
Chemical Modification of Glycosaminoglycan Polysaccharides

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
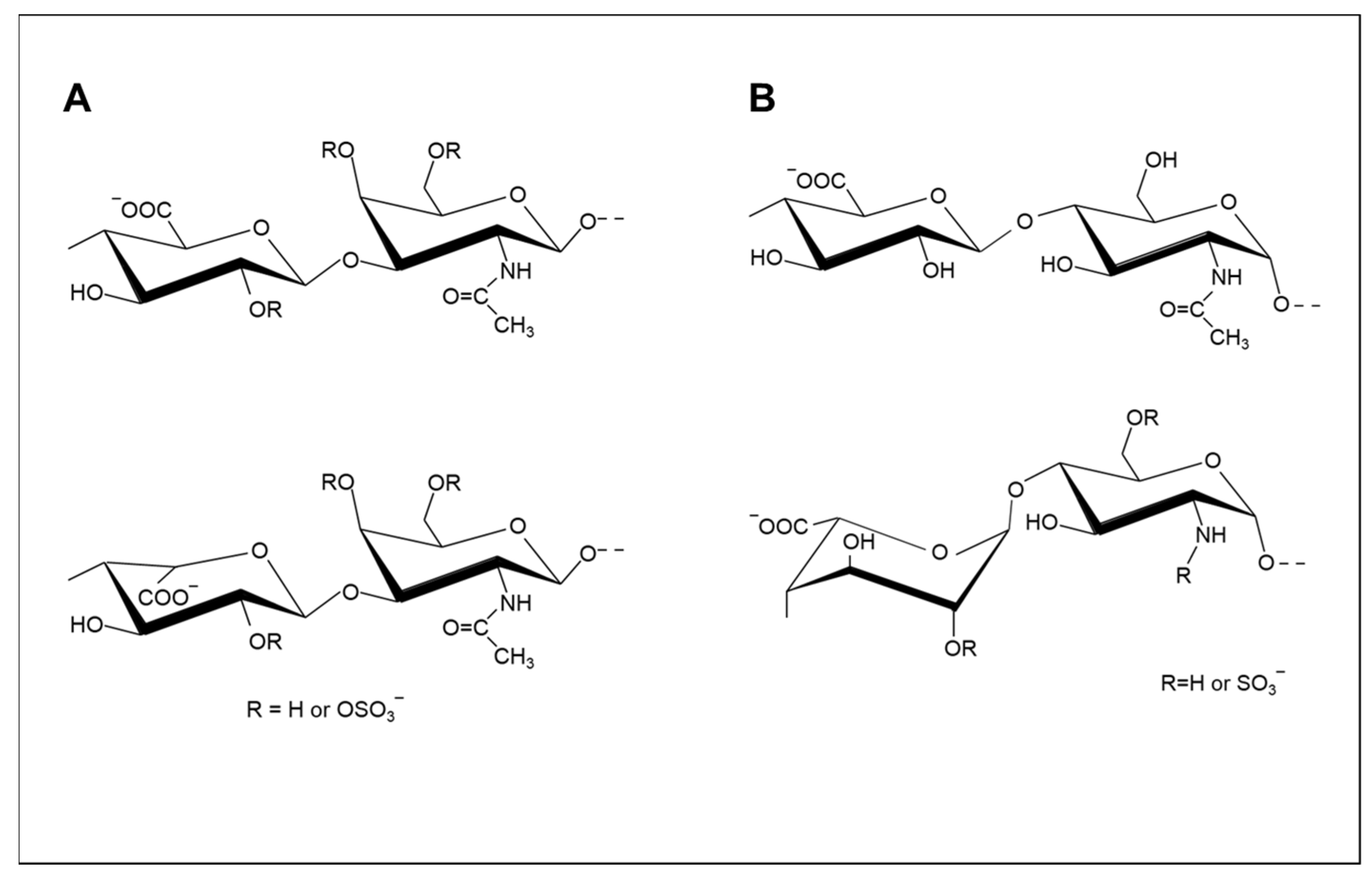
1.1. Structure and Classes of Glycosaminoglycans
1.2. General Properties
1.3. Biosynthesis
1.4. Glycosaminoglycan Interactions with Proteins
2. Chemical Modifications
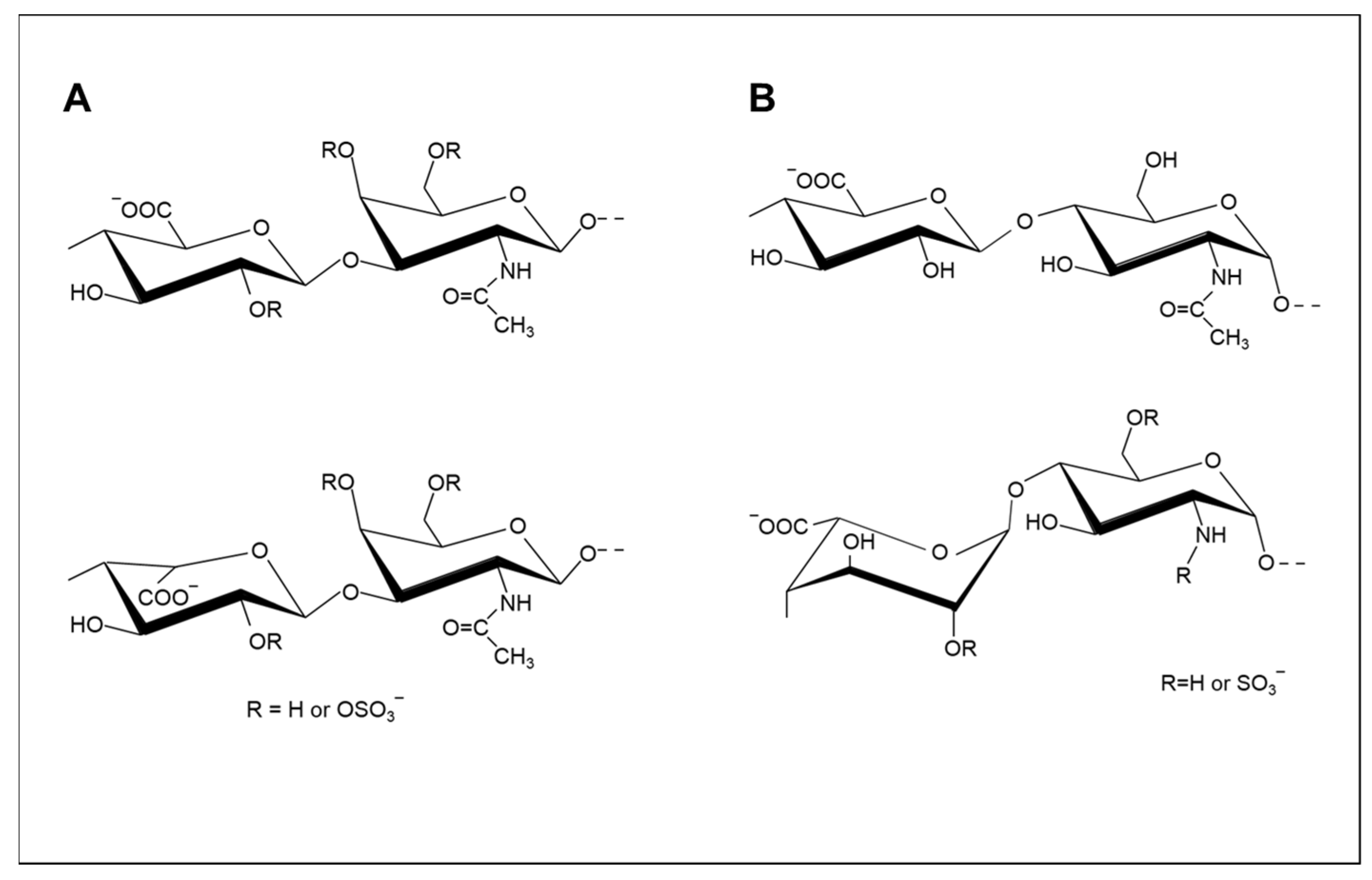
2.1. Modification of Sulfation Patterns
2.1.1. Selective Addition of Sulfate Groups
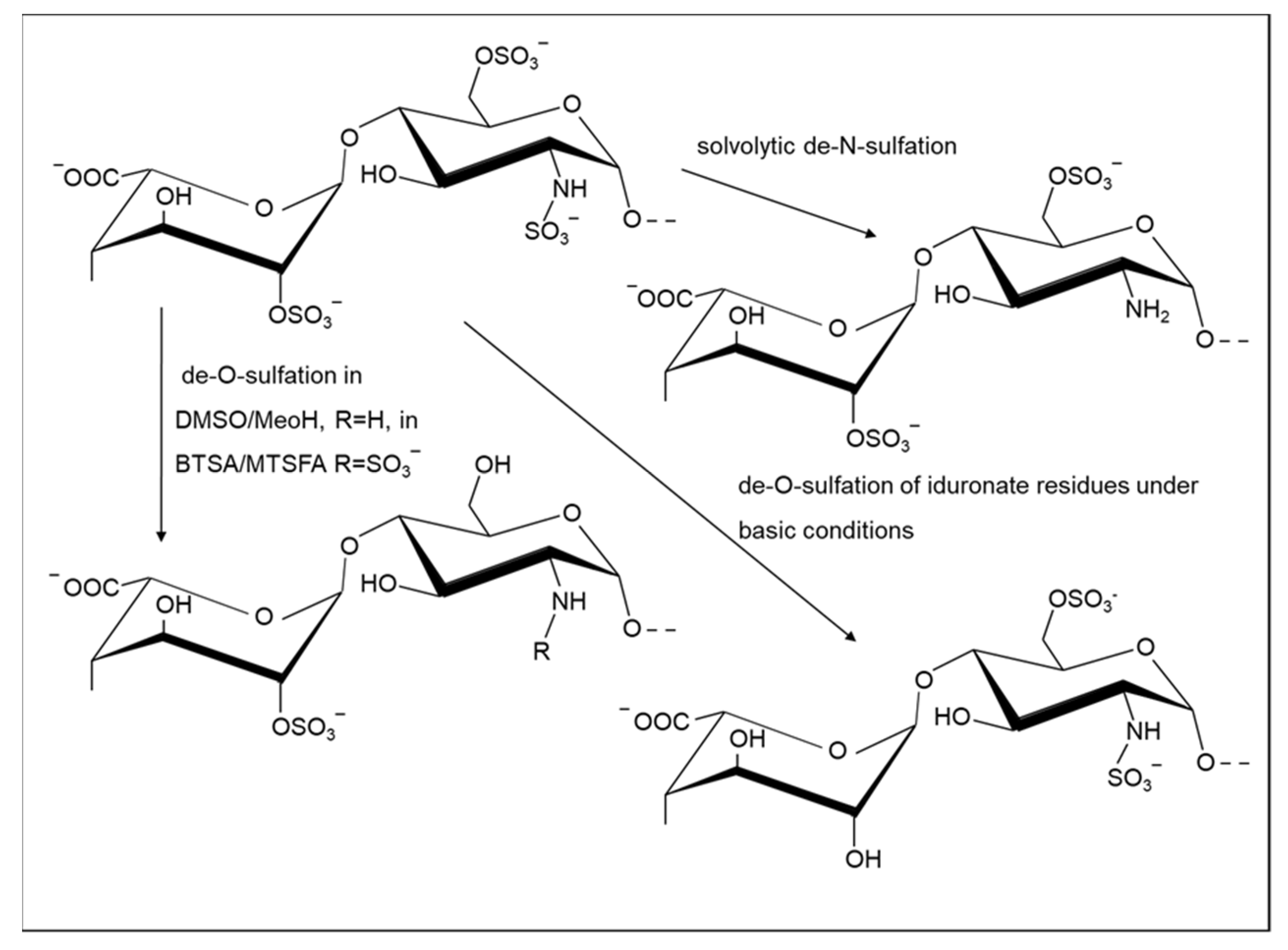
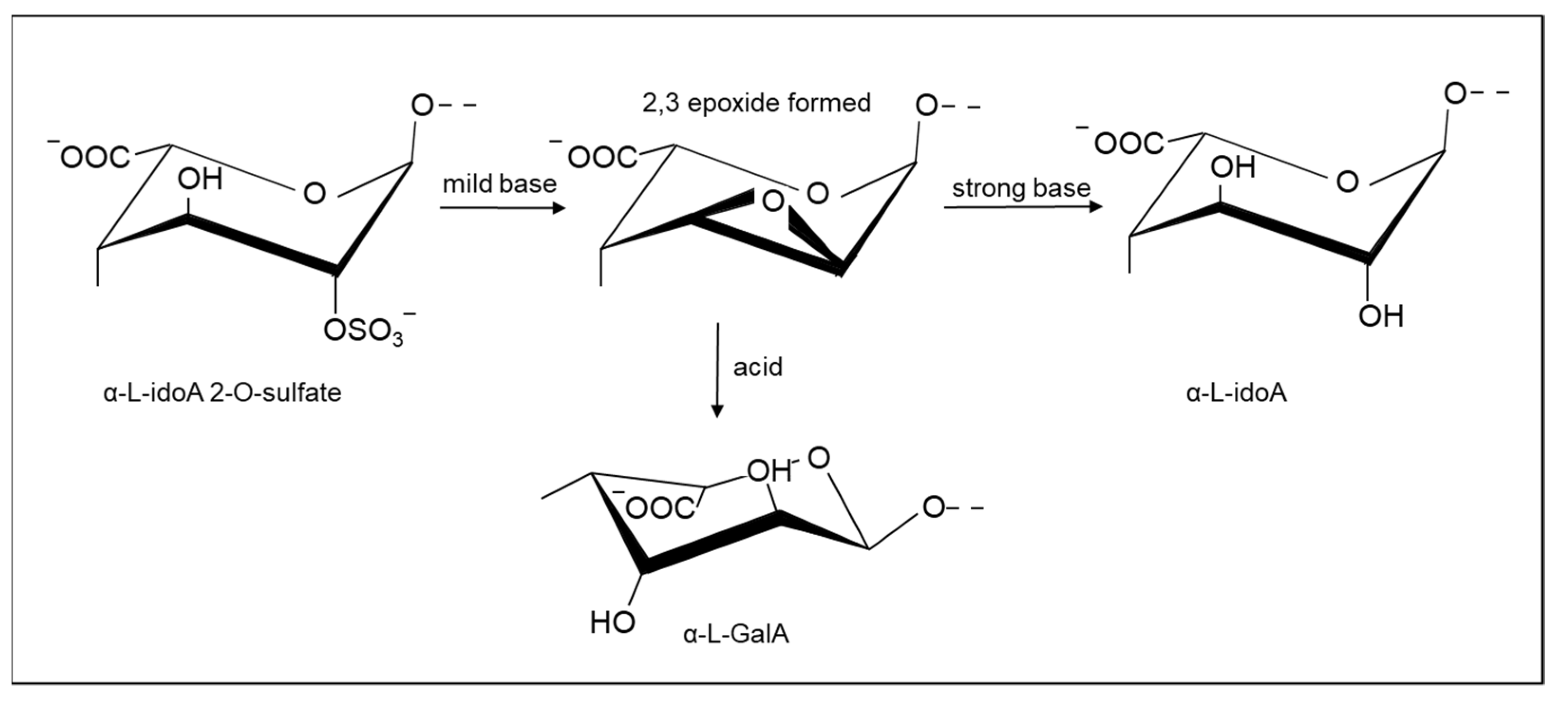
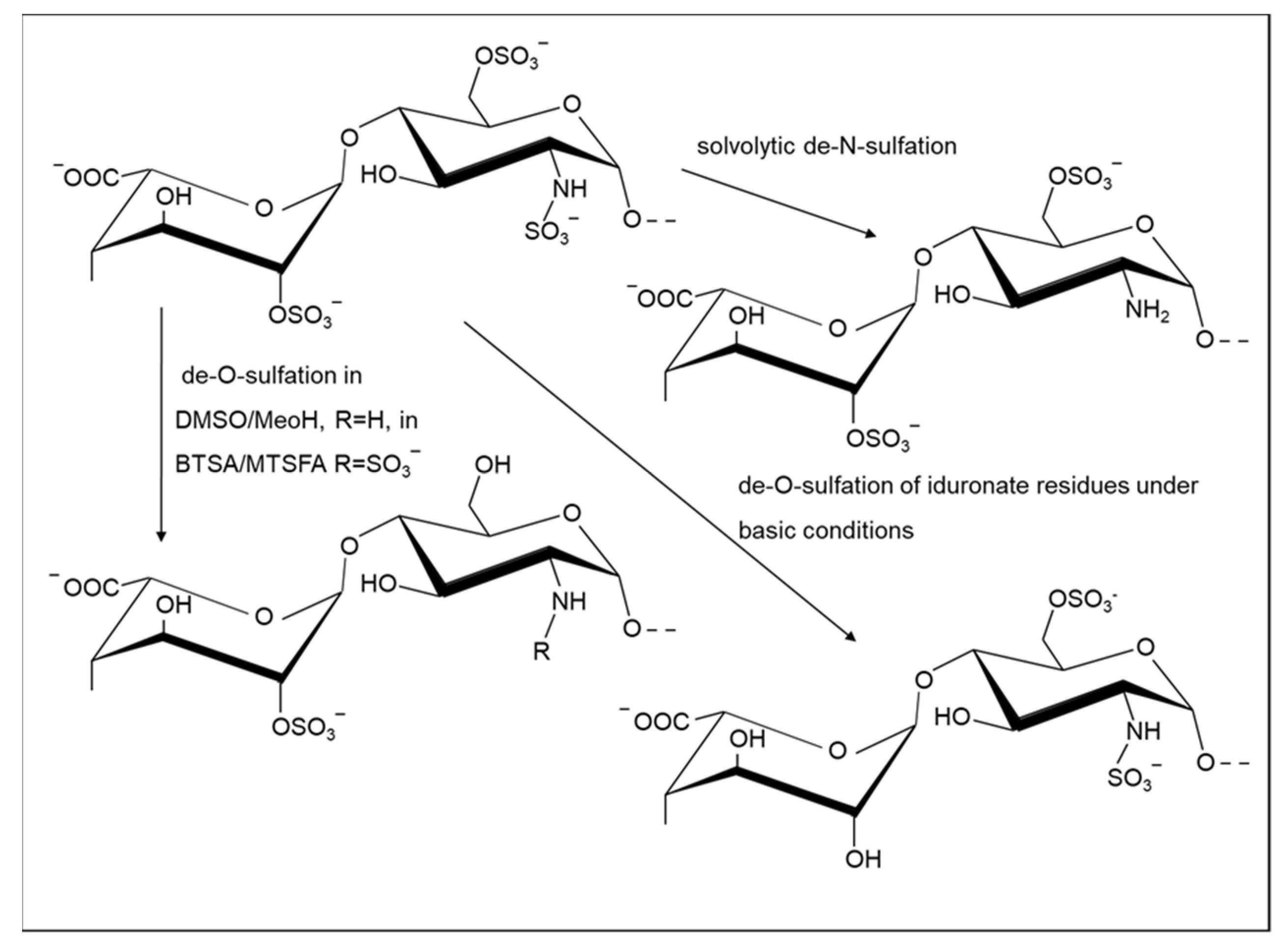
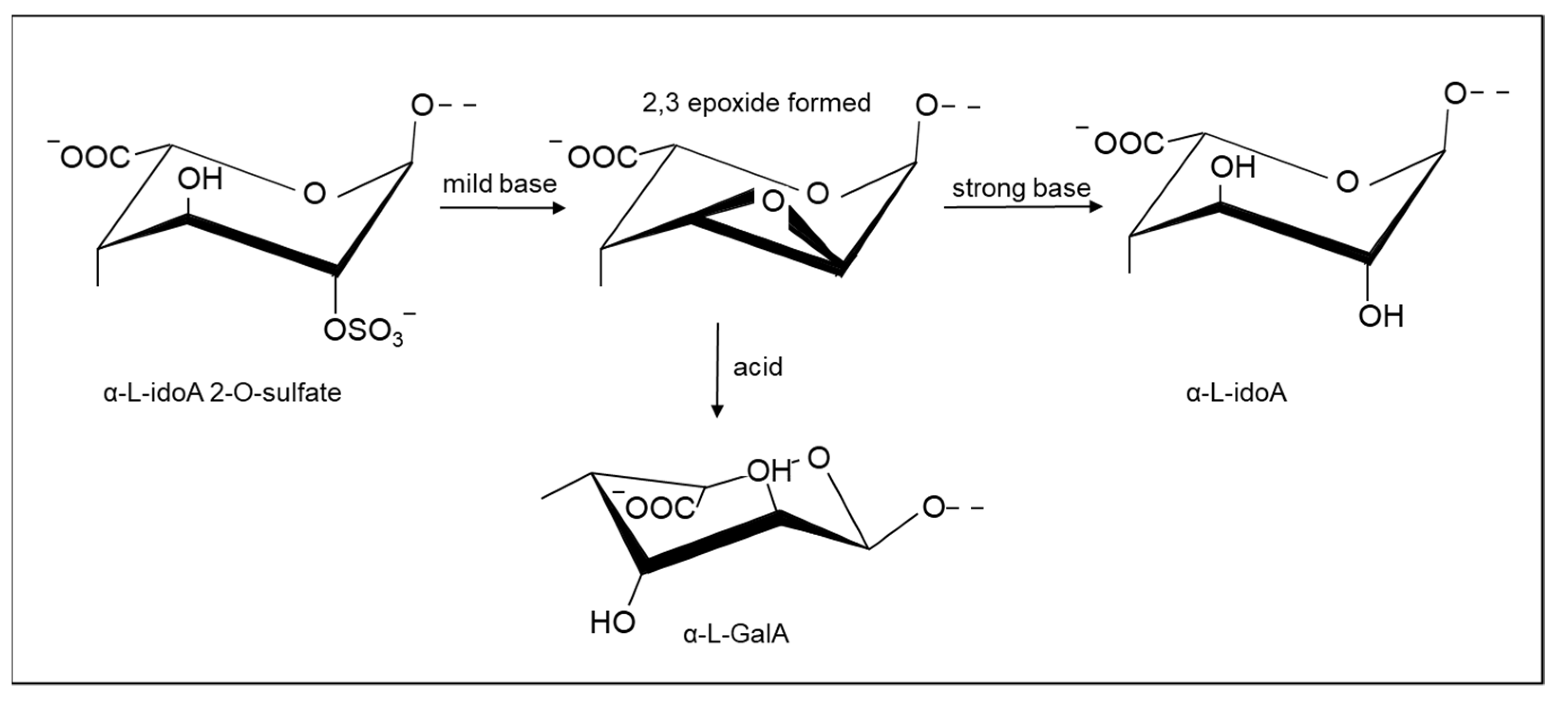
2.1.2. Selective Removal of Sulfate Groups
2.2. Modification of Acetylation
2.3. Oxidation and Reduction Reactions
2.4. Other Modifications
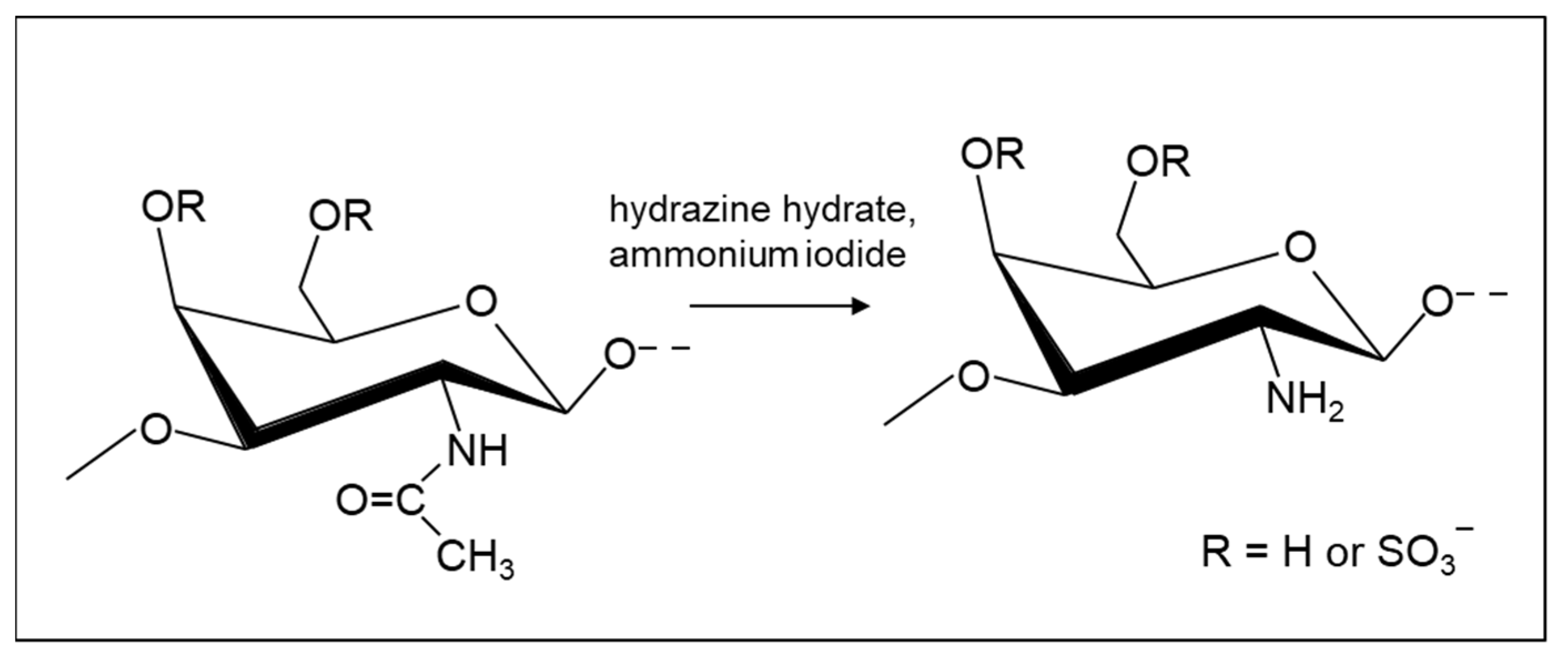
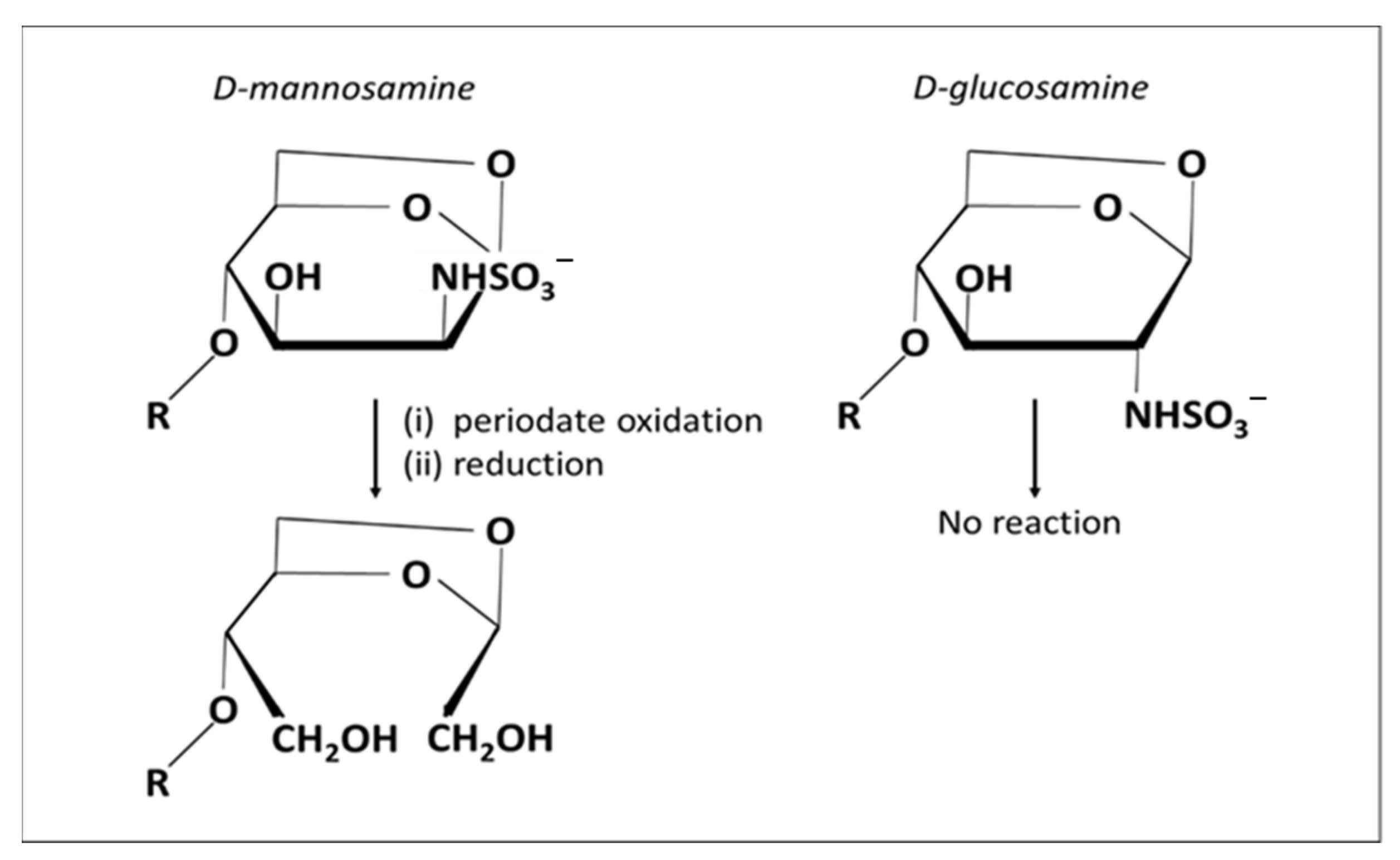
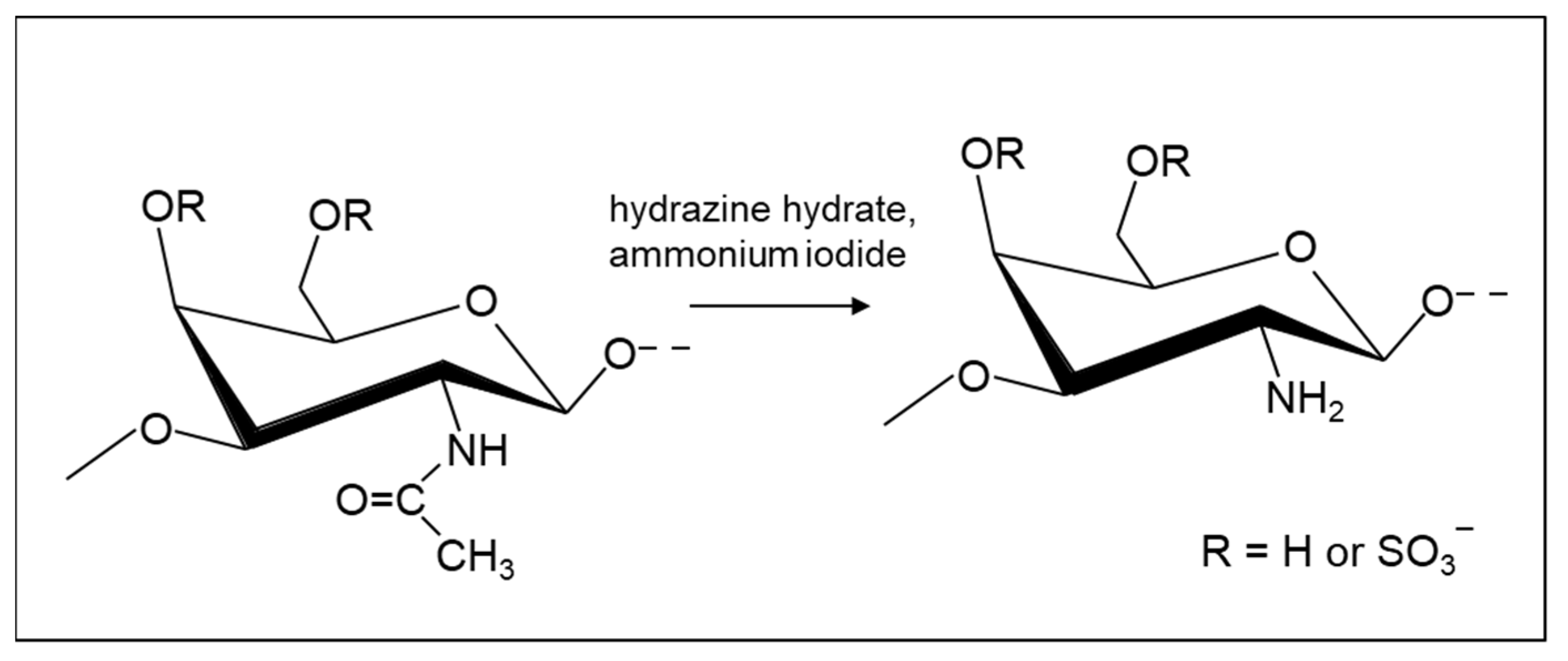
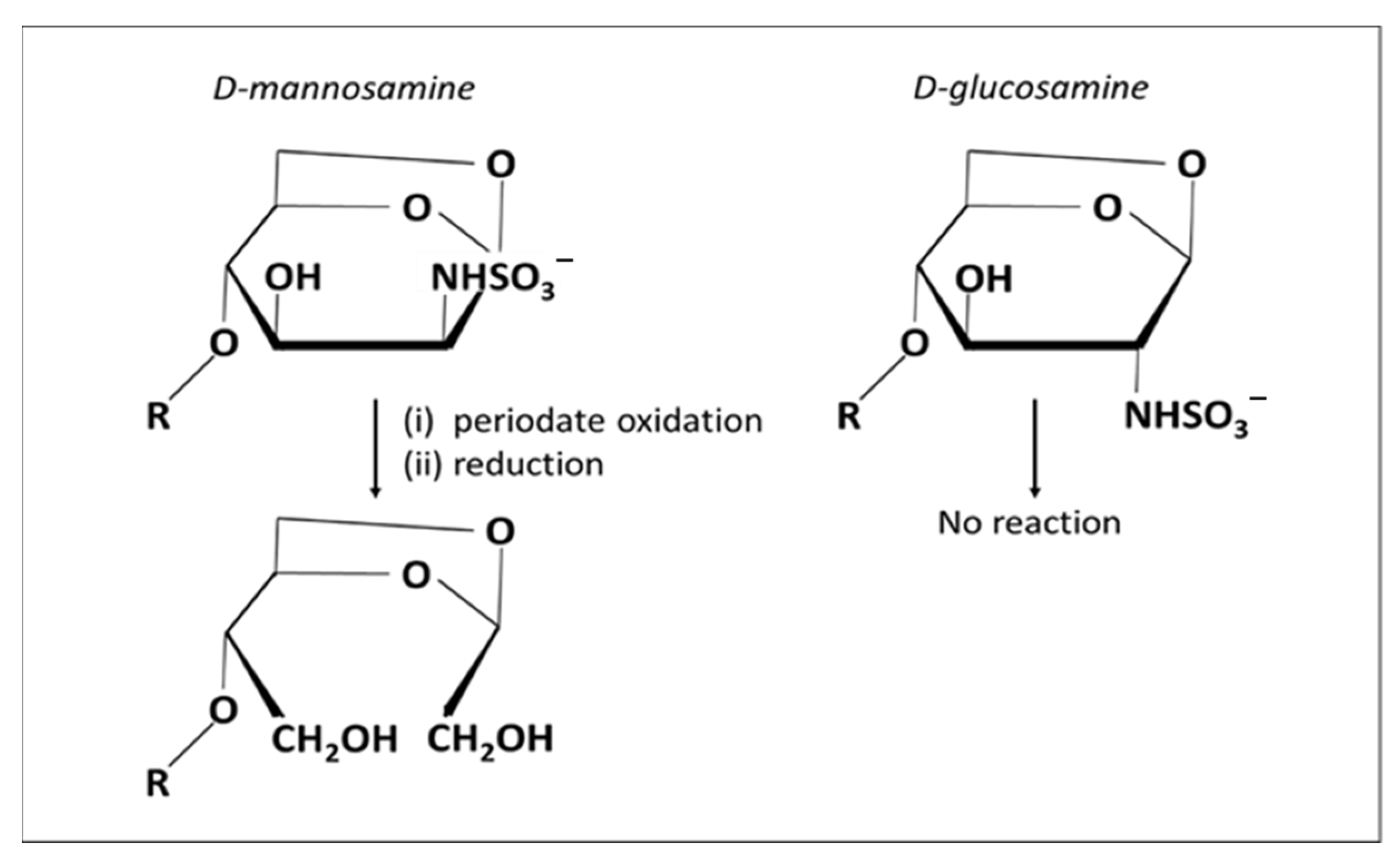
2.4.1. Amination
2.4.2. Phosphorylation
2.4.3. Derivatization of Carboxylates
2.4.4. Derivatization of Hydroxyl Groups
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeAngelis, P.L. Glycosaminoglycan polysaccharide biosynthesis and production: Today and tomorrow. Appl. Microbiol. Biotechnol. 2012, 94, 295–305. [Google Scholar] [CrossRef]
- Bedini, E.; Lavezzi, A.; Iadonisi, A. Chemical Derivatization of Sulfated Glycosaminoglycans. Eur. J. Org. Chem. 2016, 18, 3018–3042. [Google Scholar] [CrossRef]
- Karst, N.A.; Linhardt, R.J. Recent Chemical and Enzymatic Approaches to the Synthesis of Glycosaminoglycan Oligosaccharides. Curr. Med. Chem. 2003, 10, 1993–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, C.; Hattan, C.M.; Kerns, R.J. Semi-synthetic heparin derivatives: Chemical modifications of heparin beyond chain length, sulfate substitution pattern and N-sulfo/N-acetyl groups. Carbohydr. Res. 2006, 341, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Dordick, J.S.; Deangelis, P.L.; Liu, J. Enzymatic synthesis of glycosaminoglycan heparin. Semin. Thromb. Hemost. 2007, 33, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vibert, A.; Lopin-Bon, C.; Jacquinet, J.-C. Efficient and Stereocontrolled Construction of Homo- and Heterogeneously 4- and 6-Sulfated Biotinylated Chondroitin Oligomers. Eur. J. Org. Chem. 2011, 22, 4183–4204. [Google Scholar] [CrossRef]
- Guimond, S.E.; Turnbull, J.E.; Yates, E.A. Engineered bio-active polysaccharides from heparin. Macromol. Biosci. 2006, 6, 681–686. [Google Scholar] [CrossRef]
- Pavão, M.S.; Mourao, P.A.; Mulloy, B.; Tollefsen, D.M. A unique dermatan sulfate-like glycosaminoglycan from ascidian. Its structure and the effect of its unusual sulfation pattern on anticoagulant activity. J. Biol. Chem. 1995, 270, 31027–31036. [Google Scholar] [CrossRef] [Green Version]
- Ernst, S.; Langer, R.; Cooney, C.L.; Sasisekharan, R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387–444. [Google Scholar] [CrossRef]
- Volpi, N. Therapeutic Applications of Glycosaminoglycans. Curr. Med. Chem. 2006, 13, 1799–1810. [Google Scholar] [CrossRef]
- Dietrich, C.P.; Nader, H.B.; Straus, A.H. Structural Differences of Heparan Sulfates According to the Tissue and Species of Origin. Biochem. Biophys. Res. Commun. 1983, 111, 865–871. [Google Scholar] [CrossRef]
- Sasisekharan, R.; Venkataraman, G. Heparin and heparan sulfate: Biosynthesis, structure, and function. Curr. Opin. Struct. Biol. 2000, 4, 626–631. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, J.J.; Robinson, H.C. The Molecular-weight distribution of glycosaminoglycans. Biochem. J. 1973, 135, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.D.; Pantoliano, M.W.; Springer, B.A. Energetic Characterization of the Basic Fibroblast Growth Factor-Heparin Interaction: Identification of the Heparin Binding Domain. Biochemistry 1994, 33, 3831–3840. [Google Scholar] [CrossRef]
- Yang, C.; Cao, M.; Liu, H.; He, Y.; Xu, J.; Du, Y.; Liu, Y.; Wang, W.; Cui, L.; Hu, J.; et al. The high and low molecular weight forms of hyaluronan have distinct effects on CD44 clustering. J. Biol. Chem. 2012, 287, 43094–43107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, C.I.; Tully, S.E.; Sotogaku, N.; Clark, P.M.; Rawat, M.; Vaidehi, N.; Goddard III, W.A.; Nishi, A.; Hsieh-Wilson, L.C. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2006, 2, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarup, S.; Huang, W.; Mackay, T.F.; Anholt, R.R. Analysis of natural variation reveals neurogenetic networks for Drosophila olfactory behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 1017–1022. [Google Scholar] [CrossRef] [Green Version]
- Martín, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H1585–H1597. [Google Scholar] [CrossRef] [Green Version]
- Rudd, T.R.; Yates, E.A. A highly efficient tree structure for the biosynthesis of heparan sulfate accounts for the commonly observed disaccharides and suggests a mechanism for domain synthesis. Mol. BioSystems 2012, 8, 1499–1506. [Google Scholar] [CrossRef]
- Meneghetti, M.C.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef] [Green Version]
- Esko, J.D.; Zhang, L. Influence of core protein sequence on glycosaminoglycan assembly. Curr. Opin. Struct. Biol. 1996, 6, 663–670. [Google Scholar] [CrossRef]
