
Synthesis, Characterization, and Reaction Studies of Pd(II) Tripeptide Complexes


Abstract
:
1. Introduction
2. Results
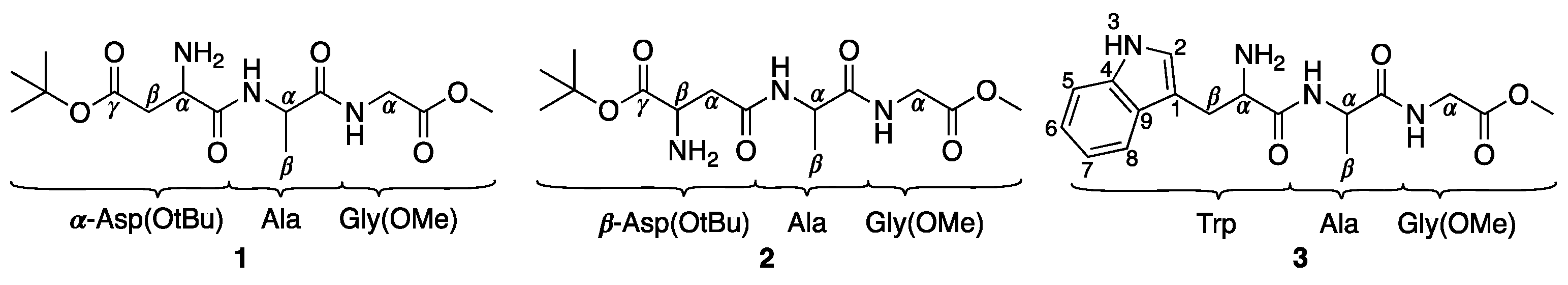
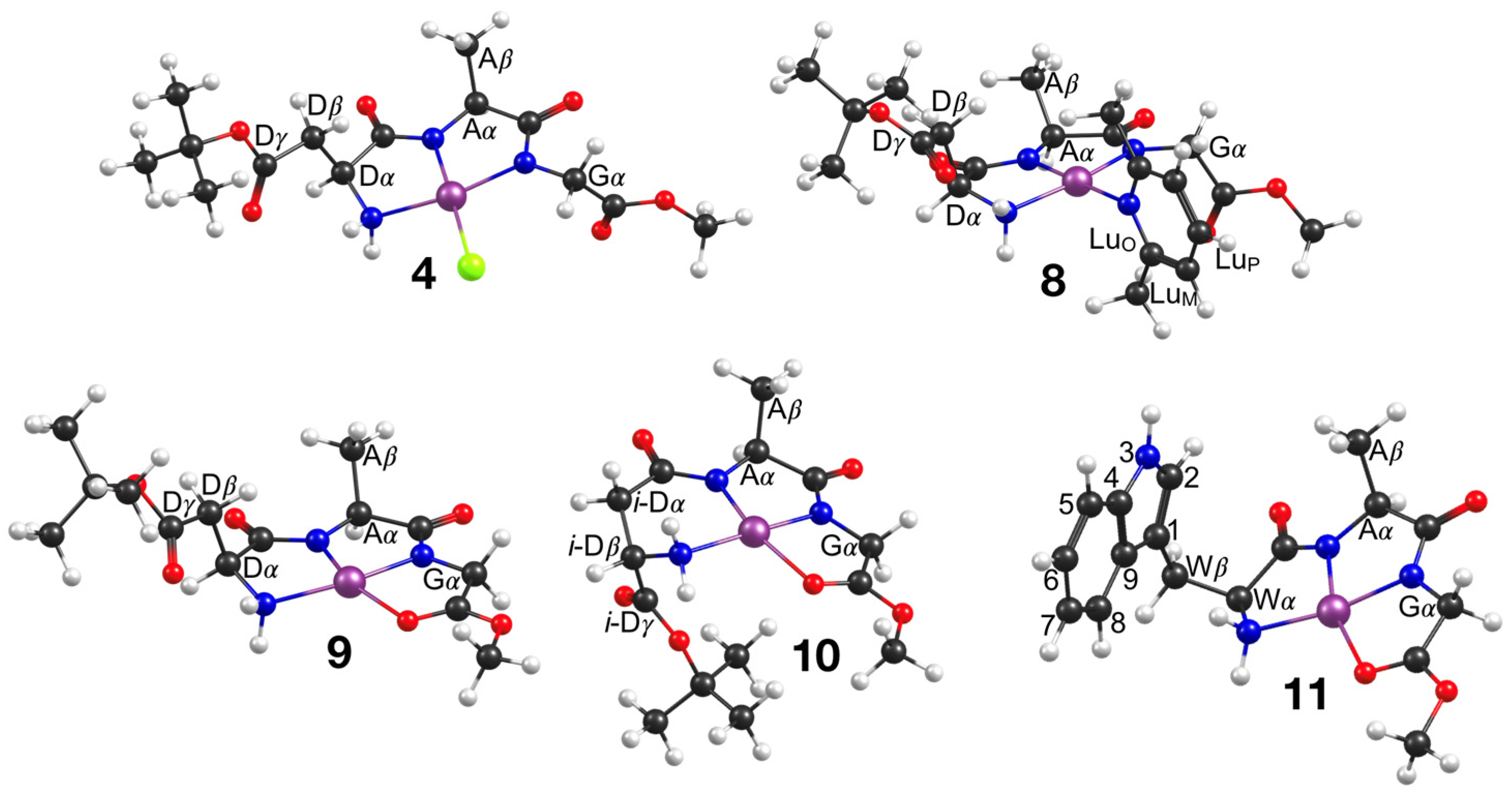
2.1. Synthesis and Cation Exchange of Mono-Anionic Complexes 4–6
Ligand Exchange Reactions
2.2. Synthesis of Neutral Complexes Tetradentate Pd(II) Complexes 9–11
Ligand Exchange Reactions
2.3. DFT Calculations
2.4. Characterization
2.4.1. H- and 13C-NMR Spectroscopy
2.4.2. Infrared Spectroscopy
2.4.3. Mass Spectrometry
2.5. Reactivity Studies
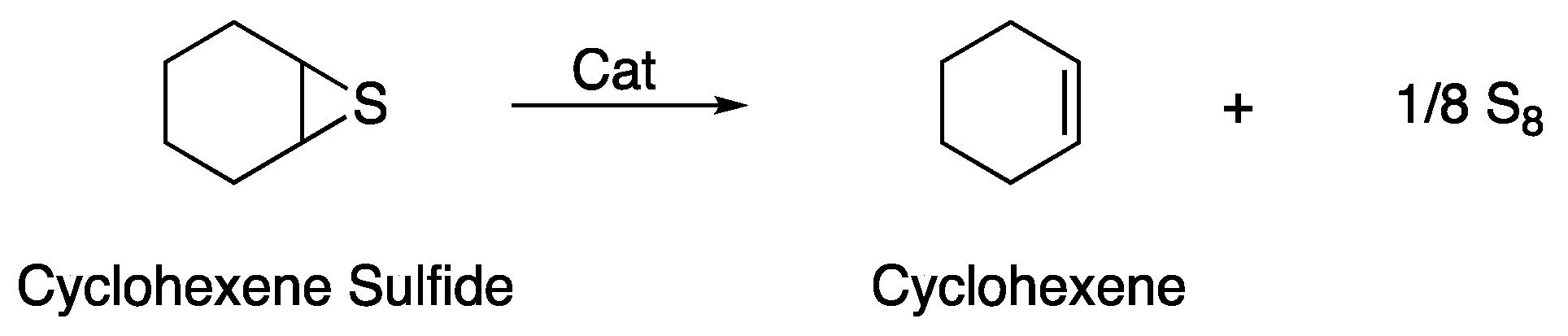
2.5.1. Sulfur Reagents
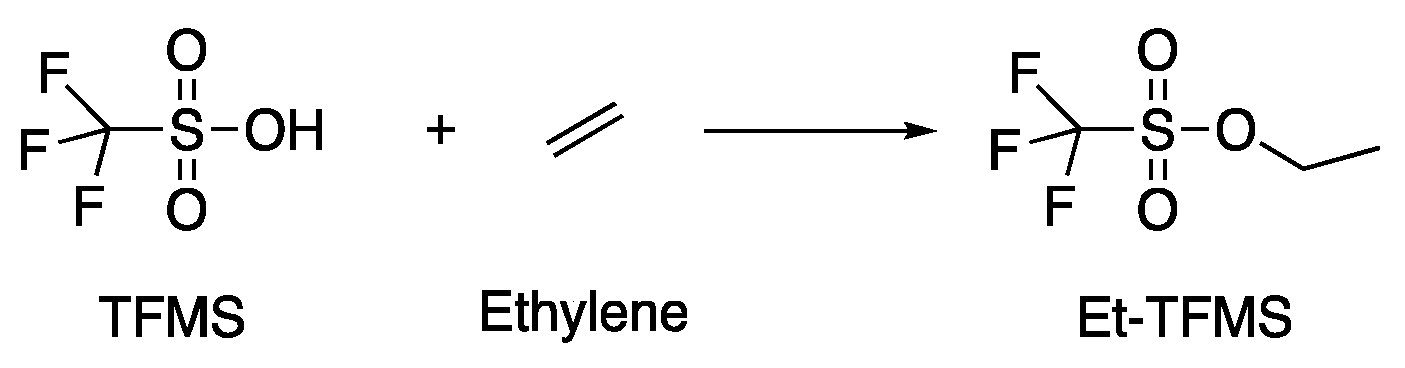
2.5.2. Olefins
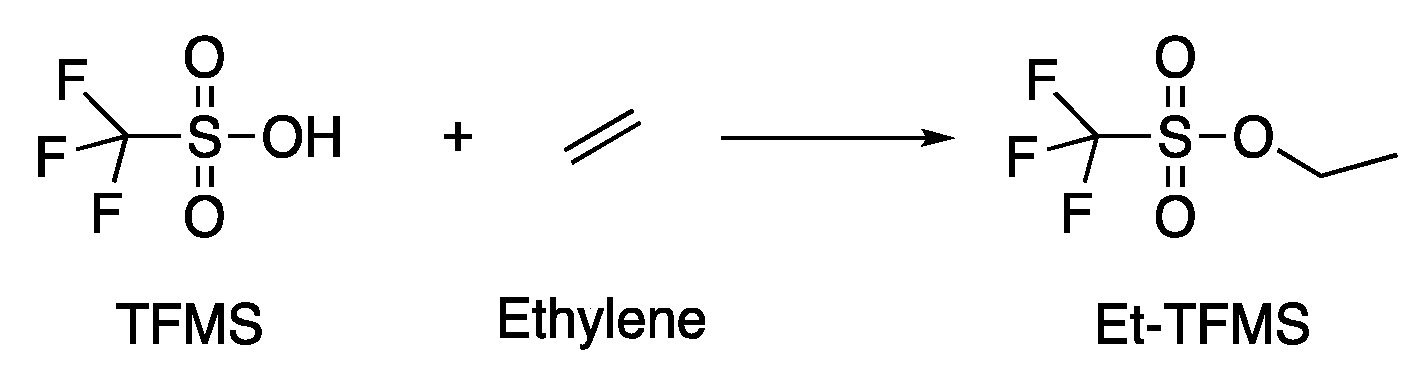
2.5.3. Derivatives of CO2 and CO
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.1.1. K[Pd{α-Asp(OtBu)AlaGly(OMe)}Cl] (4)
4.1.2. (Et3N)[Pd{α-Asp(OtBu)AlaGly(OMe)}Cl] (5)
4.1.3. (PPh4)[Pd{α-Asp(OtBu)AlaGly(OMe)}Cl] (6)
4.1.4. Pd{α-Asp(OtBu)AlaGly(OMe)}(Lu) (7)
4.1.5. Pd{α-Asp(OtBu)AlaGly(OMe)} (9)
4.1.6. Pd{β-Asp(OtBu)AlaGly(OMe)} (10)
4.1.7. Pd{TrpAlaGly(OMe)} (11)
4.2. Reaction Studies
4.2.1. Sulfur Reagents
4.2.2. Ethylene and Acids
4.3. Quantum Chemical Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kitteringham, E.; McKeon, A.M.; O’Dowd, P.; Devocelle, M.; Murphy, B.M.; Griffith, D.M. Synthesis and Characterisation of a Novel Mono Functionalisable Pt(IV) Oxaliplatin-Type Complex and Its Peptide Conjugate. Inorg. Chim. Acta 2020, 505, 119492. [Google Scholar] [CrossRef]
- Metzler-Nolte, N.; Guo, Z. Themed Issue on “Metallodrugs: Activation, Targeting, and Delivery”. Dalton Trans. 2016, 45, 12965. [Google Scholar] [CrossRef]
- Bradford, S.S.; Cowan, J.A. From Traditional Drug Design to Catalytic Metallodrugs: A Brief History of the Use of Metals in Medicine. Metallodrugs 2014, 1, 10–23. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Sadler, P.J. Approaches to the Design of Catalytic Metallodrugs. Curr. Opin. Chem. Biol. 2015, 25, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Ward, T.R. ACS Central Science Virtual Issue on Bioinspired Catalysis. ACS Cent. Sci. 2019, 5, 1732–1735. [Google Scholar] [CrossRef] [Green Version]
- Sigel, H.; Martin, R.B. Coordinating Properties of the Amide Bond. Stability and Structure of Metal Ion Complexes of Peptides and Related Ligands. Chem. Rev. 1982, 82, 385–426. [Google Scholar] [CrossRef]
- Sóvágó, I.; Ősz, K. Metal Ion Selectivity of Oligopeptides. Dalton Trans. 2006, 32, 3841–3854. [Google Scholar] [CrossRef]
- Murphy, J.M.; Powell, B.A.; Brumaghim, J.L. Stability Constants of Bio-Relevant, Redox-Active Metals with Amino Acids: The Challenges of Weakly Binding Ligands. Coord. Chem. Rev. 2020, 412, 213253. [Google Scholar] [CrossRef]
