
Cashew Nut Shell Liquid (CNSL) as a Source of Drugs for Alzheimer’s Disease

 , , and
, , and
Abstract
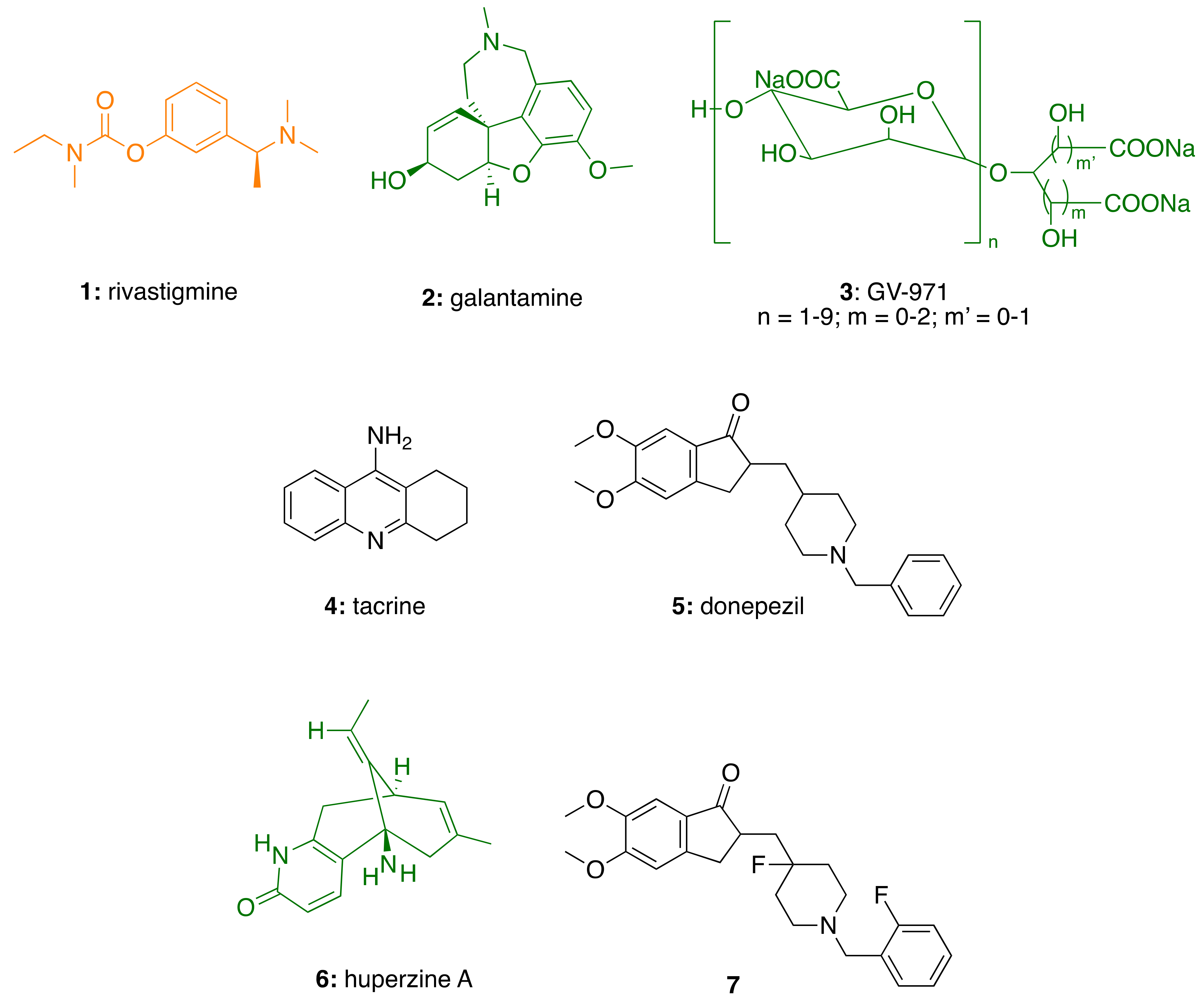
:1. Introduction
2. Relevant Alzheimer’s Disease Pathways and Their Potential as Drug Targets
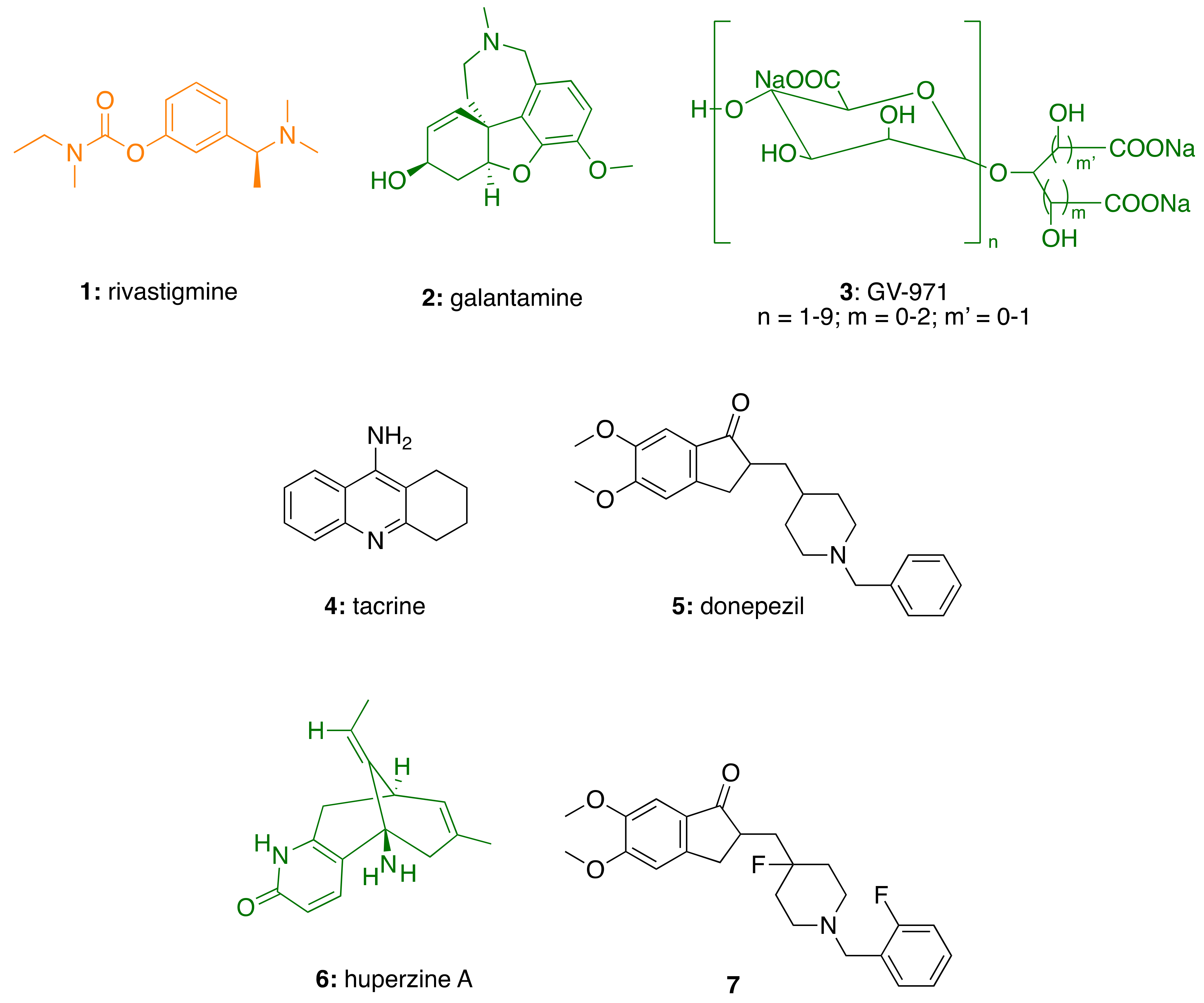
2.1. AChE in Alzheimer’s Disease
2.2. HDAC in Alzheimer’s Disease
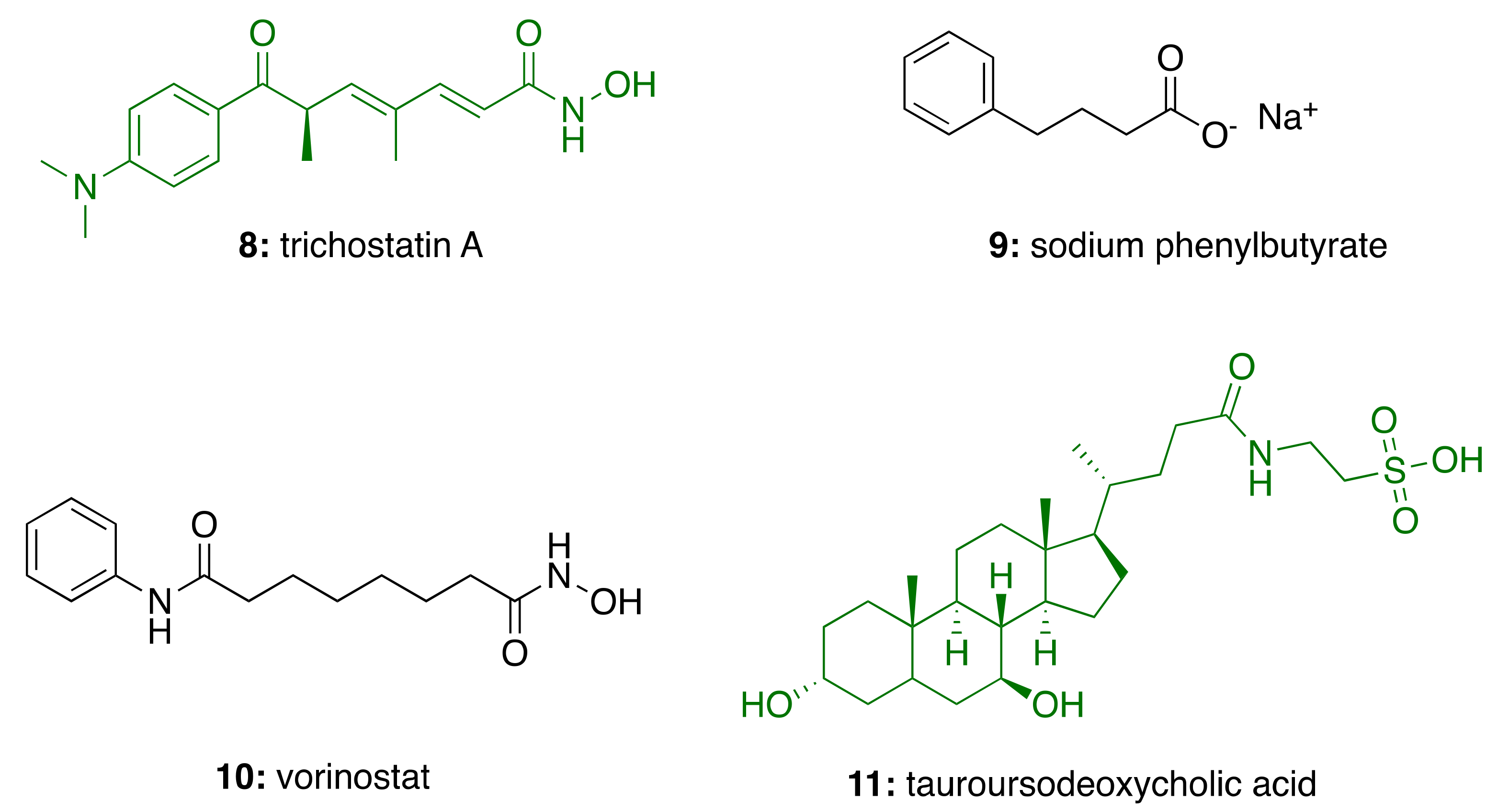
2.3. Neuroinflammation in Alzheimer’s Disease
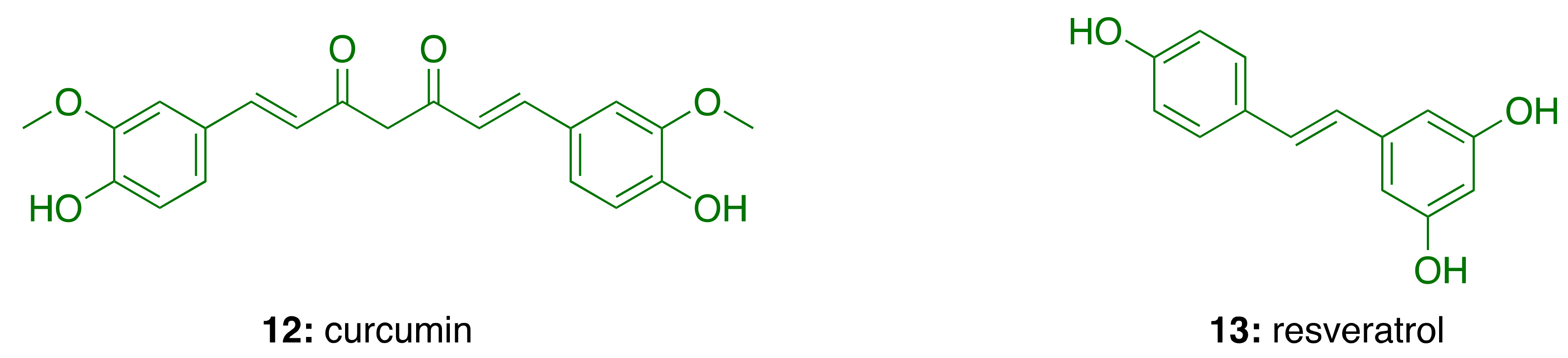
3. Cashew Nut Shell Liquid (CNSL) Components for the Discovery of Anti-AD Lead Candidates
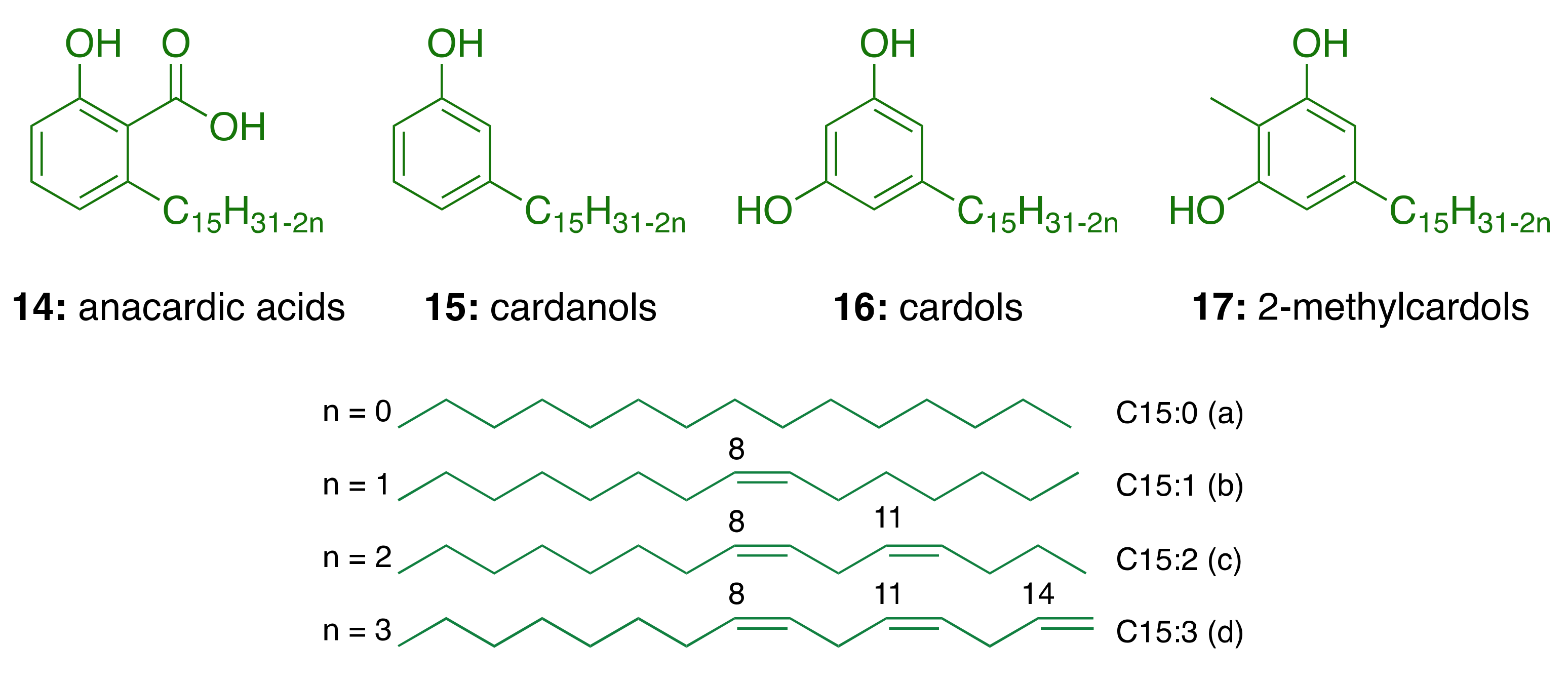
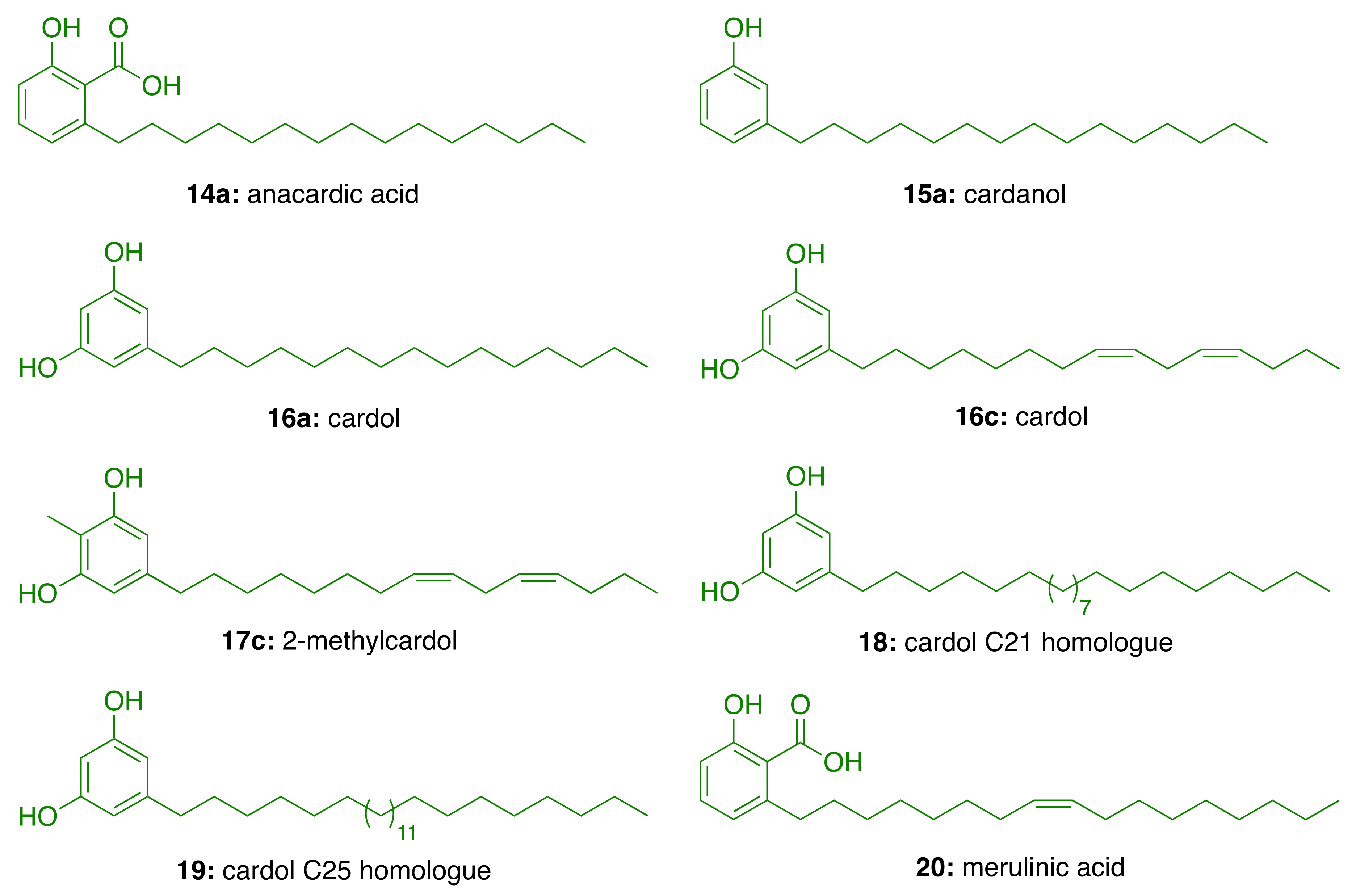
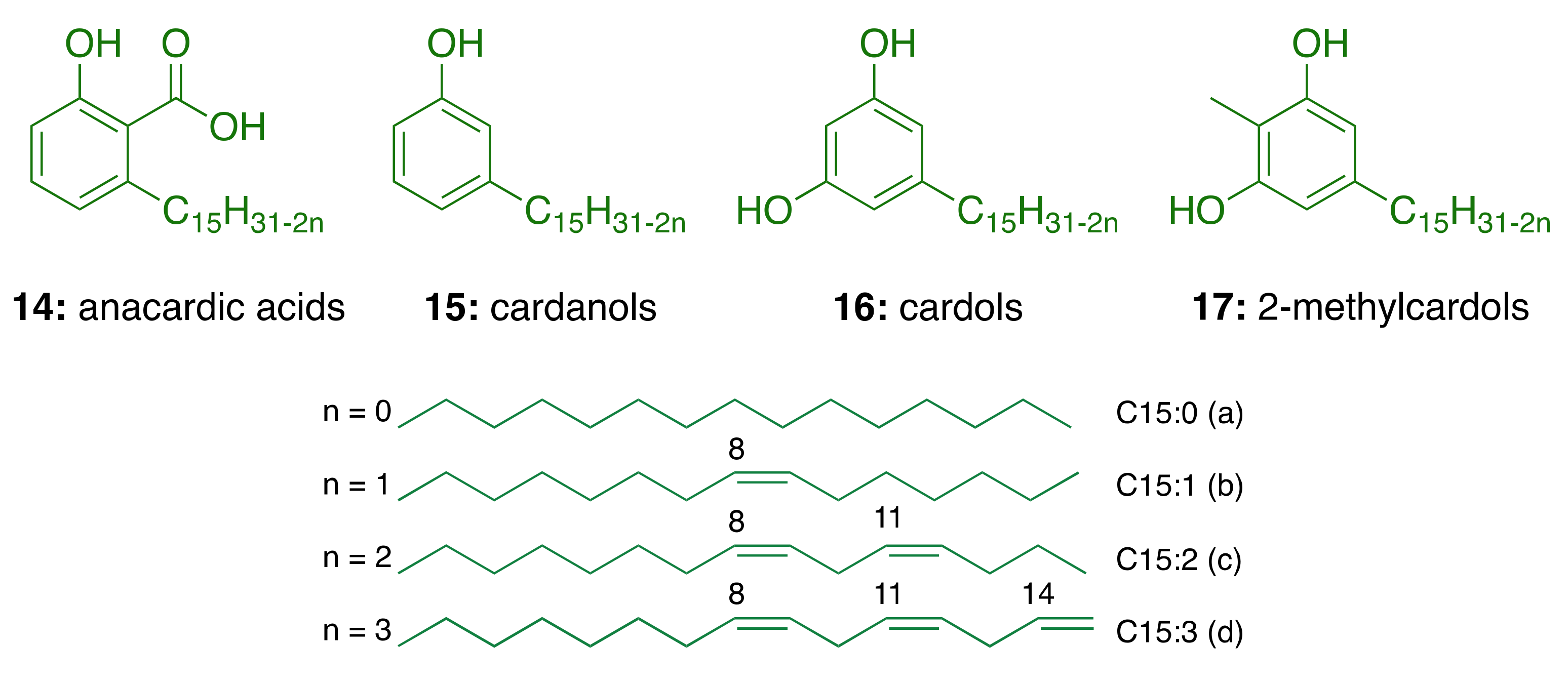
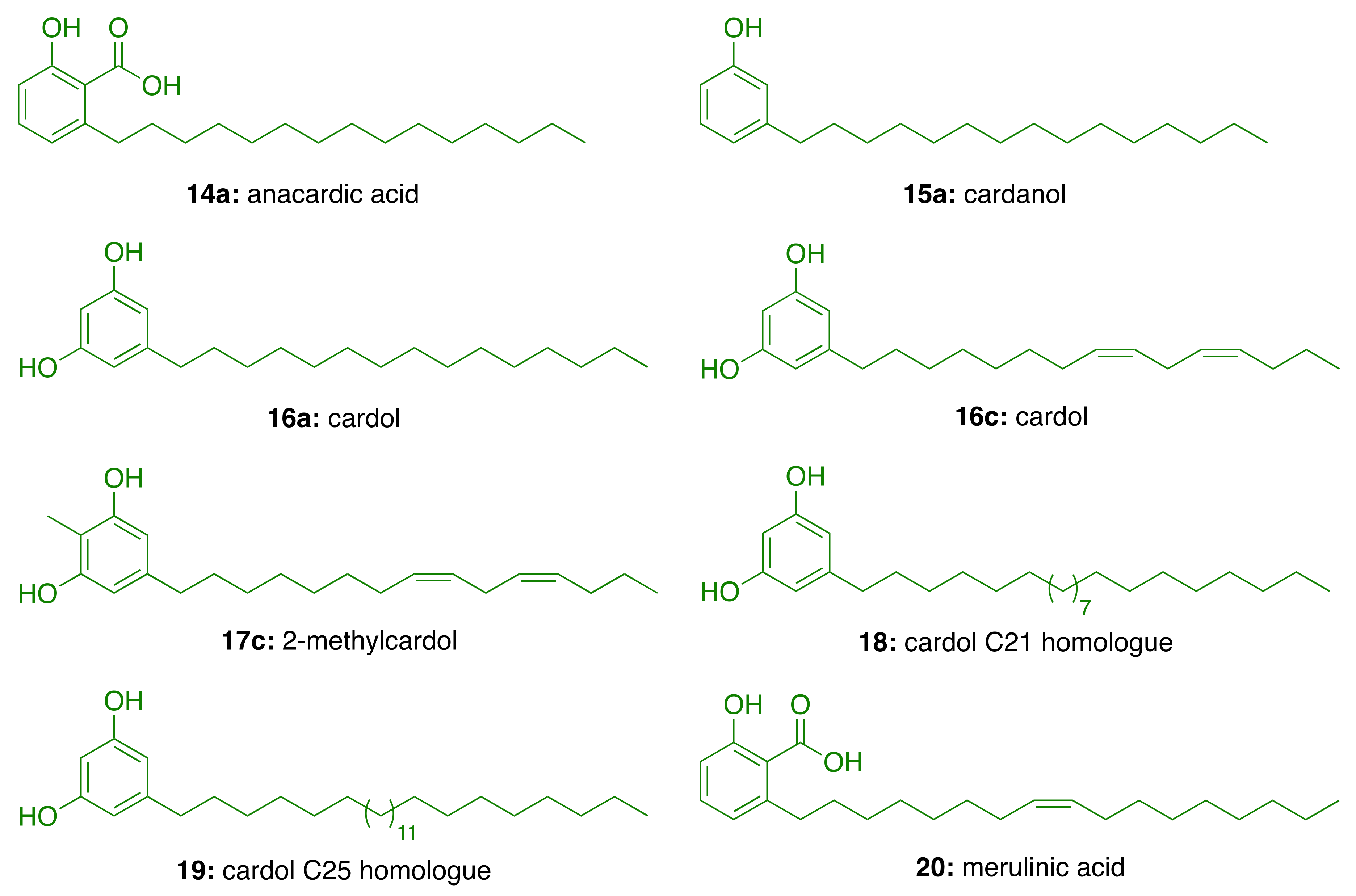
3.1. CNSL Phenolic Lipids: The Raw Material
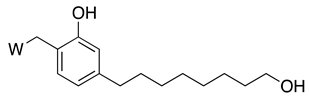
3.2. Phenolic Lipids as AChE Inhibitors
3.3. CNSL-Derived AChE Inhibitors
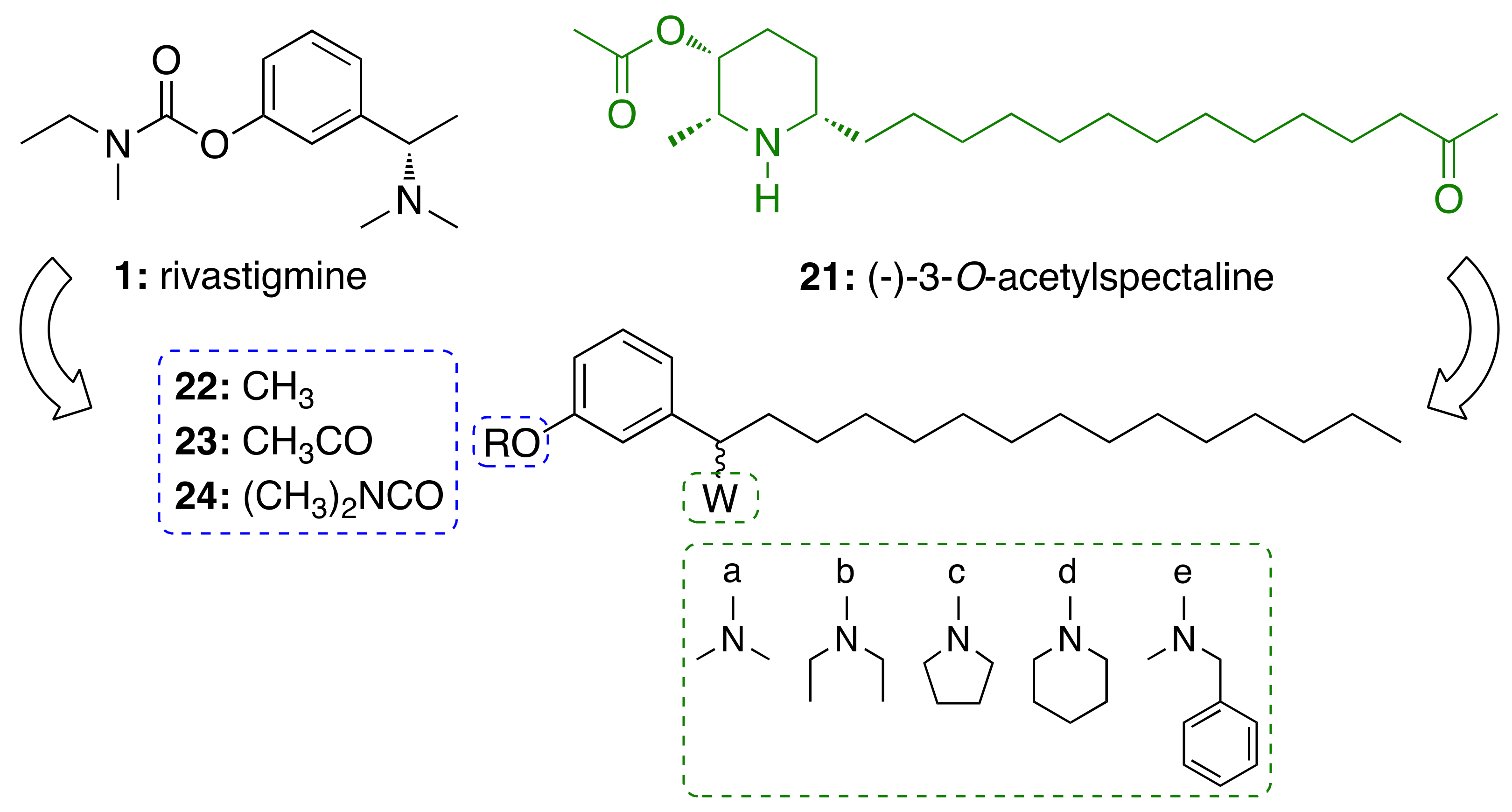
3.3.1. CNSL-Derived Rivastigmine Analogues
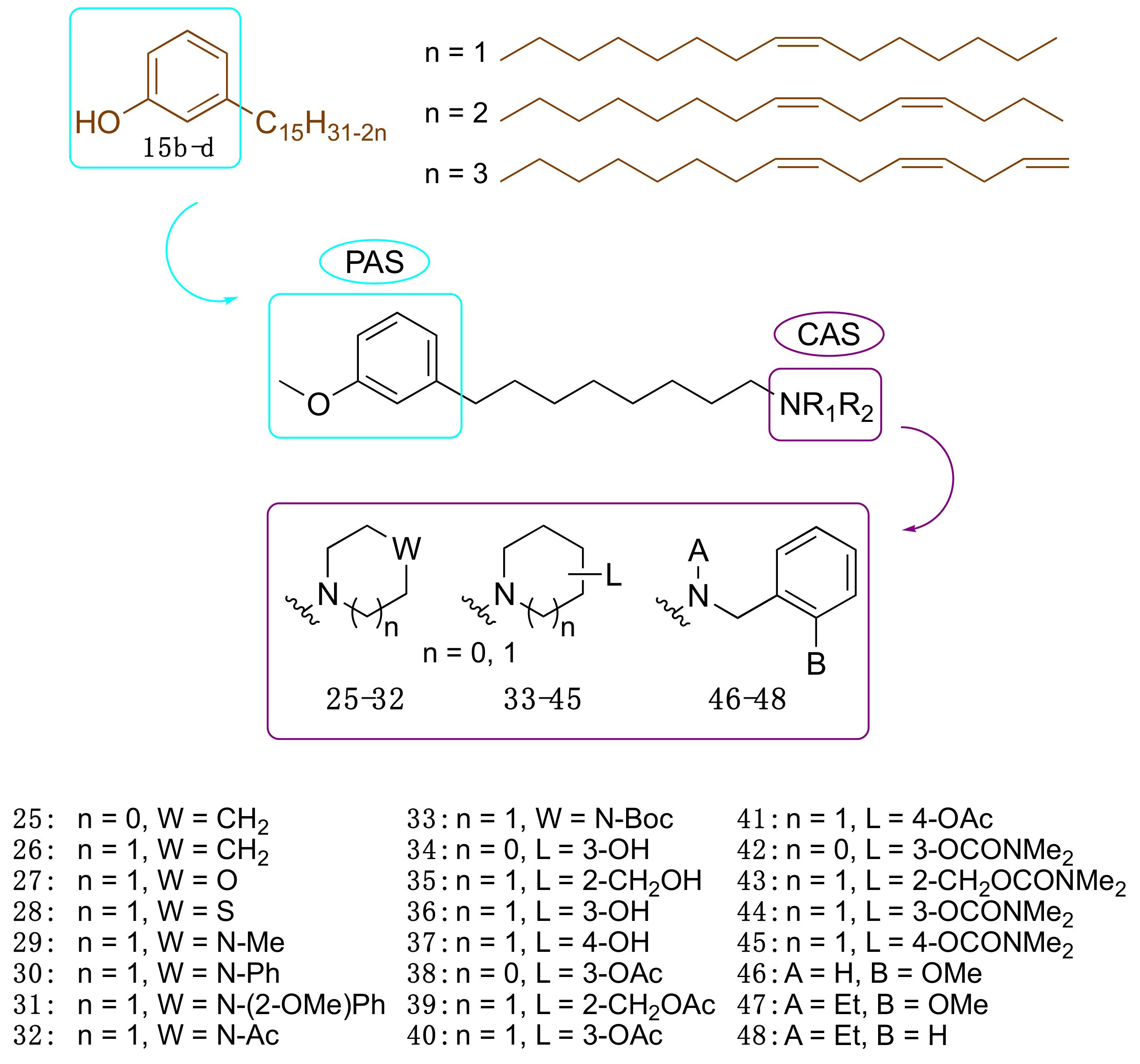
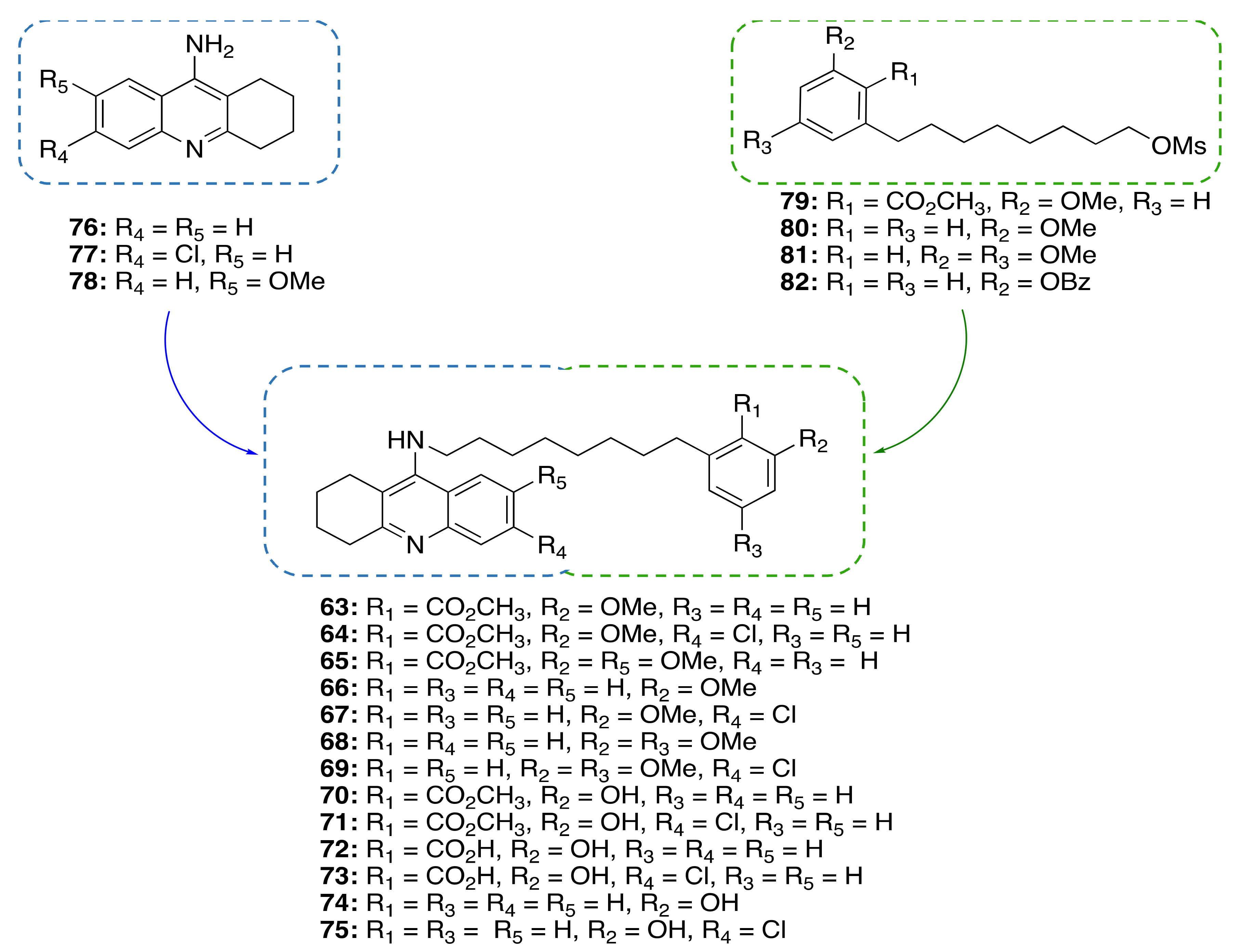
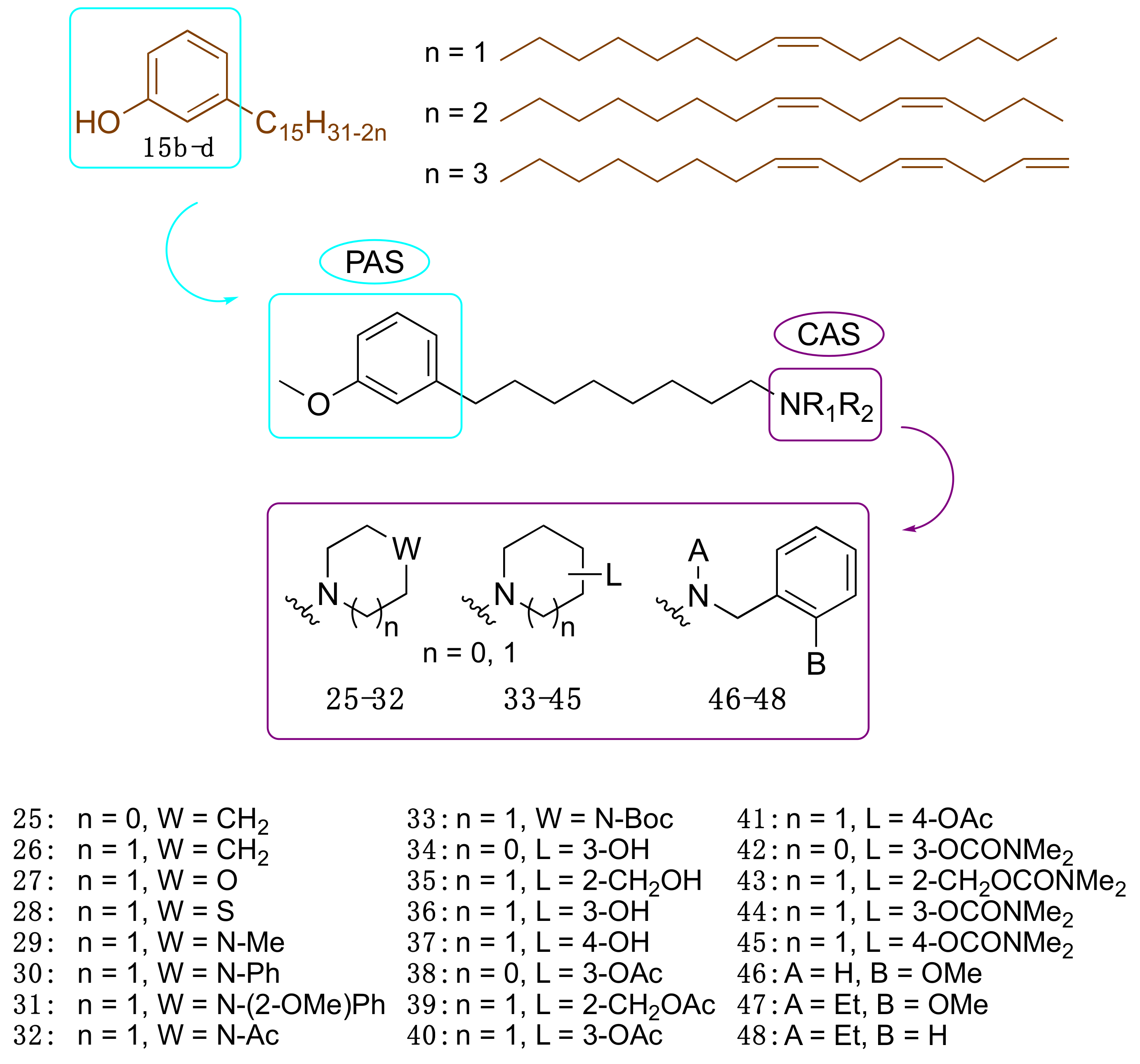
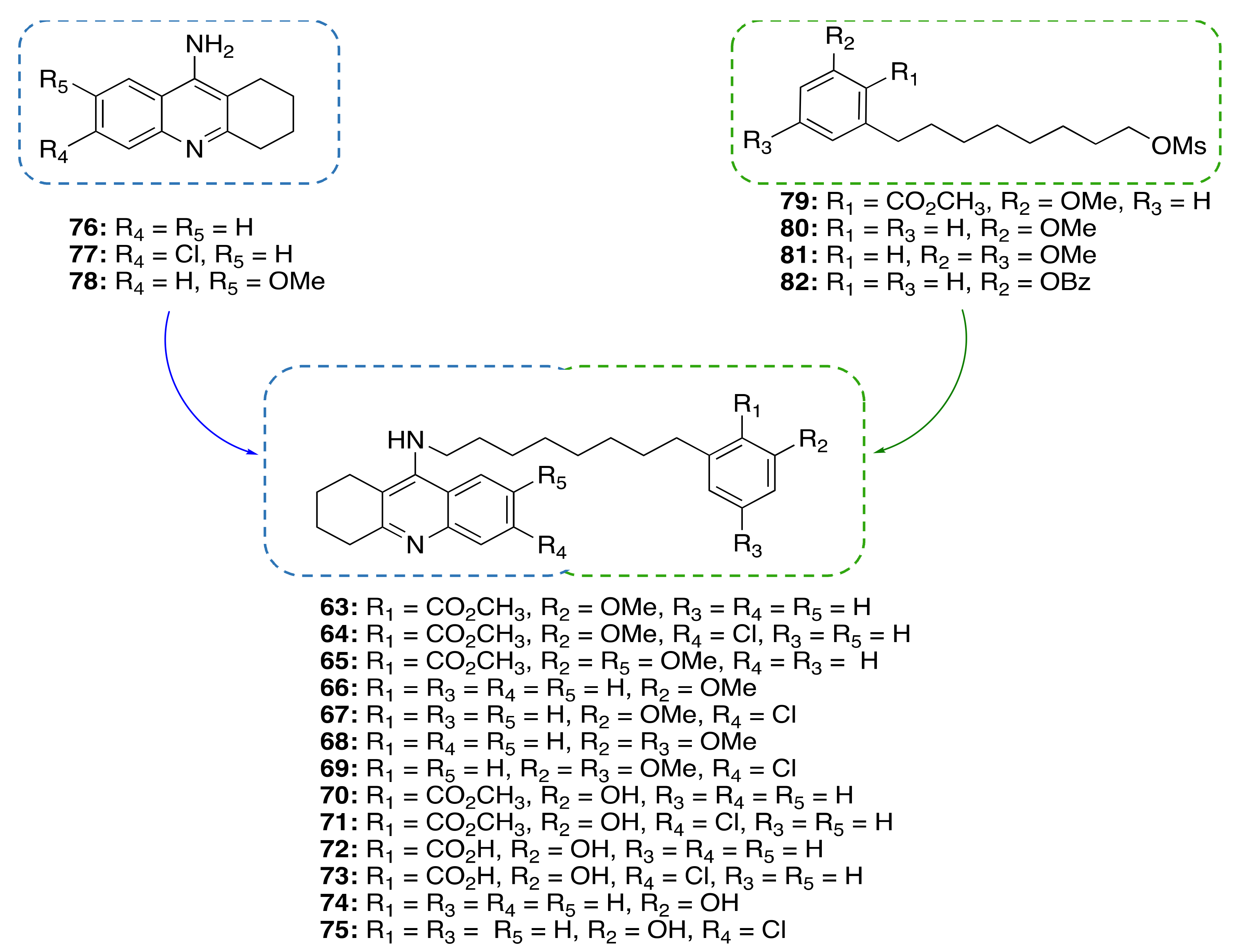
3.3.2. CNSL-Derived Dual Binding AChE Inhibitors
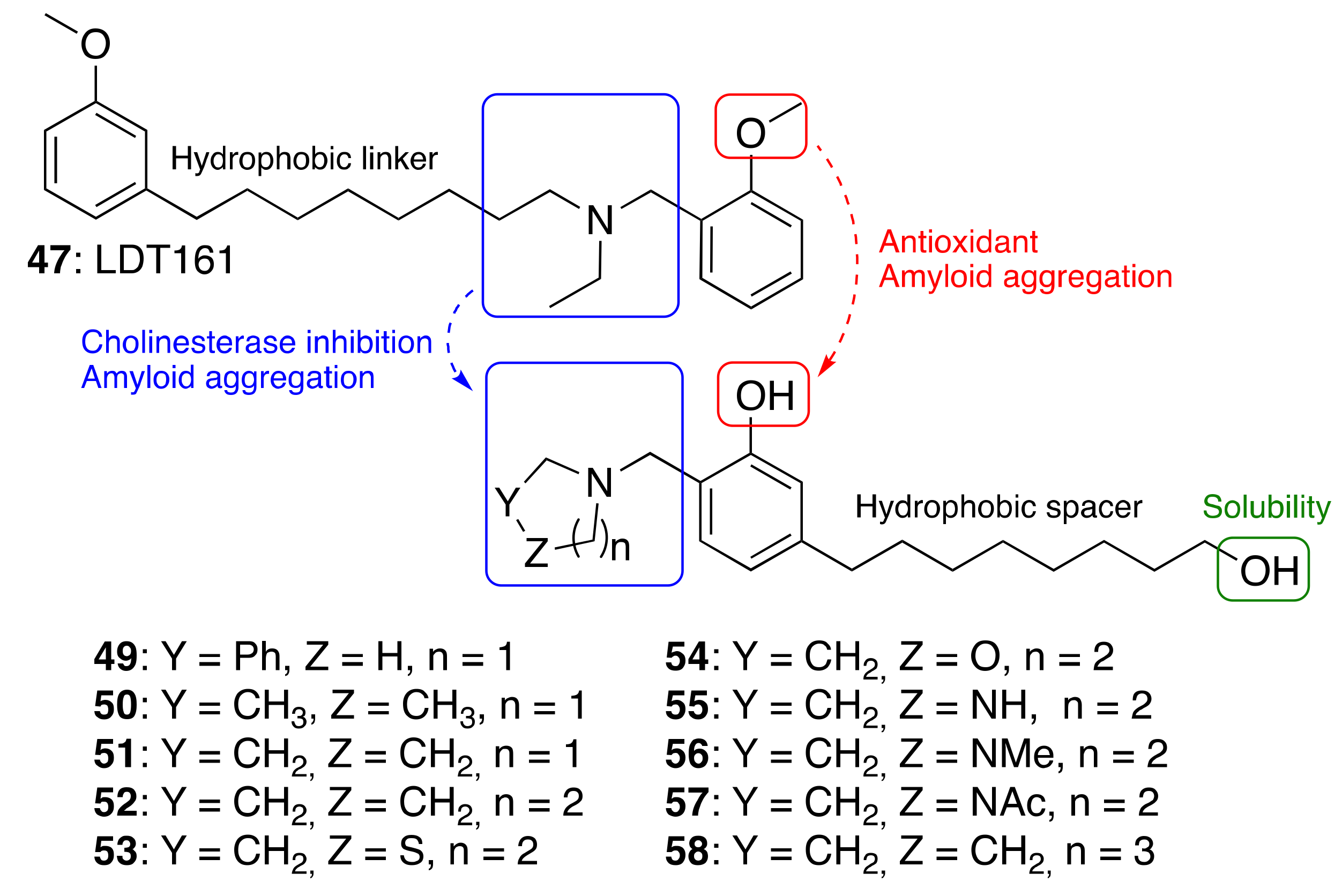
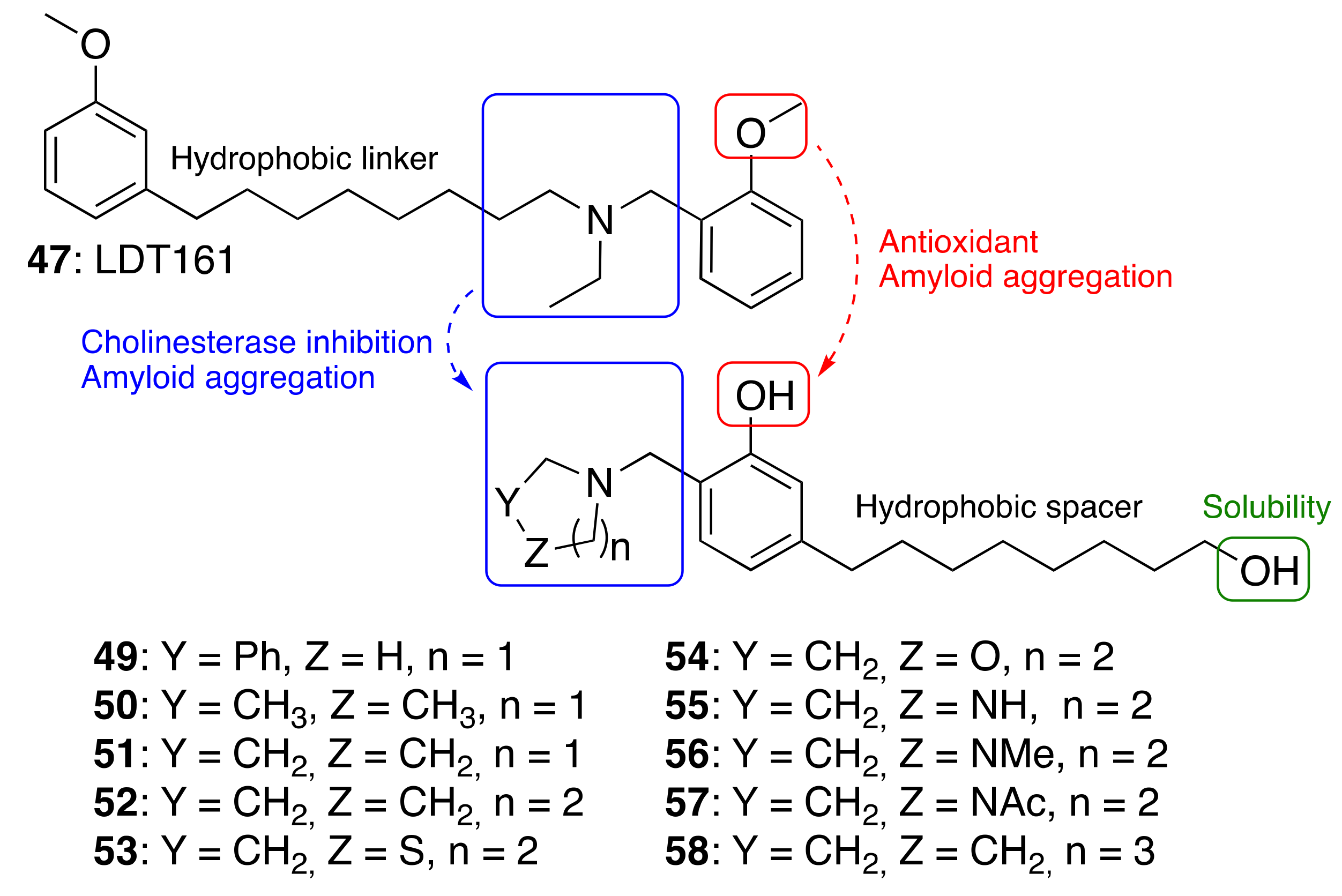
3.3.3. Cardanol-Derived Cholinesterase Inhibitors with Antioxidant and Anti-Amyloid Properties
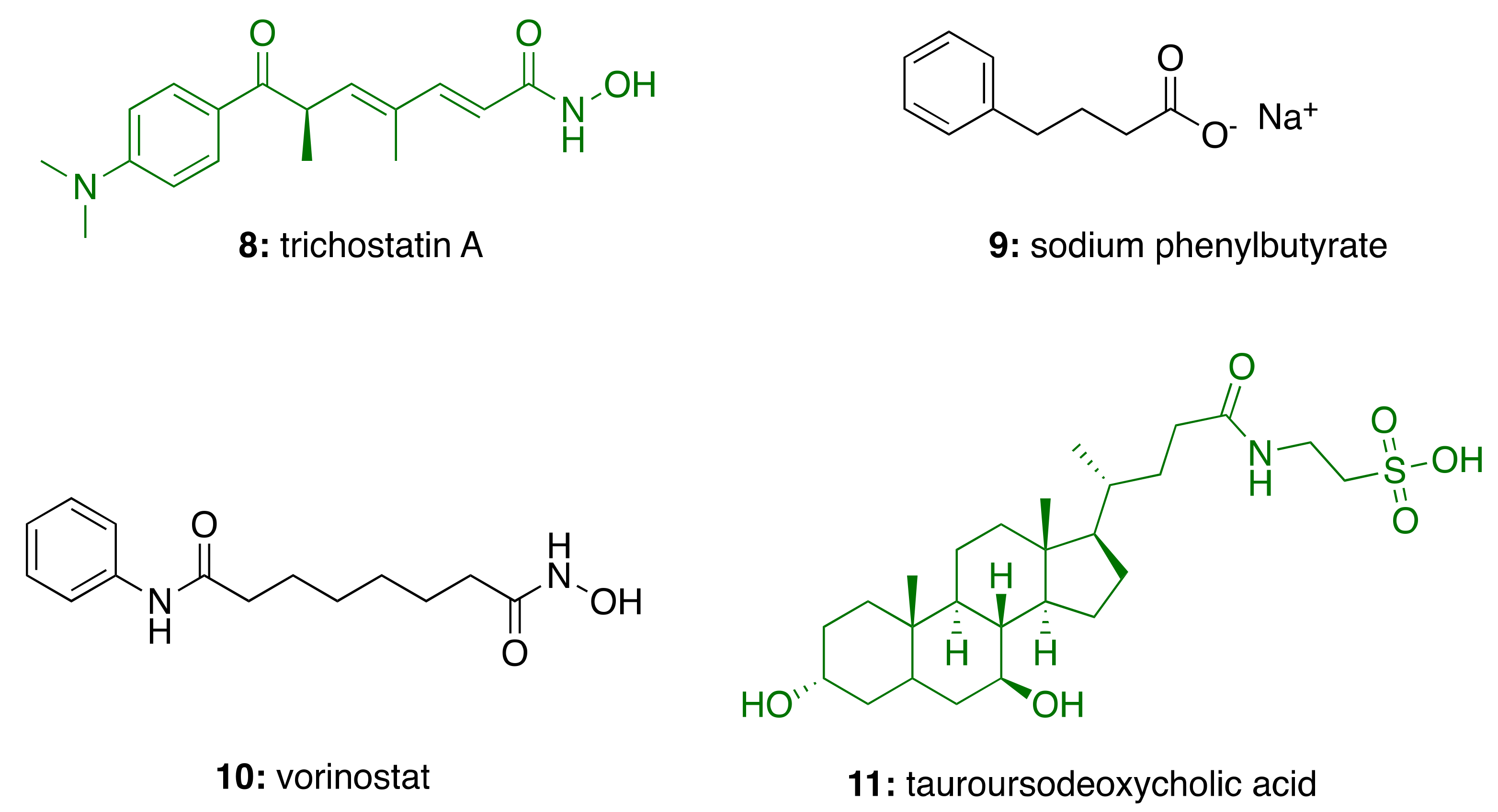
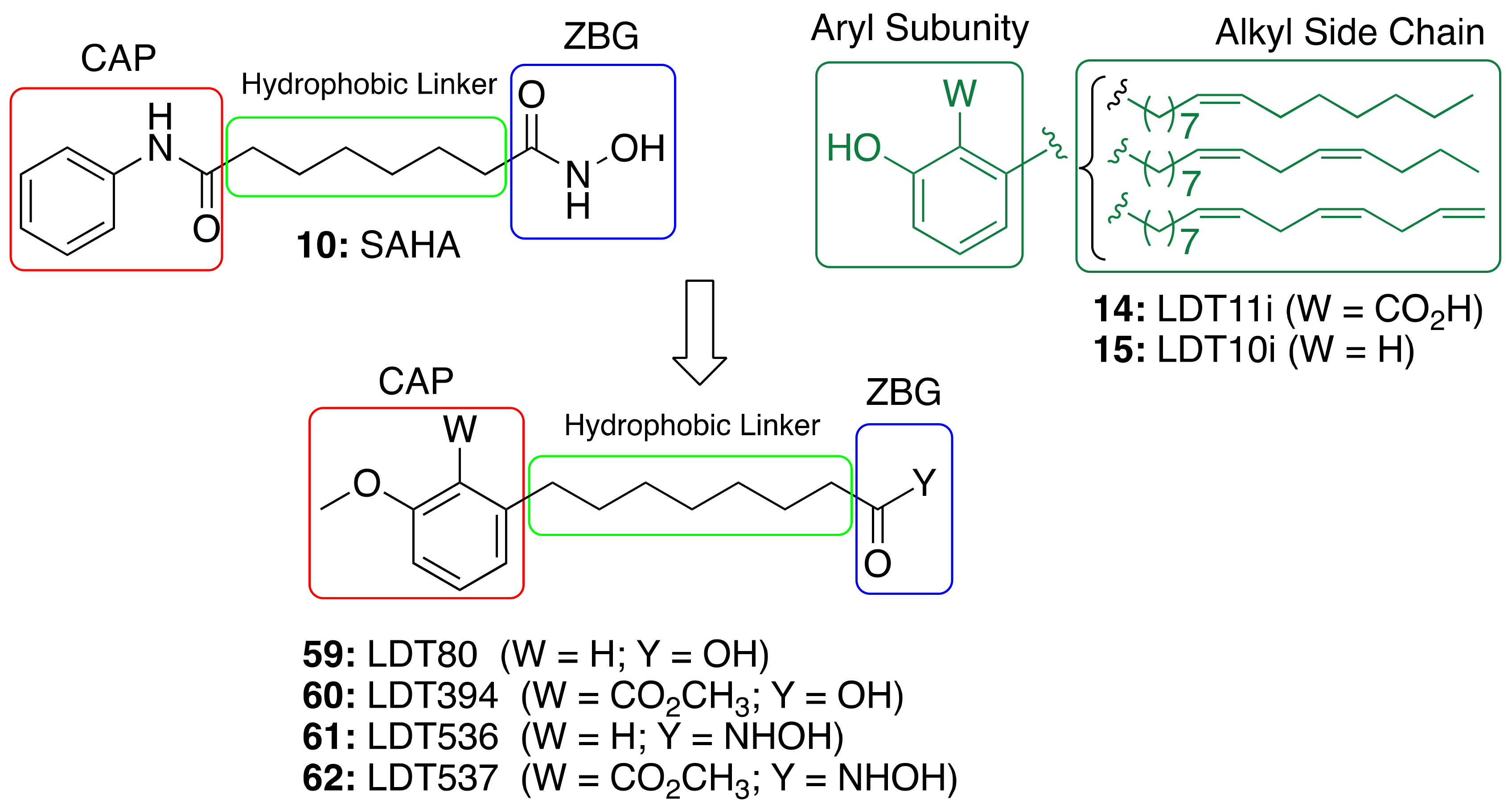
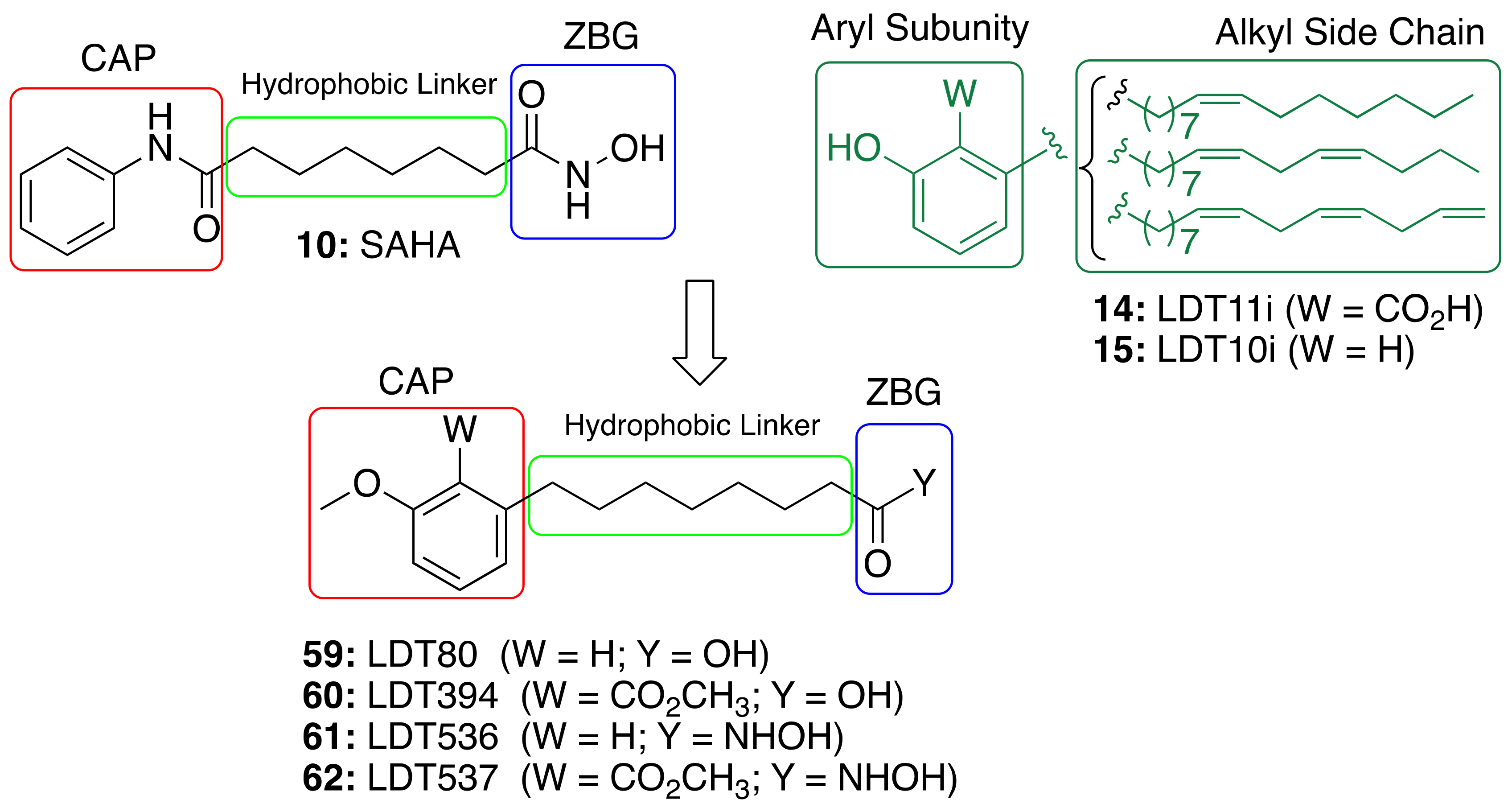
3.4. CNSL-Derived HDAC Inhibitors
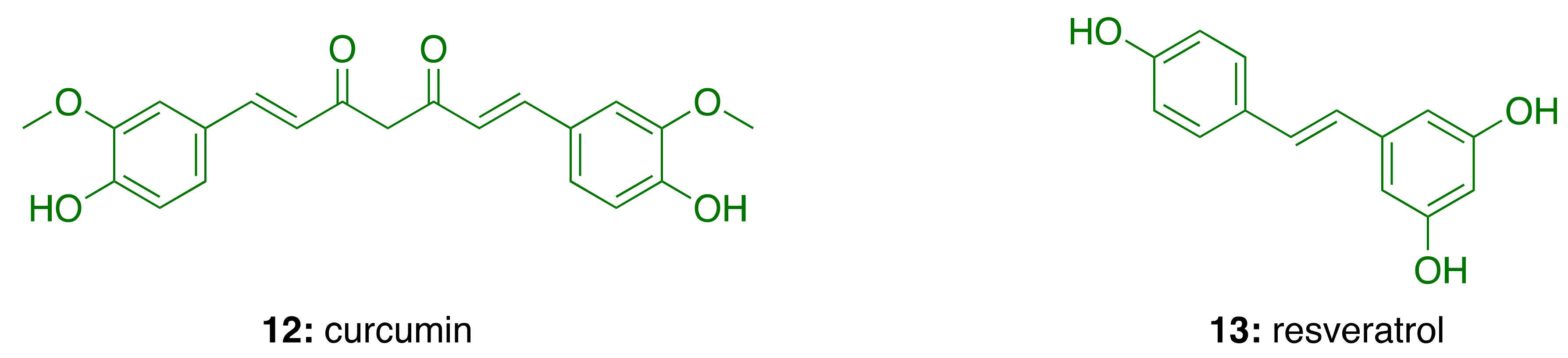
3.5. Anti-Inflammatory Activity of Phenolic Lipids from CNSL
A Framework Combination Approach towards Rationally Designed CNSL-Derived Multitarget Compounds
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef]
- Elmaleh, D.R.; Farlow, M.R.; Conti, P.S.; Tompkins, R.G.; Kundakovic, L.; Tanzi, R.E. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. J. Alzheimers Dis. 2019, 71, 715–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J. Drug Development for Psychotropic, Cognitive-Enhancing, and Disease-Modifying Treatments for Alzheimer’s Disease. J. Neuropsychiatry Clin. Neurosci. 2021, 33, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Neurodegenerative drug discovery: Building on the past, looking to the future. Future Med. Chem. 2017, 9, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Calixto, J.B. The role of natural products in modern drug discovery. Acad. Bras. Cienc. 2019, 91 (Suppl. S3), e20190105. [Google Scholar] [CrossRef]
- Bharate, S.S.; Mignani, S.; Vishwakarma, R.A. Why Are the Majority of Active Compounds in the CNS Domain Natural Products? A Critical Analysis. J. Med. Chem. 2018, 61, 10345–10374. [Google Scholar] [CrossRef]
- Howes, M.J.; Perry, E. The role of phytochemicals in the treatment and prevention of dementia. Drugs Aging 2011, 28, 439–468. [Google Scholar] [CrossRef]
