16341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, V.K.; Pawar, G.G.; Das, R.; Adhikary, A.; Kapur, M. Heteroatom-Guided, Palladium-Catalyzed Regioselective C-H Functionalization in the Synthesis of 3-Arylquinolines. Org. Lett. 2013, 15, 3310–3313. [Google Scholar] [CrossRef]
- Shang, Y.; Jie, X.; Zhao, H.; Hu, P.; Su, W. Rh(III)-Catalyzed Amide-Directed Cross-Dehydrogenative Heteroarylation of Pyridines. Org. Lett. 2014, 16, 416–419. [Google Scholar] [CrossRef]
- He, Y.; Wu, Z.; Ma, C.; Zhou, X.; Liu, X.; Wang, X.; Huang, G. Palladium-Catalyzed Selective C-H Activation: A Simple Method to Synthesize C-3 Site Arylated Quinoline Derivatives. Adv. Synth. Catal. 2016, 358, 375–379. [Google Scholar] [CrossRef]
- Zhou, J.; Li, B.; Hu, F.; Shi, B.-F. Rhodium(III)-Catalyzed Oxidative Olefination of Pyridines and Quinolines: Multigram-Scale Synthesis of Naphthyridinones. Org. Lett. 2013, 15, 3460–3463. [Google Scholar] [CrossRef]
- Li, B.-J.; Shi, Z.-J. Ir-Catalyzed Highly Selective Addition of Pyridyl C-H Bonds to Aldehydes Promoted by Triethylsilane. Chem. Sci. 2011, 2, 488–493. [Google Scholar] [CrossRef]
- Shang, M.; Sun, S.-Z.; Wang, H.-L.; Laforteza, B.N.; Dai, H.-X.; Yu, J.-Q. Exceedingly Fast Copper(II)-Promoted Ortho C-H Trifluoromethylation of Arenes Using TMSCF3. Angew. Chem. 2014, 126, 10607–10610. [Google Scholar] [CrossRef]
