
Ruthenium Olefin Metathesis Catalysts Featuring N-Heterocyclic Carbene Ligands Tagged with Isonicotinic and 4-(Dimethylamino)benzoic Acid Rests: Evaluation of a Modular Synthetic Strategy


Abstract
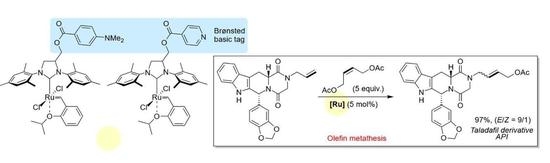
:
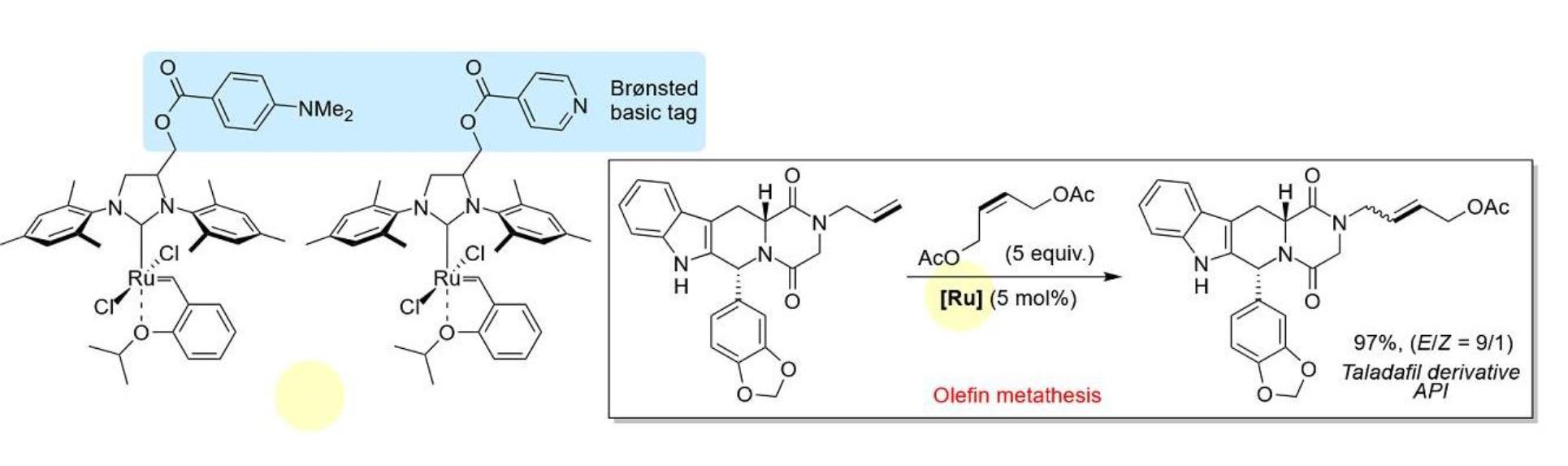{kind=link}
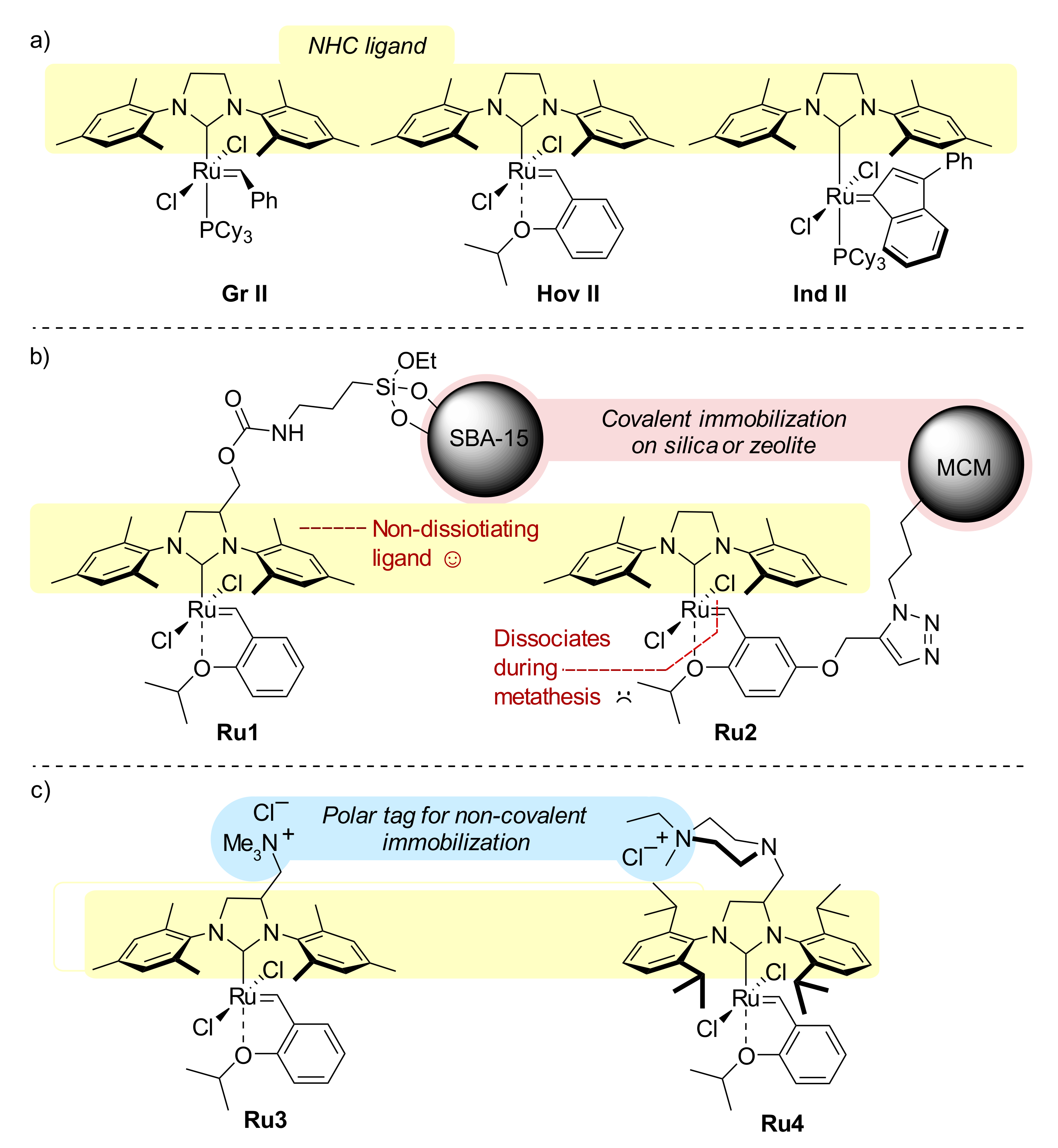{kind=link}
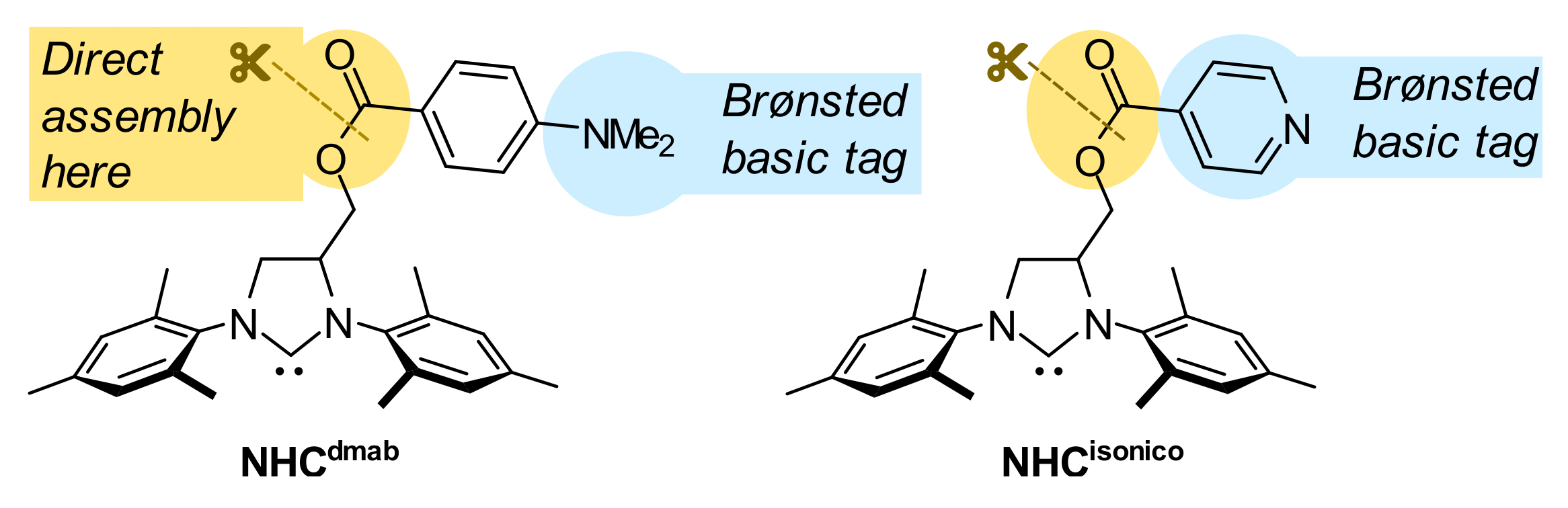{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
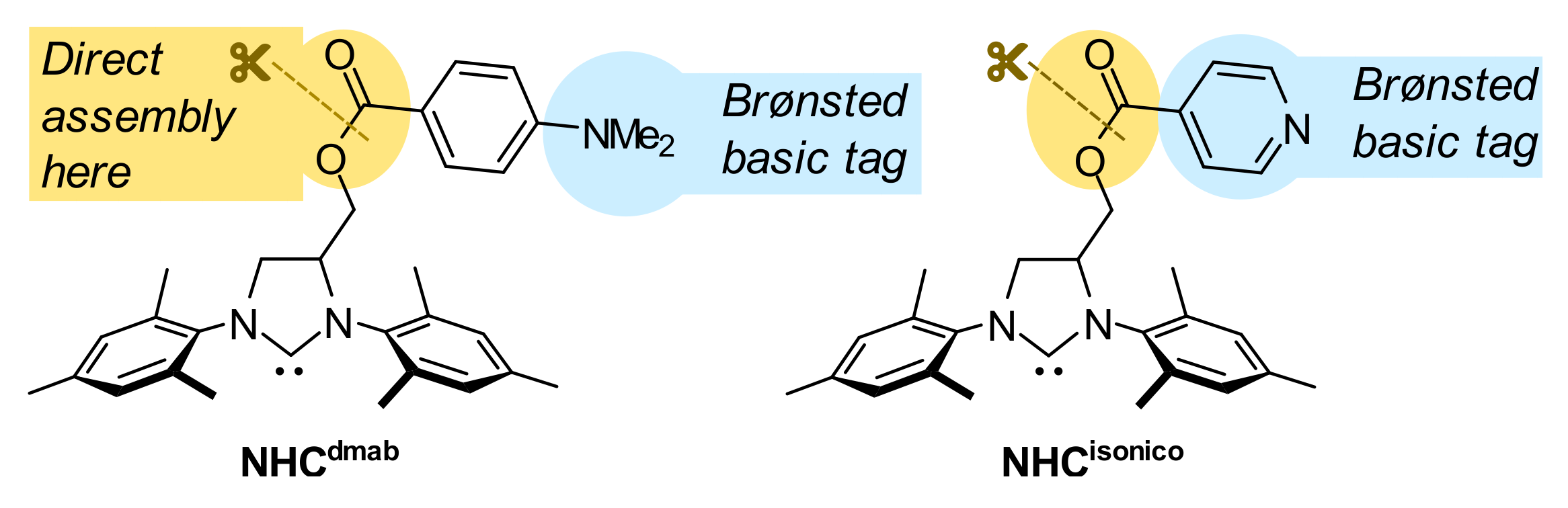
1. Introduction
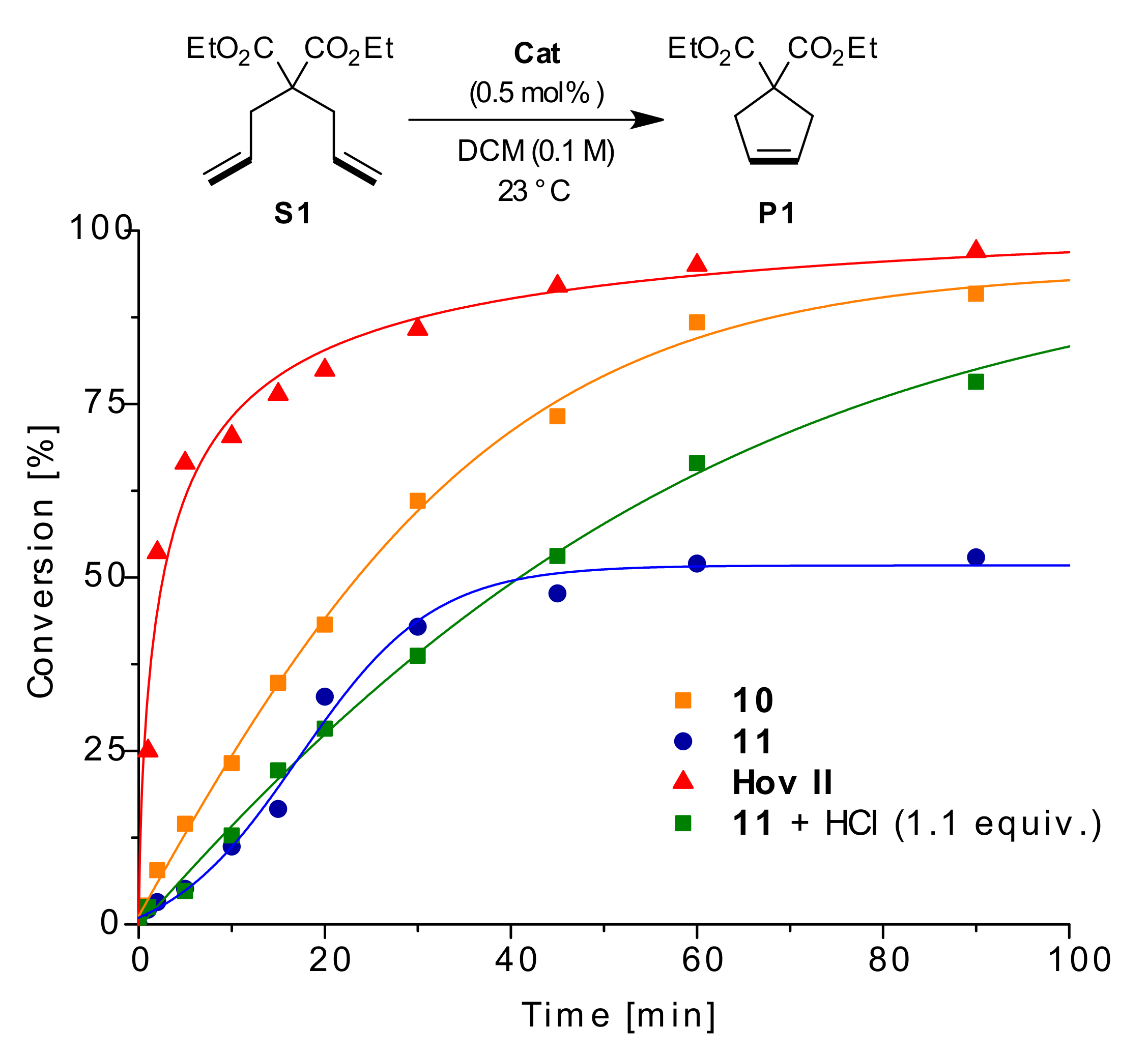
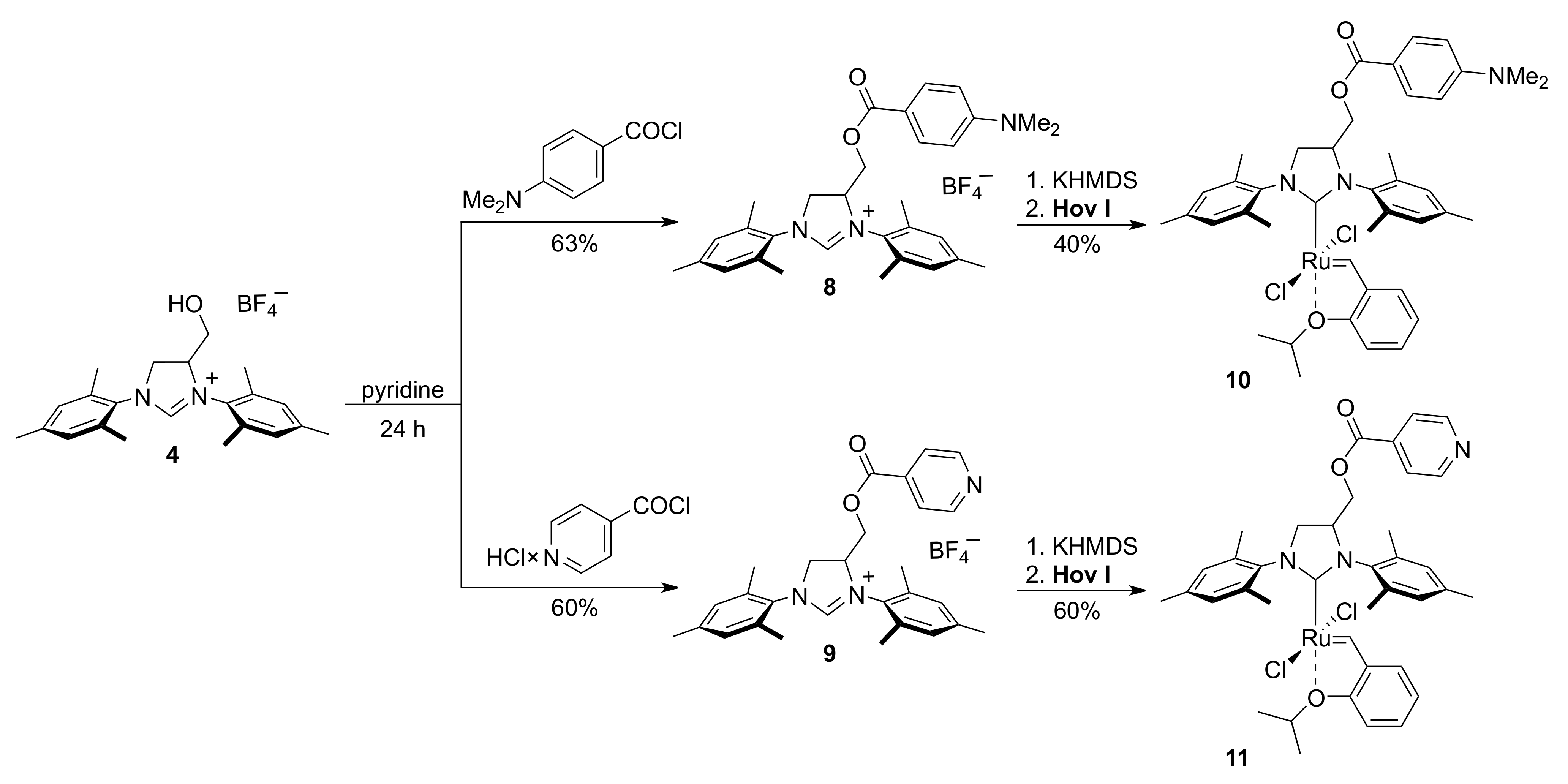
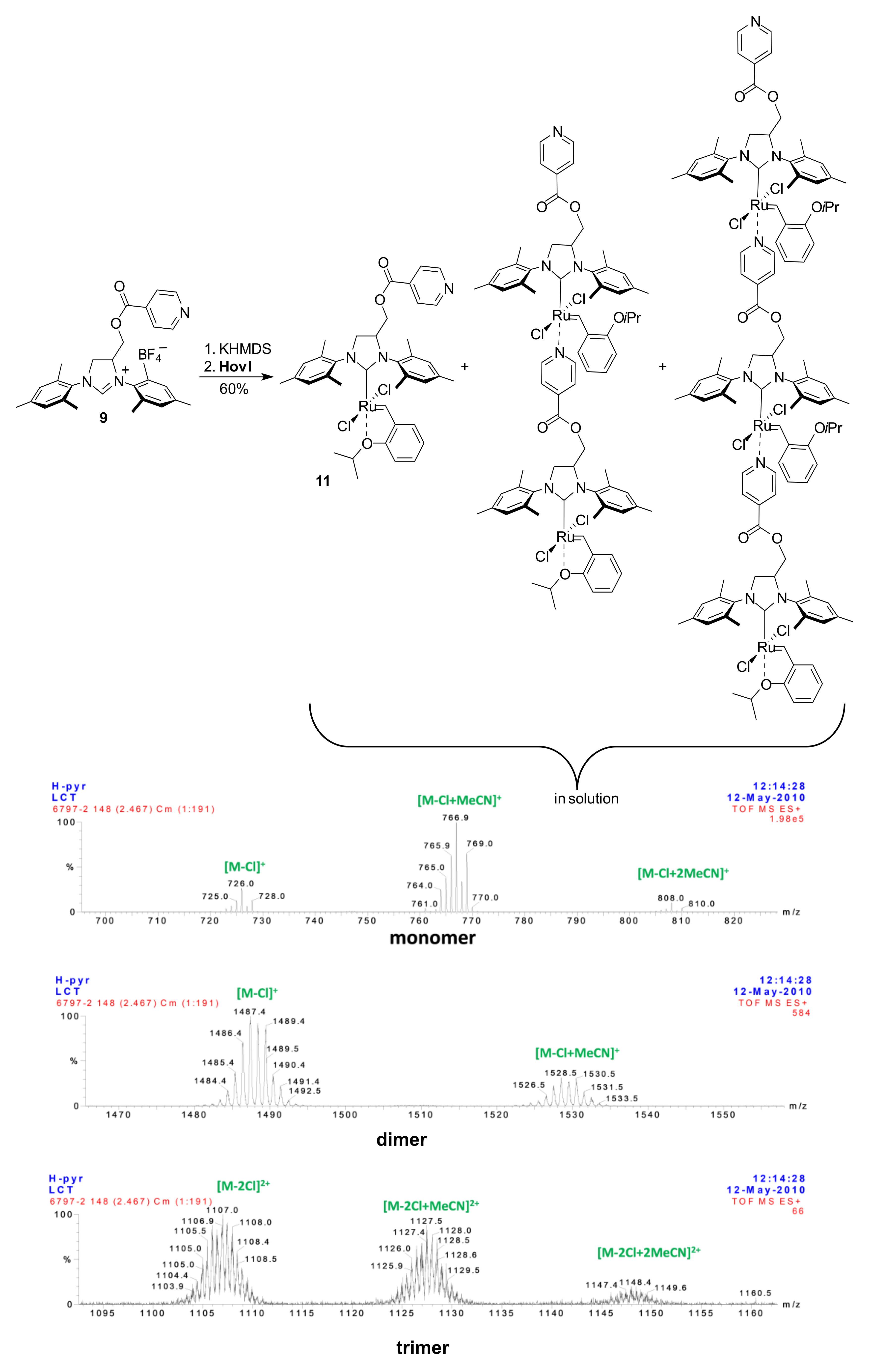
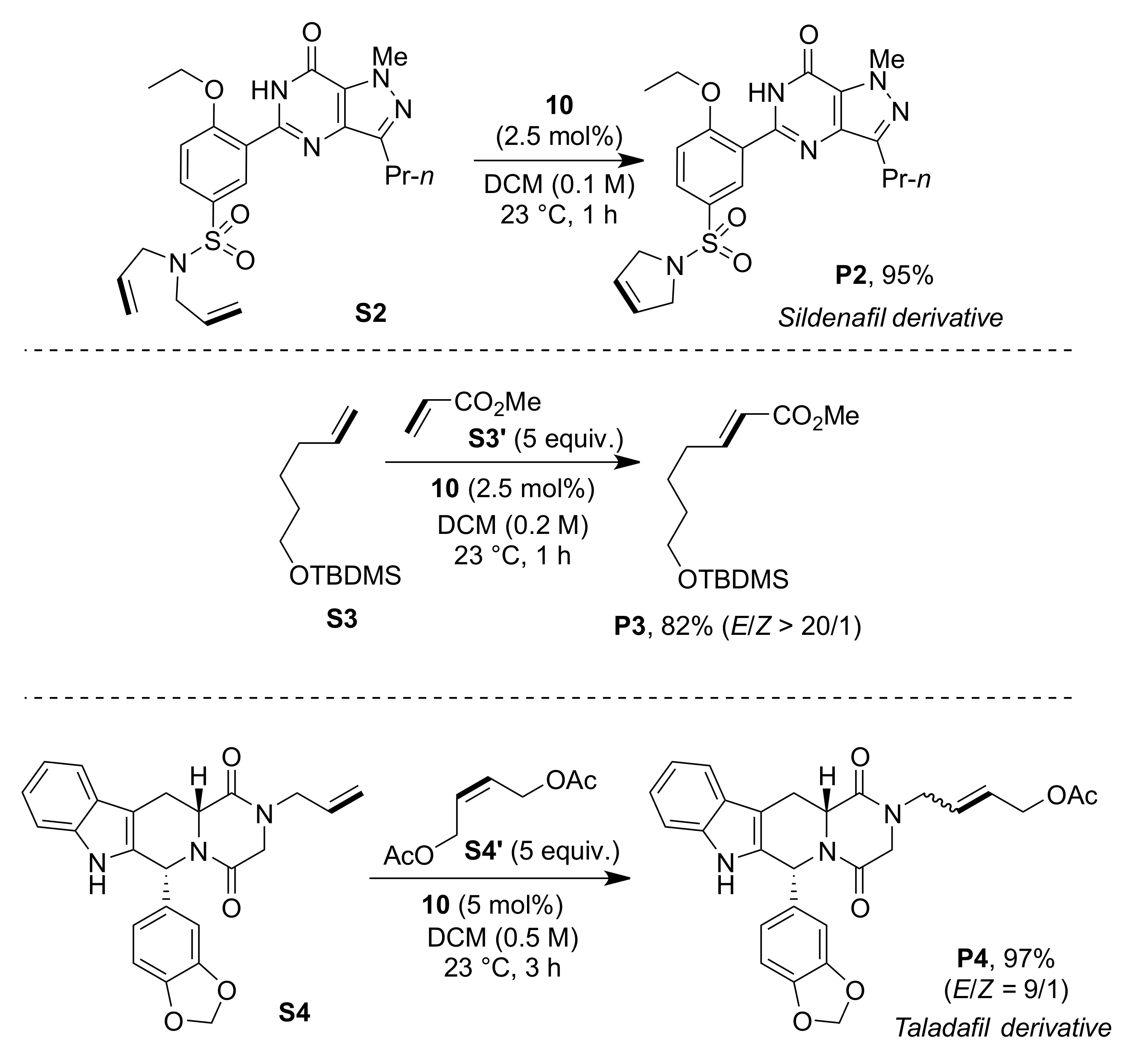
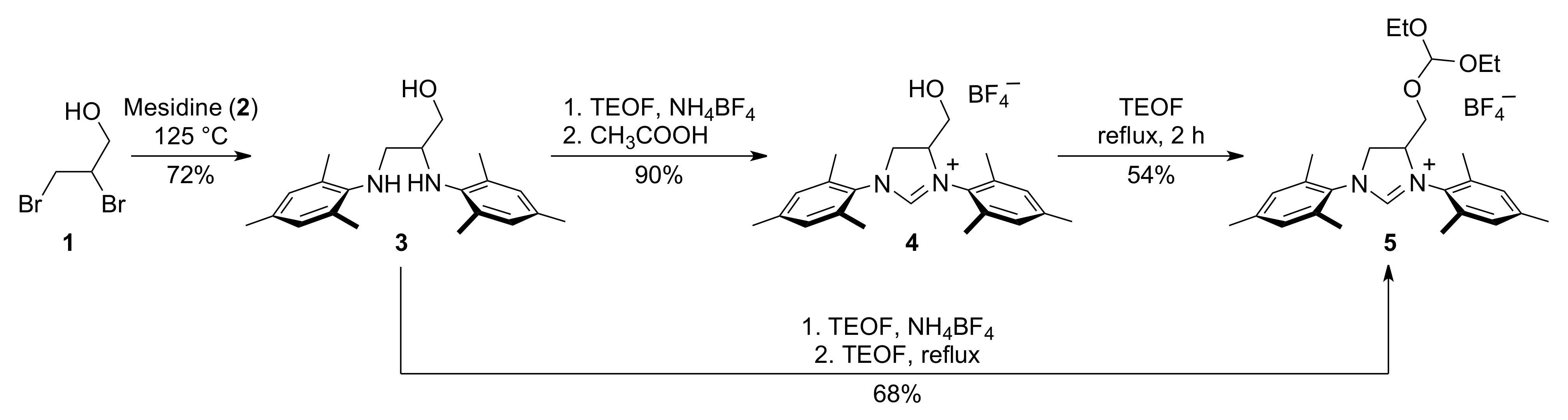
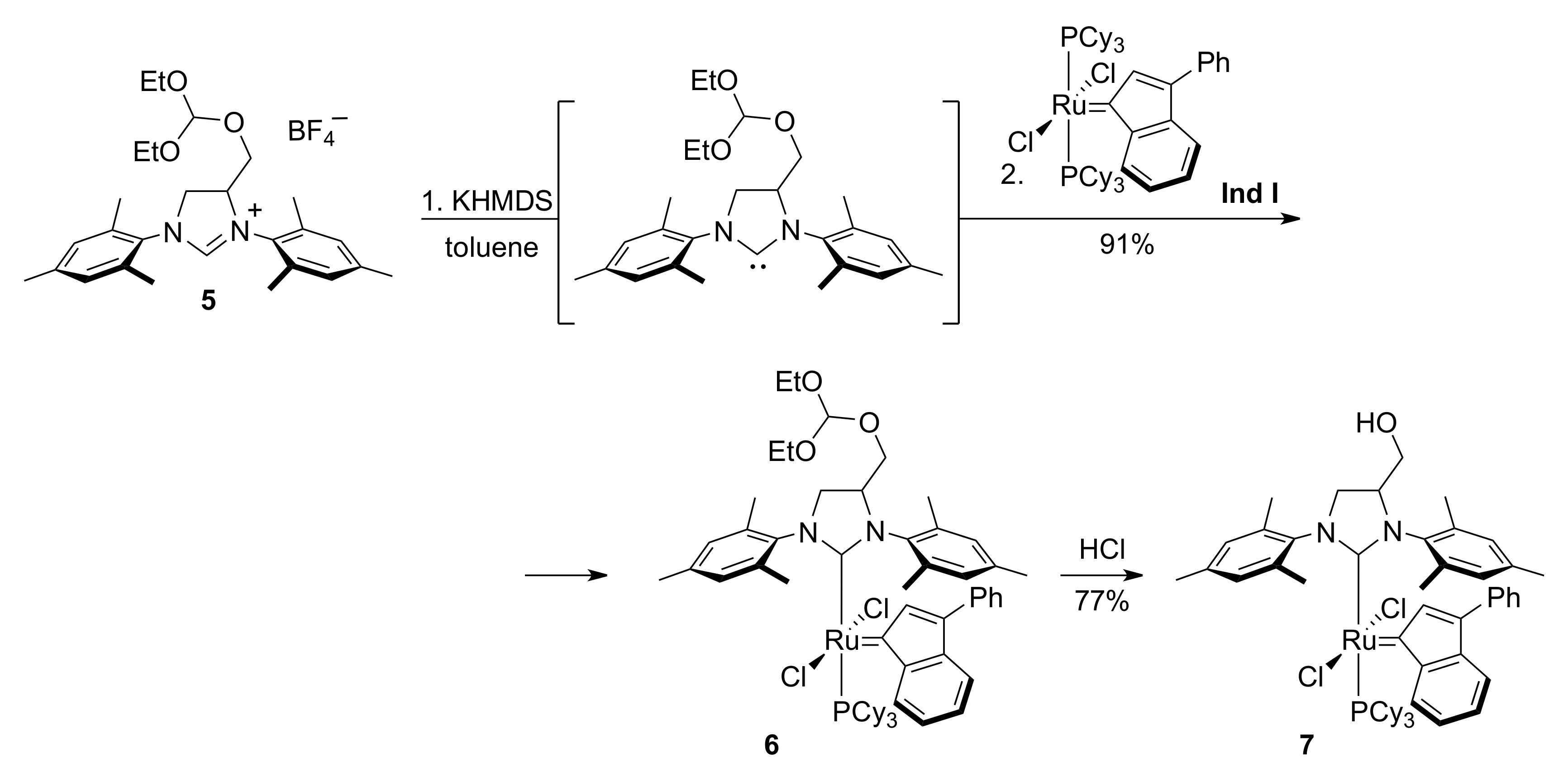
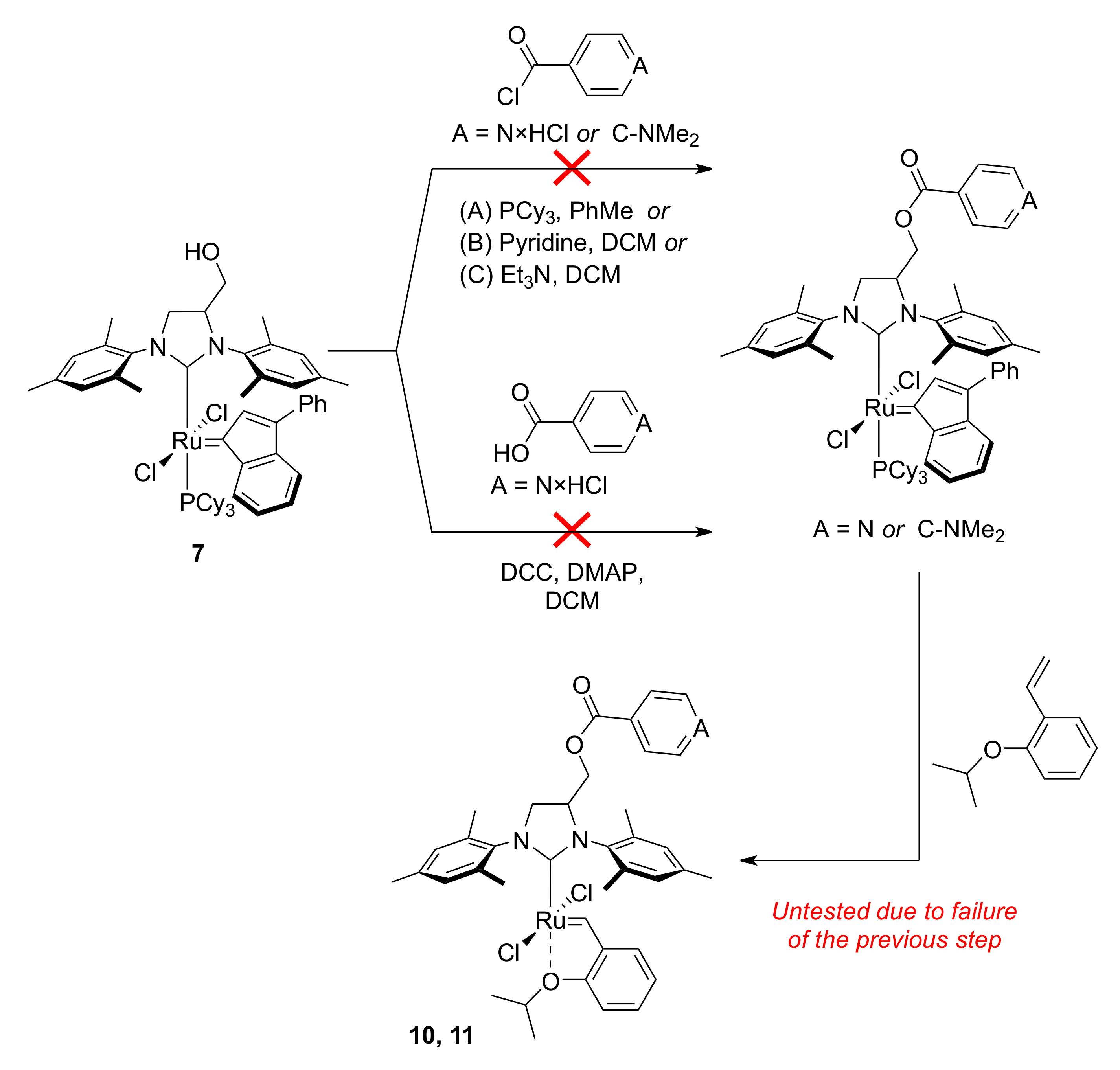
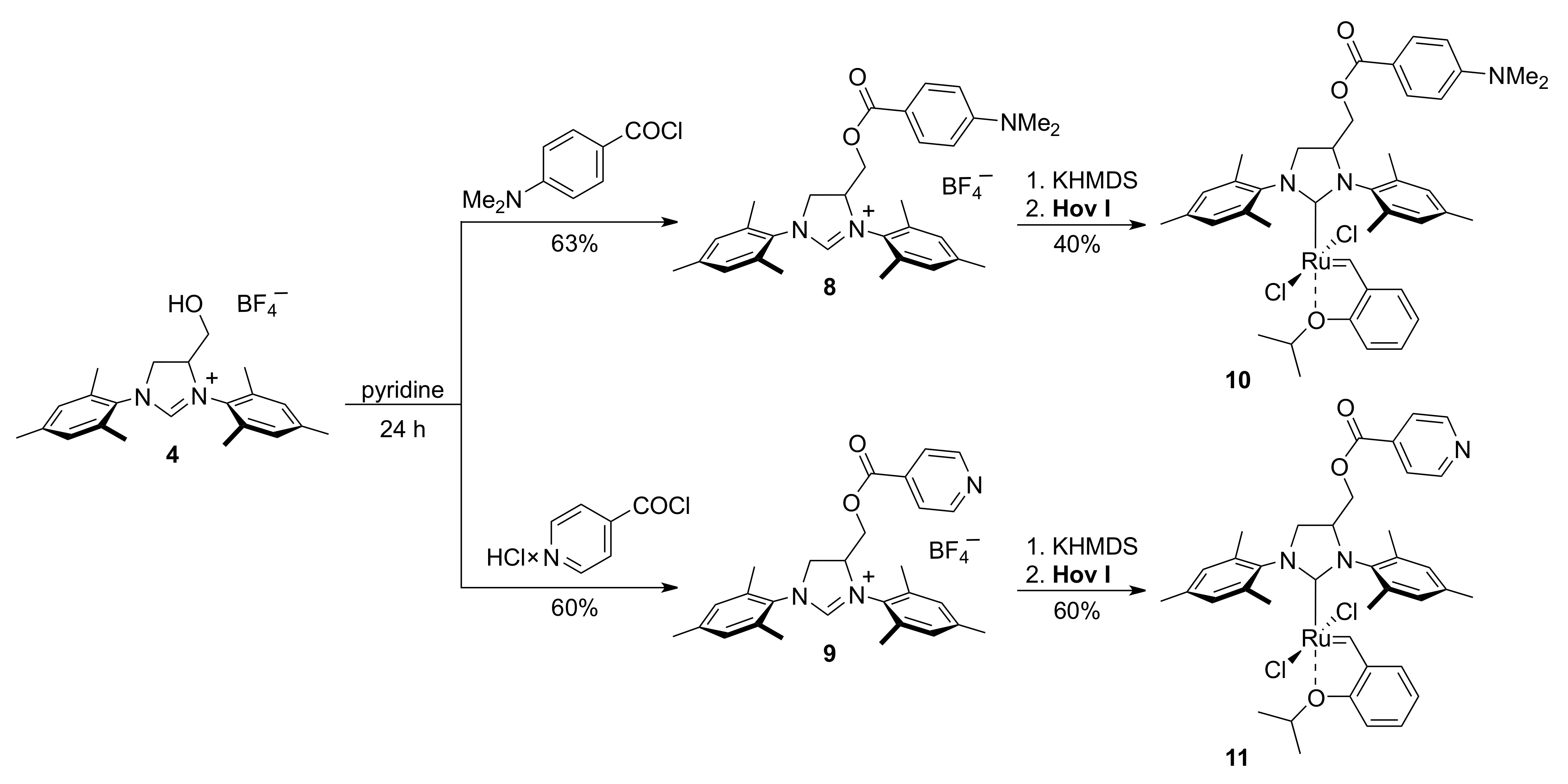
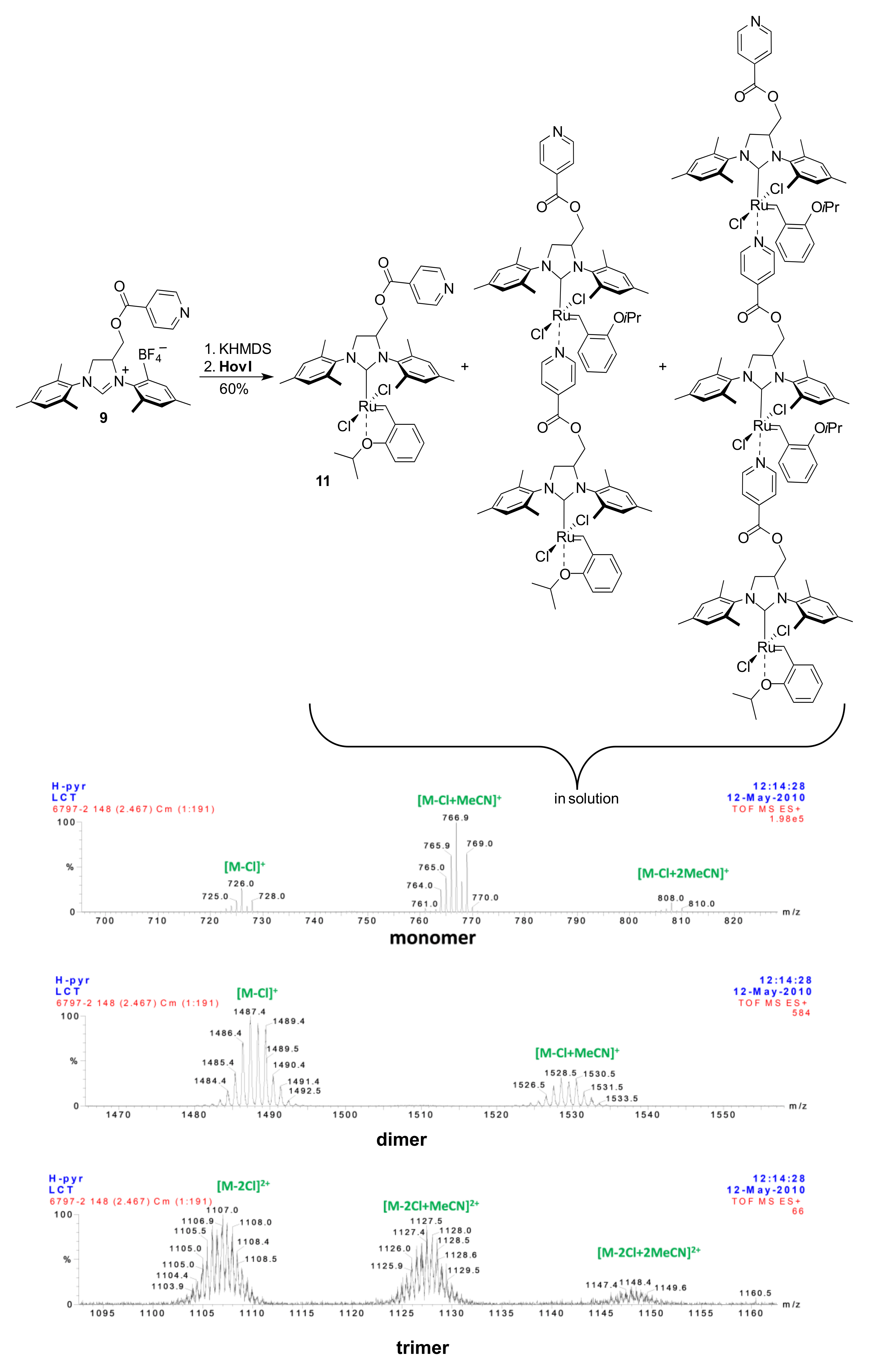
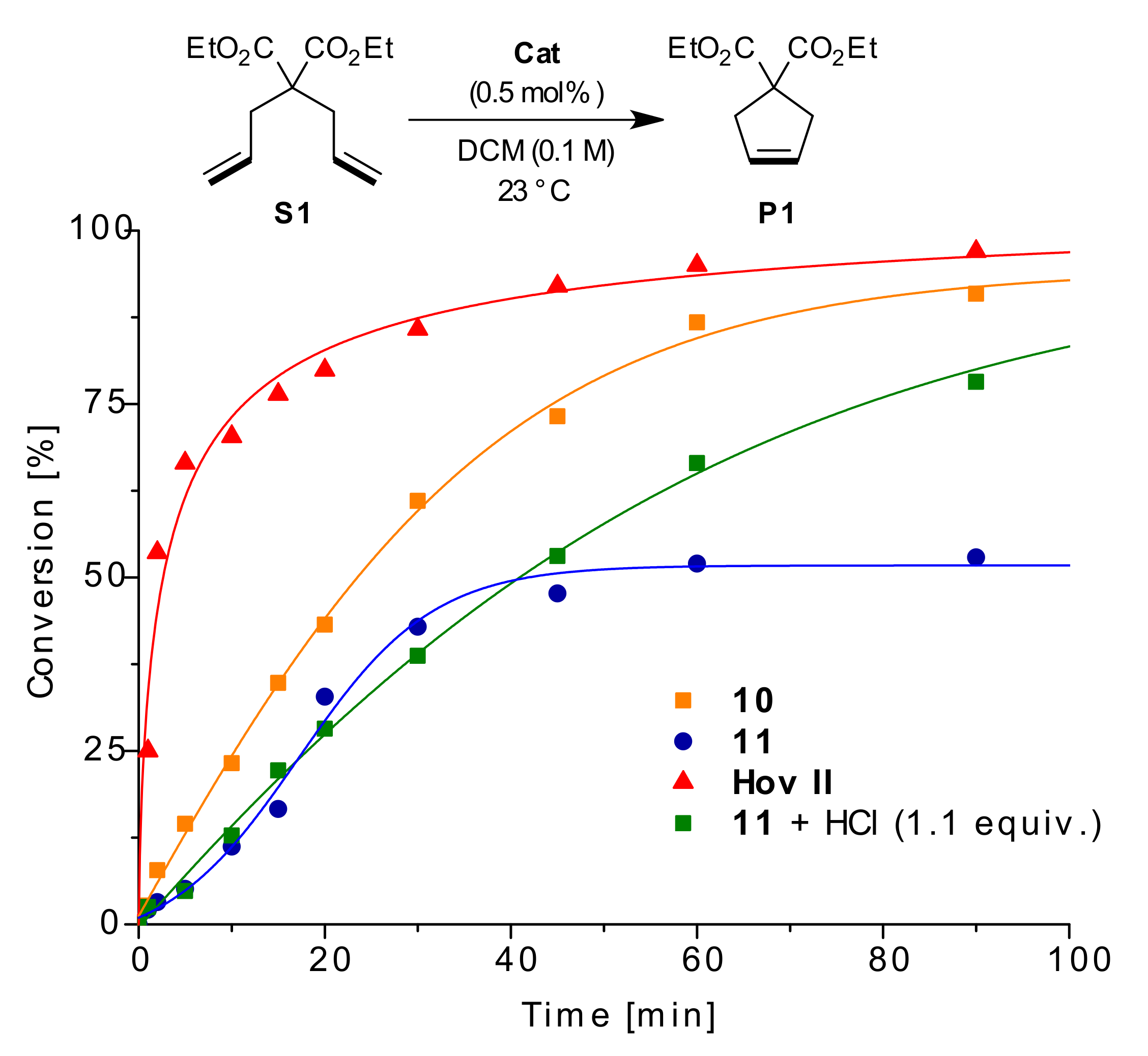
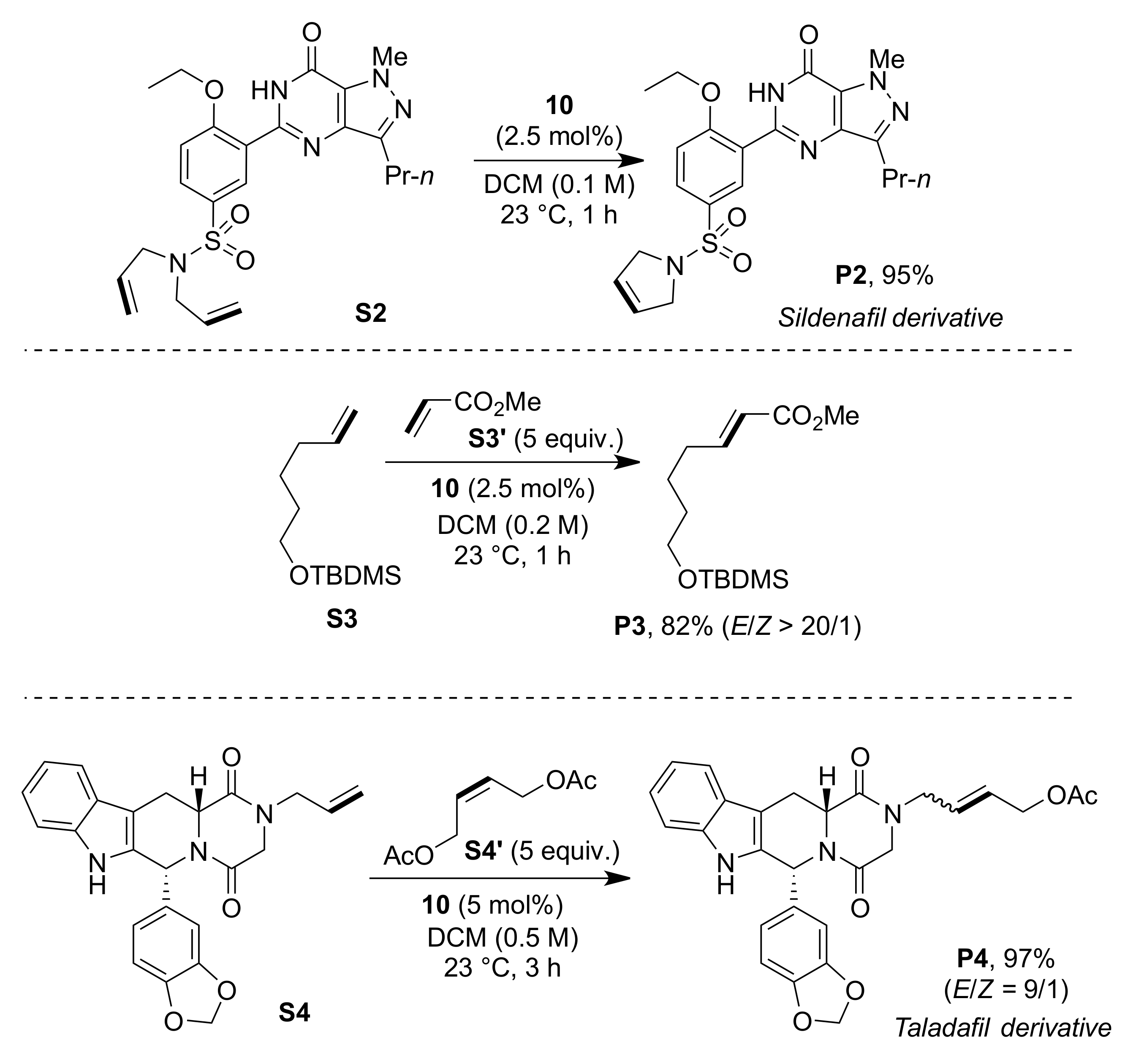
2. Results
3. Experimental Data
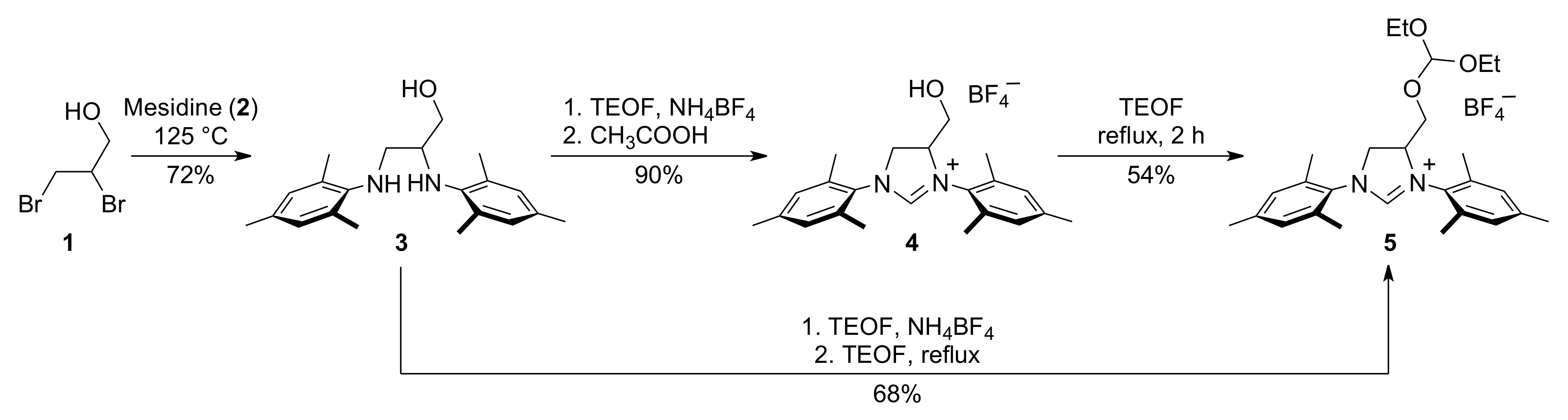
3.1. N,N′-Dimesityl-2,3-diamino-1-propanol (3)
3.2. 1,3-Bis(1-mesityl)-4,5-dihydro-4-hydroxymethyl-1H-imidazol-3-ium Tetrafluoroborate (4)
3.3. 1,3-Bis(1-mesityl)-4,5-dihydro-4-diethyloxymethoxymethyl-1H-imidazol-3-ium Tetrafluoroborate (5)
3.4. Alternative Synthesis of 1,3-bis(1-mesityl)-4,5-dihydro-4-diethyloxymethoxymethyl-1H-imidazol-3-ium Tetrafluoroborate (5)
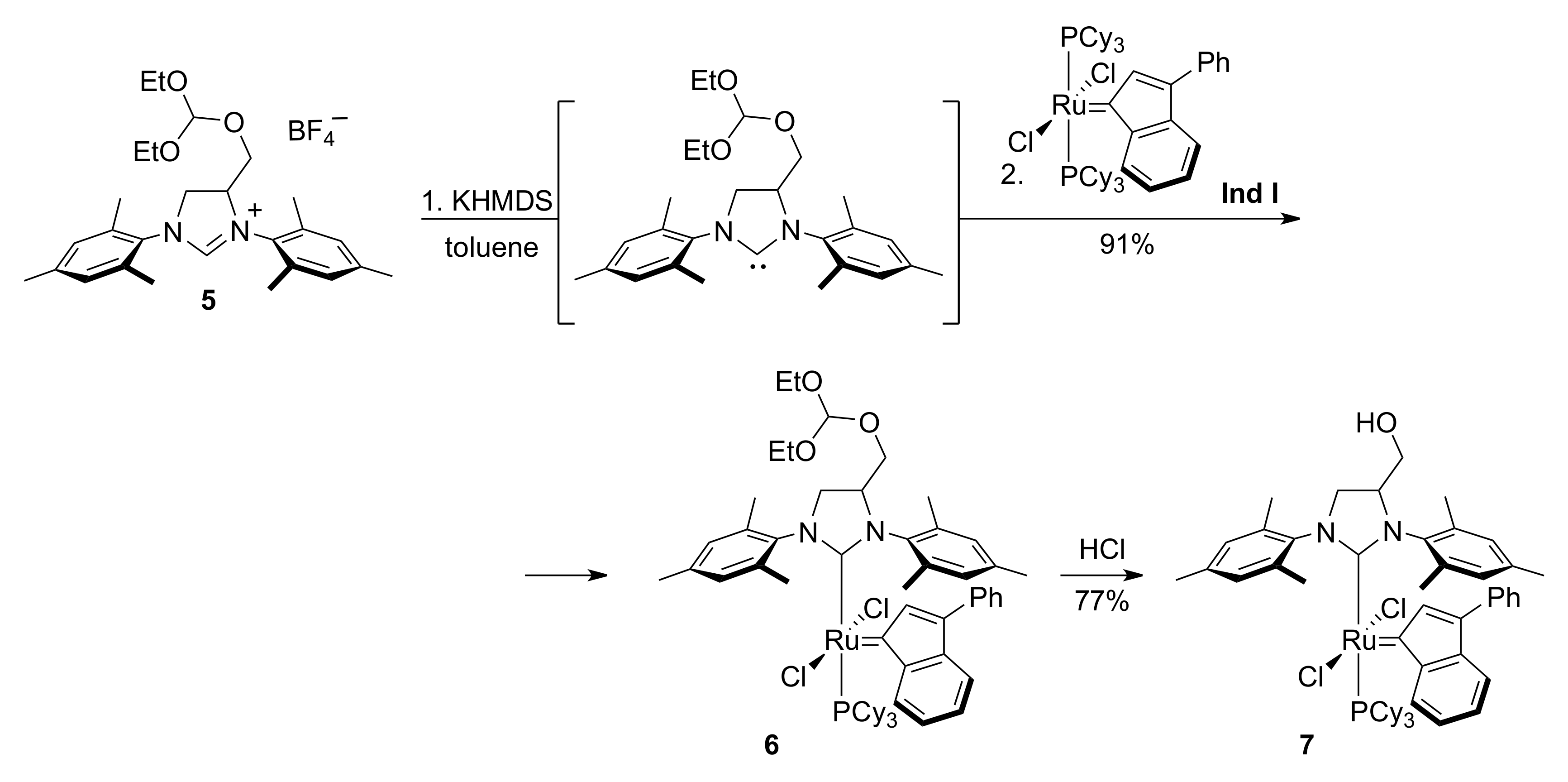
3.5. Catalyst 6
3.6. Catalyst 7
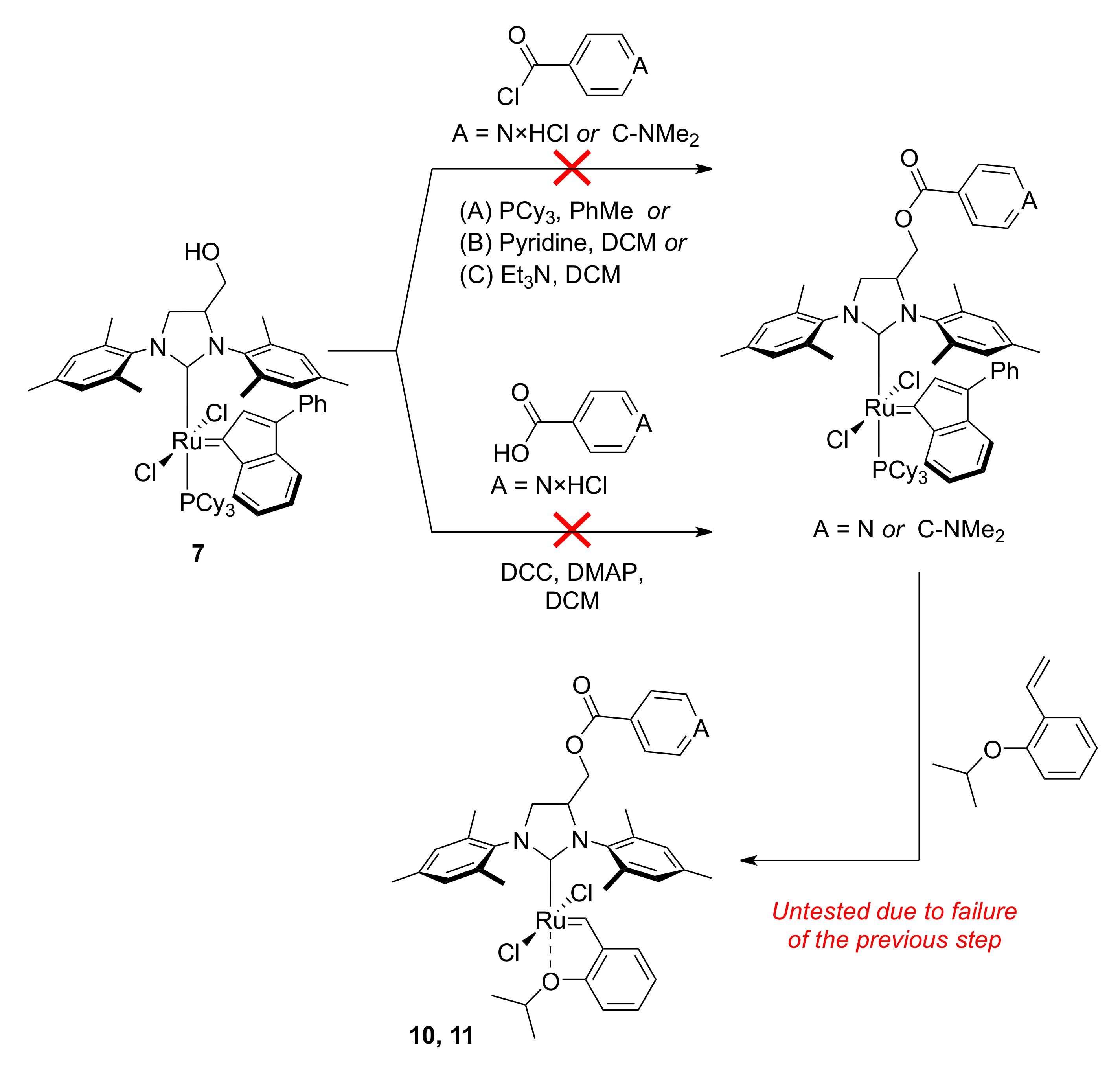
3.7. 1,3-Bis(1-mesityl)-4,5-dihydro-4-[4-(dimethylamino)benzoyloxymethyl]-1H-imidazol-3-ium Tetrafluoroborate (8)
3.8. 1,3-Bis(1-mesityl)-4,5-dihydro-4-isonicotinoyloxymethyl-1H-imidazol-3-ium Tetrafluoroborate (9)
3.9. Catalyst 10
3.10. Catalyst 11
