
Protein Adducts and Protein Oxidation as Molecular Mechanisms of Flavonoid Bioactivity


Abstract
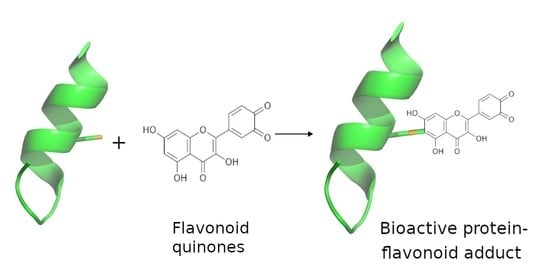
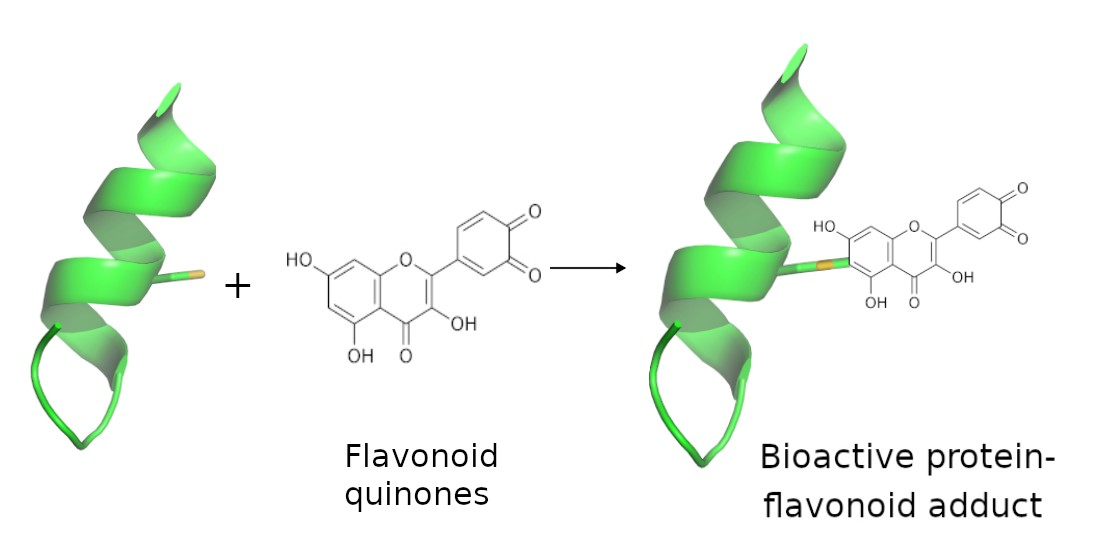{kind=link}
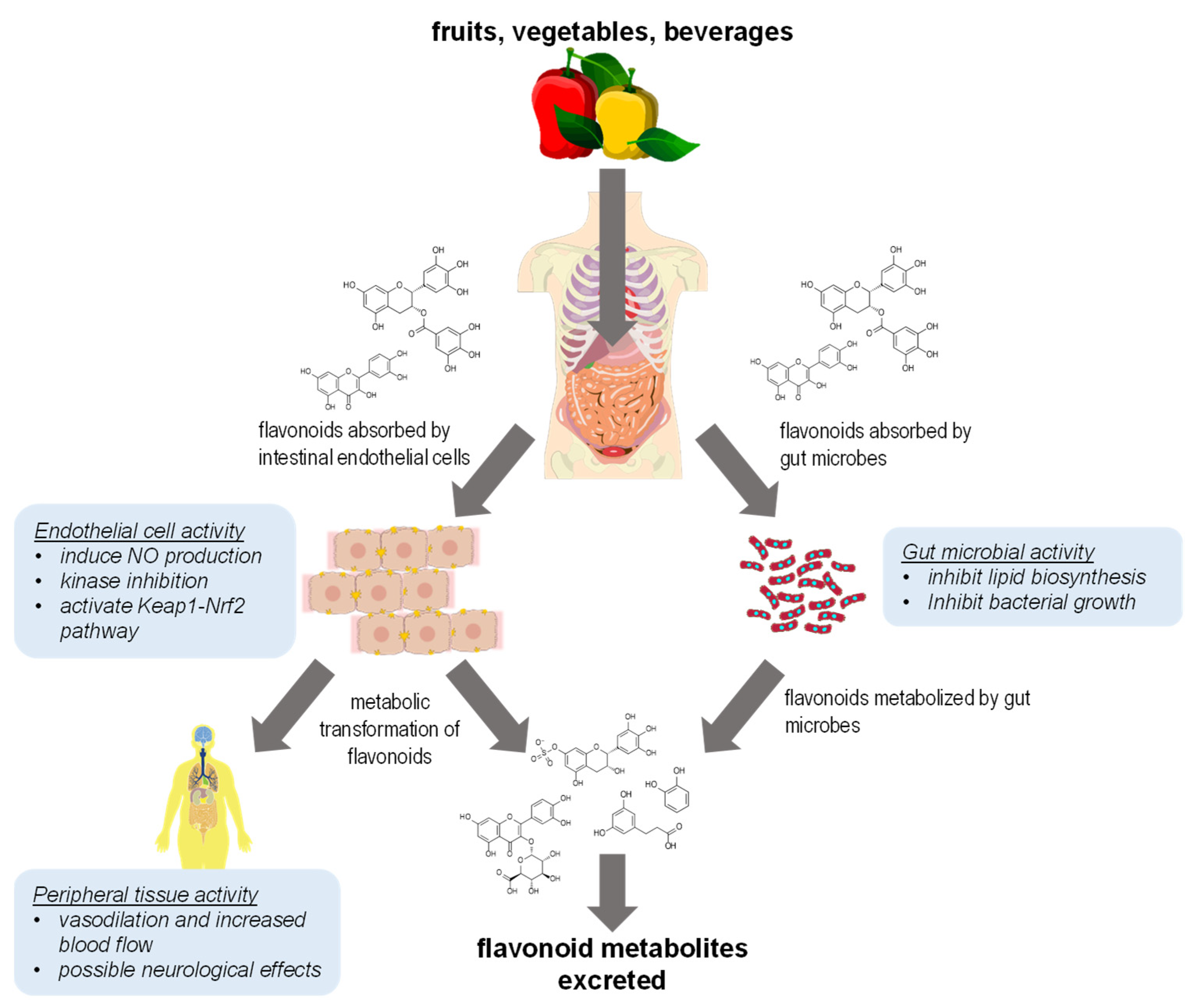{kind=link}
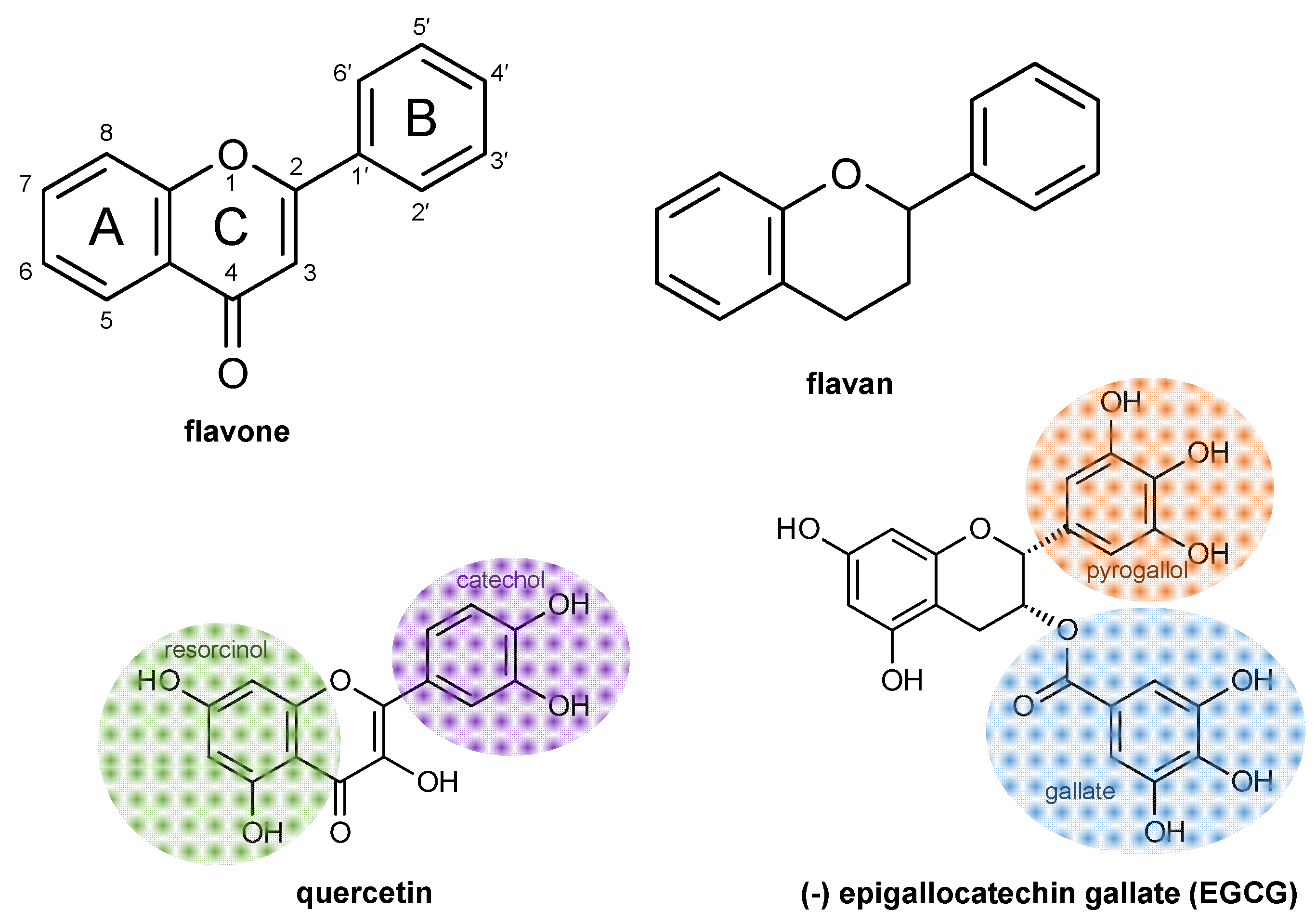{kind=link}
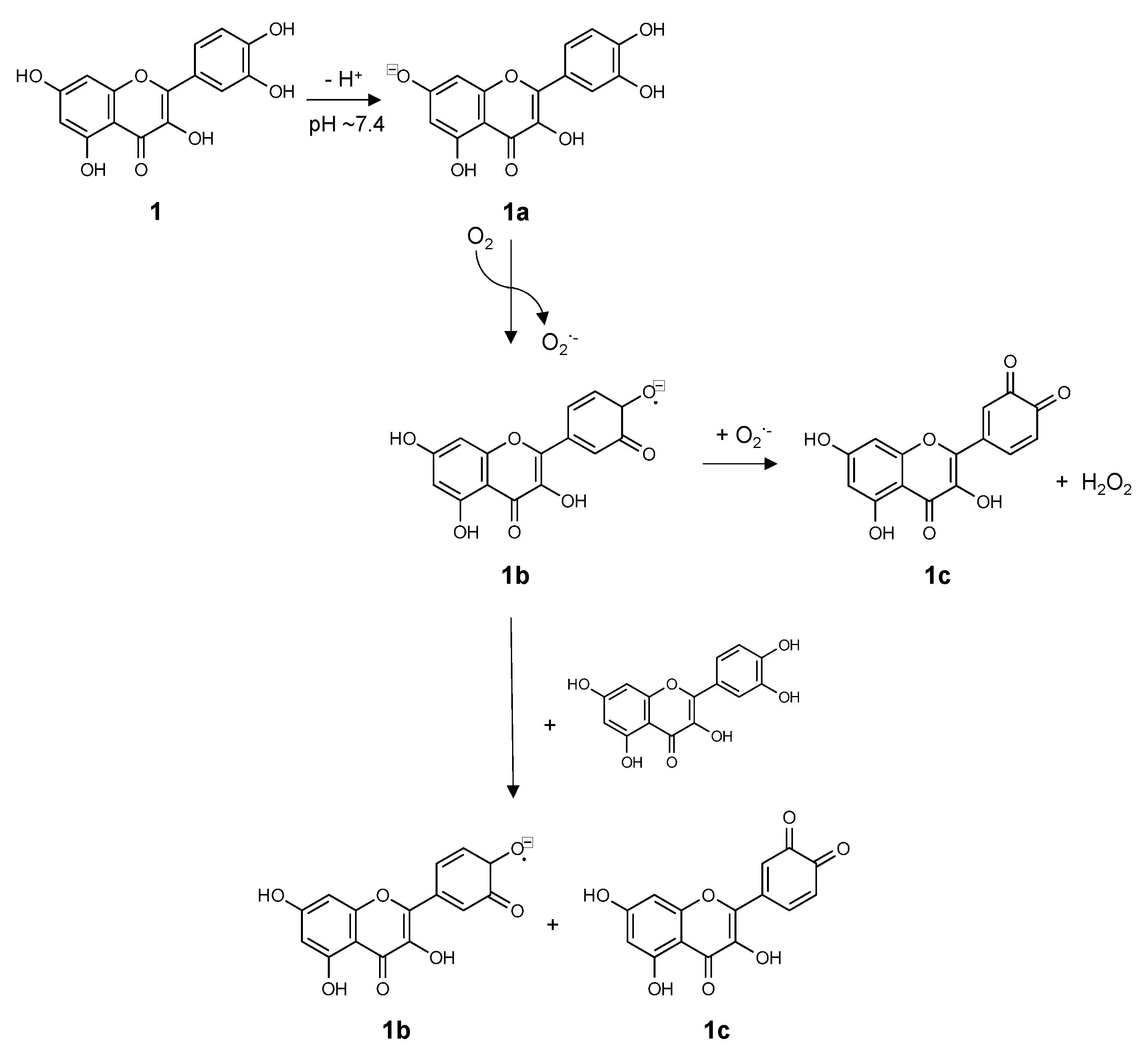{kind=link}
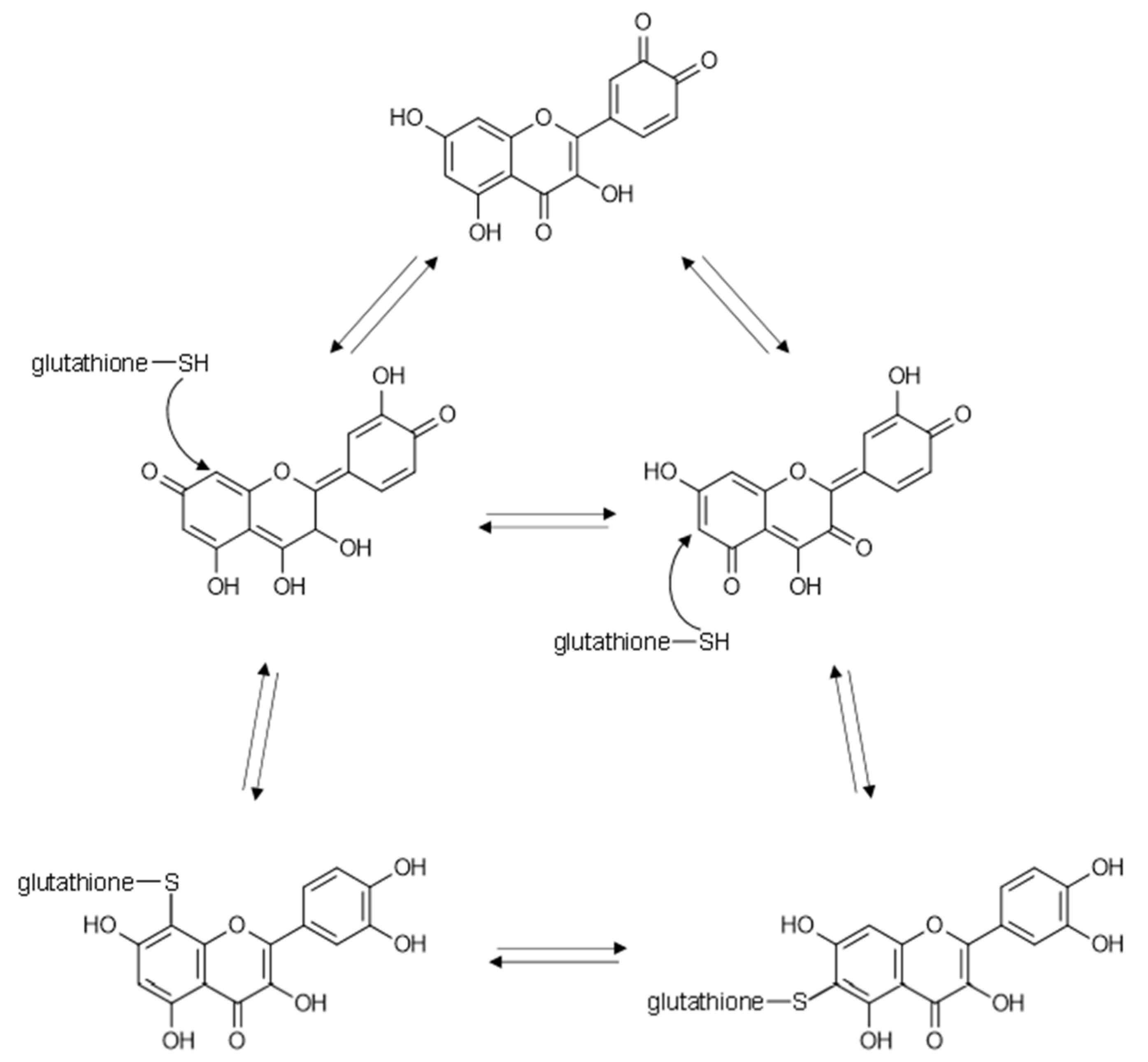{kind=link}
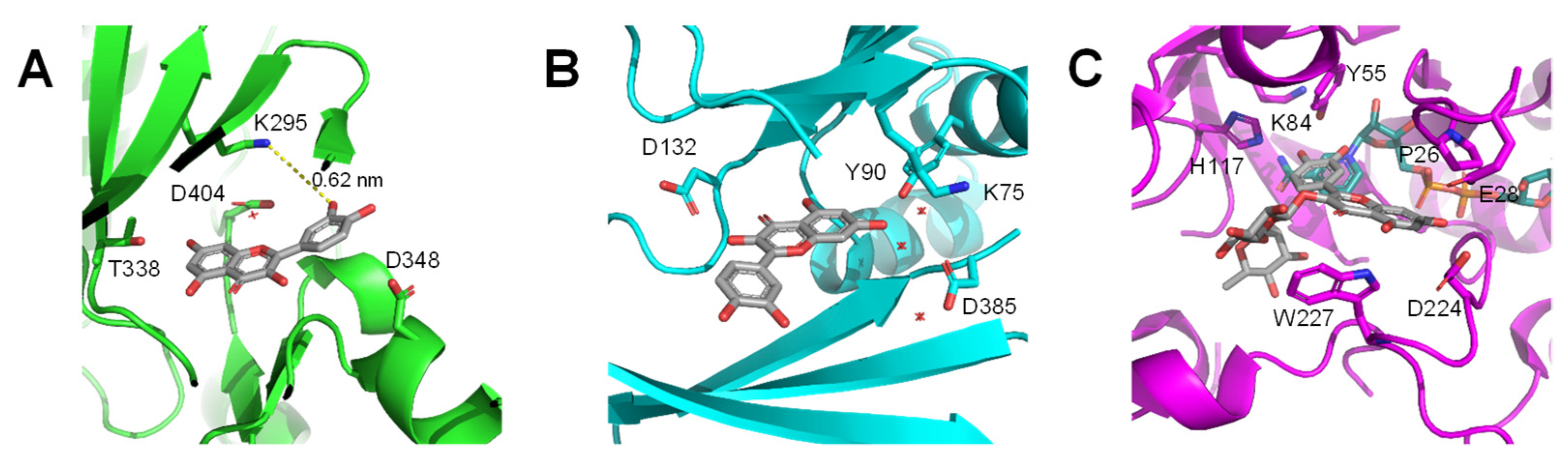{kind=link}
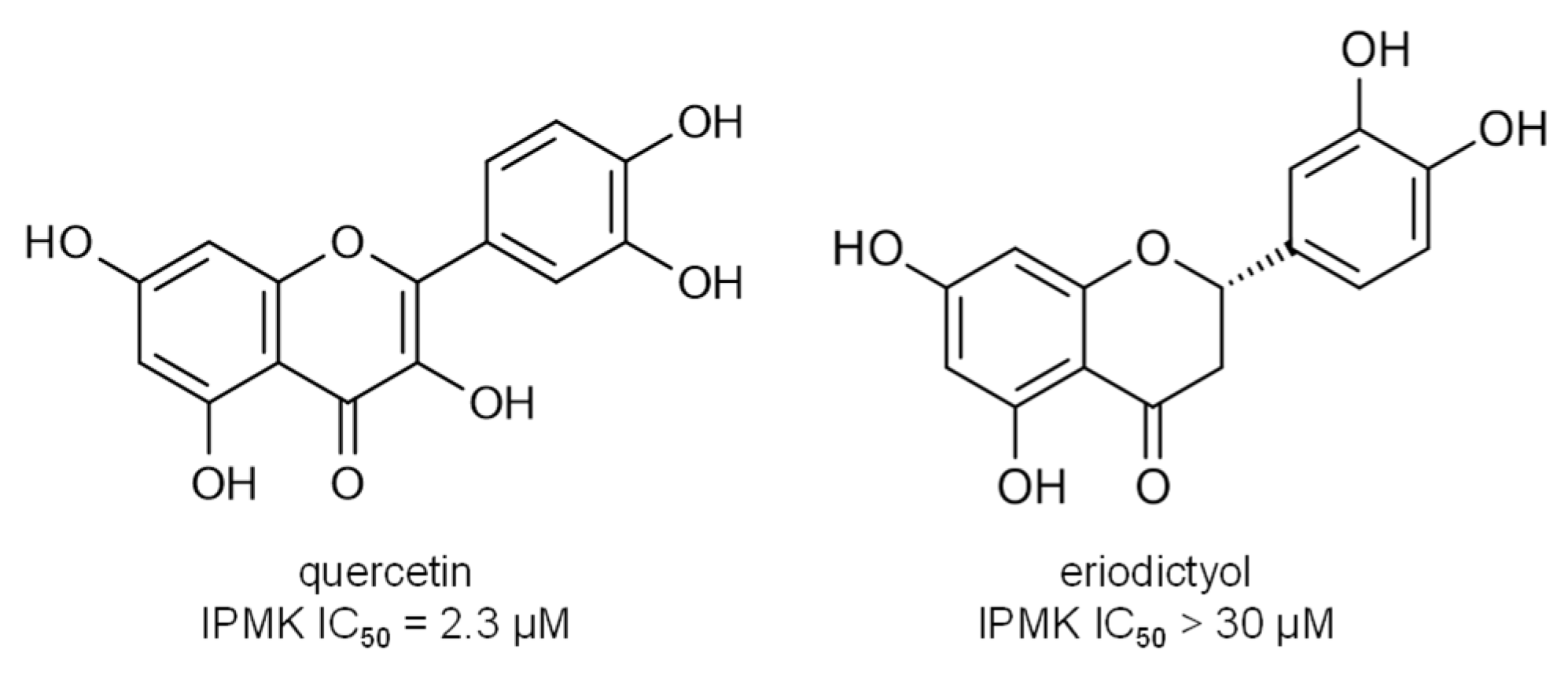{kind=link}
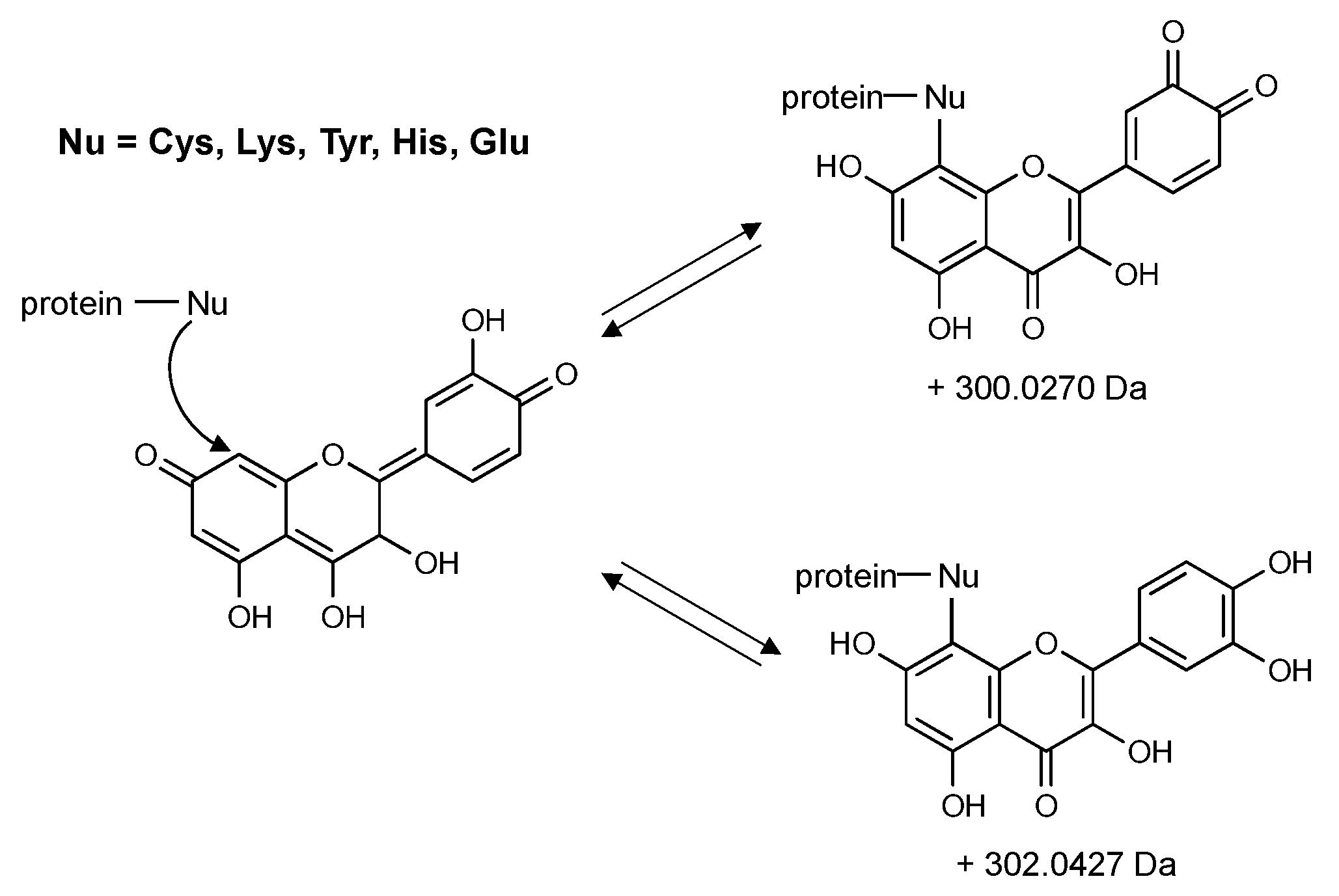{kind=link}
1. Introduction
2. Flavonoid Chemical Structures
3. Chemistry of Flavonoids in Aqueous Solutions
4. Flavonoids May Exert Bioactivity by Forming Reversible Protein Adducts
5. Reactions and Mechanisms of Protein Oxidation
6. Summary and Conclusions
- Rigorously account for the various redox properties and reactive intermediates of flavonoids in experiments using analytical methods such as NMR or chemical additives such as glutathione and ascorbic acid;
- Test precise hypotheses about protein-flavonoid interactions at specific amino acid residues;
- Look for alternative interpretations of experimental results that account for protein-flavonoid interactions and go beyond the traditional paradigm of simple, non-covalent binding.
- Avoid hypothesis or experiments that invoke the radical scavenging activity of flavonoids as a mechanistic explanation for observed bioactivity.
Funding
Conflicts of Interest
References
- Iwashina, T. The Structure and Distribution of the Flavonoids in Plants. J. Plant Res. 2000, 113, 287–299. [Google Scholar] [CrossRef]
- Weston, L.A.; Mathesius, U. Flavonoids: Their Structure, Biosynthesis and Role in the Rhizosphere, Including Allelopathy. J. Chem. Ecol. 2013, 39, 283–297. [Google Scholar] [CrossRef]
- Hodgson, J.M.; Croft, K.D. Tea Flavonoids and Cardiovascular Health. Mol. Asp. Med. 2010, 31, 495–502. [Google Scholar] [CrossRef]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical Antioxidants as Novel Neuroprotective Agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Chen, Y.C. A Review of the Dietary Flavonoid, Kaempferol on Human Health and Cancer Chemoprevention. Food Chem. 2013, 138, 2099–2107. [Google Scholar] [CrossRef]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant Therapy: Current Status and Future Prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef] [PubMed]
- Cushnie, T.P.T.; Lamb, A.J. Recent Advances in Understanding the Antibacterial Properties of Flavonoids. Int. J. Antimicrob. Agents 2011, 38, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial Activity of Flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Urquiaga, I.; Leighton, F. Plant Polyphenol Antioxidants and Oxidative Stress. Biol. Res. 2000, 33, 55–64. [Google Scholar] [CrossRef]
- Godos, J.; Vitale, M.; Micek, A.; Ray, S.; Martini, D.; Del Rio, D.; Riccardi, G.; Galvano, F.; Grosso, G. Dietary Polyphenol Intake, Blood Pressure, and Hypertension: A Systematic Review and Meta-Analysis of Observational Studies. Antioxidants 2019, 8, 152. [Google Scholar] [CrossRef]
- Lu, M.-F.; Xiao, Z.-T.; Zhang, H.-Y. Where Do Health Benefits of Flavonoids Come from? Insights from Flavonoid Targets and Their Evolutionary History. Biochem. Biophys. Res. Commun. 2013, 434, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free Radicals and Antioxidants—Quo Vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Santhakumar, A.B.; Battino, M.; Alvarez-Suarez, J.M. Dietary Polyphenols: Structures, Bioavailability and Protective Effects against Atherosclerosis. Food Chem. Toxicol. 2018, 113, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Vervoort, J.; Beekmann, K.; Baccaro, M.; Kamelia, L.; Wesseling, S.; Rietjens, I.M.C.M. Interindividual Differences in Human Intestinal Microbial Conversion of (−)-Epicatechin to Bioactive Phenolic Compounds. J. Agric. Food Chem. 2020, 68, 14168–14181. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, J.I.; Borges, G.; Momma, T.Y.; Spencer, J.P.E.; Keen, C.L.; Crozier, A.; Schroeter, H. The Metabolome of [2-14C](−)-Epicatechin in Humans: Implications for the Assessment of Efficacy, Safety and Mechanisms of Action of Polyphenolic Bioactives. Sci. Rep. 2016, 6, 29034. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Maas, R.; Troost, R.; Böger, R.H. Clinical Pharmacokinetics of Antioxidants and Their Impact on Systemic Oxidative Stress. Clin. Pharmacokinet. 2003, 42, 437–459. [Google Scholar] [CrossRef]
- Del Rio, D.; Calani, L.; Cordero, C.; Salvatore, S.; Pellegrini, N.; Brighenti, F. Bioavailability and Catabolism of Green Tea Flavan-3-Ols in Humans. Nutrition 2010, 26, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C.; Halliwell, B. Antioxidants: Molecules, Medicines, and Myths. Biochem. Biophys. Res. Commun. 2010, 393, 561–564. [Google Scholar] [CrossRef]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in Cell Culture: Rapid Generation of Hydrogen Peroxide on Addition of (−)-Epigallocatechin, (−)-Epigallocatechin Gallate, (+)-Catechin, and Quercetin to Commonly Used Cell Culture Media. Biochem. Biophys. Res. Commun. 2000, 273, 50–53. [Google Scholar] [CrossRef]
- Selma, M.V.; Espín, J.C.; Tomás-Barberán, F.A. Interaction between Phenolics and Gut Microbiota: Role in Human Health. J. Agric. Food Chem. 2009, 57, 6485–6501. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; Fernández-Quintela, A.; Milagro, F.I.; Aguirre, L.; Martínez, J.A.; Portillo, M.P. Impact of Polyphenols and Polyphenol-Rich Dietary Sources on Gut Microbiota Composition. J. Agric. Food Chem. 2013, 61, 9517–9533. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Ishihara, N.; Oura, A.; Serit, M.; Kim, M.; Yamamoto, T.; Mitsuoka, T. In Vivo Effects of Tea Polyphenol Intake on Human Intestinal Microflora and Metabolism. Biosci. Biotechnol. Biochem. 1992, 56, 588–591. [Google Scholar] [CrossRef]
- Queipo-Ortuño, M.I.; Boto-Ordóñez, M.; Murri, M.; Gomez-Zumaquero, J.M.; Clemente-Postigo, M.; Estruch, R.; Cardona Diaz, F.; Andrés-Lacueva, C.; Tinahones, F.J. Influence of Red Wine Polyphenols and Ethanol on the Gut Microbiota Ecology and Biochemical Biomarkers. Am. J. Clin. Nutr. 2012, 95, 1323–1334. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of Polyphenols on Gut Microbiota and Implications in Human Health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef]
- Lee, H.C.; Jenner, A.M.; Low, C.S.; Lee, Y.K. Effect of Tea Phenolics and Their Aromatic Fecal Bacterial Metabolites on Intestinal Microbiota. Res. Microbiol. 2006, 157, 876–884. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Rock, C.O. Evaluation of Epigallocatechin Gallate and Related Plant Polyphenols as Inhibitors of the FabG and FabI Reductases of Bacterial Type II Fatty-Acid Synthase. J. Biol. Chem. 2004, 279, 30994–31001. [Google Scholar] [CrossRef]
- Allison, B.J.; Allenby, M.C.; Bryant, S.S.; Min, J.E.; Hieromnimon, M.; Joyner, P.M. Antibacterial Activity of Fractions from Three Chumash Medicinal Plant Extracts and in Vitro Inhibition of the Enzyme Enoyl Reductase by the Flavonoid Jaceosidin. Nat. Prod. Res. 2017, 31, 707–712. [Google Scholar] [CrossRef]
- Rendeiro, C.; Rhodes, J.S.; Spencer, J.P.E. The Mechanisms of Action of Flavonoids in the Brain: Direct versus Indirect Effects. Neurochem. Int. 2015, 89, 126–139. [Google Scholar] [CrossRef]
- Rees, A.; Dodd, G.F.; Spencer, J.P.E. The Effects of Flavonoids on Cardiovascular Health: A Review of Human Intervention Trials and Implications for Cerebrovascular Function. Nutrients 2018, 10, 1852. [Google Scholar] [CrossRef]