- Shang, M.; Wang, H.-L.; Sun, S.-Z.; Dai, H.-X.; Yu, J.-Q. Cu(II)-Mediated Ortho C-H Alkynylation of (Hetero)Arenes with Terminal Alkynes. J. Am. Chem. Soc. 2014, 136, 11590–11593. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Sato, K.; Hartwig, J.F.; Ishiyama, T.; Miyaura, N. Iridium-Catalyzed C-H Coupling Reaction of Heteroaromatic Compounds with Bis(Pinacolato)Diboron: Regioselective Synthesis of Heteroarylboronates. Tetrahedron Lett. 2002, 43, 5649–5651. [Google Scholar] [CrossRef]
- Harrisson, P.; Morris, J.; Marder, T.B.; Steel, P.G. Microwave-Accelerated Iridium-Catalyzed Borylation of Aromatic C-H Bonds. Org. Lett. 2009, 11, 3586–3589. [Google Scholar] [CrossRef] [PubMed]
- Tajuddin, H.; Harrisson, P.; Bitterlich, B.; Collings, J.C.; Sim, N.; Batsanov, A.S.; Cheung, M.S.; Kawamorita, S.; Maxwell, A.C.; Shukla, L.; et al. Iridium-Catalyzed C-H Borylation of Quinolines and Unsymmetrical 1,2-Disubstituted Benzenes: Insights into Steric and Electronic Effects on Selectivity. Chem. Sci. 2012, 3, 3505–3515. [Google Scholar] [CrossRef]
- Cheng, C.; Hartwig, J.F. Iridium-Catalyzed Silylation of Aryl C-H Bonds. J. Am. Chem. Soc. 2015, 137, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Hartwig, J.F. Rhodium-Catalyzed Intermolecular C-H Silylation of Arenes with High Steric Regiocontrol. Science 2014, 343, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.M.; Murphy, J.M.; Hapke, M.; Ishiyama, T.; Miyaura, N.; Hartwig, J.F. Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc. 2005, 127, 14263–14278. [Google Scholar] [CrossRef]
- Cheng, C.; Simmons, E.M.; Hartwig, J.F. Iridium-Catalyzed, Diastereoselective Dehydrogenative Silylation of Terminal Alkenes with (TMSO)2MeSiH. Angew. Chem. Int. Ed. 2013, 52, 8984–8989. [Google Scholar] [CrossRef] [PubMed]