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Grela, K. Preface. In Olefin Metathesis: Theory and Practice; Grela, K., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. ix–x. [Google Scholar]
- Handbook of Metathesis; Grubbs, R.H.; Wenzel, A.G.; O’Leary, D.J.; Khosravi, E. (Eds.) Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2009, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Samojłowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.P.; Johns, A.M.; Grubbs, R.H. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.S.; Baslé, O.; Mauduit, M. A tutorial review of stereoretentive olefin metathesis based on ruthenium dithiolate catalysts. Beilstein J. Org. Chem. 2018, 14, 2999–3010. [Google Scholar] [CrossRef]
- Grela, K.; Kajetanowicz, A.; Szadkowska, A.; Czaban-Jóźwiak, J. Alkene Cross-Metathesis Reactions. In Organic Reactions; Weinreb, S.M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; Volume 106, pp. 1–1189. [Google Scholar]
- Lin, S.; Wang, L.; Sharma, A. Acrylic boronate: A multifunctional C3 building block for catalytic synthesis of rare organoborons and chemoselective heterobifunctional ligations. Chem. Sci. 2021, 12, 7924–7929. [Google Scholar] [CrossRef]
- Kajetanowicz, A.; Grela, K. Nitro and Other Electron Withdrawing Group Activated Ruthenium Catalysts for Olefin Metathesis Reactions. Angew. Chem. Int. Ed. 2021, 60, 13738–13756. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.S.; Maynard, H.D. Modification of proteins using olefin metathesis. Mater. Chem. Front. 2020, 4, 1040–1051. [Google Scholar] [CrossRef]
- Yelchuri, V.; Srikanth, K.; Prasad, R.B.N.; Karuna, M.S.L. Olefin metathesis of fatty acids and vegetable oils. J. Chem. Sci. 2019, 131, 39. [Google Scholar] [CrossRef] [Green Version]
- Clavier, H.; Grela, K.; Kirschning, A.; Mauduit, M.; Nolan, S.P. Sustainable Concepts in Olefin Metathesis. Angew. Chem. Int. Ed. 2007, 46, 6786–6801. [Google Scholar] [CrossRef] [PubMed]