- Malmstrom, A.; Bartolini, B.; Thelin, M.A.; Pacheco, B.; Maccarana, M. Iduronic acid in chondroitin/dermatan sulfate: Biosynthesis and biological function. J. Histochem. Cytochem. 2012, 60, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.; Kitagawa, H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol. 2000, 10, 518–527. [Google Scholar] [CrossRef]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneghetti, M.C.Z.; Gesteira Ferreira, T.; Tashima, A.K.; Chavante, S.F.; Yates, E.A.; Liu, J.; Nader, H.B.; Lima, M.A. Insights into the role of 3-O-sulfotransferase in heparan sulfate biosynthesis. Org. Biomol. Chem. 2017, 15, 6792–6799. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Free, P.; Courty, J.; Wilkinson, M.C.; Fernig, D.G. Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell. Proteom. 2009, 8, 2256–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Ori, A.; Rudd, T.R.; Uniewicz, K.A.; Ahmed, Y.A.; Guimond, S.E.; Siligardi, G.; Yates, E.A.; Fernig, D.G. Diversification of the structural determinants of fibroblast growth factor-heparin interactions: Implications for binding specificity. J. Biol. Chem. 2012, 287, 40061–40073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, C.; Yates, E.A.; Jiang, C.; Wilkinson, M.C.; Fernig, D.G. Heparin binding preference and structures in the fibroblast growth factor family parallel their evolutionary diversification. Open Biol. 2016, 6, 150275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thao, B.P. Selective Labelling of Arginine Residues in Protein Sulfated Glycosaminoglycan Binding Sites. Ph.D. Thesis, The University of Liverpool, London, UK, July 2019. [Google Scholar]
- Hricovini, M.; Hricovini, M. Solution Conformation of Heparin Tetrasaccharide. DFT Analysis of Structure and Spin(-)Spin Coupling Constants. Molecules 2018, 23, 3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nader, H.B.; Dietrich, C.P.; Buonassisi, V.; Colburn, P. Heparin sequences in the heparan sulfate chains of an endothelial cell proteoglycan. Proc. Natl. Acad. Sci. USA 1987, 84, 3565–3569. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Chiba, M.; Murata, T.; Matsuura, A. Heparan sulfate is a clearance receptor for aberrant extracellular proteins. J. Cell Biol. 2020, 219, e201911126. [Google Scholar] [CrossRef]
- Pumphrey, C.Y.; Theus, A.M.; Li, S.; Parrish, R.S.; Sanderson, R.D. Neoglycans, Carbodiimide-modified Glycosaminoglycans: A New Class of Anticancer Agents That Inhibit Cancer Cell Proliferation and Induce Apoptosis. Cancer Res. 2002, 62, 3722–3728. [Google Scholar]
- Yamada, S.; Sugahara, K. Potential Therapeutic Application of Chondroitin Sulfate/Dermatan Sulfate. Curr. Drug Discov. Technol. 2008, 5, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, S.; Wiencek, J.M.; Ren, R.X.; Linhardt, R.J. Benzoate-based room temperature ionic liquids—Thermal properties and glycosaminoglycan dissolution. Carbohydr. Polym. 2006, 63, 268–271. [Google Scholar] [CrossRef]
- Laremore, T.N.; Murugesan, S.; Park, T.-J.; Avci, F.Y.; Zagorevski, D.V.; Linhardt, R.J. Matrix-assisted laser desorption/ionisation mass spectrometric analysis of uncom-plexed highly sulfated oligosaccharides using ionic liquid matrices. Anal. Chem. 2006, 78, 1774–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laremore, T.N.; Zhang, F.; Linhardt, R.J. Ionic liquid matrix for direct UV-MALDI-TOF-MS analysis of dermatan sulfate and chondroitin sulfate oligosaccharides. Anal. Chem. 2007, 79, 1604–1610. [Google Scholar] [CrossRef] [Green Version]
- Przybylski, C.; Gonnet, F.; Bonnaffe, D.; Hersant, Y.; Lortat-Jacob, H.; Daniel, R. HABA-based ionic liquid matrices for UV-MALDI-MS analysis of heparin and heparan sulfate oligosaccharides. Glycobiology 2010, 20, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.K.; Luo, B.; Guneta, V.; Li, L.; Foo, S.E.M.; Dai, Y.; Tan, T.T.Y.; Tan, N.S.; Choong, C.; Wong, M.T.C. Supercritical carbon dioxide extracted extracellular matrix material from adipose tissue. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 75, 349–358. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, G.; Lang, X.; Li, J.P.; Li, X. Lipase-catalyzed synthesis of long-chain cellulose esters using ionic liquid mixtures as reaction media. J. Chem. Technol. Biotechnol. 2017, 92, 1203–1210. [Google Scholar] [CrossRef]
- Bayón, C.; Cortés, Á.; Berenguer, J.; Hernáiz, M.J. Highly efficient enzymatic synthesis of Galβ-(1→3)-GalNAc and Galβ-(1→3)-GlcNAc in ionic liquids. Tetrahedron 2013, 69, 4973–4978. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, S.; Deng, Y. Recent advances in ionic liquid catalysis. Green Chem. 2011, 13, 2619–2637. [Google Scholar] [CrossRef]
- Zakrzewska, M.E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Solubility of Carbohydrates in Ionic Liquids. Energy Fuels 2010, 24, 737–745. [Google Scholar] [CrossRef]
- Prasad, K.; Kaneko, Y.; Kadokawa, J. Novel gelling systems of kappa-, iota- and lambda-carrageenans and their composite gels with cellulose using ionic liquid. Macromol. Biosci. 2009, 9, 376–382. [Google Scholar] [CrossRef]
- Gericke, M.; Fardim, P.; Heinze, T. Ionic liquids--promising but challenging solvents for homogeneous derivatization of cellulose. Molecules 2002, 17, 7458–7502. [Google Scholar] [CrossRef] [Green Version]
- Olivier-Bourbigou, H.; Magna, L.; Morvan, D. Ionic liquids and catalysis: Recent progress from knowledge to applications. Appl. Catal. A Gen. 2010, 373, 1–56. [Google Scholar] [CrossRef]
- Camp, J.E. Bio-available Solvent Cyrene: Synthesis, Derivatization, and Applications. ChemSusChem 2018, 11, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Bousfield, T.W.; Pearce, K.P.R.; Nyamini, S.B.; Angelis-Dimakis, A.; Camp, J.E. Synthesis of amides from acid chlorides and amines in the bio-based solvent Cyrene™. Green Chem. 2019, 21, 3675–3681. [Google Scholar] [CrossRef]