- Kozłowski, H.; Bal, W.; Dyba, M.; Kowalik-Jankowska, T. Specific Structure–Stability Relations in Metallopeptides. Coord. Chem. Rev. 1999, 184, 319–346. [Google Scholar] [CrossRef]
- El-Sherif, A.A. Coordination chemistry of palladium (II) ternary complexes with relevant biomolecules. Stoichiometry and Research. In The Importance of Quantity in Biomedicine; IntechOpen: London, UK, 2012; pp. 80–120. [Google Scholar]
- Jószai, V.; Nagy, Z.; Ősz, K.; Sanna, D.; Di Natale, G.; La Mendola, D.; Pappalardo, G.; Rizzarelli, E.; Sóvágó, I. Transition Metal Complexes of Terminally Protected Peptides Containing Histidyl Residues. J. Inorg. Biochem. 2006, 100, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, S.E.; Nolan, J.D. Metal Chelates of Biologically Important Compounds. I. Complexes of DL-Ethionine and S-Methyl-L-Cysteine. Inorg. Chem. 1968, 7, 1447–1451. [Google Scholar] [CrossRef]
- Chandrasekharan, M.; Udupa, M.R.; Aravamudan, G. Cysteine Complexes of Palladium(II) and Platinum(II). Inorg. Chim. Acta 1973, 7, 88–90. [Google Scholar] [CrossRef]
- Pettit, L.D.; Bezer, M. Complex Formation between Palladium(II) and Amino Acids, Peptides and Related Ligands. Coord. Chem. Rev. 1985, 61, 97–114. [Google Scholar] [CrossRef]
- McAuliffe, C.A. The Infrared Spectra of Palladium(II) and Platinum(II) Complexes of (±)-Methionine. J. Chem. Soc. A 1967, 0, 641–642. [Google Scholar] [CrossRef]
- Ágoston, C.G.; Jankowska, T.K.; Sóvágó, I. Potentiometric and NMR Studies on Palladium(II) Complexes of Oligoglycines and Related Ligands with Non-Co-Ordinating Side Chains. J. Chem. Soc. Dalton Trans. 1999, 18, 3295–3302. [Google Scholar] [CrossRef]
- Wilson, E.W.; Martin, R.B. Circular Dichroism of Palladium(II) Complexes of Amino Acids and Peptides. Inorg. Chem. 1970, 9, 528–532. [Google Scholar] [CrossRef]
- Maiti, B.K.; Govil, N.; Kundu, T.; Moura, J.J.G. Designed Metal-ATCUN Derivatives: Redox- and Non-Redox-Based Applications Relevant for Chemistry, Biology, and Medicine. iScience 2020, 23, 101792. [Google Scholar] [CrossRef]
- Perinelli, M.; Guerrini, R.; Albanese, V.; Marchetti, N.; Bellotti, D.; Gentili, S.; Tegoni, M.; Remelli, M. Cu(II) Coordination to His-Containing Linear Peptides and Related Branched Ones: Equalities and Diversities. J. Inorg. Biochem. 2020, 205, 110980. [Google Scholar] [CrossRef]
- Gonzalez, P.; Bossak, K.; Stefaniak, E.; Hureau, C.; Raibaut, L.; Bal, W.; Faller, P. N-Terminal Cu-Binding Motifs (Xxx-Zzz-His, Xxx-His) and Their Derivatives: Chemistry, Biology and Medicinal Applications. Chem. Eur. J. 2018, 24, 8029–8041. [Google Scholar] [CrossRef]