- Barbaras, D.; Brozio, J.; Johannsen, I.; Allmendinger, T. Removal of Heavy Metals from Organic Reaction Mixtures: Preparation and Application of Functionalized Resins. Org. Process. Res. Dev. 2009, 13, 1068–1079. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C. Removing Ruthenium Residues from Olefin Metathesis Reaction Products. Chem. A Eur. J. 2012, 18, 8868–8880. [Google Scholar] [CrossRef] [PubMed]
- Suriboot, J.; Bazzi, H.S.; Bergbreiter, D.E. Supported Catalysts Useful in Ring-Closing Metathesis, Cross Metathesis, and Ring-Opening Metathesis Polymerization. Polymers 2016, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bek, D.; Balcar, H.; Zílková, N.z.d.; Zukal, A.t.; Horáček, M.; Čejka, J. Grubbs Catalysts Immobilized on Mesoporous Molecular Sieves via Phosphine and Pyridine Linkers. ACS Catal. 2011, 1, 709–718. [Google Scholar] [CrossRef]
- Olszewski, T.K.; Bieniek, M.; Skowerski, K. Ruthenium-Based Complexes Bearing Quaternary Ammonium Tags as Versatile Catalysts for Olefin Metathesis: From the Discovery to Practical Applications. Org. Process. Res. Dev. 2020, 24, 125–145. [Google Scholar] [CrossRef]
- Jana, A.; Grela, K. Forged and fashioned for faithfulness—Ruthenium olefin metathesis catalysts bearing ammonium tags. Chem. Commun. 2018, 54, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Buchmeiser, M.R. Polymer-Supported Well-Defined Metathesis Catalysts. Chem. Rev. 2009, 109, 303–321. [Google Scholar] [CrossRef]
- Buchmeiser, M.R. Immobilization of Olefin Metathesis Catalysts. In Olefin Metathesis: Theory and Practice. Grela, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Szczepaniak, G.; Kosinski, K.; Grela, K. Towards “cleaner” olefin metathesis: Tailoring the NHC ligand of second generation ruthenium catalysts to afford auxiliary traits. Green Chem. 2014, 16, 4474–4492. [Google Scholar] [CrossRef]