- Marco, L.; do Carmo Carreiras, M. Galanthamine, a natural product for the treatment of Alzheimer’s disease. Recent Pat. CNS Drug Discov. 2006, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef]
- Ji, H.F.; Zhang, H.Y. Multipotent natural agents to combat Alzheimer’s disease. Functional spectrum and structural features. Acta Pharm. Sin. 2008, 29, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habtemariam, S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules 2019, 24, 1519. [Google Scholar] [CrossRef] [Green Version]
- Viegas, C.; Bolzani, V.A.S.; Barreiro, E.J.; Fraga, C.A. New anti-Alzheimer drugs from biodiversity: The role of the natural acetylcholinesterase inhibitors. Mini Rev. Med. Chem. 2005, 5, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Biazotto, K.R.; de Souza Mesquita, L.M.; Neves, B.V.; Braga, A.R.C.; Tangerina, M.M.P.; Vilegas, W.; Mercadante, A.Z.; De Rosso, V.V. Brazilian Biodiversity Fruits: Discovering Bioactive Compounds from Underexplored Sources. J. Agric. Food Chem. 2019, 67, 1860–1876. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterases: New roles in brain function and in Alzheimer’s disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef]
- Chalupova, K.; Korabecny, J.; Bartolini, M.; Monti, B.; Lamba, D.; Caliandro, R.; Pesaresi, A.; Brazzolotto, X.; Gastellier, A.J.; Nachon, F.; et al. Novel tacrine-tryptophan hybrids: Multi-target directed ligands as potential treatment for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 168, 491–514. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Yu, Q.S.; Kulkarni, S.S.; Parrish, D.A.; Holloway, H.W.; Tweedie, D.; Shafferman, A.; Lahiri, D.K.; Brossi, A.; Greig, N.H. Inhibition of human acetyl- and butyrylcholinesterase by novel carbamates of (−)- and (+)-tetrahydrofurobenzofuran and methanobenzodioxepine. J. Med. Chem. 2006, 49, 2174–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Castro, A. Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2006, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Torrero, D. Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr. Med. Chem. 2008, 15, 2433–2455. [Google Scholar] [CrossRef]
- Minarini, A.; Milelli, A.; Simoni, E.; Rosini, M.; Bolognesi, M.L.; Marchetti, C.; Tumiatti, V. Multifunctional tacrine derivatives in Alzheimer’s disease. Curr. Top. Med. Chem. 2013, 13, 1771–1786. [Google Scholar] [CrossRef]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorgan. Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, Y.; Yin, W.; Li, J.; Wang, W.; Bai, F.; Xu, S.; Gong, Q.; Peng, T.; Hong, Y.; et al. Kinetics-Driven Drug Design Strategy for Next-Generation Acetylcholinesterase Inhibitors to Clinical Candidate. J. Med. Chem. 2021, 64, 1844–1855. [Google Scholar] [CrossRef]
