
Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes



Abstract
1. Introduction
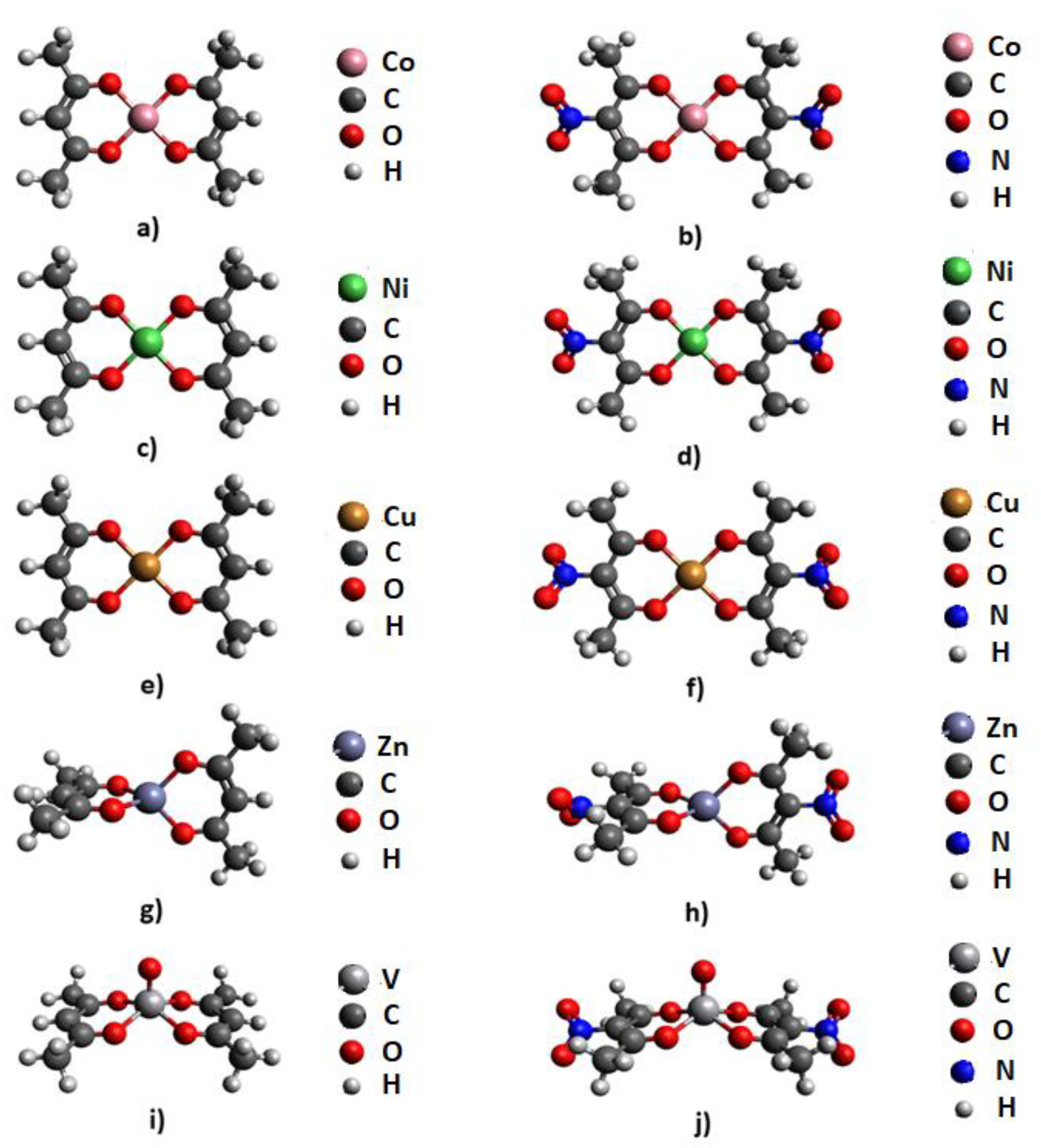
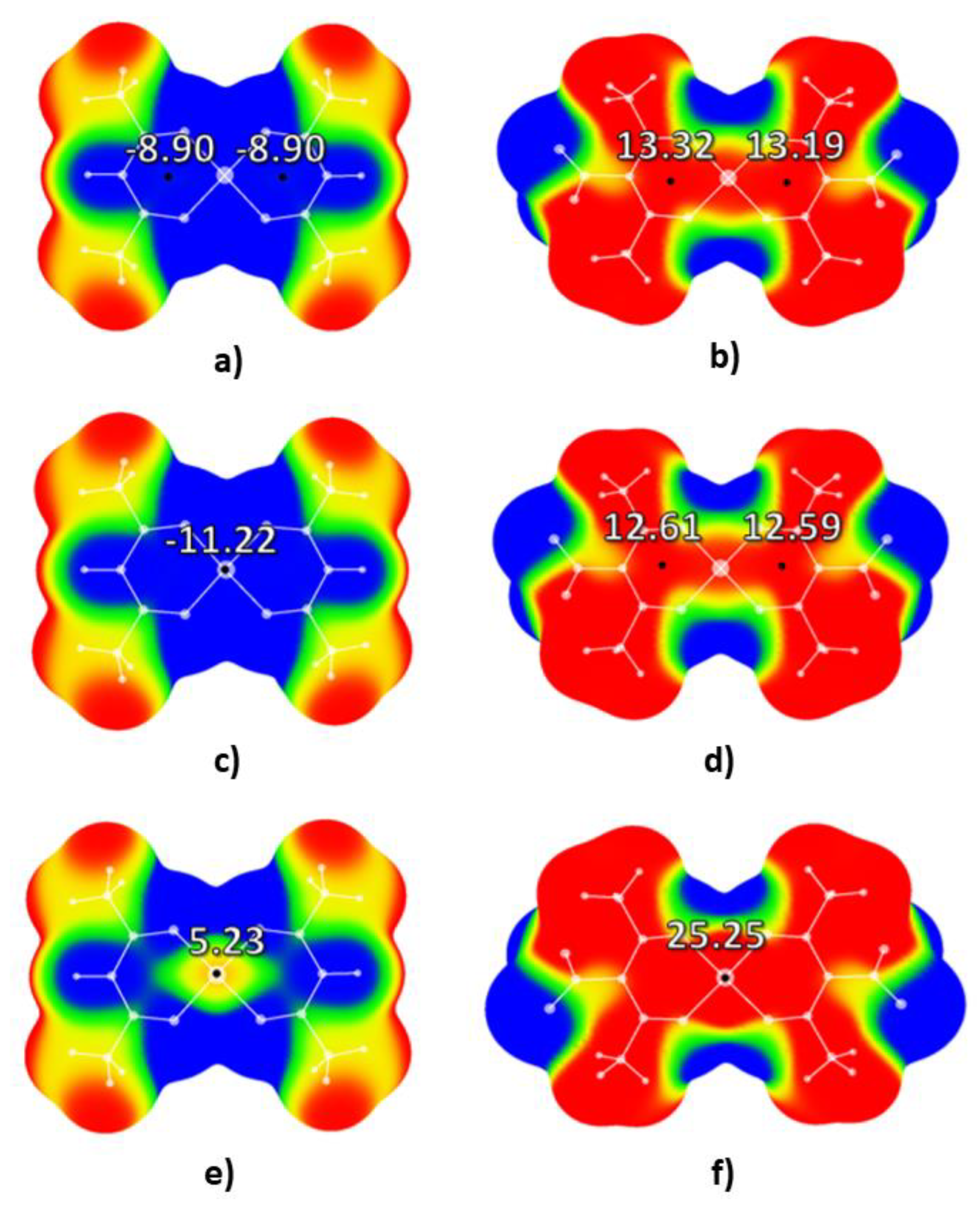
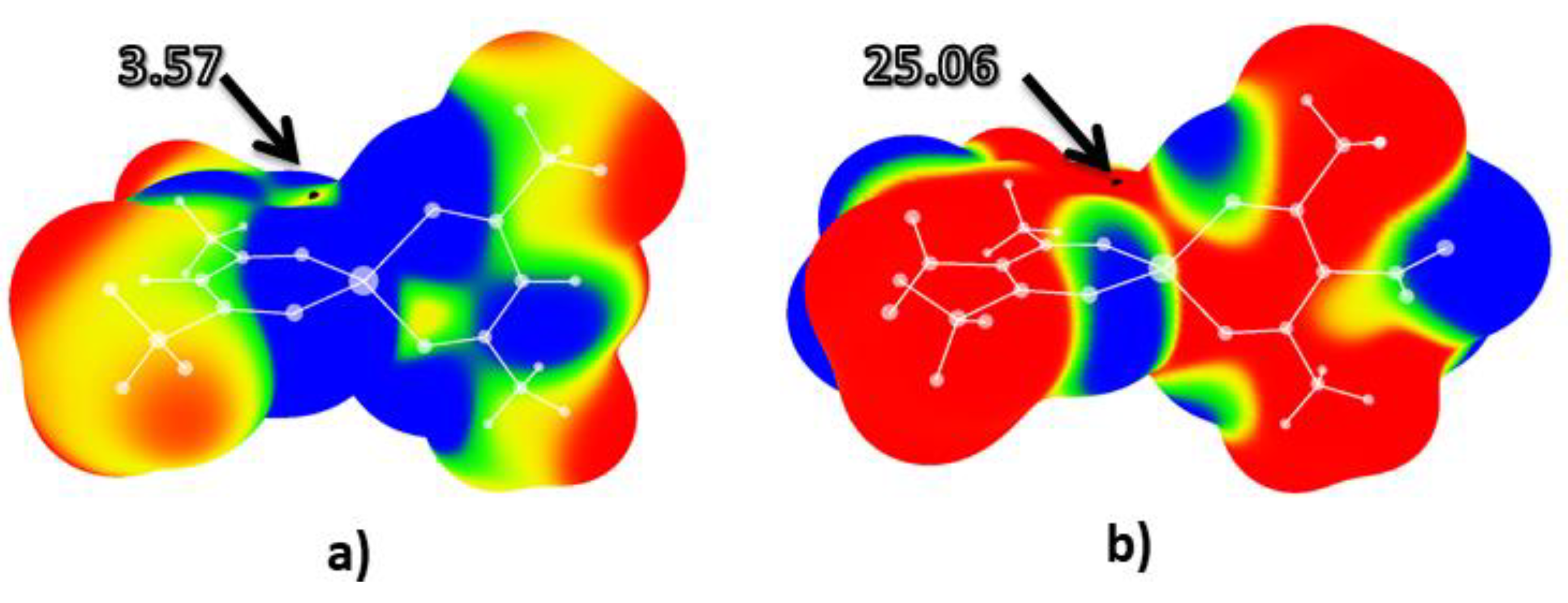
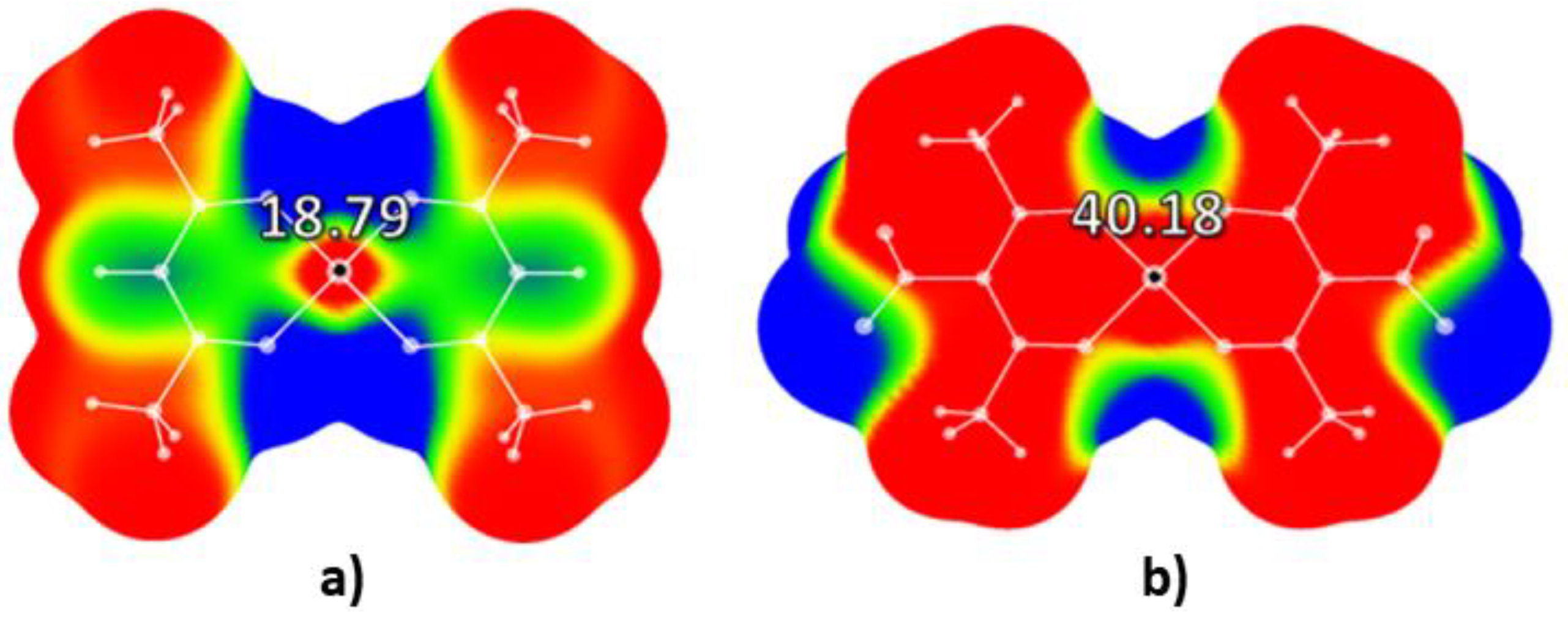
2. Results and Discussion
2.1. Molecular Electrostatic Potential Analysis
2.2. Bond Dissociation Energies Calculations
3. Discussion
4. Methodology
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Thottempudi, V.; Gao, H.; Shreeve, J.M. Trinitromethyl-Substituted 5-Nitro- or 3-Azo-1,2,4-triazoles: Synthesis, Characterization, and Energetic Properties. J. Am. Chem. Soc. 2011, 133, 6464–6471. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Some molecular/crystalline factors that affect the sensitivities of energetic materials: Molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J. Mol. Model. 2015, 21, 25. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, D. Atom Pair Contribution Method: Fast and General Procedure to Predict Molecular Formation Ethalpies. J. Chem. Inf. Model. 2018, 58, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tong, W.; Li, H.; Zhang, G.; Liu, J. Chelates with π-stacking and hydrogen-bonding interactions as safer and structurally reinforced energetic materials. Inorg. Chim. Acta 2017, 466, 405–409. [Google Scholar] [CrossRef]