- Peana, M.; Gumienna-Kontecka, E.; Piras, F.; Ostrowska, M.; Piasta, K.; Krzywoszynska, K.; Medici, S.; Zoroddu, M.A. Exploring the Specificity of Rationally Designed Peptides Reconstituted from the Cell-Free Extract of Deinococcus radiodurans toward Mn(II) and Cu(II). Inorg. Chem. 2020, 59, 4661–4684. [Google Scholar] [CrossRef]
- Vicatos, G.M.; Jackson, G.E.; Hammouda, A.N.; Bonomo, R.P.; Valora, G. Potentiometric and Spectroscopic Studies of the Complex Formation between Copper(II) and Gly-Leu-Phe or Sar-Leu-Phe Tripeptides. Polyhedron 2019, 170, 553–563. [Google Scholar] [CrossRef]
- Gavrish, S.P.; Lampeka, Y.D.; Babak, M.V.; Arion, V.B. Palladium Complexes of N,N′-Bis(2-Aminoethyl)Oxamide (H2L): Structural (PdIIL, PdII2L2, and PdIVLCl2), Electrochemical, Dynamic 1H NMR, and Cytotoxicity Studies. Inorg. Chem. 2018, 57, 1288–1297. [Google Scholar] [CrossRef]
- Gonzalez, P.; Vileno, B.; Bossak, K.; El Khoury, Y.; Hellwig, P.; Bal, W.; Hureau, C.; Faller, P. Cu(II) Binding to the Peptide Ala-His-His, a Chimera of the Canonical Cu(II)-Binding Motifs Xxx-His and Xxx-Zzz-His. Inorg. Chem. 2017, 56, 14870–14879. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Lee, J.Y.; Himes, R.A.; Thomas, G.S.; Blackburn, N.J.; Karlin, K.D. Copper–Peptide Complex Structure and Reactivity When Found in Conserved His-Xaa-His Sequences. J. Am. Chem. Soc. 2014, 136, 12532–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sóvágó, I.; Kállay, C.; Várnagy, K. Peptides as Complexing Agents: Factors Influencing the Structure and Thermodynamic Stability of Peptide Complexes. Coord. Chem. Rev. 2012, 256, 2225–2233. [Google Scholar] [CrossRef]
- Griffith, D.M.; Bíró, L.; Platts, J.A.; Müller-Bunz, H.; Farkas, E.; Buglyó, P. Synthesis and Solution Behaviour of Stable Mono-, Di- and Trinuclear Pd(II) Complexes of 2,5-Pyridinedihydroxamic Acid: X-Ray Crystal Structure of a Novel Pd(II) Hydroxamato Complex. Inorg. Chim. Acta 2012, 380, 291–300. [Google Scholar] [CrossRef]
- Gábor Ágoston, C.; Miskolczy, Z.; Nagy, Z.; Sóvágó, I. The Effect of Ring Size of Fused Chelates on the Stability Constants and Spectroscopic Properties of Nickel(II) and Palladium(II) Complexes of Peptides. Polyhedron 2003, 22, 2607–2615. [Google Scholar] [CrossRef]
- Hay, R.W.; Pujari, M.P. The Palladium(II) Promoted Hydrolysis of Methyl, Ethyl and Isopropyl Glycylglycylglycinate. Inorg. Chim. Acta 1986, 123, 47–51. [Google Scholar] [CrossRef]
- Monger, L.J.; Runarsdottir, G.R.; Suman, S.G. Directed Coordination Study of [Pd(En)(H2O)2]2+ with Hetero-Tripeptides Containing C-Terminus Methyl Esters Employing NMR Spectroscopy. J. Biol. Inorg. Chem. 2020, 25, 811–825. [Google Scholar] [CrossRef]