- Cacabelos, R. Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 673–701. [Google Scholar] [CrossRef]
- Bortolami, M.; Rocco, D.; Messore, A.; Di Santo, R.; Costi, R.; Madia, V.N.; Scipione, L.; Pandolfi, F. Acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease—A patent review (2016–present). Expert Opin. Ther. Pat. 2021, 31, 1–22. [Google Scholar] [CrossRef]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Milelli, A. Histone Deacetylase Inhibitors as Multitarget Ligands: New Players in Alzheimer’s Disease Drug Discovery? ChemMedChem 2019, 14, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.A.; Pinheiro, P.S.M.; Sagrillo, F.S.; Bolognesi, M.L.; Fraga, C.A.M. Histone deacetylases as targets for the treatment of neurodegenerative disorders: Challenges and future opportunities. Med. Res. Rev. 2020, 40, 2177–2211. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Frank, C.L.; Dobbin, M.M.; Tsunemoto, R.K.; Tu, W.; Peng, P.L.; Guan, J.S.; Lee, B.H.; Moy, L.Y.; Giusti, P.; et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 2008, 60, 803–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Cook, C.; Stankowski, J.N.; Carlomagno, Y.; Stetler, C.; Petrucelli, L. Acetylation: A new key to unlock tau’s role in neurodegeneration. Alzheimers Res. Ther. 2014, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Fan, S.J.; Huang, F.I.; Chao, H.Y.; Hsu, K.C.; Lin, T.E.; Yeh, T.K.; Lai, M.J.; Li, Y.H.; Huang, H.L.; et al. 5-Aroylindoles Act as Selective Histone Deacetylase 6 Inhibitors Ameliorating Alzheimer’s Disease Phenotypes. J. Med. Chem. 2018, 61, 7087–7102. [Google Scholar] [CrossRef] [PubMed]
- Peña-Altamira, E.; Petralla, S.; Massenzio, F.; Virgili, M.; Bolognesi, M.L.; Monti, B. Nutritional and Pharmacological Strategies to Regulate Microglial Polarization in Cognitive Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 175. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Garcia-Osta, A. Long-term phenylbutyrate administration prevents memory deficits in Tg2576 mice by decreasing Abeta. Front. Biosci. Elite Ed. 2011, 3, 1375–1384. [Google Scholar]
- Uliassi, E.; Gandini, A.; Perone, R.C.; Bolognesi, M.L. Neuroregeneration versus neurodegeneration: Toward a paradigm shift in Alzheimer’s disease drug discovery. Future Med. Chem. 2017, 9, 995–1013. [Google Scholar] [CrossRef]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.A.; Ficek, B.N.; Westbrook, R. Understanding the Role of Systemic Inflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 3340–3342. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimers Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Peña-Altamira, E.; Prati, F.; Massenzio, F.; Virgili, M.; Contestabile, A.; Bolognesi, M.L.; Monti, B. Changing paradigm to target microglia in neurodegenerative diseases: From anti-inflammatory strategy to active immunomodulation. Expert Opin. Ther. Targets 2016, 20, 627–640. [Google Scholar] [CrossRef]