- Myers, T.W.; Bjorgaard, J.A.; Brown, K.E.; Chavez, D.E.; Hanson, S.K.; Scharff, R.J.; Tretiak, S.; Veauthier, J.M. Energetic Chromophores: Low-Energy Laser Initiation in Explosive Fe(II) Tetrazine Complexes. J. Am. Chem. Soc. 2016, 138, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- Joyner, T.B. Explosive sensitivity of cobalt(III) ammine complexes. Can. J. Chem. 1969, 47, 2729–2730. [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Serushkin, V.V. Design and Combustion Behaviour of Explosive Coordination Compounds. Def. Sci. J. 2013, 46, 371–383. [Google Scholar] [CrossRef][Green Version]
- Deblitz, R.; Hrib, C.G.; Blaurock, S.; Jones, P.G.; Plenikowski, G.; Edelmann, F.T. Explosive Werner-type cobalt(III) complexes. Inorg. Chem. Front. 2014, 1, 621–640. [Google Scholar] [CrossRef]
- Wojewódka, A.; Bełzowski, J. Hydrazine complexes of transition metals as perspective explosives. Chemik 2011, 65, 20–27. [Google Scholar]
- Young, C.G.; Volaric, S. Synthesis, Iodometric Analysis, and IR Spectroscopy of the Peroxide Double Salt [Zn(NH3)4][Mo(O2)4]. J. Chem. Educ. 2020, 97, 1120–1122. [Google Scholar] [CrossRef]
- Chhabra, J.S.; Talawar, M.B.; Makashir, P.S.; Asthana, S.N.; Singh, H. Synthesis, characterization and thermal studies of (Ni/Co) metal salts of hydrazine: Potential initiatory compounds. J. Hazard. Mater. 2003, 99, 225–239. [Google Scholar] [CrossRef]