- Hostyn, S.; Maes, B.U.W.; Pieters, L.; Lemière, G.L.F.; Mátyus, P.; Hajós, G.; Dommisse, R.A. Synthesis of the Benzo-β-Carboline Isoneocryptolepine: The Missing Indoloquinoline Isomer in the Alkaloid Series Cryptolepine, Neocryptolepine and Isocryptolepine. Tetrahedron 2005, 61, 1571–1577. [Google Scholar] [CrossRef]
- Nakao, Y.; Yamada, Y.; Kashihara, N.; Hiyama, T. Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis. J. Am. Chem. Soc. 2010, 132, 13666–13668. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Shih, W.-C.; Fang, C.-H.; Li, C.-Y.; Ong, T.-G.; Yap, G.P.A. Bimetallic Nickel Aluminun Mediated Para-Selective Alkenylation of Pyridine: Direct Observation of H2,H1-Pyridine Ni(0)−Al(III) Intermediates Prior to C-H Bond Activation. J. Am. Chem. Soc. 2010, 132, 11887–11889. [Google Scholar] [CrossRef]
- Lee, W.-C.; Chen, C.-H.; Liu, C.-Y.; Yu, M.-S.; Lin, Y.-H.; Ong, T.-G. Nickel-Catalysed Para-CH Activation of Pyridine with Switchable Regioselective Hydroheteroarylation of Allylarenes. Chem. Commun. 2015, 51, 17104–17107. [Google Scholar] [CrossRef]
- Zhu, L.; Sheng, X.; Li, Y.; Lu, D.; Qiu, R.; Kambe, N. Nickel-Catalyzed Remote C4-H Arylation of 8-Aminoquinolines. Org. Lett. 2019, 21, 6785–6789. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-W.; Jiang, K.; Ding, W.; Yuan, Y.; Shuai, L.; Chen, Y.-C.; Wei, Y. Selective Remote C-H Sulfonylation of Aminoquinolines with Arylsulfonyl Chlorides via Copper Catalysis. Chem. Commun. 2015, 51, 16928–16931. [Google Scholar] [CrossRef]