- Fu, W.Y.; Wang, X.; Ip, N.Y. Targeting Neuroinflammation as a Therapeutic Strategy for Alzheimer’s Disease: Mechanisms, Drug Candidates, and New Opportunities. ACS Chem. Neurosci. 2019, 10, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Drug Discovery Foundation’s Annual Reports. Available online: https://www.alzdiscovery.org/about-addf/annual-reports (accessed on 22 February 2021).
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharm. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2020, 41, 2606–2633. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Mazzetto, S.E.; Lomonaco, D.; Mele, G. Óleo da castanha de caju: Oportunidades e desafios no contexto do desenvolvimento e sustentabilidade industrial. Quim. Nova 2009, 32, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Phani Kumar, P.; Paramashivappa, R.; Vithayathil, P.J.; Subba Rao, P.V.; Srinivasa Rao, A. Process for isolation of cardanol from technical cashew (Anacardium occidentale L.) nut shell liquid. J. Agric. Food Chem. 2002, 50, 4705–4708. [Google Scholar] [CrossRef]
- Tyman, J.H. Synthetic and Natural Phenols; Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Patel, R.N.; Bandyopadhyay, S.; Ganesh, A. Extraction of cashew (Anacardium occidentale) nut shell liquid using supercritical carbon dioxide. Bioresour. Technol. 2006, 97, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Pandey, M.K.; Ahn, K.S.; Yi, T.; Chaturvedi, M.M.; Liu, M.; Aggarwal, B.B. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood 2008, 111, 4880–4891. [Google Scholar]
- Stasiuk, M.; Kozubek, A. Biological activity of phenolic lipids. Cell Mol. Life Sci. 2010, 67, 841–860. [Google Scholar] [CrossRef] [PubMed]
- Green, I.R.; Tocoli, F.E.; Lee, S.H.; Nihei, K.; Kubo, I. Design and evaluation of anacardic acid derivatives as anticavity agents. Eur. J. Med. Chem. 2008, 43, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Swamy, B.N.; Suma, T.K.; Rao, G.V.; Reddy, G.C. Synthesis of isonicotinoylhydrazones from anacardic acid and their in vitro activity against Mycobacterium smegmatis. Eur. J. Med. Chem. 2007, 42, 420–424. [Google Scholar] [CrossRef]
- Green, I.R.; Tocoli, F.E.; Lee, S.H.; Nihei, K.; Kubo, I. Molecular design of anti-MRSA agents based on the anacardic acid scaffold. Bioorg. Med. Chem. 2007, 15, 6236–6241. [Google Scholar] [CrossRef]
- Trevisan, M.T.; Pfundstein, B.; Haubner, R.; Würtele, G.; Spiegelhalder, B.; Bartsch, H.; Owen, R.W. Characterization of alkyl phenols in cashew (Anacardium occidentale) products and assay of their antioxidant capacity. Food Chem. Toxicol. 2006, 44, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.M.; Severino, R.P.; Vieira, P.C.; Fernandes, J.B.; da Silva, M.F.; Zottis, A.; Andricopulo, A.D.; Oliva, G.; Corrêa, A.G. Anacardic acid derivatives as inhibitors of glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma cruzi. Bioorgan. Med. Chem. 2008, 16, 8889–8895. [Google Scholar] [CrossRef]