- Zhang, J.; White, G.B.; Ryan, M.D.; Hunt, A.J.; Katz, M.J. Dihydrolevoglucosenone (Cyrene) As a Green Alternative to N, N-Dimethylformamide (DMF) in MOF Synthesis. ACS Sustain. Chem. Eng. 2016, 4, 7186–7192. [Google Scholar] [CrossRef]
- Caputo, H.E.; Straub, J.E.; Grinstaff, M.W. Design, synthesis, and biomedical applications of synthetic sulphated polysaccharides. Chem. Soc. Rev. 2019, 48, 2338–2365. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, R.C.; Burkhardt, D.; Ghosh, P.; Read, R.; Cake, M.; Swain, M.V.; Murrell, G.A. Topographical analysis of the structural, biochemical and dynamic biomechanical properties of cartilage in an ovine model of osteoarthritis. Osteoarthr. Cartil. 2003, 11, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Patey, S.J.; Edwards, E.A.; Yates, E.A.; Turnbull, J.E. Heparin Derivatives as Inhibitors of BACE-1, the Alzheimer’s a-Secretase, with Reduced Activity against Factor Xa and Other Proteases. J. Med. Chem. 2006, 49, 6129–6132. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin inhibits cellular invasion by SARS-CoV-2. Thromb. Haemost. 2020, 10, 1700–1715. [Google Scholar]
- Bedini, E.; Laezza, A.; Parrilli, M.; Iadonisi, A. A review of chemical methods for the selective sulfation and desulfation of polysaccharides. Carbohydr. Polym. 2017, 174, 1224–1239. [Google Scholar] [CrossRef]
- Hintze, V.; Miron, A.; Moeller, S.; Schnabelrauch, M.; Wiesmann, H.P.; Worch, H.; Scharnweber, D. Sulfated hyaluronan and chondroitin sulfate derivatives interact differently with human transforming growth factor-beta1 (TGF-beta1). Acta Biomater. 2012, 8, 2144–2152. [Google Scholar] [CrossRef]
- Casu, B.; Grazioli, G.; Razi, N.; Guerrini, M.; Naggi, A.; Torri, G.; Oreste, P.; Tursi, T.; Zoppetti, G.; Lindahl, U. Heparin-like compounds prepared by chemical modification of capsular polysaccharide K5. Carbohydr. Res. 1994, 263, 271–284. [Google Scholar] [CrossRef]
- Gilbert, E.E. The reactions of sulfur trioxide, and its adducts, with organic compounds. Chem. Rev. 1962, 62, 549–589. [Google Scholar] [CrossRef]
- Ogamo, A.; Metori, A.; Uchiyama, H.; Nagasawa, K. Reactivity toward Chemical Sulfation of Hydroxyl Groups of Heparin. Carbohydr. Res. 1989, 193, 165–172. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, M.; Wang, J.; Zhai, G. Chondroitin sulfate-based nanocarriers for drug/gene delivery. Carbohydr. Polym. 2015, 133, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, W.; Yang, T.; Zhang, X.; Zuo, Y.; Tian, J.; Yao, J.; Zhang, J.; Lei, Z. Catalytic synthesis of sulfated polysaccharides I: Characterization of chemical structure. Int. J. Biol. Macromol. 2015, 74, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bacilieri, M.; Naggi, A.; Ceol, M.; Schleicher, E.D.; Tosetto, E.; Comoli, M.; Torri, G.; Moro, S.; Palumbo, M.; Gambaro, G. Inhibitory effects of glycosaminoglycans on basal and stimulated transforming growth factor-beta1 expression in mesangial cells: Biochemical and structural considerations. Glycobiology 2011, 21, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Naggi, A.; Torri, G.; Casu, B.; Pangrazzi, J.; Abbadini, M.; Zametta, M.; Donati, M.B.; Lansen, J.; Maffrand, J.P. “Supersulfated” Heparin Fragments, A New Type of Low-Molecular Weight Heparin. Biochem. Pharmacol. 1987, 36, 1895–1900. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Linhardt, R.J. Lessons learned from the contamination of heparin. Nat. Prod. Rep. 2009, 26, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Ramacciotti, E.; Clark, M.; Sadeghi, N.; Hoppensteadt, D.; Thethi, I.; Gomes, M.; Fareed, J. Review: Contaminants in heparin: Review of the literature, molecular profiling, and clinical implications. Clin. Appl. Thromb./Hemost. 2011, 17, 126–135. [Google Scholar] [CrossRef]
- Chen, T.; Li, B.; Li, Y.; Zhao, C.; Shen, J.; Zhang, H. Catalytic synthesis and antitumor activities of sulfated polysaccharide from Gynostemma pentaphyllum Makino. Carbohydr. Polym. 2011, 83, 554–560. [Google Scholar] [CrossRef]
- Chopin, N.; Sinquin, C.; Ratiskol, J.; Zykwinska, A.; Weiss, P.; Cerantola, S.; Le Bideau, J.; Colliec-Jouault, S. A Direct Sulfation Process of a Marine Polysaccharide in Ionic Liquid. BioMed Res. Int. 2015, 2015, 508656. [Google Scholar] [CrossRef]
- Merceron, C.; Portron, S.; Vignes-Colombeix, C.; Rederstorff, E.; Masson, M.; Lesoeur, J.; Sourice, S.; Sinquin, C.; Colliec-Jouault, S.; Weiss, P.; et al. Pharmacological modulation of human mesenchymal stem cell chondrogenesis by a chemically oversulfated polysaccharide of marine origin: Potential application to cartilage regenerative medicine. Stem Cells 2012, 30, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Zoppetti, G.; Oreste, P. Process for the Preparation of Chondroitin Sulfates from K4 Polysaccharide and Obtained Products. United States Patent No. US 6,777,398 B2, 17 August 2004. [Google Scholar]
- Laezza, A.; De Castro, C.; Parrilli, M.; Bedini, E. Inter vs. intraglycosidic acetal linkages control sulfation pattern in semi-synthetic chondroitin sulfate. Carbohydr. Polym. 2014, 112, 546–555. [Google Scholar] [CrossRef]
- de Araújo, C.A.; Noseda, M.D.; Cipriani, T.R.; Goncalves, A.G.; Duarte, M.E.; Ducatti, D.R. Selective sulfation of carrageenans and the influence of sulfate regiochemistry on anticoagulant properties. Carbohydr. Polym. 2013, 91, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Maza, S.; de Paz, J.L.; Nieto, P.M. Microwave-assisted sulfonation of heparin oligosaccharides. Tetrahedron Lett. 2011, 52, 441–443. [Google Scholar] [CrossRef]
- Xu, P.; Laval, S.; Guo, Z.; Yu, B. Microwave-assisted simultaneous O, N-sulfonation in the synthesis of heparin-like oligosaccharides. Org. Chem. Front. 2016, 3, 103–109. [Google Scholar] [CrossRef]