- Zhang, Z.; Tanaka, K.; Yu, J.-Q. Remote Site-Selective C-H Activation Directed by a Catalytic Bifunctional Template. Nature 2017, 543, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Achar, T.K.; Biswas, J.P.; Porey, S.; Pal, T.; Ramakrishna, K.; Maiti, S.; Maiti, D. Palladium-Catalyzed Template Directed C-5 Selective Olefination of Thiazoles. J. Org. Chem. 2019, 84, 8315–8321. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, K.; Biswas, J.P.; Jana, S.; Achar, T.K.; Porey, S.; Maiti, D. Coordination Assisted Distal C-H Alkylation of Fused Heterocycles. Angew. Chem. Int. Ed. 2019, 58, 13808–13812. [Google Scholar] [CrossRef] [PubMed]
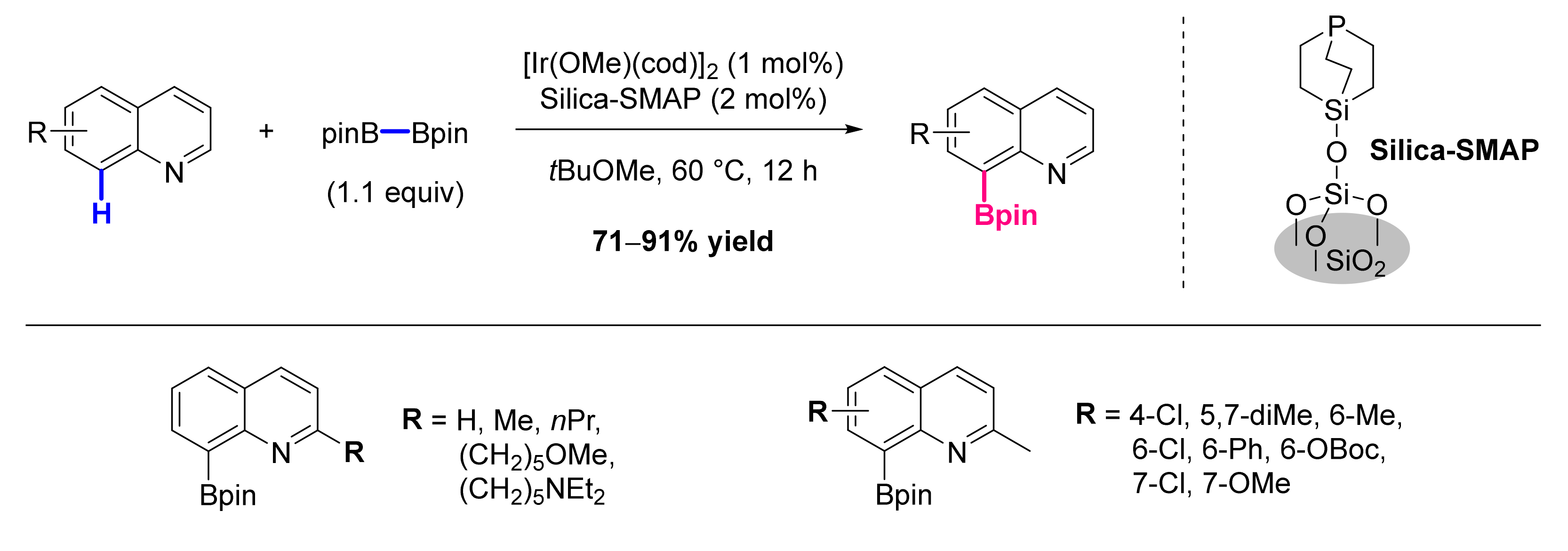
- Konishi, S.; Kawamorita, S.; Iwai, T.; Steel, P.G.; Marder, T.B.; Sawamura, M. Site-Selective C-H Borylation of Quinolines at the C8 Position Catalyzed by a Silica-Supported Phosphane-Iridium System. Chem. Asian J. 2014, 9, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Dong, G. Ortho vs Ipso: Site-Selective Pd and Norbornene-Catalyzed Arene C-H Amination Using Aryl Halides. J. Am. Chem. Soc. 2013, 135, 18350–18353. [Google Scholar] [CrossRef]
- Kwak, J.; Kim, M.; Chang, S. Rh(NHC)-Catalyzed Direct and Selective Arylation of Quinolines at the 8-Position. J. Am. Chem. Soc. 2011, 133, 3780–3783. [Google Scholar] [CrossRef]
- Shin, K.; Park, S.-W.; Chang, S. Cp*Ir(III)-Catalyzed Mild and Broad C-H Arylation of Arenes and Alkenes with Aryldiazonium Salts Leading to the External Oxidant-Free Approach. J. Am. Chem. Soc. 2015, 137, 8584–8592. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.E.; Lakey-Beitia, J.; Atesin, A.C.; Ateşin, T.A.; Chavez, G.; Arman, H.D.; Larionov, O.V. Palladium-Catalyzed C8-Selective C-H Arylation of Quinoline N-Oxides: Insights into the Electronic, Steric and Solvation Effects on the Site-Selectivity by Mechanistic and DFT Computational Studies. ACS Catal. 2015, 5, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
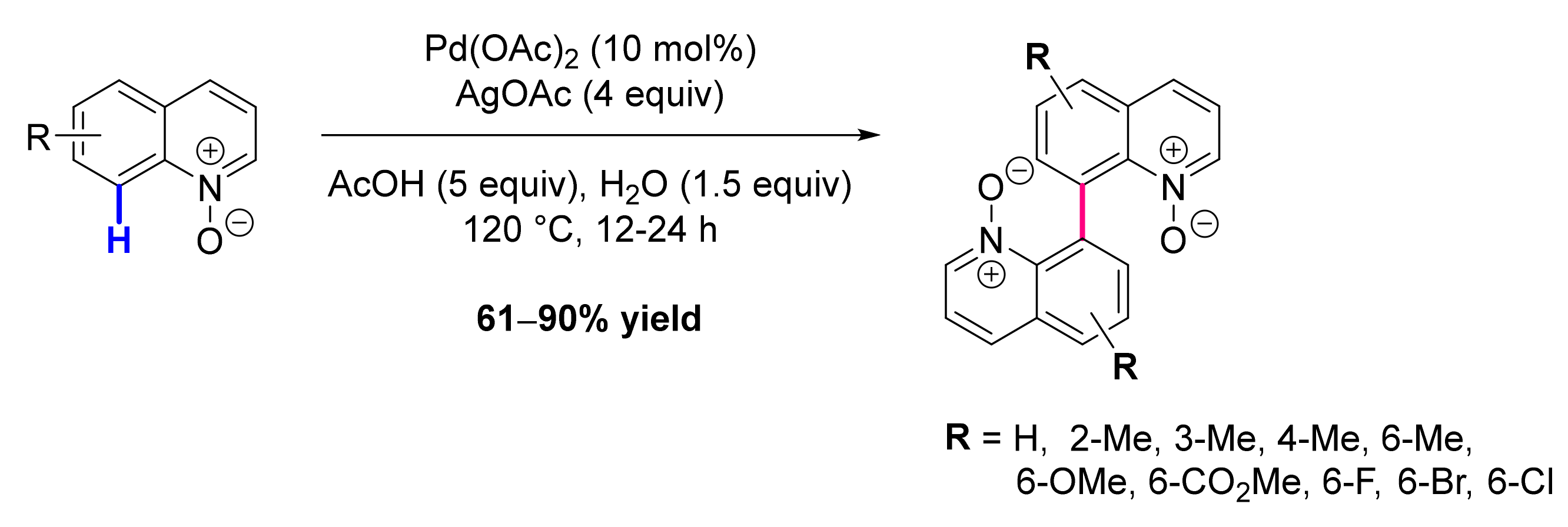
- Stephens, D.E.; Lakey-Beitia, J.; Chavez, G.; Ilie, C.; Arman, H.D.; Larionov, O.V. Experimental and Mechanistic Analysis of the Palladium-Catalyzed Oxidative C8-Selective C-H Homocoupling of Quinoline N-Oxides. Chem. Commun. 2015, 51, 9507–9510. [Google Scholar] [CrossRef] [Green Version]
- Fuchi, Y.; Sakuma, M.; Ohyama, K.; Hagihara, R.; Kohno, M.; Hamada, K.; Mizutani, A.; Karasawa, S. Selective Synthesis of Substituted Amino-Quinoline Derivatives by C-H Activation and Fluorescence Evaluation of Their Lipophilicity-Responsive Properties. Sci. Rep. 2019, 9, 17723. [Google Scholar] [CrossRef]
- Shibata, T.; Matsuo, Y. Directed C-H Alkenylation of Quinoline N-Oxides at the C-8 Position Using a Cationic Rhodium(I) Catalyst. Adv. Synth. Catal. 2014, 356, 1516–1520. [Google Scholar] [CrossRef]
- Jeong, J.; Patel, P.; Hwang, H.; Chang, S. Rhodium(III)-Catalyzed C-C Bond Formation of Quinoline N-Oxides at the C-8 Position under Mild Conditions. Org. Lett. 2014, 16, 4598–4601. [Google Scholar] [CrossRef]
- Sharma, R.; Kumar, R.; Kumar, I.; Sharma, U. RhIII-Catalyzed Dehydrogenative Coupling of Quinoline N-Oxides with Alkenes: N-Oxide as Traceless Directing Group for Remote C-H Activation. Eur. J. Org. Chem. 2015, 2015, 7519–7528. [Google Scholar] [CrossRef]