- Freitas, R.F.; Prokopczyk, I.M.; Zottis, A.; Oliva, G.; Andricopulo, A.D.; Trevisan, M.T.; Vilegas, W.; Silva, M.G.; Montanari, C.A. Discovery of novel Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase inhibitors. Bioorg. Med. Chem. 2009, 17, 2476–2482. [Google Scholar] [CrossRef]
- Paramashivappa, R.; Phani Kumar, P.; Subba Rao, P.V.; Srinivasa Rao, A. Synthesis of sildenafil analogues from anacardic acid and their phosphodiesterase-5 inhibition. J. Agric. Food Chem. 2002, 50, 7709–7713. [Google Scholar] [CrossRef]
- Paramashivappa, R.; Phani Kumar, P.; Subba Rao, P.V.; Srinivasa Rao, A. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657–660. [Google Scholar] [CrossRef]
- Rekowski, M.; Giannis, A. Histone acetylation modulation by small molecules: A chemical approach. Biochim. Biophys. Acta 2010, 1799, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, G.; Castellano, S.; Vicidomini, C.; Rotili, D.; Nebbioso, A.; Miceli, M.; Altucci, L.; Mai, A. Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg. Med. Chem. Lett. 2008, 18, 2788–2792. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Rotili, D.; Tarantino, D.; Nebbioso, A.; Castellano, S.; Sbardella, G.; Tini, M.; Altucci, L. Identification of 4-hydroxyquinolines inhibitors of p300/CBP histone acetyltransferases. Bioorg. Med. Chem. Lett. 2009, 19, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Chandregowda, V.; Kush, A.; Reddy, G.C. Synthesis of benzamide derivatives of anacardic acid and their cytotoxic activity. Eur. J. Med. Chem. 2009, 44, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Hundt, J.; Li, Z.; Liu, Q. The inhibitory effects of anacardic acid on hepatitis C virus life cycle. PLoS ONE 2015, 10, e0117514. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Heitel, P.; Kalinowsky, L.; Merk, D. Opportunities and Challenges for Fatty Acid Mimetics in Drug Discovery. J. Med. Chem. 2017, 60, 5235–5266. [Google Scholar] [CrossRef] [PubMed]
- Stasiuk, M.; Bartosiewicz, D.; Kozubek, A. Inhibitory effect of some natural and semisynthetic phenolic lipids upon acetylcholinesterase activity. Food Chem. 2008, 108, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Stasiuk, M.; Janiszewska, A.; Kozubek, A. Phenolic lipids affect the activity and conformation of acetylcholinesterase from Electrophorus electricus (Electric eel). Nutrients 2014, 6, 1823–1831. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Kronman, C.; Flashner, Y.; Leitner, M.; Segall, Y.; Ariel, N.; Cohen, S.; Velan, B.; Shafferman, A. Dissection of the human acetylcholinesterase active center determinants of substrate specificity. Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket. J. Biol. Chem. 1993, 268, 17083–17095. [Google Scholar] [CrossRef]