- Chen, S.; Vasquez, L.; Noll, B.C.; Rakowski DuBois, M. Synthesis and Characterization of Mononuclear Indoline Complexes. Studies of σ and π Bonding Modes. Organometallics 1997, 16, 1757–1764. [Google Scholar] [CrossRef]
- Sundberg, R.J. Electrophilic Substitution Reactions of Indoles. In Heterocyclic Scaffolds II; Gribble, G.W., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2010; Volume 26, pp. 47–115. ISBN 978-3-642-15732-5. [Google Scholar]
- Singh, M.P.; Saleem, F.; Pal, R.S.; Singh, A.K. Palladacycles Having Normal and Spiro Chelate Rings Designed from Bi- and Tridentate Ligands with an Indole Core: Structure, Synthesis and Applications as Catalysts. New J. Chem. 2017, 41, 11342–11352. [Google Scholar] [CrossRef]
- Kaminskaia, N.V.; Ullmann, G.M.; Fulton, D.B.; Kostić, N.M. Spectroscopic, Kinetic, and Mechanistic Study of a New Mode of Coordination of Indole Derivatives to Platinum(II) and Palladium(II) Ions in Complexes. Inorg. Chem. 2000, 39, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Jack Mccormick, B.; Jaynes, E.N.; Kaplan, R.I.; Clark, H.C.; Ruddick, J.D. Dichloro(ethylenediamine)palladium(II). In Inorganic Syntheses; Cotton, F.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 216–218. ISBN 978-0-470-13244-9. [Google Scholar]
- Walton, R.A. The Infra-Red Spectra of Complexes of Palladium (II) and Platinum (II) Halides with Methyl Phenyl Cyanides. Spectrochim. Acta 1965, 21, 1795–1801. [Google Scholar] [CrossRef]
- Rochon, F.D.; Melanson, R.; Howard-Lock, H.E.; Lock, C.J.L.; Turner, G. The Vibrational Spectra, Crystal and Molecular Structure of Bis(Acetonitrile)Dichloroplatinum(II). Can. J. Chem. 1984, 62, 860–869. [Google Scholar] [CrossRef]
- Schreiner, B.; Robl, C.; Wagner-Schuh, B.; Beck, W. Metal Complexes of Biologically Important Ligands, CLXXIV [1]. Palladium(II) and Platinum(II) Complexes with Schiff Bases from 2-(Diphenylphosphino)Benzaldehyde and α-Amino Acid Esters. Z. Nat. B 2010, 65, 503–510. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th ed.; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-0-471-74339-2. [Google Scholar]
- Zhu, L.; Kostic, N.M. Toward Artificial Metallopeptidases: Mechanisms by Which Platinum(II) and Palladium(II) Complexes Promote Selective, Fast Hydrolysis of Unactivated Amide Bonds in Peptides. Inorg. Chem. 1992, 31, 3994–4001. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Souza, B.C.; Paiva, R.E.F.; Bergamini, F.R.G.; Gomes, A.F.; Gozzo, F.C.; Lustri, W.R.; Formiga, A.L.B.; Rigatto, G.; Corbi, P.P. Synthesis, Spectroscopic Characterization, DFT Studies, and Initial Antibacterial Assays in Vitro of a New Palladium(II) Complex with Tryptophan. J. Coord. Chem. 2012, 65, 1700–1711. [Google Scholar] [CrossRef]