- Hübner, S.; de Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Mennecke, K.; Grela, K.; Kunz, U.; Kirschning, A. Immobilisation of the Grubbs III Olefin Metathesis Catalyst with Polyvinyl Pyridine (PVP). Synlett 2005, 2005, 2948–2952. [Google Scholar] [CrossRef]
- Pastva, J.; Skowerski, K.; Czarnocki, S.J.; Žilková, N.; Čejka, J.; Bastl, Z.; Balcar, H. Ru-Based Complexes with Quaternary Ammonium Tags Immobilized on Mesoporous Silica as Olefin Metathesis Catalysts. ACS Catal. 2014, 4, 3227–3236. [Google Scholar] [CrossRef]
- Michrowska, A.; Mennecke, K.; Kunz, U.; Kirschning, A.; Grela, K. A New Concept for the Noncovalent Binding of a Ruthenium-Based Olefin Metathesis Catalyst to Polymeric Phases: Preparation of a Catalyst on Raschig Rings. J. Am. Chem. Soc. 2006, 128, 13261–13267. [Google Scholar] [CrossRef]
- Kirschning, A.; Harmrolfs, K.; Mennecke, K.; Messinger, J.; Schön, U.; Grela, K. Homo- and heterogeneous Ru-based metathesis catalysts in cross-metathesis of 15-allylestrone—towards 17β-hydroxysteroid dehydrogenase type 1 inhibitors. Tetrahedron Lett. 2008, 49, 3019–3022. [Google Scholar] [CrossRef]
- Chołuj, A.; Nogaś, W.; Patrzałek, M.; Krzesiński, P.; Chmielewski, M.J.; Kajetanowicz, A.; Grela, K. Preparation of Ruthenium Olefin Metathesis Catalysts Immobilized on MOF, SBA-15, and 13X for Probing Heterogeneous Boomerang Effect. Catalysts 2020, 10, 438. [Google Scholar] [CrossRef]
- Liu, G.; Wu, B.; Zhang, J.; Wang, X.; Shao, M.; Wang, J. Controlled Reversible Immobilization of Ru Carbene on Single-Walled Carbon Nanotubes: A New Strategy for Green Catalytic Systems Based on a Solvent Effect on π−π Interaction. Inorg. Chem. 2009, 48, 2383–2390. [Google Scholar] [CrossRef]