- Haskell-Ramsay, C.F.; Schmitt, J.; Actis-Goretta, L. The Impact of Epicatechin on Human Cognition: The Role of Cerebral Blood Flow. Nutrients 2018, 10, 986. [Google Scholar] [CrossRef]
- Bondonno, C.P.; Croft, K.D.; Ward, N.; Considine, M.J.; Hodgson, J.M. Dietary Flavonoids and Nitrate: Effects on Nitric Oxide and Vascular Function. Nutr. Rev. 2015, 73, 216–235. [Google Scholar] [CrossRef]
- Kanakis, C.D.; Nafisi, S.; Rajabi, M.; Shadaloi, A.; Tarantilis, P.A.; Polissiou, M.G.; Bariyanga, J.; Tajmir-Riahi, H.A. Structural Analysis of DNA and RNA Interactions with Antioxidant Flavonoids. Spectroscopy 2009, 23, 29–43. [Google Scholar] [CrossRef]
- Nafisi, S.; Hashemi, M.; Rajabi, M.; Tajmir-Riahi, H.A. DNA Adducts with Antioxidant Flavonoids: Morin, Apigenin, and Naringin. DNA Cell Biol. 2008, 27, 433–442. [Google Scholar] [CrossRef]
- Ohshima, H.; Yoshie, Y.; Auriol, S.; Gilibert, I. Antioxidant and Pro-Oxidant Actions of Flavonoids: Effects on DNA Damage Induced by Nitric Oxide, Peroxynitrite and Nitroxyl Anion. Free Radic. Biol. Med. 1998, 25, 1057–1065. [Google Scholar] [CrossRef]
- Tarahovsky, Y.S.; Kim, Y.A.; Yagolnik, E.A.; Muzafarov, E.N. Flavonoid–Membrane Interactions: Involvement of Flavonoid–Metal Complexes in Raft Signaling. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1235–1246. [Google Scholar] [CrossRef]
- Selvaraj, S.; Krishnaswamy, S.; Devashya, V.; Sethuraman, S.; Krishnan, U.M. Influence of Membrane Lipid Composition on Flavonoid–Membrane Interactions: Implications on Their Biological Activity. Prog. Lipid Res. 2015, 58, 1–13. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Erlejman, A.G.; Verstraeten, S.V.; Keen, C.L.; Fraga, C.G. Flavonoid-Membrane Interactions: A Protective Role of Flavonoids at the Membrane Surface? Clin. Dev. Immunol. 2005, 12, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dangles, O.; Dufour, C. Flavonoid–Protein Binding Processes and their Potential Impact on Human Health. In Recent Advances in Polyphenol Research; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2008; pp. 67–87. ISBN 978-1-4443-0240-0. [Google Scholar]
- Williams, R.J.; Spencer, J.P.E.; Rice-Evans, C. Flavonoids: Antioxidants or Signalling Molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Walle, T. Absorption and Metabolism of Flavonoids. Free Radic. Biol. Med. 2004, 36, 829–837. [Google Scholar] [CrossRef]
- Hasni, I.; Bourassa, P.; Hamdani, S.; Samson, G.; Carpentier, R.; Tajmir-Riahi, H.-A. Interaction of Milk α- and β-Caseins with Tea Polyphenols. Food Chem. 2011, 126, 630–639. [Google Scholar] [CrossRef]
- Kanakis, C.D.; Hasni, I.; Bourassa, P.; Tarantilis, P.A.; Polissiou, M.G.; Tajmir-Riahi, H.-A. Milk β-Lactoglobulin Complexes with Tea Polyphenols. Food Chem. 2011, 127, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Tajmir-Riahi, H.A.; Subirade, M. Interaction of β-Lactoglobulin with Resveratrol and Its Biological Implications. Biomacromolecules 2008, 9, 50–56. [Google Scholar] [CrossRef]
- Bourassa, P.; Kanakis, C.D.; Tarantilis, P.; Pollissiou, M.G.; Tajmir-Riahi, H.A. Resveratrol, Genistein, and Curcumin Bind Bovine Serum Albumin. J. Phys. Chem. B 2010, 114, 3348–3354. [Google Scholar] [CrossRef]
- Bourassa, P.; Bariyanga, J.; Tajmir-Riahi, H.A. Binding Sites of Resveratrol, Genistein, and Curcumin with Milk α- and β-Caseins. J. Phys. Chem. B 2013, 117, 1287–1295. [Google Scholar] [CrossRef]