- De Paula, A.A.N.; Martins, J.B.L.; Gargano, R.; dos Santos, M.L.; Romeiro, L.A.S. Electronic structure calculations toward new potentially AChE inhibitors. Chem. Phys. Lett. 2007, 446, 304–308. [Google Scholar] [CrossRef]
- De Paula, A.A.; Martins, J.B.; dos Santos, M.L.; Nascente, L.E.C.; Romeiro, L.A.; Areas, T.F.; Vieira, K.S.; Gambôa, N.F.; Castro, N.G.; Gargano, R. New potential AChE inhibitor candidates. Eur. J. Med. Chem. 2009, 44, 3754–3759. [Google Scholar] [CrossRef]
- Lemes, L.F.N.; de Andrade Ramos, G.; de Oliveira, A.S.; da Silva, F.M.R.; de Castro Couto, G.; da Silva Boni, M.; Guimarães, M.J.R.; Souza, I.N.O.; Bartolini, M.; Andrisano, V.; et al. Cardanol-derived AChE inhibitors: Towards the development of dual binding derivatives for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 108, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Melchiorre, C. From dual binding site acetylcholinesterase inhibitors to multi-target-directed ligands (MTDLs): A step forward in the treatment of Alzheimer’s disease. Mini Rev. Med. Chem. 2008, 8, 960–967. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew. Chem. Int. Ed. Engl. 2007, 46, 3689–3692. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Belluti, F.; Gobbi, S.; Bisi, A. Hybrid-based multi-target ligands for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 2011, 11, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Bartolini, M.; Tarozzi, A.; Morroni, F.; Lizzi, F.; Milelli, A.; Minarini, A.; Rosini, M.; Hrelia, P.; Andrisano, V.; et al. Multitargeted drugs discovery: Balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg. Med. Chem. Lett. 2011, 21, 2655–2658. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, C.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V. Polyamines in drug discovery: From the universal template approach to the multitarget-directed ligand design strategy. J. Med. Chem. 2010, 53, 5906–5914. [Google Scholar] [CrossRef]
- De Abreu Silva, M.; Sette, C.D.A.B.; Kiametis, A.S.; Romeiro, L.A.S.; Gargano, R. Molecular modeling of cardanol-derived AChE inhibitors. Chem. Phys. Lett. 2019, 731, 136591. [Google Scholar] [CrossRef]
- De Andrade Ramos, G.; Souza de Oliveira, A.; Bartolini, M.; Naldi, M.; Liparulo, I.; Bergamini, C.; Uliassi, E.; Wu, L.; Fraser, P.E.; Abreu, M.; et al. Discovery of sustainable drugs for Alzheimer’s disease: Cardanol-derived cholinesterase inhibitors with antioxidant and anti-amyloid properties. RSC Med. Chem. 2021, 12, 1154–1163. [Google Scholar] [CrossRef]