- Nasrallah, H.; Germain, S.; Queval, P.; Bouvier, C.; Mauduit, M.; Crévisy, C.; Schulz, E. Non covalent immobilization of pyrene-tagged ruthenium complexes onto graphene surfaces for recycling in olefin metathesis reactions. J. Mol. Catal. A: Chem. 2016, 425, 136–146. [Google Scholar] [CrossRef]
- Cabrera, J.; Padilla, R.; Bru, M.; Lindner, R.; Kageyama, T.; Wilckens, K.; Balof, S.L.; Schanz, H.-J.; Dehn, R.; Teles, J.H.; et al. Linker-Free, Silica-Bound Olefin-Metathesis Catalysts: Applications in Heterogeneous Catalysis. Chem. A Eur. J. 2012, 18, 14717–14724. [Google Scholar] [CrossRef] [PubMed]
- Skowerski, K.; Wierzbicka, C.; Szczepaniak, G.; Gulajski, L.; Bieniek, M.; Grela, K. Easily removable olefin metathesis catalysts. Green Chem. 2012, 14, 3264–3268. [Google Scholar] [CrossRef]
- Van Berlo, B.; Houthoofd, K.; Sels, B.F.; Jacobs, P.A. Silica Immobilized Second Generation Hoveyda-Grubbs: A Convenient, Recyclable and Storageable Heterogeneous Solid Catalyst. Adv. Synth. Catal. 2008, 350, 1949–1953. [Google Scholar] [CrossRef]
- Kirschning, A.; Gułajski, Ł.; Mennecke, K.; Meyer, A.; Busch, T.; Grela, K. Highly Active Ammonium-Tagged Olefin-Metathesis Catalyst for Simplified Purification. Synlett 2008, 2008, 2692–2696. [Google Scholar] [CrossRef]
- Solodenko, W.; Doppiu, A.; Frankfurter, R.; Vogt, C.; Kirschning, A. Silica Immobilized Hoveyda Type Pre-Catalysts: Convenient and Reusable Heterogeneous Catalysts for Batch and Flow Olefin Metathesis. Aust. J. Chem. 2013, 66, 183–191. [Google Scholar] [CrossRef]
- Michrowska, A.; Gulajski, L.; Grela, K. A simple and practical phase-separation approach to the recycling of a homogeneous metathesis catalyst. Chem. Commun. 2006, 8, 841–843. [Google Scholar] [CrossRef]