- Liu, Y.; Cai, Y.; Ying, D.; Fu, Y.; Xiong, Y.; Le, X. Ovalbumin as a Carrier to Significantly Enhance the Aqueous Solubility and Photostability of Curcumin: Interaction and Binding Mechanism Study. Int. J. Biol. Macromol. 2018, 116, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Mao, F.; Yang, F.; Zhao, Y.; Zhang, C.; Yamamoto, K. Interaction of Dietary Polyphenols with Bovine Milk Proteins: Molecular Structure-Affinity Relationship and Influencing Bioactivity Aspects. Mol. Nutr. Food Res. 2011, 55, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Kaldas, M.I.; Walle, U.K.; van der Woude, H.; McMillan, J.M.; Walle, T. Covalent Binding of the Flavonoid Quercetin to Human Serum Albumin. J. Agric. Food Chem. 2005, 53, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F.; Bikádi, Z.; Simonyi, M. Probing the Binding of the Flavonoid, Quercetin to Human Serum Albumin by Circular Dichroism, Electronic Absorption Spectroscopy and Molecular Modelling Methods. Biochem. Pharmacol. 2003, 65, 447–456. [Google Scholar] [CrossRef]
- Murray, N.J.; Williamson, M.P.; Lilley, T.H.; Haslam, E. Study of the Interaction between Salivary Proline-rich Proteins and a Polyphenol by 1H-NMR Spectroscopy. Eur. J. Biochem. 1994, 219, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Wróblewski, K.; Muhandiram, R.; Chakrabartty, A.; Bennick, A. The Molecular Interaction of Human Salivary Histatins with Polyphenolic Compounds. Eur. J. Biochem. 2001, 268, 4384–4397. [Google Scholar] [CrossRef]
- Canon, F.; Paté, F.; Meudec, E.; Marlin, T.; Cheynier, V.; Giuliani, A.; Sarni-Manchado, P. Characterization, Stoichiometry, and Stability of Salivary Protein–Tannin Complexes by ESI-MS and ESI-MS/MS. Anal. Bioanal. Chem. 2009, 395, 2535. [Google Scholar] [CrossRef] [PubMed]
- Siebert, K.J. Effects of Protein−Polyphenol Interactions on Beverage Haze, Stabilization, and Analysis. J. Agric. Food Chem. 1999, 47, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Siebert, K.J.; Troukhanova, N.V.; Lynn, P.Y. Nature of Polyphenol–Protein Interactions. J. Agric. Food Chem. 1996, 44, 80–85. [Google Scholar] [CrossRef]
- Charlton, A.J.; Baxter, N.J.; Khan, M.L.; Moir, A.J.G.; Haslam, E.; Davies, A.P.; Williamson, M.P. Polyphenol/Peptide Binding and Precipitation. J. Agric. Food Chem. 2002, 50, 1593–1601. [Google Scholar] [CrossRef]
- Atrahimovich, D.; Vaya, J.; Khatib, S. The Effects and Mechanism of Flavonoid–RePON1 Interactions. Structure–Activity Relationship Study. Bioorg. Med. Chem. 2013, 21, 3348–3355. [Google Scholar] [CrossRef] [PubMed]
- Atrahimovich, D.; Vaya, J.; Tavori, H.; Khatib, S. Glabridin Protects Paraoxonase 1 from Linoleic Acid Hydroperoxide Inhibition via Specific Interaction: A Fluorescence-Quenching Study. J. Agric. Food Chem. 2012, 60, 3679–3685. [Google Scholar] [CrossRef]
- Xiao, J.; Kai, G.; Ni, X.; Yang, F.; Chen, X. Interaction of Natural Polyphenols with α-Amylase in Vitro: Molecular Property–Affinity Relationship Aspect. Mol. Biosyst. 2011, 7, 1883–1890. [Google Scholar] [CrossRef]
- Xu, W.; Shao, R.; Xiao, J. Is There Consistency between the Binding Affinity and Inhibitory Potential of Natural Polyphenols as α-Amylase Inhibitors? Crit. Rev. Food Sci. Nutr. 2016, 56, 1630–1639. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Tomé, S.M.; Oliveira, E.F.T.; Viegas, M.F.; Araújo, A.N.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; et al. Evaluation of a Flavonoids Library for Inhibition of Pancreatic α-Amylase towards a Structure–Activity Relationship. J. Enzym. Inhib. Med. Chem. 2019, 34, 577–588. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.F.T.; Sousa, J.L.C.; Tomé, S.M.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. α-Glucosidase Inhibition by Flavonoids: An in Vitro and in Silico Structure–Activity Relationship Study. J. Enzym. Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Van de Ven, W.J.M.; Verbiest, T.; Koeckelberghs, G.; Chen, C.; Cui, Y.; Vermorken, A.J.M. Polyphenols Can Inhibit Furin in Vitro as a Result of the Reactivity of Their Auto-Oxidation Products to Proteins. Curr. Med. Chem. 2013, 20, 840–850. [Google Scholar]