- Hemshekhar, M.; Sebastin Santhosh, M.; Kemparaju, K.; Girish, K.S. Emerging roles of anacardic acid and its derivatives: A pharmacological overview. Basic Clin. Pharm. Toxicol. 2012, 110, 122–132. [Google Scholar] [CrossRef]
- Soares Romeiro, L.A.; da Costa Nunes, J.L.; de Oliveira Miranda, C.; Simões Heyn Roth Cardoso, G.; de Oliveira, A.S.; Gandini, A.; Kobrlova, T.; Soukup, O.; Rossi, M.; Senger, J.; et al. Novel Sustainable-by-Design HDAC Inhibitors for the Treatment of Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10, 671–676. [Google Scholar] [CrossRef]
- De Souza, M.Q.; Teotônio, I.M.S.N.; de Almeida, F.C.; Heyn, G.S.; Alves, P.S.; Romeiro, L.A.S.; Pratesi, R.; de Medeiros Nóbrega, Y.K.; Pratesi, C.B. Molecular evaluation of anti-inflammatory activity of phenolic lipid extracted from cashew nut shell liquid (CNSL). BMC Complement. Altern. Med. 2018, 18, 181. [Google Scholar] [CrossRef]
- Gomes Júnior, A.L.; Islam, M.T.; Nicolau, L.A.D.; de Souza, L.K.M.; Araújo, T.S.L.; Lopes de Oliveira, G.A.; de Melo Nogueira, K.; da Silva Lopes, L.; Medeiros, J.R.; Mubarak, M.S.; et al. Anti-Inflammatory, Antinociceptive, and Antioxidant Properties of Anacardic Acid in Experimental Models. ACS Omega 2020, 5, 19506–19515. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Tinarelli, C.; Schulz, R.J.; Polidori, M.C. Nutraceuticals in cognitive impairment and Alzheimer’s disease. Front. Pharmacol. 2014, 5, 147. [Google Scholar] [CrossRef] [Green Version]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; Taskforce, I.N.P.S. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Percentage (%) | |||
|---|---|---|---|---|
| Anacardic Acids | Cardanols | Cardols | 2-Methylcardols | |
| Saturated (15:0) | 2.2–3.0 | 3.9–4.4 | 0.2–2.7 | 0.9–1.3 |
| Monoene (15:1) | 25.0–33.3 | 21.6–32.2 | 8.4–15.2 | 16.3–25.3 |
| Diene (15:2) | 17.8–32.1 | 15.4–18.2 | 24.2–28.9 | 20.6–24.4 |
| Triene (15:3) | 36.3–50.4 | 45.1–59.0 | 36.5–67.2 | 49.8–62.2 |
| Compound | IC50 (μM) |
|---|---|
| Anacardic acid (14a) | 3.0 ± 0.2 |
| Cardanol (15a) | 4.0 ± 0.2 |
| Cardol (16a) | >94 |
| Cardol (16c) | 3.5 ± 0.2 |
| 2-Methycardol (17c) | 5.0 ± 0.4 |
| Cardol C21 homologue (18) | 44 ± 0.5 |
| Cardol C25 homologue (19) | 44 ± 4.2 |
| Merulinic acid (20) | >94 |
| Compound | EeAChE IC50(μM) a | eqBChE IC50 (μM) a | SI b | EeAChE Ki (μM) c | |
|---|---|---|---|---|---|
| 25 | LDT148 | 19.6 | 16.4 | 0.8 | 22.4 (3) |
| 40 | LDT149 | 16.1 | 19.0 | 1.2 | 19.0 (3) |
| 42 | LDT154 | 13.7 | 22.8 | 1.7 | 8.6 (2) |
| 44 | LDT150 | 14.3 | 23.5 | 1.7 | 22.4 (3) |
| 46 | LDT167 | 17.2 | 3.1 | 0.2 | 17.0 (3) |
| 47 | LDT161 | 6.6 | 5.0 | 0.8 | 24.4 (3) |
| 48 | LDT160 | 16.1 | 8.0 | 0.5 | 10.4 (2) |
 | |||||
|---|---|---|---|---|---|
| Compounds | % Inhibition hAChE a | IC50 hAChE (μM) ± SEM a | % Inhibition hBChE a | IC50 hBChE (μM) ± SEM a | HORAC b |