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. High Performance, Low Sensitivity: Conflicting or Compatible? Propellants Explos. Pyrotech. 2016, 41, 414–425. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Detonation Performance and Sensitivity: A Quest for Balance. Adv. Quantum Chem. 2014, 69, 1–30. [Google Scholar]
- Murray, J.S.; Lane, P.; Politzer, P. Effects of strongly electron-attracting components on molecular surface electrostatic potentials: Application to predicting impact sensitivities of energetic molecules. Mol. Phys. 1998, 93, 187–194. [Google Scholar] [CrossRef]
- Rice, B.M.; Hare, J.J. A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules. J. Phys. Chem. A 2002, 106, 1770–1783. [Google Scholar] [CrossRef]
- Hammerl, A.; Klapötke, T.M.; Nöth, H.; Warchhold, M. Synthesis, Structure, Molecular Orbital and Valence Bond Calculations for Tetrazole Azide. CHN7. Propellants Explos. Pyrotech. 2003, 28, 165–173. [Google Scholar] [CrossRef]
- Hammerl, A.; Klapötke, T.M.; Mayer, P.; Weigand, J.J.; Holl, G. Synthesis, Structure, Molecular Orbital Calculations and Decomposition Mechanism for Tetrazolylazide CHN7, its Phenyl Derivative PhCN7 and Tetrazolylpentazole CHN9. Propellants Explos. Pyrotech. 2005, 30, 17–26. [Google Scholar] [CrossRef]