- Jang, S.-W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A Selective TrkB Agonist with Potent Neurotrophic Activities by 7,8-Dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef]
- Proença, C.; Oliveira, A.; Freitas, M.; Ribeiro, D.; Sousa, J.L.C.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. Structural Specificity of Flavonoids in the Inhibition of Human Fructose 1,6-Bisphosphatase. J. Nat. Prod. 2020, 83, 1541–1552. [Google Scholar] [CrossRef]
- Tanaka, T.; Ishii, T.; Mizuno, D.; Mori, T.; Yamaji, R.; Nakamura, Y.; Kumazawa, S.; Nakayama, T.; Akagawa, M. (−)-Epigallocatechin-3-Gallate Suppresses Growth of AZ521 Human Gastric Cancer Cells by Targeting the DEAD-Box RNA Helicase P68. Free Radic. Biol. Med. 2011, 50, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Spickett, C.M.; Pitt, A.R. Protein Oxidation: Role in Signalling and Detection by Mass Spectrometry. Amino Acids 2012, 42, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine Oxidation and Reduction in Proteins. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840. [Google Scholar] [CrossRef]
- Dröge, W.; Schipper, H.M. Oxidative Stress and Aberrant Signaling in Aging and Cognitive Decline. Aging Cell 2007, 6, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Li, C.-W. Detection and Characterization of Catechol Quinone-Derived Protein Adducts Using Biomolecular Mass Spectrometry. Front. Chem. 2019, 7, 571. [Google Scholar] [CrossRef]
- Fisher, A.A.; Labenski, M.T.; Malladi, S.; Gokhale, V.; Bowen, M.E.; Milleron, R.S.; Bratton, S.B.; Monks, T.J.; Lau, S.S. Quinone Electrophiles Selectively Adduct “Electrophile Binding Motifs” within Cytochrome c. Biochemistry 2007, 46, 11090–11100. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Higashi, Y.; Nakabayashi, R. The Origin and Evolution of Plant Flavonoid Metabolism. Front. Plant Sci. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Harborne, J.B.; Williams, C.A. Advances in Flavonoid Research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef]
- Veitch, N.C.; Grayer, R.J. Flavonoids and Their Glycosides, Including Anthocyanins. Nat. Prod. Rep. 2011, 28, 1626–1695. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly)Phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects Against Chronic Diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of Hydroxyl Groups: An Overlooked Influence on Antiradical Properties of Flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M.G. Flavonoids as Antioxidants. J. Am. Chem. Soc. 1994, 116, 4846–4851. [Google Scholar] [CrossRef]
- Mezzetti, A.; Protti, S.; Lapouge, C.; Cornard, J.-P. Protic Equilibria as the Key Factor of Quercetin Emission in Solution. Relevance to Biochemical and Analytical Studies. Phys. Chem. Chem. Phys. 2011, 13, 6858–6864. [Google Scholar] [CrossRef] [PubMed]
- Janeiro, P.; Oliveira Brett, A.M. Catechin Electrochemical Oxidation Mechanisms. Anal. Chim. Acta 2004, 518, 109–115. [Google Scholar] [CrossRef]
- Brett, A.M.O.; Ghica, M.-E. Electrochemical Oxidation of Quercetin. Electroanalysis 2003, 15, 1745–1750. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yamazaki, S.; Kano, K.; Ikeda, T. Kinetic Analysis and Mechanistic Aspects of Autoxidation of Catechins. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2002, 1569, 35–44. [Google Scholar] [CrossRef]
- Furuno, K.; Akasako, T.; Sugihara, N. The Contribution of the Pyrogallol Moiety to the Superoxide Radical Scavenging Activity of Flavonoids. Biol. Pharm. Bull. 2002, 25, 19–23. [Google Scholar] [CrossRef]
- Belli, S.; Rossi, M.; Molasky, N.; Middleton, L.; Caldwell, C.; Bartow-McKenney, C.; Duong, M.; Chiu, J.; Gibbs, E.; Caldwell, A.; et al. Effective and Novel Application of Hydrodynamic Voltammetry to the Study of Superoxide Radical Scavenging by Natural Phenolic Antioxidants. Antioxidants 2019, 8, 14. [Google Scholar] [CrossRef]