- Bontchev, P.R.; Boneva, M.; Arnaudov, M.; Nefedov, V.I. Palladium(II) Complexes of Hydrazides of Aspartic and Glutamic Acids. Inorg. Chim. Acta 1984, 81, 75–81. [Google Scholar] [CrossRef]
- Schreiner, B.; Beck, W. Coordination of the Ester Group—Hydrido-Rhodium(III) and Iridium(III) Complexes of Orthometallated Diphenylmethylene Glycine Esters [1]. Z. Anorg. Allg. Chem. 2010, 636, 499–505. [Google Scholar] [CrossRef]
- Liu, J.; Deng, Y.; Lin, C.; Lei, A. “Push Effect” of Sulfur Coordination: Promoting the Breaking of C(Sp2)–I Bond by Pincer Thioimido-Pd(II) Complexes. Chem. Sci. 2012, 3, 1211. [Google Scholar] [CrossRef]
- Silvano, S.; Carrozza, C.F.; de Angelis, A.R.; Tritto, I.; Boggioni, L.; Losio, S. Synthesis of Sulfur-Rich Polymers: Copolymerization of Cyclohexene Sulfide and Carbon Disulfide Using Chromium Complexes. Macromolecules 2020, 53, 8837–8846. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis: A Practical Approach, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2015; ISBN 978-3-527-33165-9. [Google Scholar]
- Wang, F.; Chen, C. A Continuing Legend: The Brookhart-Type α-Diimine Nickel and Palladium Catalysts. Polym. Chem. 2019, 10, 2354–2369. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.-Y.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Palladium(II) Complexes Containing P∼N∼O Donors. Ligand Effect of Tridentate versus Bidentate Coordination on the Oligomerization of Ethylene. Organometallics 2002, 21, 3203–3207. [Google Scholar] [CrossRef]
- Howells, R.D.; Mc Cown, J.D. Trifluoromethanesulfonic Acid and Derivatives. Chem. Rev. 1977, 77, 69–92. [Google Scholar] [CrossRef]
- Gramstad, T.; Haszeldine, R.N. 806. Perfluoroalkyl Derivatives of Sulphur. Part VII. Alkyl Trifluoromethanesulphonates as Alkylating Agents, Trifluoromethanesulphonic Anhydride as a Promoter for Esterification, and Some Reactions of Trifluoromethanesulphonic. Acid. J. Chem. Soc. 1957, 4069. [Google Scholar] [CrossRef]
- Núñez Magro, A.A.; Robb, L.-M.; Pogorzelec, P.J.; Slawin, A.M.Z.; Eastham, G.R.; Cole-Hamilton, D.J. Highly Selective Formation of Unsaturated Esters or Cascade Reactions to α,ω-Diesters by the Methoxycarbonylation of Alkynes Catalysed by Palladium Complexes of 1,2-Bis(Ditertbutylphosphinomethyl)Benzene. Chem. Sci. 2010, 1, 723. [Google Scholar] [CrossRef]
- Suleiman, R.; Tijani, J.; El Ali, B. Palladium(II)-Catalyzed Catalytic Aminocarbonylation and Alkoxycarbonylation of Terminal Alkynes: Regioselectivity Controlled by the Nucleophiles. Appl. Organometal. Chem. 2009, 24, 38–46. [Google Scholar] [CrossRef]