- Skowerski, K.; Białecki, J.; Czarnocki, S.J.; Żukowska, K.; Grela, K. Effective immobilisation of a metathesis catalyst bearing an ammonium-tagged NHC ligand on various solid supports. Beilstein J. Org. Chem. 2016, 12, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Diallo, A.K.; Boisselier, E.; Liang, L.; Ruiz, J.; Astruc, D. Dendrimer-Induced Molecular Catalysis in Water: The Example of Olefin Metathesis. Chem. A Eur. J. 2010, 16, 11832–11835. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Diallo, A.K.; Gatard, S.; Liang, L.; Ornelas, C.; Martinez, V.; Méry, D.; Ruiz, J. Olefin metathesis in nano-sized systems. Beilstein J. Org. Chem. 2011, 7, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Balcar, H.; Čejka, J. SBA-15 as a Support for Effective Olefin Metathesis Catalysts. Catalysts 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Chołuj, A.; Zieliński, A.; Grela, K.; Chmielewski, M.J. Metathesis@MOF: Simple and Robust Immobilization of Olefin Metathesis Catalysts inside (Al)MIL-101-NH2. ACS Catal. 2016, 6, 6343–6349. [Google Scholar] [CrossRef]
- Chołuj, A.; Krzesiński, P.; Ruszczyńska, A.; Bulska, E.; Kajetanowicz, A.; Grela, K. Noncovalent Immobilization of Cationic Ruthenium Complex in a Metal–Organic Framework by Ion Exchange Leading to a Heterogeneous Olefin Metathesis Catalyst for Use in Green Solvents. Organometallics 2019, 38, 3397–3405. [Google Scholar] [CrossRef]
- Chołuj, A.; Karczykowski, R.; Chmielewski, M.J. Simple and Robust Immobilization of a Ruthenium Olefin Metathesis Catalyst Inside MOFs by Acid–Base Reaction. Organometallics 2019, 38, 3392–3396. [Google Scholar] [CrossRef]
- Skowerski, K.; Pastva, J.; Czarnocki, S.J.; Janoscova, J. Exceptionally Stable and Efficient Solid Supported Hoveyda-Type Catalyst. Org. Process. Res. Dev. 2015, 19, 872–877. [Google Scholar] [CrossRef]
- Skowerski, K.; Szczepaniak, G.; Wierzbicka, C.; Gułajski, Ł.; Bieniek, M.; Grela, K. Highly active catalysts for olefin metathesis in water. Catal. Sci. Technol. 2012, 2, 2424–2427. [Google Scholar] [CrossRef]