- Jørgensen, L.V.; Madsen, H.L.; Thomsen, M.K.; Dragsted, L.O.; Skibsted, L.H. Regeneration of Phenolic Antioxidants from Phenoxyl Radicals: An ESR and Electrochemical Study of Antioxidant Hierarchy. Free Radic. Res. 1999, 30, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kotani, A.; Arai, K.; Kusu, F. Estimation of the Antioxidant Activities of Flavonoids from Their Oxidation Potentials. Anal. Sci. 2001, 17, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, H.P.; Kaufman, A.D.; Lunte, C.E. Electrochemistry of Catechol-Containing Flavonoids. J. Pharm. Biomed. Anal. 1994, 12, 325–334. [Google Scholar] [CrossRef]
- Jørgensen, L.V.; Cornett, C.; Justesen, U.; Skibsted, L.H.; Dragsted, L.O. Two-Electron Electrochemical Oxidation of Quercetin and Kaempferol Changes Only the Flavonoid C-Ring. Free Radic. Res. 1998, 29, 339–350. [Google Scholar] [CrossRef]
- Awad, H.M.; Boersma, M.G.; Vervoort, J.; Rietjens, I.M.C.M. Peroxidase-Catalyzed Formation of Quercetin Quinone Methide–Glutathione Adducts. Arch. Biochem. Biophys. 2000, 378, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Boersma, M.G.; Vervoort, J.; Szymusiak, H.; Lemanska, K.; Tyrakowska, B.; Cenas, N.; Segura-Aguilar, J.; Rietjens, I.M.C.M. Regioselectivity and Reversibility of the Glutathione Conjugation of Quercetin Quinone Methide. Chem. Res. Toxicol. 2000, 13, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Awad, H.M.; Boersma, M.G.; Boeren, S.; van der Woude, H.; van Zanden, J.; van Bladeren, P.J.; Vervoort, J.; Rietjens, I.M.C.M. Identification of O-Quinone/Quinone Methide Metabolites of Quercetin in a Cellular in Vitro System. FEBS Lett. 2002, 520, 30–34. [Google Scholar] [CrossRef]
- Lee-Hilz, Y.Y.; Boerboom, A.-M.J.F.; Westphal, A.H.; van Berkel, W.J.H.; Aarts, J.M.M.J.G.; Rietjens, I.M.C.M. Pro-Oxidant Activity of Flavonoids Induces EpRE-Mediated Gene Expression. Chem. Res. Toxicol. 2006, 19, 1499–1505. [Google Scholar] [CrossRef]
- Boerboom, A.-M.J.F.; Vermeulen, M.; van der Woude, H.; Bremer, B.I.; Lee-Hilz, Y.Y.; Kampman, E.; van Bladeren, P.J.; Rietjens, I.M.C.M.; Aarts, J.M.M.J.G. Newly Constructed Stable Reporter Cell Lines for Mechanistic Studies on Electrophile-Responsive Element-Mediated Gene Expression Reveal a Role for Flavonoid Planarity. Biochem. Pharmacol. 2006, 72, 217–226. [Google Scholar] [CrossRef]
- Muzolf-Panek, M.; Gliszczyńska-Świgło, A.; de Haan, L.; Aarts, J.M.M.J.G.; Szymusiak, H.; Vervoort, J.M.; Tyrakowska, B.; Rietjens, I.M.C.M. Role of Catechin Quinones in the Induction of EpRE-Mediated Gene Expression. Chem. Res. Toxicol. 2008, 21, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Galati, G.; Lin, A.; Sultan, A.M.; O’Brien, P.J. Cellular and in Vivo Hepatotoxicity Caused by Green Tea Phenolic Acids and Catechins. Free Radic. Biol. Med. 2006, 40, 570–580. [Google Scholar] [CrossRef]
- Kejík, Z.; Kaplánek, R.; Masařík, M.; Babula, P.; Matkowski, A.; Filipenský, P.; Veselá, K.; Gburek, J.; Sýkora, D.; Martásek, P.; et al. Iron Complexes of Flavonoids-Antioxidant Capacity and Beyond. Int. J. Mol. Sci. 2021, 22, 646. [Google Scholar] [CrossRef]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant Activity of Polyphenols, Flavonoids, Anthocyanins and Carotenoids: Updated Review of Mechanisms and Catalyzing Metals. Phytother. Res. PTR 2016, 30, 1379–1391. [Google Scholar] [CrossRef]
- Halliwell, B. Are Polyphenols Antioxidants or Pro-Oxidants? What Do We Learn from Cell Culture and in Vivo Studies? Arch. Biochem. Biophys. 2008, 476, 107–112. [Google Scholar] [CrossRef]