- Naderi, A.; Koschella, A.; Heinze, T.; Shih, K.C.; Nieh, M.P.; Pfeifer, A.; Chang, C.C.; Erlandsson, J. Sulfoethylated nanofibrillated cellulose: Production and properties. Carbohydr. Polym. 2017, 169, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Brendler, E.; Gebauer, K.; Gruner, M.; Fischer, S. Synthesis and characterization of low sulfoethylated cellulose. Carbohydr. Polym. 2011, 83, 616–622. [Google Scholar] [CrossRef]
- Gill, D.M.; Male, L.; Jones, A.M. Sulfation made simple: A strategy for synthesising sulfated molecules. Chem. Commun. 2019, 55, 4319–4322. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, A.M.; Gill, D.M.; Tsang, C.W.; Jones, A.M. Chemical Methods for N- and O-Sulfation of Small Molecules, Amino Acids and Peptides. ChemBioChem 2020, 21, 938–942. [Google Scholar] [CrossRef]
- Lundin, L.; Larsson, H.; Kreuger, J.; Kanda, S.; Lindahl, U.; Salmivirta, M.; Claesson-Welsh, L. Selectively desulfated heparin inhibits fibroblast growth factor-induced mitogenicity and angiogenesis. J. Biol. Chem. 2000, 275, 24653–24660. [Google Scholar] [CrossRef] [Green Version]
- Naggi, A.; Torri, G.; Casu, B.; Oreste, P.; Zoppetti, G.; Li, J.P.; Lindahl, U. Toward a biotechnological heparin through combined chemical and enzymatic modification of the Escherichia coli K5 polysaccharide. Semin. Thromb. Hemost. 2001, 27, 437–443. [Google Scholar] [CrossRef]
- Yates, E.A.; Santini, F.; Guerrini, M.; Naggi, A.; Torri, G.; Casu, B. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydr. Res. 1996, 294, 15–27. [Google Scholar] [CrossRef]
- Kantor, T.G.; Schubert, M. A Method for the Desulfation of Chondroitin Sulfate. J. Am. Chem. Soc. 1975, 79, 152–153. [Google Scholar] [CrossRef]
- Lim, J.J.; Temenoff, J.S. The effect of desulfation of chondroitin sulfate on interactions with positively charged growth factors and upregulation of cartilaginous markers in encapsulated MSCs. Biomaterials 2013, 34, 5007–5018. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Nagasawa, K. Selective N-Desulfation of Heparin with Dimethyl Sulfoxide containing Water or Methanol. Carbohydr. Res. 1976, 46, 87–95. [Google Scholar] [CrossRef]
- Nagasawa, K.; Inoue, Y.; Kamata, T. Solvolytic Desulfation of Glycosaminoglyuronan Sulfates with Dimethyl Sulfoxide Containing Water or Methanol. Carbohydr. Res. 1977, 58, 47–55. [Google Scholar] [CrossRef]
- Naggi, A.; De Cristofano, B.; Bisio, A.; Torri, G.; Casu, B. Generation of anti-factor Xa active, 3-O-sulfated glucosamine-rich sequences by controlled desulfation of oversulfated heparins. Carbohydr. Res. 2001, 336, 283–290. [Google Scholar] [CrossRef]
- Baumann, H.; Scheen, H.; Huppertz, B.; Keller, R. Novel regio- and stereoselective O-6-desulfation of the glucosamine moiety of heparin with N-methylpyrrolidinone-water or N, N-dimethylformamide-water mixtures. Carbohydr. Res. 1998, 308, 381–388. [Google Scholar] [CrossRef]
- Kozlowski, A.M.; Yates, E.A.; Roubroeks, J.P.; Tommeraas, K.; Smith, A.M.; Morris, G.A. Hydrolytic Degradation of Heparin in Acidic Environments: Nuclear Magnetic Resonance Reveals Details of Selective Desulfation. ACS Appl. Mater. Interfaces 2021, 13, 5551–5563. [Google Scholar] [CrossRef]
- Baumann, H.; Faust, V. Concepts for improved regioselective placement of O-sulfo, N-sulfo, N-acetyl, and N-carboxymethyl groups in chitosan derivatives. Carbohydr. Res. 2001, 331, 43–57. [Google Scholar] [CrossRef]
- Becher, J.; Möller, S.; Weiss, D.; Schiller, J.; Schnabelrauch, M. Synthesis of New Regioselectively Sulfated Hyaluronans for Biomedical Application. Macromol. Symp. 2010, 296, 446–452. [Google Scholar] [CrossRef]
- Chaidedgumjorn, A.; Suzuki, A.; Toyoda, H.; Toida, T.; Imanari, T.; Linhardt, R.J. Conductivity detection for molecular mass estimation of per-O-sulfonated glycosaminoglycans separated by high-performance size-exclusion chromatography. J. Chromatogr. A 2002, 959, 95–102. [Google Scholar] [CrossRef]
- Kariya, Y.; Kyogashima, M.; Suzuki, K.; Isomura, T.; Sakamoto, T.; Horie, K.; Ishihara, M.; Takano, R.; Kamei, K.; Hara, S. Preparation of completely 6-O-desulfated heparin and its ability to enhance activity of basic fibroblast growth factor. J. Biol. Chem. 2000, 275, 25949–25958. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Takano, R.; Kamei-Hayashi, K.; Hara, S. A novel regioselective desulfation of polysaccharide sulfates: Specific 6-O-desulfation with N,O-bis(trimethylsilyl)acetamide. Carbohydr. Res. 1993, 241, 209–215. [Google Scholar] [CrossRef]
- Takano, R.; Kanda, T.; Hayashi, K.; Yoshida, K.; Hara, S. Desulfation of Sulfated Carbohydrates Mediated by Silylating Reagents. J. Carbohydr. Chem. 1995, 14, 885–888. [Google Scholar] [CrossRef]
- Skidmore, M.A.; Dumax-Vorzet, A.F.; Guimond, S.E.; Rudd, T.R.; Edwards, E.A.; Turnbull, J.E.; Craig, A.G.; Yates, E.A. Disruption of Rosetting in Plasmodium falciparum Malaria with Chemically Modified Heparin and Low Molecular Weight Derivatives Possessing Reduced Anticoagulant and Other Serine Protease Inhibition Activities. J. Med. Chem. 2008, 51, 1453–1458. [Google Scholar] [CrossRef]
- Pomin, V.H.; Valente, A.P.; Pereira, M.S.; Mourao, P.A. Mild acid hydrolysis of sulfated fucans: A selective 2-desulfation reaction and an alternative approach for preparing tailored sulfated oligosaccharides. Glycobiology 2005, 15, 1376–1385. [Google Scholar] [CrossRef]