- Katafuchi, Y.; Fujihara, T.; Iwai, T.; Terao, J.; Tsuji, Y. Palladium-Catalyzed Hydroesterification of Alkynes Employing Aryl Formates without the Use of External Carbon Monoxide. Adv. Synth. Catal. 2011, 353, 475–482. [Google Scholar] [CrossRef]
- Dong, K.; Sang, R.; Liu, J.; Razzaq, R.; Franke, R.; Jackstell, R.; Beller, M. Palladium-Catalyzed Carbonylation of sec- and tert-Alcohols. Angew. Chem. Int. Ed. 2017, 56, 6203–6207. [Google Scholar] [CrossRef]
- Sang, R.; Schneider, C.; Razzaq, R.; Neumann, H.; Jackstell, R.; Beller, M. Palladium-Catalyzed Carbonylations of Highly Substituted Olefins Using CO-Surrogates. Org. Chem. Front. 2020, 7, 3681–3685. [Google Scholar] [CrossRef]
- Razinkov, D.; Ingvarsdottir, H.I.; Kvaran, Á.; Jonsdottir, S.; Suman, S.G. Coordination Properties of Non-Rigid Phosphinoyldithioformate Complexes of the [Mo2O2(µ-S)2]2+ Cation in Catalytic Sulfur Transfer Reactions with Thiiranes. Catalysts 2021, 11, 593. [Google Scholar] [CrossRef]
- Takacs, J.; Jiang, X. The Wacker Reaction and Related Alkene Oxidation Reactions. COC 2003, 7, 369–396. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Amsterdam, The Netherlands; Boston, MA, USA, 2003; ISBN 978-0-7506-7571-0. [Google Scholar]
- Carretero, J.C.; Arrayás, R.G. Dichloro Bis(acetonitrile) Palladium. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2008; p. rn00908. ISBN 978-0-471-93623-7. [Google Scholar]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Shi, G.; Dang, Y.; Pan, T.; Liu, X.; Liu, H.; Li, S.; Zhang, L.; Zhao, H.; Li, S.; Han, J.; et al. Unexpectedly Enhanced Solubility of Aromatic Amino Acids and Peptides in an Aqueous Solution of Divalent Transition-Metal Cations. Phys. Rev. Lett. 2016, 117, 238102. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An Overlap Fitted Chain of Spheres Exchange Method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- York, D.M.; Karplus, M. A Smooth Solvation Potential Based on the Conductor-Like Screening Model. J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar] [CrossRef]
- Garcia-Ratés, M.; Neese, F. Effect of the Solute Cavity on the Solvation Energy and Its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020, 41, 922–939. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ratés, M.; Neese, F. Efficient Implementation of the Analytical Second Derivatives of Hartree–Fock and Hybrid DFT Energies within the Framework of the Conductor-like Polarizable Continuum Model. J. Comput. Chem. 2019, 40, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Bykov, D.; Petrenko, T.; Izsák, R.; Kossmann, S.; Becker, U.; Valeev, E.; Neese, F. Efficient Implementation of the Analytic Second Derivatives of Hartree–Fock and Hybrid DFT Energies: A Detailed Analysis of Different Approximations. Mol. Phys. 2015, 113, 1961–1977. [Google Scholar] [CrossRef]