- Gökçınar, E.; Klapötke, T.M.; Bellamy, A.J. Computational study on 2,6-diamino-3,5-dinitropyrazine and its 1-oxide and 1,4-dioxide derivatives. J. Mol. Struct. (THEOCHEM) 2010, 953, 18–23. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Nordheiter, A.; Stierstorfer, J. Synthesis and reactivity of an unexpected highly sensitive 1-carboxymethyl-3-diazonio-5-nitrimino-1,2,4-triazole. New J. Chem. 2012, 36, 1463–1468. [Google Scholar] [CrossRef]
- Li, H.; Shu, Y.; Gao, S.; Chen, L.; Ma, Q.; Ju, X.J. Easy methods to study the smart energetic TNT/CL-20 co-crystal. J. Mol. Model. 2013, 19, 4909–4917. [Google Scholar] [CrossRef] [PubMed]
- Janjić, G.V.; Milosavljevic, M.D.; Veljković, D.Ž.; Zarić, S.D. Prediction of strong O–H/M hydrogen bonding between water and square-planar Ir and Rh complexes. Phys. Chem. Chem. Phys. 2017, 19, 8657–8660. [Google Scholar] [CrossRef] [PubMed]
- Kretić, D.S.; Radovanović, J.I.; Veljković, D.Ž. Can the sensitivity of energetic materials be tuned by using hydrgen bonds? Another look at the role of hydrogen bonding in the design of high energetic compounds. Phys. Chem. Chem. Phys. 2021, 23, 7472–7479. [Google Scholar] [CrossRef]
- Rice, B.M.; Sahu, S.; Owens, F.J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J. Mol. Struct. (THEOCHEM) 2002, 583, 69–72. [Google Scholar] [CrossRef]