- Patrzałek, M.; Piątkowski, J.; Kajetanowicz, A.; Grela, K. Anion Metathesis in Facile Preparation of Olefin Metathesis Catalysts Bearing a Quaternary Ammonium Chloride Tag. Synlett 2019, 30, 1981–1987. [Google Scholar] [CrossRef] [Green Version]
- Milewski, M.; Kajetanowicz, A.; Grela, K. Improved preparation of an olefin metathesis catalyst bearing quaternary ammonium tag (FixCat) and its use in ethenolysis and macrocyclization reactions after immobilization on metal-organic framework (MOF). Arkivoc 2021, 73–84. [Google Scholar] [CrossRef]
- Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups; Pearson, A.L.; Roush, W.J. (Eds.) John Wiley and Sons Ltd.: London, UK, 1999; pp. 42–44. [Google Scholar]
- Monge-Marcet, A.; Pleixats, R.; Cattoën, X.; Wong Chi Man, M. Catalytic applications of recyclable silica immobilized NHC–ruthenium complexes. Tetrahedron 2013, 69, 341–348. [Google Scholar] [CrossRef]
- Sauer, D.F.; Bocola, M.; Broglia, C.; Arlt, M.; Zhu, L.-L.; Brocker, M.; Schwaneberg, U.; Okuda, J. Hybrid Ruthenium ROMP Catalysts Based on an Engineered Variant of β-Barrel Protein FhuA ΔCVFtev: Effect of Spacer Length. Chem. Asian J. 2015, 10, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K. Immobilizable N-Heterocyclic Carbene Metal Complexes with Alkoxysilyl Groups. WO2007017047A1, 2007. [Google Scholar]
- Mayr, M.; Wang, D.; Kröll, R.; Schuler, N.; Prühs, S.; Fürstner, A.; Buchmeiser, M.R. Monolithic Disk-Supported Metathesis Catalysts for Use in Combinatorial Chemistry. Adv. Synth. Catal. 2005, 347, 484–492. [Google Scholar] [CrossRef]
- Ritter, T.; Hejl, A.; Wenzel, A.G.; Funk, T.W.; Grubbs, R.H. A Standard System of Characterization for Olefin Metathesis Catalysts. Organometallics 2006, 25, 5740–5745. [Google Scholar] [CrossRef] [Green Version]