- Walle, T.; Vincent, T.S.; Walle, U.K. Evidence of Covalent Binding of the Dietary Flavonoid Quercetin to DNA and Protein in Human Intestinal and Hepatic Cells. Biochem. Pharmacol. 2003, 65, 1603–1610. [Google Scholar] [CrossRef]
- Gutzeit, H.O.; Henker, Y.; Kind, B.; Franz, A. Specific Interactions of Quercetin and Other Flavonoids with Target Proteins Are Revealed by Elicited Fluorescence. Biochem. Biophys. Res. Commun. 2004, 318, 490–495. [Google Scholar] [CrossRef]
- Anraku, M.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Redox Properties of Serum Albumin. Biochim. Biophys. Acta 2013, 1830, 5465–5472. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic Targeting of the NRF2 and KEAP1 Partnership in Chronic Diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Yu, J.; Wang, S.; Copeland, L.; Wang, S. Revealing the Mechanisms of Starch Amylolysis Affected by Tea Catechins Using Surface Plasmon Resonance. Int. J. Biol. Macromol. 2020, 145, 527–534. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin, and Staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Kim, J.; Albu, T.V.; Vaughn, A.R.; Kang, S.M.; Carver, E.A.; Stickle, D.M. A Comparison Study on Ribonuclease A Modifications Induced by Substituted P-Benzoquinones. Bioorg. Chem. 2015, 59, 106–116. [Google Scholar] [CrossRef]
- Nicolis, S.; Monzani, E.; Pezzella, A.; Ascenzi, P.; Sbardella, D.; Casella, L. Neuroglobin Modification by Reactive Quinone Species. Chem. Res. Toxicol. 2013, 26, 1821–1831. [Google Scholar] [CrossRef]
- Ishii, T.; Mori, T.; Tanaka, T.; Mizuno, D.; Yamaji, R.; Kumazawa, S.; Nakayama, T.; Akagawa, M. Covalent Modification of Proteins by Green Tea Polyphenol (–)-Epigallocatechin-3-Gallate through Autoxidation. Free Radic. Biol. Med. 2008, 45, 1384–1394. [Google Scholar] [CrossRef]
- Mori, T.; Ishii, T.; Akagawa, M.; Nakamura, Y.; Nakayama, T. Covalent Binding of Tea Catechins to Protein Thiols: The Relationship between Stability and Electrophilic Reactivity. Biosci. Biotechnol. Biochem. 2010, 74, 2451–2456. [Google Scholar] [CrossRef]
- Liu, G.; Eggler, A.L.; Dietz, B.M.; Mesecar, A.D.; Bolton, J.L.; Pezzuto, J.M.; van Breemen, R.B. Screening Method for the Discovery of Potential Cancer Chemoprevention Agents Based on Mass Spectrometric Detection of Alkylated Keap1. Anal. Chem. 2005, 77, 6407–6414. [Google Scholar] [CrossRef]
- Tu, T.; Giblin, D.; Gross, M.L. Structural Determinant of Chemical Reactivity and Potential Health Effects of Quinones from Natural Products. Chem. Res. Toxicol. 2011, 24, 1527–1539. [Google Scholar] [CrossRef]
- Bolton, J.L. Quinone Methide Bioactivation Pathway: Contribution to Toxicity and/or Cytoprotection? Curr. Org. Chem. 2014, 18, 61–69. [Google Scholar] [CrossRef]
- Sicheri, F.; Moarefi, I.; Kuriyan, J. Crystal Structure of the Src Family Tyrosine Kinase Hck. Nature 1997, 385, 602–609. [Google Scholar] [CrossRef]
- Gu, C.; Stashko, M.A.; Puhl-Rubio, A.C.; Chakraborty, M.; Chakraborty, A.; Frye, S.V.; Pearce, K.H.; Wang, X.; Shears, S.B.; Wang, H. Inhibition of Inositol Polyphosphate Kinases by Quercetin and Related Flavonoids: A Structure–Activity Analysis. J. Med. Chem. 2019, 62, 1443–1454. [Google Scholar] [CrossRef]
- Komoto, J.; Yamada, T.; Watanabe, K.; Takusagawa, F. Crystal Structure of Human Prostaglandin F Synthase (AKR1C3). Biochemistry 2004, 43, 2188–2198. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Li, H.; Grigoryan, H.; Funk, W.E.; Williams, E.R. Adductomics: Characterizing Exposures to Reactive Electrophiles. Toxicol. Lett. 2012, 213, 83–90. [Google Scholar] [CrossRef]
- Carlsson, H.; Törnqvist, M. An Adductomic Approach to Identify Electrophiles In Vivo. Basic Clin. Pharmacol. Toxicol. 2017, 121, 44–54. [Google Scholar] [CrossRef]