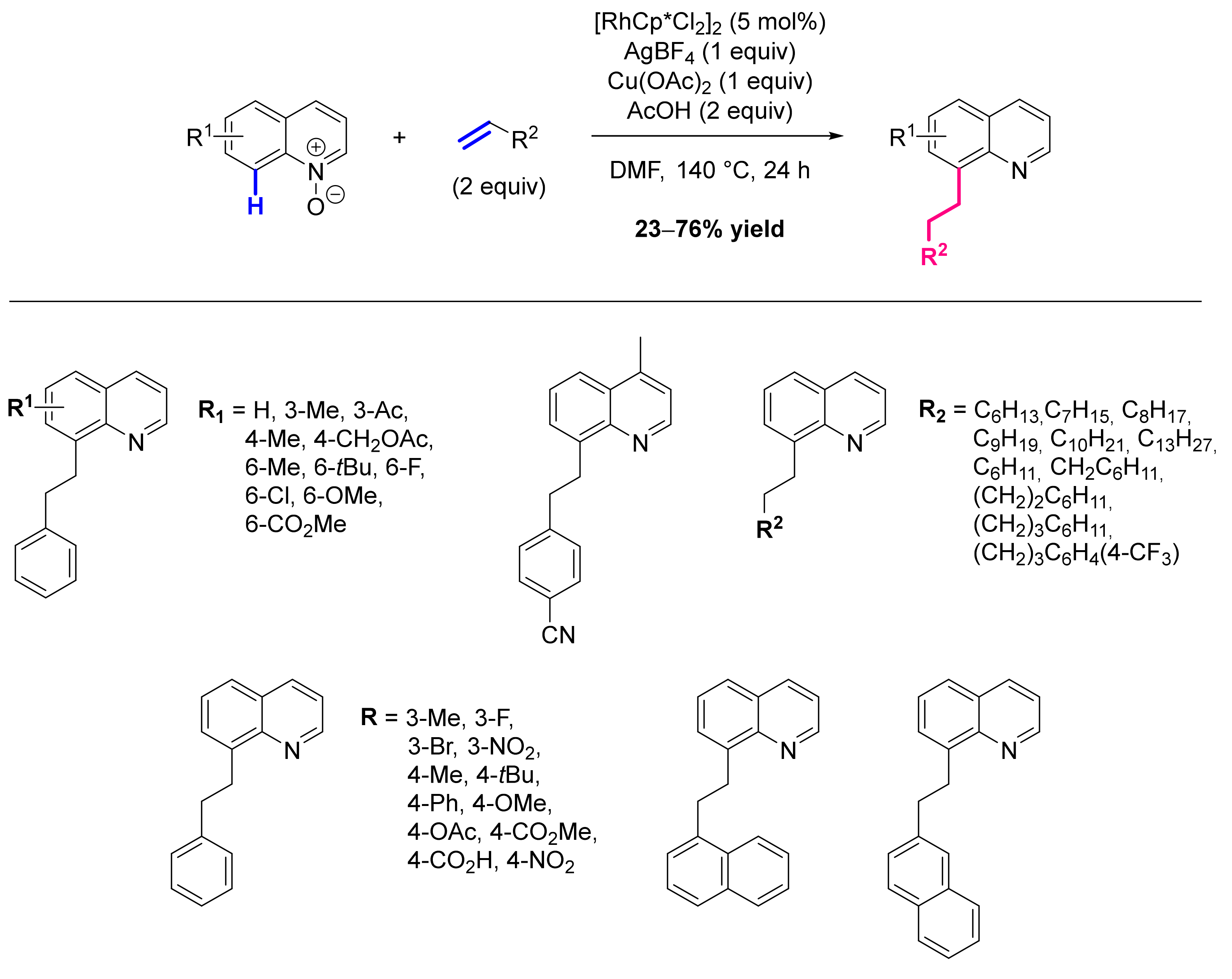
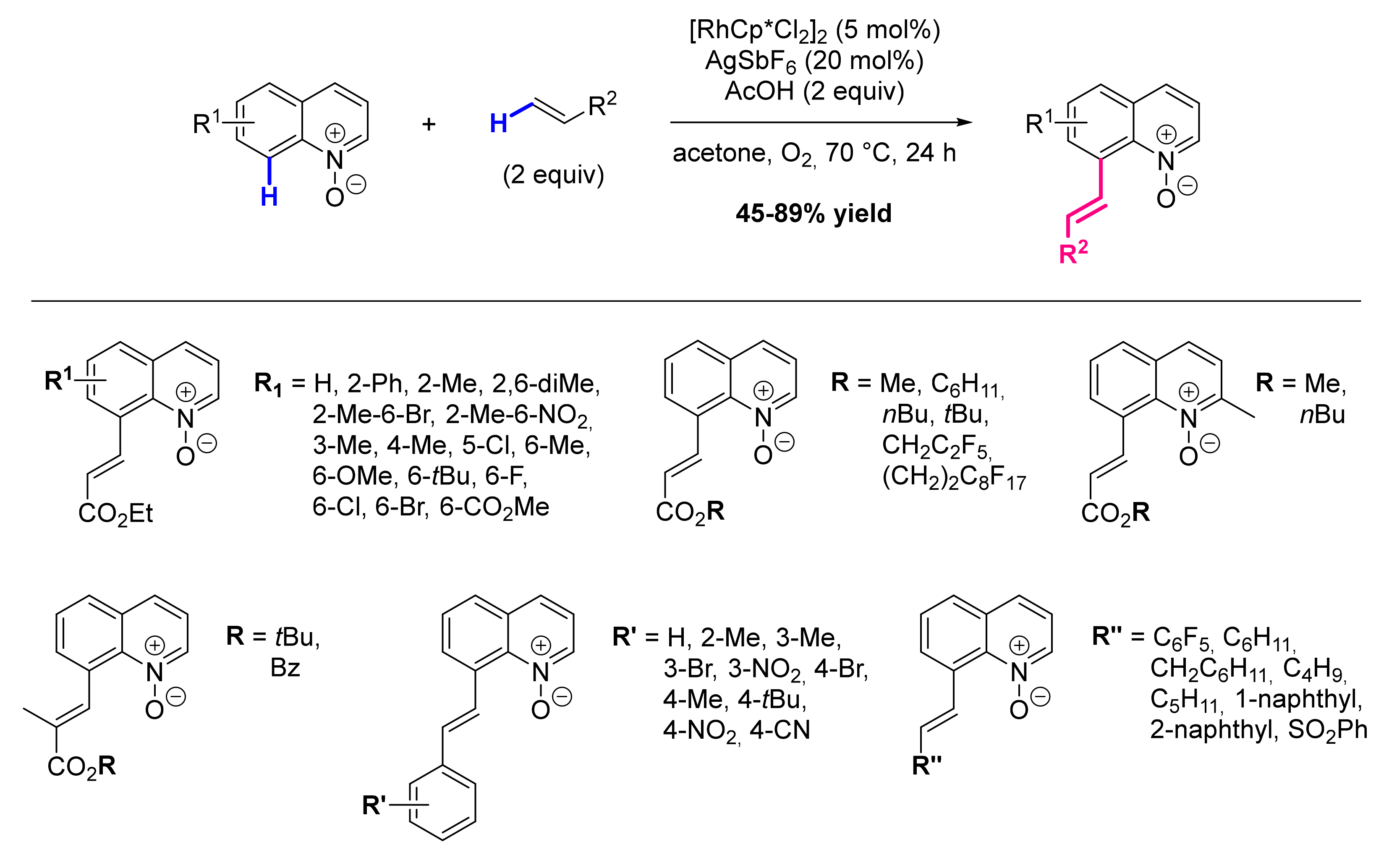
- Sharma, R.; Kumar, I.; Kumar, R.; Sharma, U. Rhodium-Catalyzed Remote C-8 Alkylation of Quinolines with Activated and Unactivated Olefins: Mechanistic Study and Total Synthesis of EP4 Agonist. Adv. Synth. Catal. 2017, 359, 3022–3028. [Google Scholar] [CrossRef]
- Sharma, R.; Kumar, R.; Sharma, U. Rh/O2-Catalyzed C8 Olefination of Quinoline N-Oxides with Activated and Unactivated Olefins. J. Org. Chem. 2019, 84, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
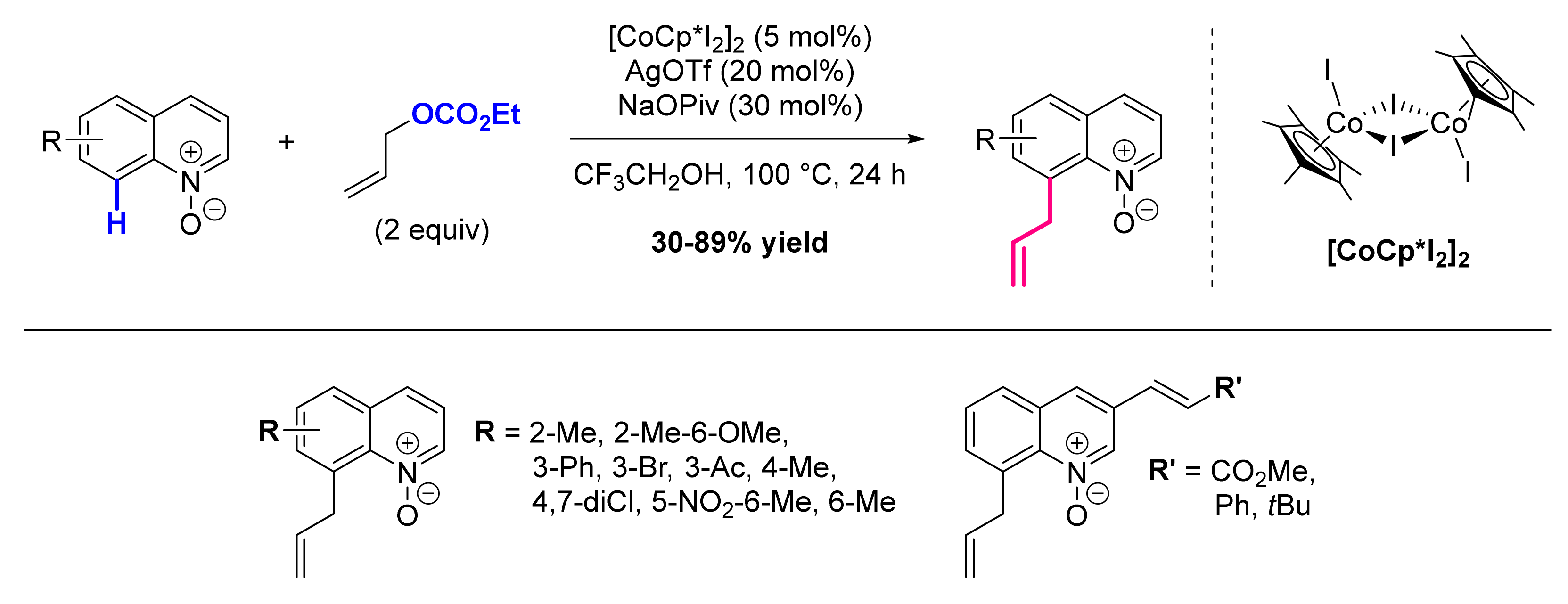
- Kalsi, D.; Laskar, R.A.; Barsu, N.; Premkumar, J.R.; Sundararaju, B. C-8-Selective Allylation of Quinoline: A Case Study of β-Hydride vs β-Hydroxy Elimination. Org. Lett. 2016, 18, 4198–4201. [Google Scholar] [CrossRef]
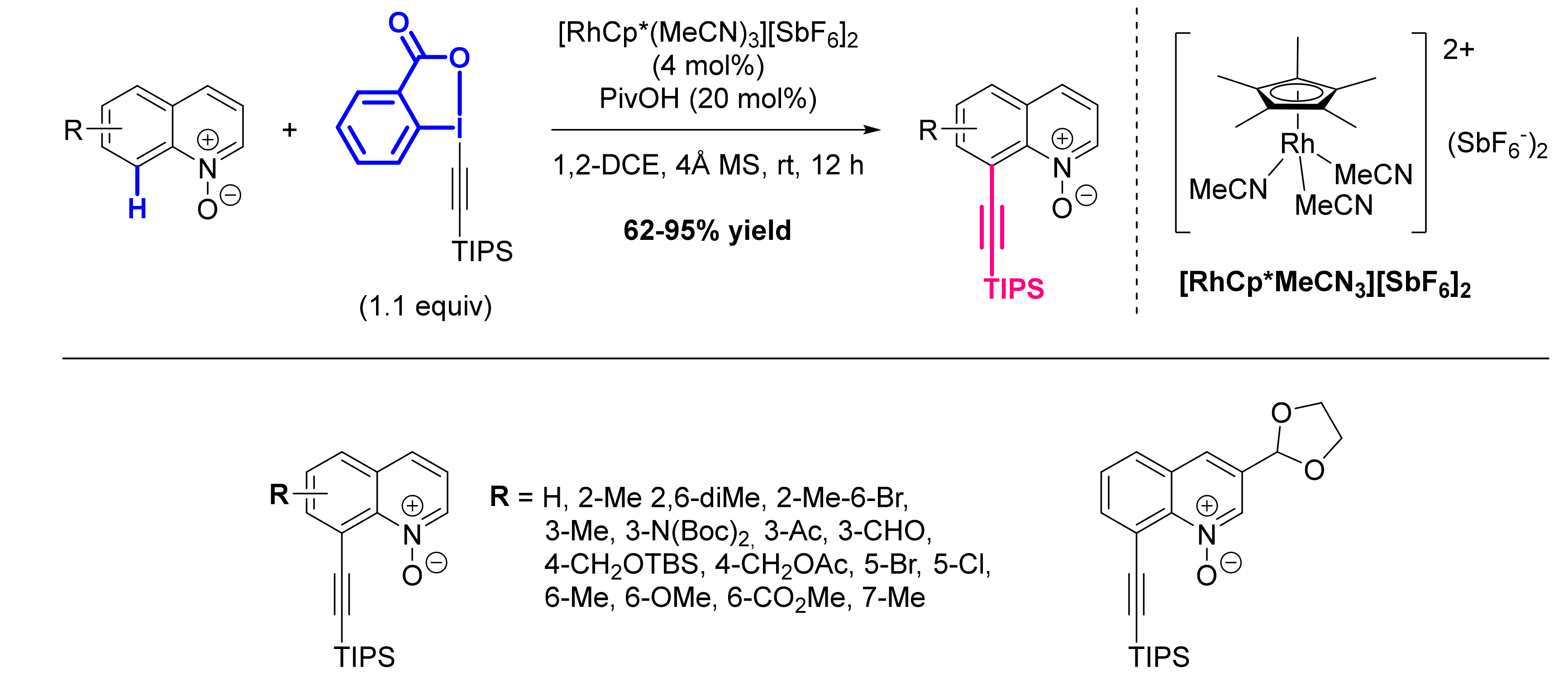
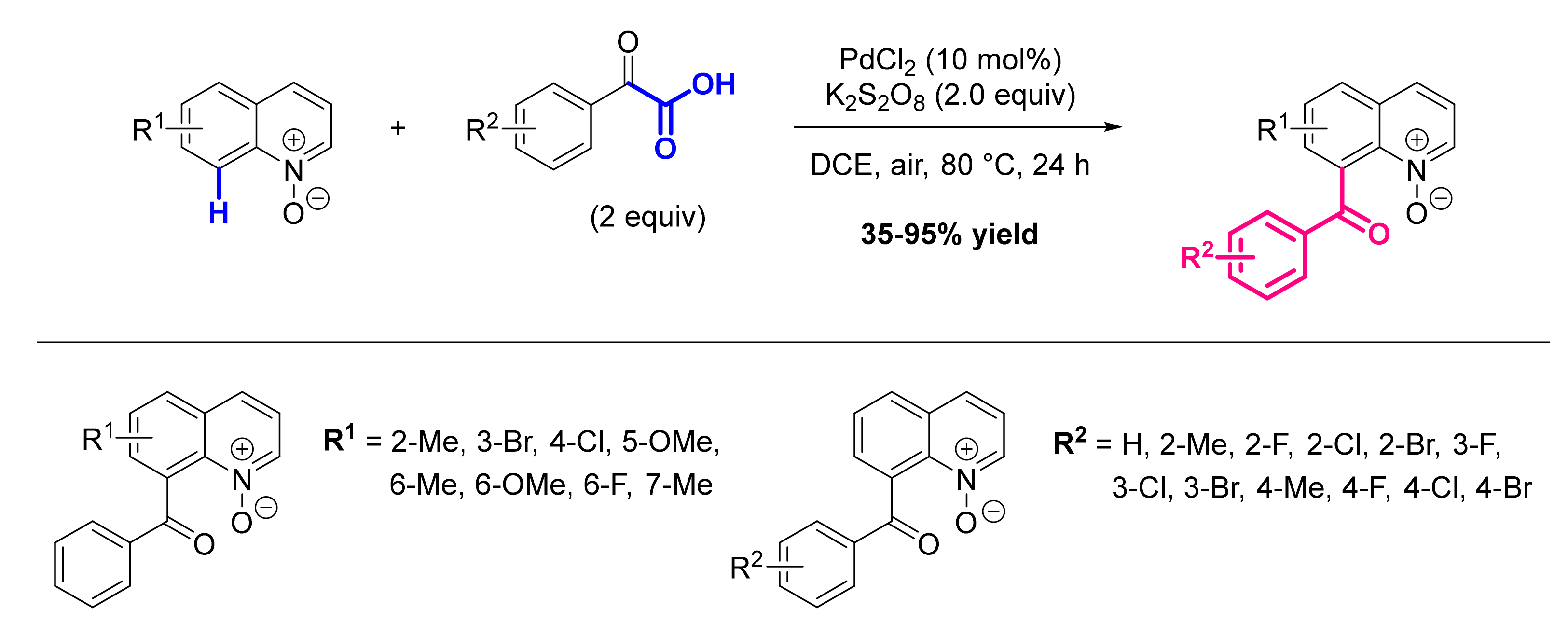
- Chen, X.; Cui, X.; Wu, Y. C8-Selective Acylation of Quinoline N-Oxides with α-Oxocarboxylic Acids via Palladium-Catalyzed Regioselective C-H Bond Activation. Org. Lett. 2016, 18, 3722–3725. [Google Scholar] [CrossRef]
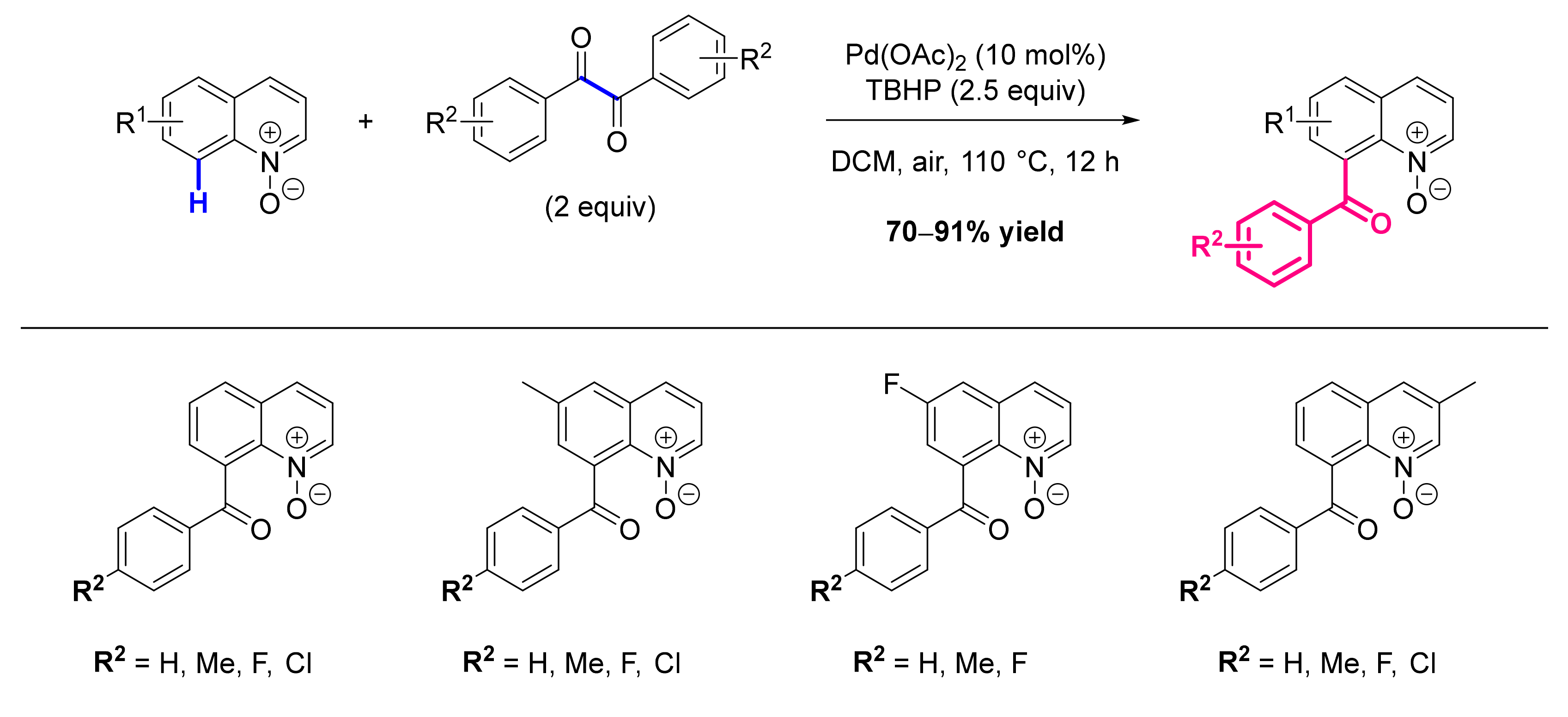
- Li, G.-H.; Dong, D.-Q.; Yu, X.-Y.; Wang, Z.-L. Direct Synthesis of 8-Acylated Quinoline N-Oxides via Palladium-Catalyzed Selective C-H Activation and C(Sp2)-C(Sp2) Cleavage. New J. Chem. 2019, 43, 1667–1670. [Google Scholar] [CrossRef]
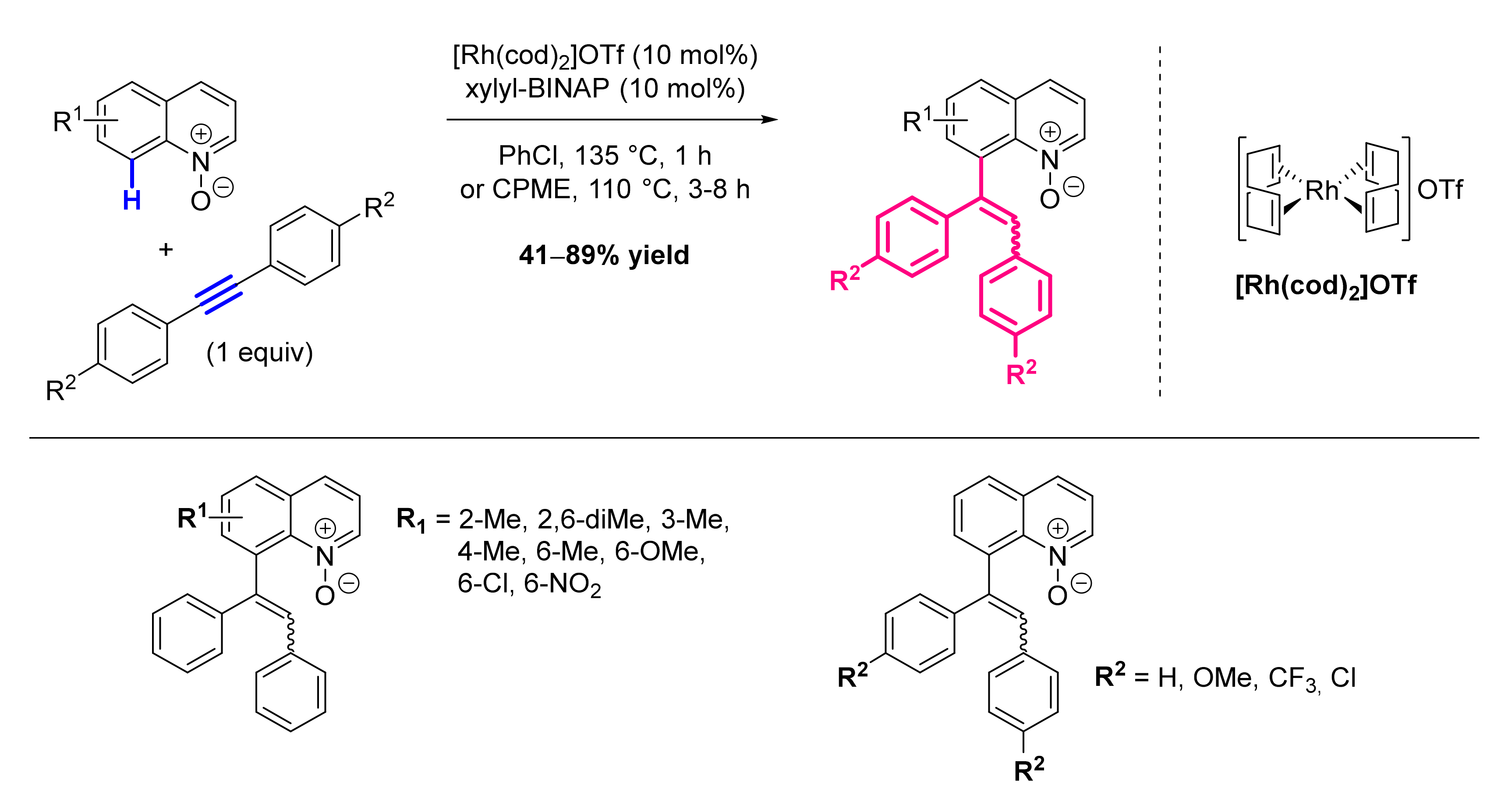