- Shevchenko, N.M.; Anastyuk, S.D.; Menshova, R.V.; Vishchuk, O.S.; Isakov, V.I.; Zadorozhny, P.A.; Sikorskaya, T.V.; Zvyagintseva, T.N. Further studies on structure of fucoidan from brown alga Saccharina gurjanovae. Carbohydr. Polym. 2015, 121, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Jaseja, M.; Rej, R.N.; Sauriol, F.; Perlin, A.S. Novel regio- and stereoselective modifications of heparin in alkaline solution. Nuclear magnetic resonance spectroscopic evidence. Can. J. Chem. 1989, 67, 1449–1456. [Google Scholar] [CrossRef]
- Piani, S.; Casu, B.; Marchi, E.G.; Torri, G.; Ungarelli, F. Alkali-Induced Optical Rotation Changes in Heparins and Heparan Sulfates, and Their Relation to Iduronic Acid-Containing Sequences. J. Carbohydr. Chem. 1993, 12, 507–521. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, Y.-T.; Xu, Z.-B.; Qu, J. Hot Water-Promoted Ring-Opening of Epoxides and Aziridines by Water and Other Nucleopliles. J. Org. Chem. 2008, 73, 2270–2274. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Guerrini, M.; Guglieri, S.; Naggi, A.; Perez, M.; Torri, G.; Cassinelli, G.; Robatti, D.; Carminati, P.; Giannini, G.; et al. Undersulfated and Glycol-Split Heparins Endowed with Antiangiogenic Activity. J. Med. Chem. 2004, 47, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Santini, F.; Bisio, A.; Guerrini, M.; Yates, E.A. Modifications under basic conditions of the minor sequences of heparin containing 2,3 or 2,3,6 sulfated D-glucosamine residues. Carbohydr. Res. 1997, 302, 103–108. [Google Scholar] [CrossRef]
- Yates, E.A.; Santini, F.; Bisio, A.; Cosentino, C. Evidence for a heparin derivative containing an N-sulfated aziridine ring that retains high anti-factor Xa activity. Carbohydr. Res. 1997, 298, 335–340. [Google Scholar] [CrossRef]
- Yamada, S.; Watanabe, M.; Sugahara, K. Covnersion of N-sulfated glucosamine to N-sulfated mannosamine in an unsaturated heparin disaccharide by non-enzymatic, base-catalyzed C-2 epimerization during enzymatic oligosaccharide preparation. Carbohydr. Res. 1998, 309, 261–268. [Google Scholar] [CrossRef]
- Toida, T.; Vlahov, I.R.; Smith, A.E.; Hileman, R.E.; Linhardt, R.J. C-2 Epimerization of N-Acetylglucosamine in an Oligosaccharide Derived from Heparan Sulfate. J. Carbohydr. Chem. 1996, 15, 351–360. [Google Scholar] [CrossRef]
- Pisano, C.; Cervoni, M.L.; Chiarucci, I. Antiangiogenic and Antitumoral Activity of Novel Heparin Derivatives Devoid of Anticoagulant Effects. In Proceedings of the National Cancer Institute-European Organization for Research and Treatment of Cancer-American Association for Cancer Research Symposium (NCI-EORTC-AACR), Frankfurt, Germany, 19–22 November 2002. Abstract A229. [Google Scholar]
- Naik, S.; Bhattacharjya, G.; Kavala, V.R.; Patel, B.K. Mild and eco-firendly chemoselective acylation of amines in aqueous medium. ARKIVOC 2004, 1, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Patey, S.J.; Edwards, E.A.; Yates, E.A.; Turnbull, J.E. Engineered heparins: Novel beta-secretase inhibitors as potential Alzheimer’s disease therapeutics. Neurodegener. Dis. 2008, 5, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kerns, R.J. Diversity-oriented chemical modification of heparin: Identification of charge-reduced N-acyl heparin derivatives having increased selectivity for heparin-binding proteins. Bioorg. Med. Chem. 2006, 14, 2300–2313. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Bandmann, H.; Schrader, T. A fluorescent polymeric heparin sensor. Chem. A Eur. J. 2007, 13, 7701–7707. [Google Scholar] [CrossRef]
- Mukhopadhyay, B.; Ravindranathan-Kartha, K.P.; Russell, D.A.; Field, R.A. Streamlined Synthesis of Per-O-acetylated Sugars, Glycosyl Iodides, or Thioglycosides from Unprotected Reducing Sugars. J. Org. Chem. 2004, 69, 7758–7760. [Google Scholar] [CrossRef] [PubMed]
- Phukan, K.; Ganguly, M.; Devi, N. Mild and Useful Method for N-Acylation of Amines. Synth. Commun. 2009, 39, 2694–2701. [Google Scholar] [CrossRef]
- Lima, M.A.; Cavalheiro, R.P.; Viana, G.M.; Meneghetti, M.C.Z.; Rudd, T.R.; Skidmore, M.A.; Powell, A.K.; Yates, E.A. 19F-labelled glycosaminoglycan probes for solution NMR and non-linear (CARS) microscopy. Glycoconj. J. 2017, 34, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Liebert, T.; Schöbitz, M.; Schaller, J.; Meister, F.; Günther, W.; Heinze, T. Interactions of Ionic Liquids with Polysaccharides 1. Unexpected Acetylation of Cellulose with 1-Ethyl-3-methylimidazolium Acetate. Macromol. Rapid Commun. 2007, 28, 2311–2317. [Google Scholar] [CrossRef]
- Zhang, Z.; Jin, F.; Wu, Z.; Jin, J.; Li, F.; Wang, Y.; Wang, Z.; Tang, S.; Wu, C.; Wang, Y. O-acylation of chitosan nanofibers by short-chain and long-chain fatty acids. Carbohydr. Polym. 2017, 177, 203–209. [Google Scholar] [CrossRef]
- Gao, N.; Wu, M.; Liu, S.; Lian, W.; Li, Z.; Zhao, J. Preparation and characterization of O-acylated fucosylated chondroitin sulfate from sea cucumber. Mar. Drugs 2012, 10, 1647–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitou, M.; Coudert, C.; Level, M.; Lormeau, J.-C.; Zuber, M.; Simenel, C.; Fournier, J.-P.; Choay, J. Selectively O-acylated glycosamionglycan derivatives. Carbohydr. Res. 1992, 236, 107–119. [Google Scholar] [CrossRef]
- Bârzu, T.; Level, M.; Petitou, M.; Lormeau, J.-C.; Choay, J. Preparation and Anti-HIV Activity of O-Acylated Heparin and Dermatan Sulfate Derivatives with Low Anticoagulant Effect. J. Med. Chem. 1993, 36, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Jäger, M.; Minnaard, A.J. Regioselective modification of unprotected glycosides. Chem. Commun. 2016, 52, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Mishiro, K.; Ueda, Y.; Fujimori, Y.; Furuta, T.; Kawabata, T. Total Synthesis of Ellagitannins through Regioselective Sequential Functionalization of Unprotected Glucose. Angew. Chem. Int. Ed. 2015, 54, 6177–6180. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Linseis, M.; Winter, R.F.; Schmidt, R.R. Regioselective Acylation of Diols and Triols: The Cyanide Effect. J. Am. Chem. Soc. 2016, 138, 6002–6009. [Google Scholar] [CrossRef]