- Törnqvist, M.; Fred, C.; Haglund, J.; Helleberg, H.; Paulsson, B.; Rydberg, P. Protein Adducts: Quantitative and Qualitative Aspects of Their Formation, Analysis and Applications. J. Chromatogr. B 2002, 778, 279–308. [Google Scholar] [CrossRef]
- Labenski, M.T.; Fisher, A.A.; Lo, H.-H.; Monks, T.J.; Lau, S.S. Protein Electrophile-Binding Motifs: Lysine-Rich Proteins Are Preferential Targets of Quinones. Drug Metab. Dispos. 2009, 37, 1211–1218. [Google Scholar] [CrossRef]
- Luo, Y.; Eggler, A.L.; Liu, D.; Liu, G.; Mesecar, A.D.; van Breemen, R.B. Sites of Alkylation of Human Keap1 by Natural Chemoprevention Agents. J. Am. Soc. Mass Spectrom. 2007, 18, 2226–2232. [Google Scholar] [CrossRef]
- Hu, C.; Nikolic, D.; Eggler, A.L.; Mesecar, A.D.; van Breemen, R.B. Screening for Natural Chemoprevention Agents That Modify Human Keap1. Anal. Biochem. 2012, 421, 108–114. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.-F. Oxidative Stress, Protein Damage and Repair in Bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Oxidation and Aging. Science 1992, 257, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein Oxidation in Aging, Disease, and Oxidative Stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and Pathology of Radical-Mediated Protein Oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical Proteomics Reveals New Targets of Cysteine Sulfinic Acid Reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef]
- Jacob, C.; Holme, A.L.; Fry, F.H. The Sulfinic Acid Switch in Proteins. Org. Biomol. Chem. 2004, 2, 1953–1956. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine Residues as Endogenous Antioxidants in Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef]
- Chao, C.-C.; Ma, Y.-S.; Stadtman, E.R. Modification of Protein Surface Hydrophobicity and Methionine Oxidation by Oxidative Systems. Proc. Natl. Acad. Sci. USA 1997, 94, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan-Giri, A.; Byun, J.; Pennathur, S. Quantitative Analysis of Amino Acid Oxidation Markers by Tandem Mass Spectrometry. In Methods in Enzymology; Conn, P.M., Ed.; The Unfolded Protein Response and Cellular Stress, Part C; Academic Press: Cambridge, MA, USA, 2011; Chapter 5; Volume 491, pp. 73–89. [Google Scholar]
- Roeser, J.; Bischoff, R.; Bruins, A.P.; Permentier, H.P. Oxidative Protein Labeling in Mass-Spectrometry-Based Proteomics. Anal. Bioanal. Chem. 2010, 397, 3441–3455. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Detection of Oxidized and Glycated Proteins in Clinical Samples Using Mass Spectrometry—A User’s Perspective. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 818–829. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of Protein Modification by Oxidants. Free Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M. Mass Spectrometry-Based Methods for Identifying Oxidized Proteins in Disease: Advances and Challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a Universal Identification Algorithm Optimized for High Accuracy Tandem Mass Spectra. J. Proteome Res. 2014, 13, 3679–3684. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joyner, P.M. Protein Adducts and Protein Oxidation as Molecular Mechanisms of Flavonoid Bioactivity. Molecules 2021, 26, 5102. https://doi.org/10.3390/molecules26165102
Joyner PM. Protein Adducts and Protein Oxidation as Molecular Mechanisms of Flavonoid Bioactivity. Molecules. 2021; 26(16):5102. https://doi.org/10.3390/molecules26165102
Chicago/Turabian StyleJoyner, P. Matthew. 2021. "Protein Adducts and Protein Oxidation as Molecular Mechanisms of Flavonoid Bioactivity" Molecules 26, no. 16: 5102. https://doi.org/10.3390/molecules26165102
APA StyleJoyner, P. M. (2021). Protein Adducts and Protein Oxidation as Molecular Mechanisms of Flavonoid Bioactivity. Molecules, 26(16), 5102. https://doi.org/10.3390/molecules26165102