- Wang, K.; Zeng, D.; Zhang, J.; Cui, Y.; Zhang, T.; Jin, X.; Li, Z. Controllable explosion: Fine-tuning the sensitivity of high-energy complexes. Dalton Trans. 2015, 44, 12497–12501. [Google Scholar] [CrossRef]
- Djordjevic, C. Aluminium(Ill) and Gallium(III) Tris-”f-Nitroacetylacetonates. Preparation and Infrared Spectra. Croat. Chem. Acta 1963, 35, 129–134. [Google Scholar]
- Chen, Y.; Wang, B.; Huang, W.; Zhang, X.; Wang, G.; Leonardi, M.J.; Huang, Y.; Lu, Z.; Marks, T.J.; Facchetti, A. Nitroacetylacetone as a Cofuel for the Combustion Synthesis of High-Performance Indium–Gallium–Zinc Oxide Transistors. Chem. Mater. 2018, 30, 3323–3329. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bulat, F.A.; Toro-Labbe, A.; Brinck, T.; Murray, J.S.; Politzer, P.J. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Kuklja, M.M. Decomposition mechanisms and kinetics of novel energetic molecules BNFF-1 and ANFF-1: Quantum-chemical modeling. Molecules 2013, 18, 8500–8517. [Google Scholar] [CrossRef] [PubMed]
- Houthuijs, K.J.; Martens, J.; Arranja, A.G.; Berden, G.; Nijsen, J.F.W.; Oomens, J. Characterization of holmium(III)-acetylacetonate complexes derived from therapeutic microspheres by infrared ion spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 15716–15722. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | E (complex) 1 | E (complex radical) 1 | E (NO2 radical) 1 | BDE 2 |