- Zhang, X.; Qi, Z.; Li, X. Rhodium(III)-Catalyzed C-C and C-O Coupling of Quinoline N-Oxides with Alkynes: Combination of C-H Activation with O-Atom Transfer. Angew. Chem. Int. Ed. Engl. 2014, 53, 10794–10798. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Qi, Z.; Qi, X.; Li, X.; Lan, Y. The Mechanism of N-O Bond Cleavage in Rhodium-Catalyzed C-H Bond Functionalization of Quinoline N-Oxides with Alkynes: A Computational Study. Chemistry 2015, 21, 10131–10137. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Park, Y.; Chang, S. Rh(III)-Catalyzed Traceless Coupling of Quinoline N-Oxides with Internal Diarylalkynes. J. Org. Chem. 2014, 79, 9899–9906. [Google Scholar] [CrossRef]
- Barsu, N.; Sen, M.; Premkumar, J.R.; Sundararaju, B. Cobalt(III) Catalyzed C-8 Selective C-H and C-O Coupling of Quinoline N-Oxide with Internal Alkynes via C-H Activation and Oxygen Atom Transfer. Chem. Commun. 2016, 52, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Kumar, R.; Kumar, R.; Upadhyay, P.; Sahal, D.; Sharma, U. Rh(III)-Catalyzed C(8)-H Functionalization of Quinolines via Simultaneous C-C and C-O Bond Formation: Direct Synthesis of Quinoline Derivatives with Antiplasmodial Potential. J. Org. Chem. 2018, 83, 12702–12710. [Google Scholar] [CrossRef] [PubMed]
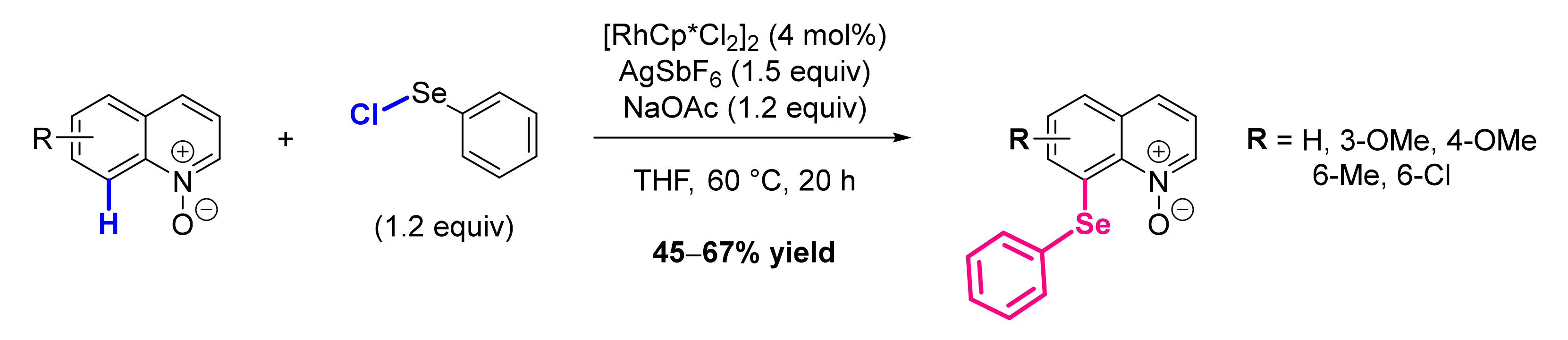
- Yu, S.; Wan, B.; Li, X. Rh(III)-Catalyzed Selenylation of Arenes with Selenenyl Chlorides/Diselenides via C-H Activation. Org. Lett. 2015, 17, 58–61. [Google Scholar] [CrossRef] [PubMed]
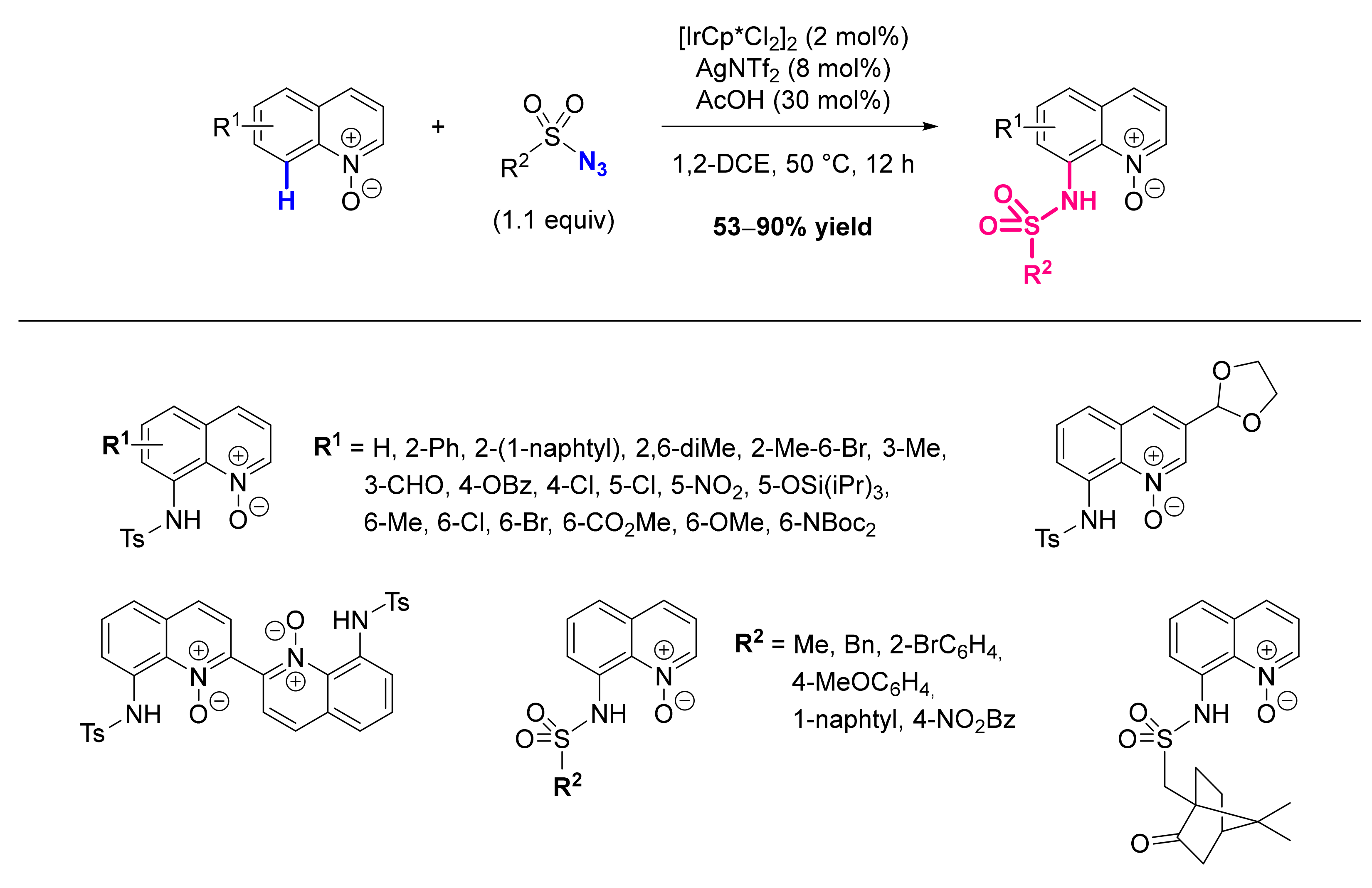
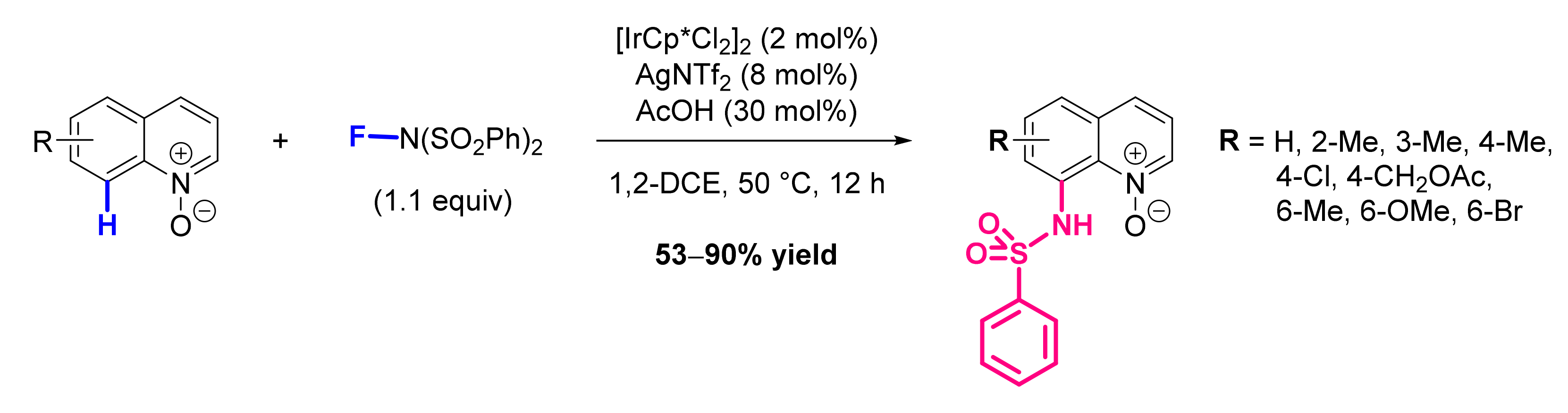