- Peri, F.; Cipolla, L.; Nicotra, F. Tin-mediated regioselective acylation of unprotected sugars on solid phase. Tetrahedron Lett. 2000, 41, 8587–8590. [Google Scholar] [CrossRef]
- Herradón, B.; Morcuende, A.; Valverde, S. Microwave Accelerated Organic Transformations: Dibutylstannylene Acetal Mediated Selective Acylation of Polyols and Amino Alcohols using Catalytic A Mounts of Dibutyltin Oxide. Influence of the Solvent and the Power Output on the Selectivity. Synlett 1995, 5, 455–458. [Google Scholar] [CrossRef]
- Sultane, P.R.; Mete, T.B.; Bhat, R.G. Chemoselective N-deacetylation under mild conditions. Org. Biomol. Chem. 2014, 12, 261–264. [Google Scholar] [CrossRef]
- Shimizu, Y.; Noshita, M.; Mukai, Y.; Morimoto, H.; Ohshima, T. Cleavage of unactivated amide bonds by ammonium salt-accelerated hydrazinolysis. Chem. Commun. 2014, 50, 12623–12625. [Google Scholar] [CrossRef] [PubMed]
- Welsh, E.R.; Schauer, C.L.; Qadri, S.B.; Price, R.R. Chitosan Cross-Linking with a Water-Soluble, Blocked Diisocyanate. 1. Solid State. Biomacromolecules 2002, 3, 1370–1374. [Google Scholar] [CrossRef]
- D’Amelio, N.; Esteban, C.; Coslovi, A.; Feruglio, L.; Uggeri, F.; Villegas, M.; Benegas, J.; Paoletti, S.; Donati, I. Insight into the molecular properties of Chitlac, a chitosan derivative for tissue engineering. J. Phys. Chem. B 2013, 117, 13578–13587. [Google Scholar] [CrossRef]
- Fransson, L.-Å. Periodate Oxidation of the D-Glucuronic Acid Resodies in Heparan Sulphate and Heparin. Carbohydr. Res. 1987, 62, 235–244. [Google Scholar] [CrossRef]
- Islam, T.; Butler, M.; Sikkander, S.A.; Toida, T.; Linhardt, R.J. Further evidence that periodate cleavage of heparin occurs primarily through the antithrombin binding site. Carbohydr. Res. 2002, 337, 2239–2243. [Google Scholar] [CrossRef]
- Casu, B.; Diamantini, G.; Fedeli, G.; Mantovani, M.; Oreste, P.; Pescador, R.; Porta, R.; Prino, G.; Torri, G.; Zoppetti, G. Retention of antilipemic activity by periodate-oxidized non-anticoagulant heparins. Arzneimittelforschung 1986, 36, 637–642. [Google Scholar]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopalco, L.; Ciccomascolo, F.; Lanza, P.; Zoppetti, G.; Caramazza, I.; Leoni, F.; Beratta, A.; Siccardi, A.G. Anti-HIV Type 1 Properties of Chemically Modified Heparins with Diminished Anticoagulant Activity. AIDS Res. Hum. Retrovir. 1994, 10, 787–793. [Google Scholar] [CrossRef]
- Alekseeva, A.; Casu, B.; Torri, G.; Pierro, S.; Naggi, A. Profiling glycol-split heparins by high-performance liquid chromatography/mass spectrometry analysis of their heparinase-generated oligosaccharides. Anal. Biochem. 2013, 434, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Alekseeva, A.; Elli, S.; Cosentino, C.; Torri, G.; Naggi, A. Susceptibility of enoxaparin reducing end amino sugars to periodate oxidation. Carbohydr. Res. 2014, 400, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Veraldi, N.; Hughes, A.J.; Rudd, T.R.; Thomas, H.B.; Edwards, S.W.; Hadfield, L.; Skidmore, M.A.; Siligardi, G.; Cosentino, C.; Shute, J.K.; et al. Heparin derivatives for the targeting of multiple activities in the inflammatory response. Carbohydr. Polym. 2015, 117, 400–407. [Google Scholar] [CrossRef]
- Bernhard, J.C.; Panitch, A. Synthesis and characterization of an aggrecan mimic. Acta Biomater. 2012, 8, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Dawlee, S.; Sugandhi, A.; Balakrishnan, B.; Labarre, D.; Jayakrishnan, A. Oxidized Chondroitin Sulfate-Cross-Linked Gelatin Matrixes: A New Class of Hydrogels. Biomacromolecules 2005, 6, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Bobula, T.; Buffa, R.; Procházková, P.; Vágnerová, H.; Moravcová, V.; Šuláková, R.; Židek, O.; Velebný, V. One-pot synthesis of α,β-unsaturated polyaldehyde of chondroitin sulfate. Carbohydr. Polym. 2016, 136, 1002–1009. [Google Scholar] [CrossRef]
- Panagos, C.; Thomson, D.; Bavington, C.D.; Uhrín, D. Structural characterisation of oligosaccharides obtained by Fenton-type radical depolymerisation of dermatan sulfate. Carbohydr. Polym. 2012, 87, 2086–2092. [Google Scholar] [CrossRef]
- Pierre, G.; Punta, C.; Delattre, C.; Melone, L.; Dubessay, P.; Fiorati, A.; Pastori, N.; Galante, Y.M.; Michaud, P. TEMPO-mediated oxidation of polysaccharides: An ongoing story. Carbohydr. Polym. 2017, 165, 71–85. [Google Scholar] [CrossRef] [Green Version]
- de Nooy, A.E.J.; Besemer, A.C.; van Bekkum, H. Highly selective nitroxyl radical-mediated oxidation of primary alcohol groups in water-soluble glucans. Carbohydr. Res. 1995, 269, 89–98. [Google Scholar] [CrossRef]
- Bragd, P.L.; Besemer, A.C.; van Bekkum, H. Bromide-free TEMPO-mediated oxidation of primary alcohol groups in starch and methyl a-D-glucopyranoside. Carbohydr. Res. 2000, 328, 355–363. [Google Scholar] [CrossRef]
- Jaušovec, D.; Vogrinčič, R.; Kokol, V. Introduction of aldehyde vs. carboxylic groups to cellulose nanofibers using laccase/TEMPO mediated oxidation. Carbohydr. Polym. 2015, 116, 74–85. [Google Scholar] [CrossRef]
- Parikka, K.; Nikkila, I.; Pitkanen, L.; Ghafar, A.; Sontag-Strohm, T.; Tenkanen, M. Laccase/TEMPO oxidation in the production of mechanically strong arabinoxylan and glucomannan aerogels. Carbohydr. Polym. 2017, 175, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Moseley, R.; Waddington, R.; Evans, P.; Halliwell, B.; and Embrey, G. The chemical modification of glycosaminoglycan structure by oxygen-derived species in vitro. Biochem. Biophys. Acta 1995, 245–252. [Google Scholar] [CrossRef]