|---|---|---|---|---|
| Co(acac-NO2)2 | −2482.339456 | −2277.113539 | −205.13191 | 58.99 |
| Ni(acac-NO2)2 | −2607.885082 | −2402.656789 | −205.13191 | 60.48 |
| Cu(acac-NO2)2 | −2740.057538 | −2534.830809 | −205.13191 | 59.50 |
| Zn(acac-NO2)2 | −2878.928600 | −2673.704112 | −205.13191 | 58.09 |
| VO(acac-NO2)2 | −2118.925282 | −1913.668804 | −205.13191 | 78.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretić, D.S.; Veljković, I.S.; Đunović, A.B.; Veljković, D.Ž. Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes. Molecules 2021, 26, 5438. https://doi.org/10.3390/molecules26185438
Kretić DS, Veljković IS, Đunović AB, Veljković DŽ. Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes. Molecules. 2021; 26(18):5438. https://doi.org/10.3390/molecules26185438
Chicago/Turabian StyleKretić, Danijela S., Ivana S. Veljković, Aleksandra B. Đunović, and Dušan Ž. Veljković. 2021. "Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes" Molecules 26, no. 18: 5438. https://doi.org/10.3390/molecules26185438
APA StyleKretić, D. S., Veljković, I. S., Đunović, A. B., & Veljković, D. Ž. (2021). Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes. Molecules, 26(18), 5438. https://doi.org/10.3390/molecules26185438