- Monsigny, L.; Piątkowski, J.; Trzybiński, D.; Wozniak, K.; Nienałtowski, T.; Kajetanowicz, A.; Grela, K. Activated Hoveyda-Grubbs Olefin Metathesis Catalysts Derived from a Large Scale Produced Pharmaceutical Intermediate—Sildenafil Aldehyde. Adv. Synth. Catal. 2021. [Google Scholar] [CrossRef]
- Mayr, M.; Buchmeiser, M.R.; Wurst, K. Synthesis of a Silica-Based Heterogeneous Second Generation Grubbs Catalyst. Adv. Synth. Catal. 2002, 344, 712–719. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnocki, S.; Monsigny, L.; Sienkiewicz, M.; Kajetanowicz, A.; Grela, K. Ruthenium Olefin Metathesis Catalysts Featuring N-Heterocyclic Carbene Ligands Tagged with Isonicotinic and 4-(Dimethylamino)benzoic Acid Rests: Evaluation of a Modular Synthetic Strategy. Molecules 2021, 26, 5220. https://doi.org/10.3390/molecules26175220
Czarnocki S, Monsigny L, Sienkiewicz M, Kajetanowicz A, Grela K. Ruthenium Olefin Metathesis Catalysts Featuring N-Heterocyclic Carbene Ligands Tagged with Isonicotinic and 4-(Dimethylamino)benzoic Acid Rests: Evaluation of a Modular Synthetic Strategy. Molecules. 2021; 26(17):5220. https://doi.org/10.3390/molecules26175220
Chicago/Turabian StyleCzarnocki, Stefan, Louis Monsigny, Michał Sienkiewicz, Anna Kajetanowicz, and Karol Grela. 2021. "Ruthenium Olefin Metathesis Catalysts Featuring N-Heterocyclic Carbene Ligands Tagged with Isonicotinic and 4-(Dimethylamino)benzoic Acid Rests: Evaluation of a Modular Synthetic Strategy" Molecules 26, no. 17: 5220. https://doi.org/10.3390/molecules26175220
APA StyleCzarnocki, S., Monsigny, L., Sienkiewicz, M., Kajetanowicz, A., & Grela, K. (2021). Ruthenium Olefin Metathesis Catalysts Featuring N-Heterocyclic Carbene Ligands Tagged with Isonicotinic and 4-(Dimethylamino)benzoic Acid Rests: Evaluation of a Modular Synthetic Strategy. Molecules, 26(17), 5220. https://doi.org/10.3390/molecules26175220