- Brown, K.J.; Hendry, I.A.; Parrish, C.R. Evidence that carboxyl-reduced heparin fails to potentiate acidic fibroblast growth factor activity due to an inability to interact with cell surface heparin receptors. Exp. Cell Res. 1995, 217, 132–139. [Google Scholar] [CrossRef]
- Gildersleeve, J.C.; Oyelaran, O.; Simpson, J.T.; Allred, B. Improved Procedure for Direct Coupling of Carbohydrates to Proteins via Reductive Amination. Bioconj. Chem. 2008, 19, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- Simi, C.K.; Abraham, T.E. Physico chemical properties of aminated tamarind xyloglucan. Colloids Surf. B Biointerfaces 2010, 81, 513–520. [Google Scholar] [CrossRef]
- Koshida, S.; Suda, Y.; Arano, A.; Sobel, M.; Kusumoto, S. An efficient method for the assembly of sulfated oligosaccharides using reductive amination. Tetrahedron Lett. 2001, 42, 1293–1296. [Google Scholar] [CrossRef]
- Esposito, E.; Vlodavsky, I.; Barash, U.; Roscilli, G.; Milazzo, F.M.; Giannini, G.; Naggi, A. Novel N-acetyl-Glycol-split heparin biotin-conjugates endowed with anti-heparanase activity. Eur. J. Med. Chem. 2020, 186, 111831. [Google Scholar] [CrossRef]
- Guerry, A.; Bernard, J.; Samain, E.; Fleury, E.; Cottaz, S.; Halila, S. Aniline-catalyzed reductive amination as a powerful method for the preparation of reducing end-“clickable” chitooligosaccharides. Bioconj. Chem. 2013, 24, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Gulberti, S.; Lattard, V.; Fondeur, M.; Jacquinet, J.C.; Mulliert, G.; Netter, P.; Magdalou, J.; Ouzzine, M.; Fournel-Gigleux, S. Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human β1,4-galactosyltransferase 7 (GalT-I) and β1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 2005, 280, 1417–1425. [Google Scholar] [PubMed] [Green Version]
- Oshima, T.; Taguchi, S.; Ohe, K.; Baba, Y. Phosphorylated bacterial cellulose for adsorption of proteins. Carbohydr. Polym. 2011, 83, 953–958. [Google Scholar] [CrossRef]
- Feng, H.; Fan, J.; Yang, S.; Zhao, X.; Yi, X. Antiviral activity of phosphorylated Radix Cyathulae officinalis polysaccharide against Canine Parvovirus in vitro. Int. J. Biol. Macromol. 2017, 99, 511–518. [Google Scholar] [CrossRef]
- Song, Y.; Ni, Y.; Hu, X.; Li, Q. Effect of phosphorylation on antioxidant activities of pumpkin (Cucurbita pepo, Lady godiva) polysaccharide. Int. J. Biol. Macromol. 2015, 81, 41–48. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Yao, Q.; Zhao, M.; Qi, H. Phosphorylation of low-molecular-weight polysaccharide from Enteromorpha linza with antioxidant activity. Carbohydr. Polym. 2013, 96, 371–375. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Y.; Li, Y.; Wei, Q.; Hu, L.; Yan, T.; Feng, R.; Yan, L.; Du, B. Phosphorylated chitosan/CoFe2O4 composite for the efficient removal of Pb(II) and Cd(II) from aqueous solution: Adsorption performance and mechanism studies. J. Mol. Liq. 2019, 277, 181–188. [Google Scholar] [CrossRef]
- Nishi, N.; Maekita, Y.; Nishimura, S.; Hasegawa, O.; Tokura, S. Highly phosphorylated derivatives of chitin and chitosan as new functional polymers: Metal binding property of the insolubilized materials. Int. J. Biol. Macromol. 1987, 9, 109–114. [Google Scholar] [CrossRef]
- Jayakumar, R.; Selvamurugan, N.; Nair, S.V.; Tokura, S.; Tamura, H. Preparative methods of phosphorylated chitin and chitosan—An overview. Int. J. Biol. Macromol. 2008, 43, 221–225. [Google Scholar] [CrossRef]
- Dadhich, P.; Das, B.; Dhara, S. Microwave assisted rapid synthesis of N-methylene phosphonic chitosan via Mannich-type reaction. Carbohydr. Polym. 2015, 133, 345–352. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques, 2nd ed.; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Bergman, K.; Elvingson, C.; Hilborn, J.; Svensk, G.; Bowden, T. Hyaluronic Acid Derivatives Prepared in Aqueous Media by Triazine-Activated Amidation. Biomacromolecules 2007, 8, 2190–2195. [Google Scholar] [CrossRef]
- D’Este, M.; Eglin, D.; Alini, M. A systematic analysis of DMTMM vs EDC/NHS for ligation of amines to hyaluronan in water. Carbohydr. Polym. 2014, 108, 239–246. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Lan, J.; Kang, Y.; Zhang, T.; Ding, Y.; Zhang, X.; Lu, L. Reduction-sensitive CD44 receptor-targeted hyaluronic acid derivative micelles for doxorubicin delivery. Int. J. Nanomed. 2018, 13, 4361–4378. [Google Scholar] [CrossRef] [Green Version]
- Freudenberg, U.; Sommer, J.U.; Levental, K.R.; Welzel, P.B.; Zieris, A.; Chwalek, K.; Schneider, K.; Prokoph, S.; Prewitz, M.; Dockhorn, R.; et al. Using Mean Field Theory to Guide Biofunctional Materials Design. Adv. Funct. Mater. 2012, 22, 1391–1398. [Google Scholar] [CrossRef]
- Freudenberg, U.; Hermann, A.; Welzel, P.B.; Stirl, K.; Schwarz, S.C.; Grimmer, M.; Zieris, A.; Panyanuwat, W.; Zschoche, S.; Meinhold, D.; et al. A star-PEG-heparin hydrogel platform to aid cell replacement therapies for neurodegenerative diseases. Biomaterials 2009, 30, 5049–5060. [Google Scholar] [CrossRef]
- Palazon, F.; Benavides, C.M.; Leonard, D.; Souteyrand, E.; Chevolot, Y.; Cloarec, J.P. Carbodiimide/NHS derivatization of COOH-terminated SAMs: Activation or byproduct formation? Langmuir 2014, 30, 4545–4550. [Google Scholar] [CrossRef]
- Akaji, K.; Barker, G.; Bonewald, L.F. Supplement. Houben-Weyl Methods of Organic Chemistry, 4th ed.; Georg Thieme Verlag: Stuttgart, Germany, 2004; Volume 22a, pp. 517–534. [Google Scholar]