| Gallic Acid Equivalents | |||||
| 49 | 19.4 ± 7.1 | ND | 68.3 ± 1.3 | 6.74 ± 0.7 | 3.70 ± 0.05 |
| 50 | 12.4 ± 1.2 | ND | 50.5 ±0.7 | 17.5 ± 3.5 | 5.49 ± 0.58 |
| 51 | 31.1 ± 2.8 | 47.6 ± 4.1 | 59.0 ± 0.8 | 13.3 ± 0.5 | 4.88 ± 0.05 |
| 52 | 40.5 ± 1.8 | 30.0 ± 2.6 | 73.5 ± 0.4 | 6.12 ± 0.8 | 4.37 ± 0.54 |
| 53 | 12.1 ± 0.8 | ND | 16.6 ± 2.4 | ND | 4.38 ± 0.23 |
| 54 | <5 | ND | <10 | ND | 9.73 ± 0.86 |
| 55 | <10 | ND | 17.7 ± 3.1 | ND | 1.49 ± 0.20 |
| 56 | <5 | ND | 10.6 ± 2.1 | ND | 4.80 ± 0.49 |
| 57 | <10 | ND | <5 | ND | 4.52 ± 0.64 |
| 58 | <10 | 785 ± 42 | 77.1 ± 0.2 | 4.62 ± 0.14 | 3.50 ± 0,23 |
| 47 | 5.65 ± 0.48 | n.a. | |||
| Ferulic acid | 4.04 ± 0.51 | ||||
| Compound | HDAC1 IC50 ± SEM [nM] | HDAC6 IC50 ± SEM [nM] | |
|---|---|---|---|
| 9 | Vorinostat | 119 | 53 |
| 60 | LDT394 | 12% * | 12% * |
| 61 | LDT536 | 316.2 ± 37 | 190.1 ± 38.2 |
| 62 | LDT537 | 774.7 ± 14.4 | 215.4 ± 28.6 |
| Compound | In Vitro Effects | In Vivo Effects | Refs. |
|---|---|---|---|
| Anacardic acid (14) | AChE inhibition antioxidant anti-inflammatory | anti-inflammatory | [76,92,93] |
| Cardanol (15) | AChE inhibition | - | [76] |
| Cardol (16) | AChE inhibition | - | [76] |
| 2-Methycardol (17) | AChE inhibition | - | [76] |
| CNSL-derived rivastigmine analogues (22–24) | AChE inhibition | - | [79,80] |
| CNSL-derived dual binding AChE inhibitors (25–48) | AChE inhibition | - | [81] |
| Cardanol-derived cholinesterase inhibitors with antioxidant and anti-amyloid properties (49–58) | ChE inhibition antioxidant anti-amyloid | - | [89] |
| CNSL-derived HDAC inhibitors (59–62) | HDAC inhibition immunomodulatory | - | [91] |
| CNSL-derived compounds-tacrine hybrids (63–75) | ChE inhibition anti-neuroinflammatory neuroprotective | - | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uliassi, E.; de Oliveira, A.S.; de Camargo Nascente, L.; Romeiro, L.A.S.; Bolognesi, M.L. Cashew Nut Shell Liquid (CNSL) as a Source of Drugs for Alzheimer’s Disease. Molecules 2021, 26, 5441. https://doi.org/10.3390/molecules26185441
Uliassi E, de Oliveira AS, de Camargo Nascente L, Romeiro LAS, Bolognesi ML. Cashew Nut Shell Liquid (CNSL) as a Source of Drugs for Alzheimer’s Disease. Molecules. 2021; 26(18):5441. https://doi.org/10.3390/molecules26185441
Chicago/Turabian StyleUliassi, Elisa, Andressa Souza de Oliveira, Luciana de Camargo Nascente, Luiz Antonio Soares Romeiro, and Maria Laura Bolognesi. 2021. "Cashew Nut Shell Liquid (CNSL) as a Source of Drugs for Alzheimer’s Disease" Molecules 26, no. 18: 5441. https://doi.org/10.3390/molecules26185441
APA StyleUliassi, E., de Oliveira, A. S., de Camargo Nascente, L., Romeiro, L. A. S., & Bolognesi, M. L. (2021). Cashew Nut Shell Liquid (CNSL) as a Source of Drugs for Alzheimer’s Disease. Molecules, 26(18), 5441. https://doi.org/10.3390/molecules26185441