- Jensen, F. Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding. J. Chem. Theory Comput. 2015, 11, 132–138. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Angle | 4 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| NH2-Pd-Nala | 82.84 | 82.71 | 82.97 | 95.01 | 82.61 |
| Nala-Pd-Ngly | 81.92 | 82.11 | 83.58 | 83.17 | 83.68 |
| Ngly-Pd-X | 99.54 | 98.35 | 80.47 | 80.99 | 80.32 |
| X-Pd-NH2 | 95.70 | 96.88 | 112.90 | 100.54 | 113.38 |
| Bond Length | |||||
| NH2-Pd | 2.061 | 2.060 | 2.077 | 2.064 | 2.073 |
| Nala-Pd | 1.940 | 1.942 | 1.916 | 1.964 | 1.911 |
| Ngly-Pd | 1.996 | 1.996 | 1.937 | 1.936 | 1.938 |
| X-Pd | 2.355 | 2.070 | 2.142 | 2.109 | 2.142 |
| Chemical Shift (ppm) | 4 | 7 | 8 | 8 + Δ * | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|
| PyOrtho | - | 8.73 | - | - | - | - | - |
| Py/LuMeta | - | 7.57 | 7.33 | 7.02 | - | - | - |
| Py/LuPara | - | 7.99 | 7.78 | 7.54 | - | - | - |
| N-H2 | 4.20 3.86 | 4.96 4.38 | 4.76 4.15 | 5.03 4.54 | 5.04 4.55 | 4.63 4.48 | 4.96 4.24 |
| α-CAla | 3.54 | 3.66 | 3.66 | 3.68 | 3.68 | 3.89 | 3.78 |
| α-CGly | 3.64 | 3.52 | 3.14 2.91 | 3.86 | 3.87 | 3.62 | 3.96 |
| α-CTrp/Asp | 3.40 | 3.44 | 3.54 | 3.53 | 3.52 | 2.33 | 3.40 |
| OCH3 Gly | 3.50 | 3.34 | 3.34 | 3.55 | 3.55 | 3.55 | 3.55 |
| Lu CH3 | - | - | 3.18 | 2.41 | - | - | - |
| β-CTrp/Asp | 2.45 2.35 | 2.61 2.40 | 2.57 2.41 | 2.63 2.46 | 2.61 2.45 | 3.18 | 3.28 2.87 |
| OC(CH3)3 Asp | 1.40 | 1.40 | 1.41 | 1.42 | 1.42 | 1.44 | - |
| β-CAla | 1.15 | 1.23 | 1.23 | 1.20 | 1.20 | 1.08 | 1.24 |
| Chemical Shift (ppm) | 4 | 8 * | 9 | 10 | 11 |
|---|---|---|---|---|---|
| C=OTrp/Asp | 170.14 | 169.93 | 169.79 | 170.75 | 172.93 |
| C=OAla | 185.17 | 185.45 | 177.03 | 176.26 | 178.24 |
| γ-C=OAsp | 175.91 | 175.9 | 172.89 | 172.58 | - |
| C=OGly | 172.34 | 171.71 | 186.47 | n.d. | 186.55 |
| OC(CH3)3 Asp | 79.93 | 80.14 | 80.47 | 82.32 | - |
| OCH3 Gly | 50.52 | 50.79 | 50.96 | 50.92 | 50.93 |
| α-CTrp/Asp | 56.55 | 56.50 | 55.81 | 40.16 | 59.58 |
| α-CAla | 57.05 | 57.13 | 56.60 | 58.87 | 56.50 |
| α-CGly | 46.49 | 45.84 | 46.79 | 44.73 | 46.86 |
| β-CTrp/Asp | 39.86 | 39.73 | 39.94 | 54.7 | 29.93 |
| OC(CH3)3 Asp | 27.79 | 27.79 | 27.79 | 27.58 | - |
| β-CAla | 19.43 | 19.78 | 19.80 | 20.64 | 19.92 |
| Chemical Shift (ppm) | 4 (obs) | 4 (calc) | 8 (obs) | 8 (calc) | 9 (obs) | 9 (calc) | 10 (obs) | 10 (calc) | 11 (obs) | 11 (calc) |
|---|---|---|---|---|---|---|---|---|---|---|