- Toida, T. Method for Producing Alkyl-Esterified Glycosaminoglycan. United States Patent No. US 2006/0172967 A1, 3 August 2006. [Google Scholar]
- Hirano, K.; Sakai, S.; Ishikawa, T.; Avci, F.Y.; Linhardt, R.J.; Toida, T. Preparation of the methyl ester of hyaluronan and its enzymatic degradation. Carbohydr. Res. 2005, 340, 2297–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimkovic, I.; Mendichi, R.; Kelnar, I.; Filip, J.; Hricovini, M. Cationization of heparin for film applications. Carbohydr. Polym. 2015, 115, 551–558. [Google Scholar] [CrossRef]
- Siahaan, P.; Mentari, N.C.; Wiedyanto, U.O.; Hudiyanti, D.; Hildayani, S.Z.; Laksitorini, M.D. The Optimum Conditions of Carboxymethyl Chitosan Synthesis on Drug Delivery Application and Its Release of Kinetics Study. Indones. J. Chem. 2017, 17, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Prestwich, G.; Zhang, J.; Kennedy, T.P.; Rao, N. Use of Alkylated Semi-Synthetic Glycosaminoglycosan Ethers for the Treatment of Inflammation. United States Patent No. US 9,549,945 B2, 24 January 2017. [Google Scholar]
- Callegaro, L.; Renier, D. Sulphated Hyaluronic Acid for Treating Degenerative Osteoarthritis. United States Patent No. US 2009/0197807 A1, 6 August 2009. [Google Scholar]
- Venbrocks, R.; Roth, A.; Mueller, P.-J.; Moeller, S.; Ozegowski, J.; Peschel, G. Use of Hyaluronic Acid Derivatives for Inhibiting Inflammatory Arthritis. US Patent Number US 2007/0054878 A1, 8 March 2007. [Google Scholar]
- Dell, A.; Khoo, K.-H.; Panico, M.; McDowell, R.A.; Etienne, A.T.; Reason, A.J.; Morris, H.R. FAB-MS and ES-MS of Glycoproteins. In Glycobiology: A Practical Approach; Fukuda, M., Kobata, A., Eds.; Oxford University Press: New York, NY, USA, 1993; pp. 187–222. [Google Scholar]
- Huang, R.; Pomin, V.H.; Sharp, J.S. LC-MS(n) analysis of isomeric chondroitin sulfate oligosaccharides using a chemical derivatization strategy. J. Amermican Soc. Mass Spectrom. 2011, 22, 1577–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Borges, C.R. A spin column-free approach to sodium hydroxide-based glycan permethylation. Analyst 2017, 142, 2748–2759. [Google Scholar] [CrossRef] [PubMed]
- Ciucanu, I.; Kerek, F. A Simple and Rapid Permathylation Method for the Permethylation of Carbohydrates. Carbohydr. Res. 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Sims, I.M.; Carnachan, S.M.; Bell, T.J.; Hinkley, S.F.R. Methylation analysis of polysaccharides: Technical advice. Carbohydr. Polym. 2018, 188, 1–7. [Google Scholar] [CrossRef]
- Cushnie, T.P.; Lamb, A.J. Recent advances in understanding the antibacterial properties of flavonoids. Int. J. Antimicrob. Agents 2011, 38, 99–107. [Google Scholar] [CrossRef]
- Sales, M.S.; Roy, A.; Antony, L.; Banu, S.K.; Jeyaraman, S.; Manikkam, R. Octyl gallate and gallic acid isolated from Terminalia bellarica regulates normal cell cycle in human breast cancer cell lines. Biomed. Pharmacother. 2018, 103, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- de Cordova, C.A.; Locatelli, C.; Assuncao, L.S.; Mattei, B.; Mascarello, A.; Winter, E.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Octyl and dodecyl gallates induce oxidative stress and apoptosis in a melanoma cell line. Toxicol. Vitr. 2011, 25, 2025–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Li, Q.; Dong, F.; Feng, Y.; Guo, Z. Phenolic antioxidants-functionalized quaternized chitosan: Synthesis and antioxidant properties. Int. J. Biol. Macromol. 2013, 53, 77–81. [Google Scholar] [CrossRef]
- Spizzirri, U.G.; Parisi, O.I.; Iemma, F.; Cirillo, G.; Puoci, F.; Curcio, M.; Picci, N. Antioxidant–polysaccharide conjugates for food application by eco-friendly grafting procedure. Carbohydr. Polym. 2010, 79, 333–340. [Google Scholar] [CrossRef]
- Ballell, L.; Joosten, J.A.F.; Maate, F.A.; Liskamp, R.M.J.; Pieters, R.J. Microwave-assisted, tin-mediated, regioselective 3-O-alkylation of galactosides. Tetrahedron Lett. 2004, 45, 6685–6687. [Google Scholar] [CrossRef]
- Hamer, G.K.; Perlin, A.S. A 13C-N.M.R. Spectral Study of Chondroitin Sulfates A, B, and C: Evidence of Heterogeneity. Carbohydr. Res. 1976, 49, 37–48. [Google Scholar] [CrossRef]
- Huckerby, T.N.; Lauder, R.M.; Brown, G.M.; Nieduszynski, I.A.; Anderson, K.; Boocock, J.; Sandall, P.L.; Weeks, S.D. Characterization of oligosaccharides from the chondroitin sulfates. Eur. J. Biochem. 2001, 268, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Huckerby, T.N.; Brown, G.M.; Nieduszynski, I.A. 13C-NMR spectroscopy of keratan sulphates. Eur. J. Biochem. 1998, 251, 991–997. [Google Scholar] [CrossRef]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palhares, L.C.G.F.; London, J.A.; Kozlowski, A.M.; Esposito, E.; Chavante, S.F.; Ni, M.; Yates, E.A. Chemical Modification of Glycosaminoglycan Polysaccharides. Molecules 2021, 26, 5211. https://doi.org/10.3390/molecules26175211
Palhares LCGF, London JA, Kozlowski AM, Esposito E, Chavante SF, Ni M, Yates EA. Chemical Modification of Glycosaminoglycan Polysaccharides. Molecules. 2021; 26(17):5211. https://doi.org/10.3390/molecules26175211
Chicago/Turabian StylePalhares, Lais C. G. F., James A. London, Aleksandra M. Kozlowski, Emiliano Esposito, Suely F. Chavante, Minghong Ni, and Edwin A. Yates. 2021. "Chemical Modification of Glycosaminoglycan Polysaccharides" Molecules 26, no. 17: 5211. https://doi.org/10.3390/molecules26175211
APA StylePalhares, L. C. G. F., London, J. A., Kozlowski, A. M., Esposito, E., Chavante, S. F., Ni, M., & Yates, E. A. (2021). Chemical Modification of Glycosaminoglycan Polysaccharides. Molecules, 26(17), 5211. https://doi.org/10.3390/molecules26175211