- Hwang, H.; Kim, J.; Jeong, J.; Chang, S. Regioselective Introduction of Heteroatoms at the C-8 Position of Quinoline N-Oxides: Remote C-H Activation Using N-Oxide as a Stepping Stone. J. Am. Chem. Soc. 2014, 136, 10770–10776. [Google Scholar] [CrossRef]
- Dhiman, A.K.; Gupta, S.S.; Sharma, R.; Kumar, R.; Sharma, U. Rh(III)-Catalyzed C(8)-H Activation of Quinoline N-Oxides: Regioselective C-Br and C-N Bond Formation. J. Org. Chem. 2019, 84, 12871–12880. [Google Scholar] [CrossRef] [PubMed]










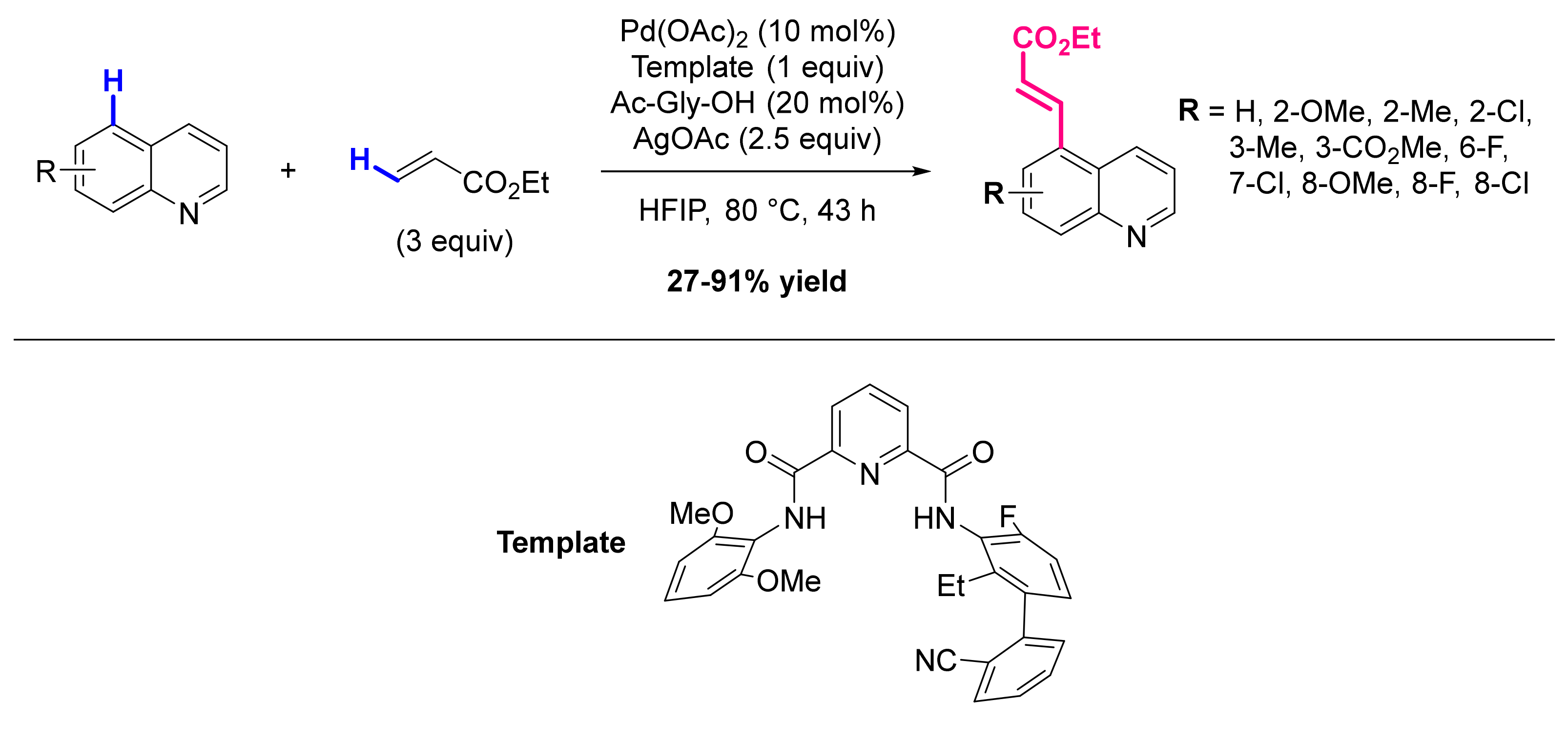
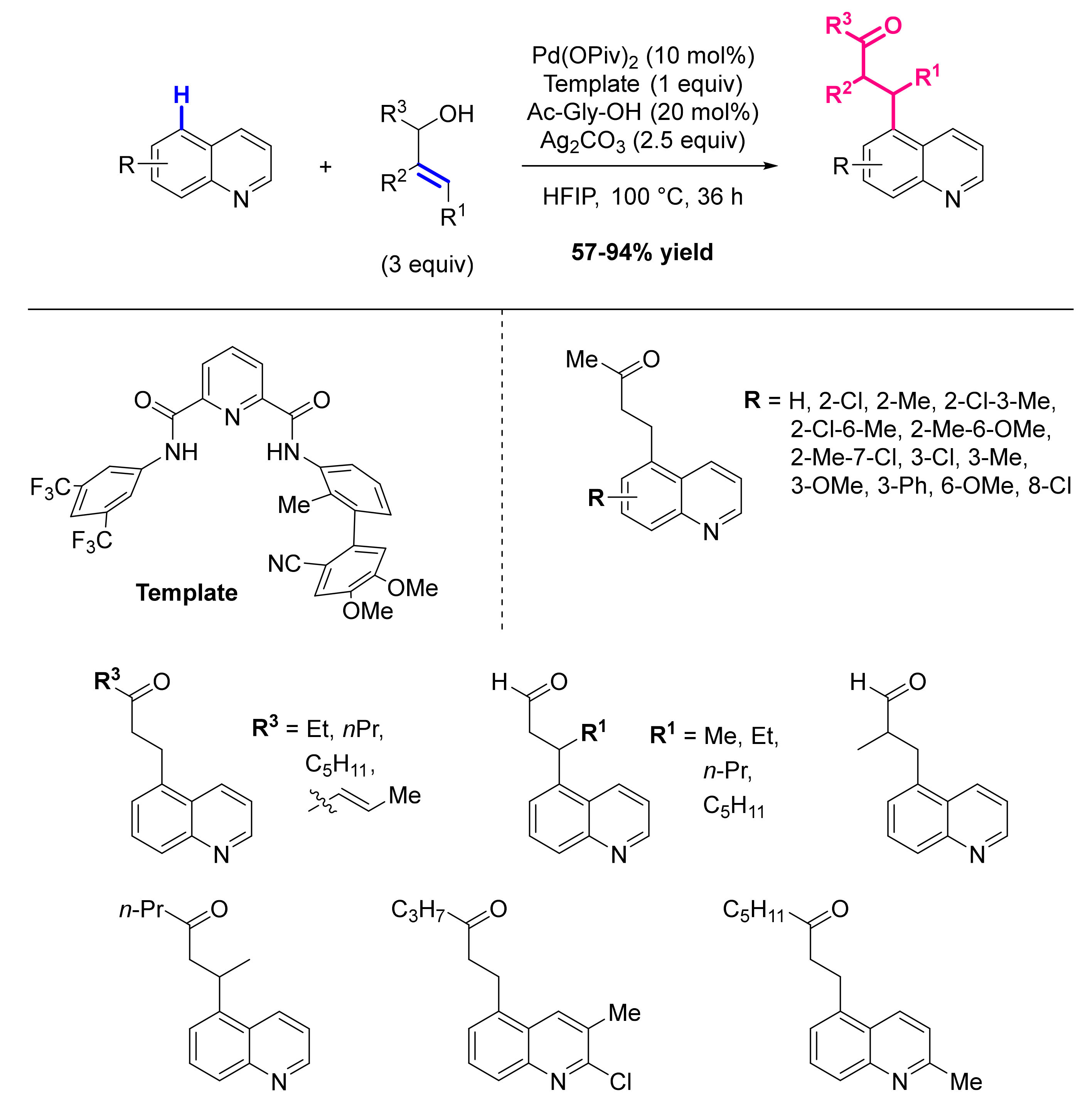
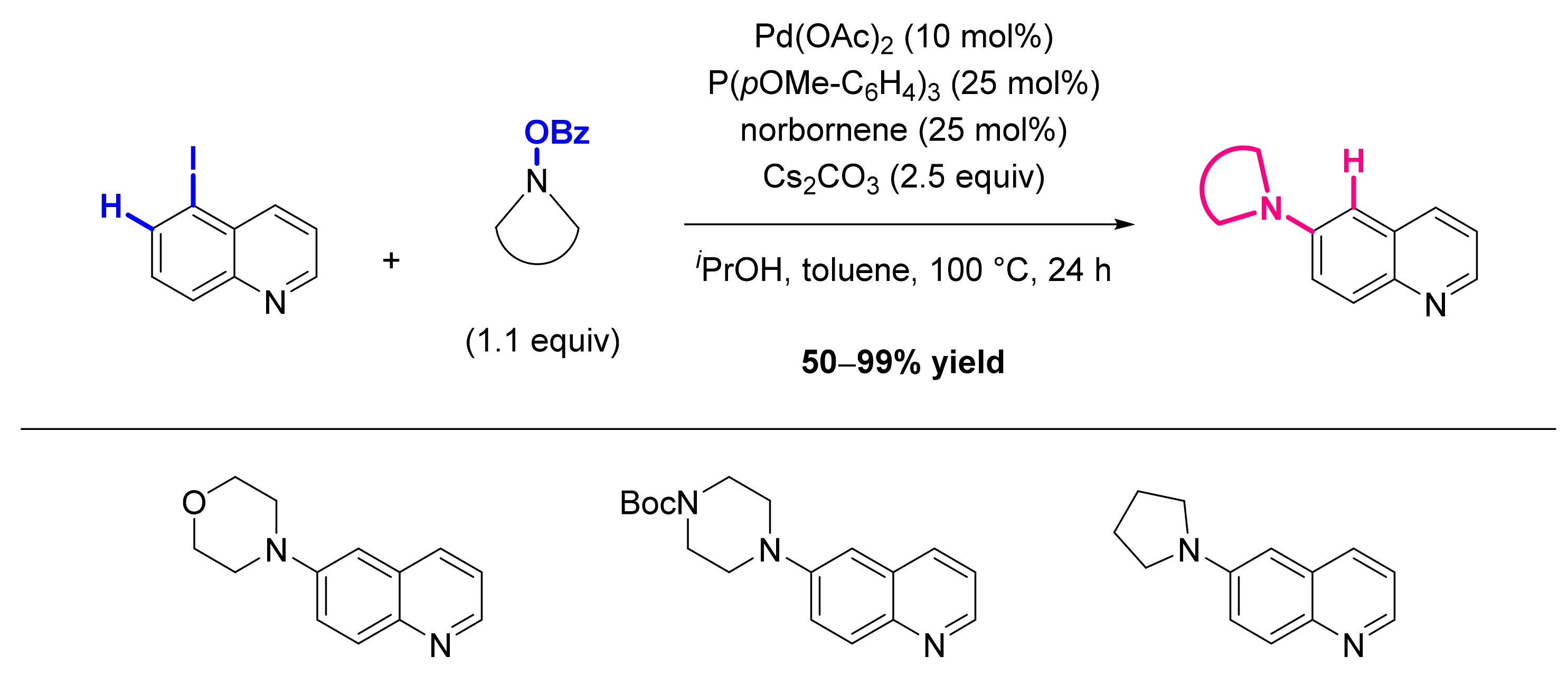
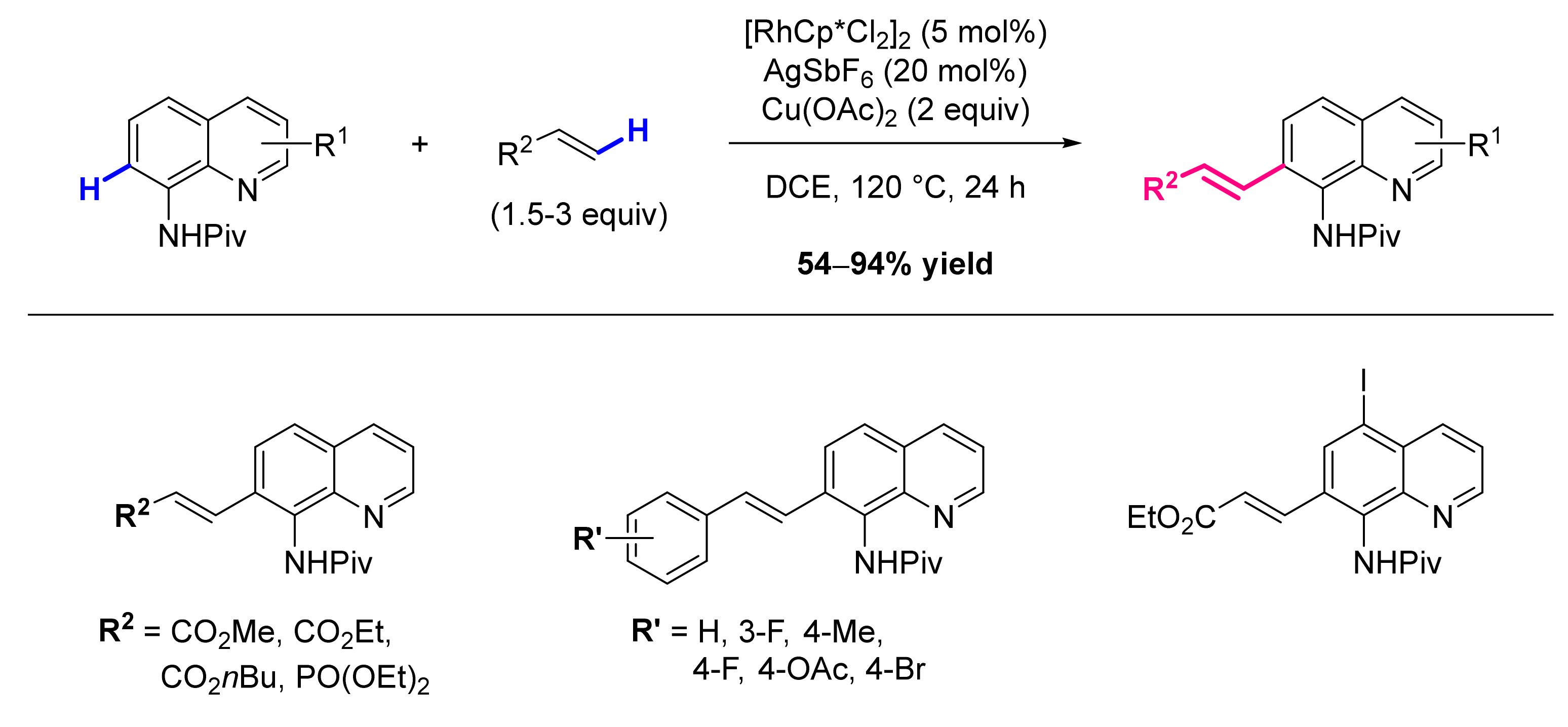









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corio, A.; Gravier-Pelletier, C.; Busca, P. Regioselective Functionalization of Quinolines through C-H Activation: A Comprehensive Review. Molecules 2021, 26, 5467. https://doi.org/10.3390/molecules26185467
Corio A, Gravier-Pelletier C, Busca P. Regioselective Functionalization of Quinolines through C-H Activation: A Comprehensive Review. Molecules. 2021; 26(18):5467. https://doi.org/10.3390/molecules26185467
Chicago/Turabian StyleCorio, Alessandra, Christine Gravier-Pelletier, and Patricia Busca. 2021. "Regioselective Functionalization of Quinolines through C-H Activation: A Comprehensive Review" Molecules 26, no. 18: 5467. https://doi.org/10.3390/molecules26185467
APA StyleCorio, A., Gravier-Pelletier, C., & Busca, P. (2021). Regioselective Functionalization of Quinolines through C-H Activation: A Comprehensive Review. Molecules, 26(18), 5467. https://doi.org/10.3390/molecules26185467