| C=OTrp/Asp | 170.14 | 170.82 | 169.93 | 171.07 | 169.79 | 171.39 | 170.75 | 165.3 | 172.93 | 169.9 |
| C=OAla | 185.17 | 181.86 | 185.45 | 182.5 | 177.03 | 177.63 | 176.26 | 176.5 | 178.24 | 177.11 |
| γ-C=OAsp | 175.91 | 171.72 | 175.9 | 174.71 | 172.89 | 172.31 | 172.58 | 172.4 | - | - |
| C=OGly | 172.34 | 171.34 | 171.71 | 173.3 | 186.47 | 186.23 | n.d. | 187.8 | 186.55 | 185.82 |
| Frequency (cm−1) | 1 | 2 | 3 | 4 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|
| ν(NH2) ν(NH) | 3370 3314 | 3375 * 3304 | 3370 * 3297 | 3234 3138 | 3223 3100 | 3245 3129 | 3239 3121 | 3286 3235 |
| ν(C=O) | ||||||||
| OtBu | 1725 | 1743 | - | 1723 | 1732 | 1729 | 1732 | - |
| OMe | 1759 * | 1757 * | 1749 | 1748 * | 1750 * | 1541 | 1551 | 1554 |
| Amide I | 1670 | 1659 | 1655 | 1585 1570 | 1604 1582 | 1608 1584 | 1590 1567 | 1598 1569 |
| ν(C-O) | ||||||||
| OtBu | 1250 | 1254 | - | 1254 | 1250 | 1253 | 1258 | - |
| OMe | 1213 | 1212 | 1213 | 1210 | 1210 | 1205 | 1207 | 1207 |
| ν(O-C) | ||||||||
| OtBu | 1155 | 1155 | - | 1148 | 1147 | 1145 | 1155 | - |
| ν(M-N) | 1250 | 1254 | - | 573 548 | 568 548 | 573 548 | 573 533 | 568 534 |
| Complex | ν(C=O) OtBu | ν(C=O) OMe | ν(C=O) Amide I | ν(C=O) Amide I |
|---|---|---|---|---|
| 4 Obs | 1723 | 1748 | 1585 | 1570 |
| DFT | 1767 | 1782 | 1659 | 1639 |
| Δ | 44 | 34 | 74 | 69 |
| 8 Obs | 1732 | 1750 | 1604 | 1582 |
| DFT | 1749 | 1790 | 1664 | 1642 |
| Δ | 17 | 40 | 60 | 60 |
| 9 Obs | 1729 | 1541 | 1608 | 1584 |
| DFT | 1757 | 1677 | 1666 | 1652 |
| Δ | 28 | 136 | 58 | 68 |
| 10 Obs | 1732 | 1551 | 1590 | 1567 |
| DFT | 1784 | 1674 | 1653 | 1653 |
| Δ | 52 | 123 | 63 | 86 |
| 11 Obs | - | 1554 | 1598 | 1569 |
| DFT | - | 1652 | 1677 | 1662 |
| Δ | - | 98 | 79 | 93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monger, L.J.; Razinkov, D.; Bjornsson, R.; Suman, S.G. Synthesis, Characterization, and Reaction Studies of Pd(II) Tripeptide Complexes. Molecules 2021, 26, 5169. https://doi.org/10.3390/molecules26175169
Monger LJ, Razinkov D, Bjornsson R, Suman SG. Synthesis, Characterization, and Reaction Studies of Pd(II) Tripeptide Complexes. Molecules. 2021; 26(17):5169. https://doi.org/10.3390/molecules26175169
Chicago/Turabian StyleMonger, Lindsey J., Dmitrii Razinkov, Ragnar Bjornsson, and Sigridur G. Suman. 2021. "Synthesis, Characterization, and Reaction Studies of Pd(II) Tripeptide Complexes" Molecules 26, no. 17: 5169. https://doi.org/10.3390/molecules26175169
APA StyleMonger, L. J., Razinkov, D., Bjornsson, R., & Suman, S. G. (2021). Synthesis, Characterization, and Reaction Studies of Pd(II) Tripeptide Complexes. Molecules, 26(17), 5169. https://doi.org/10.3390/molecules26175169