
The Raw Milk Microbiota from Semi-Subsistence Farms Characteristics by NGS Analysis Method

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
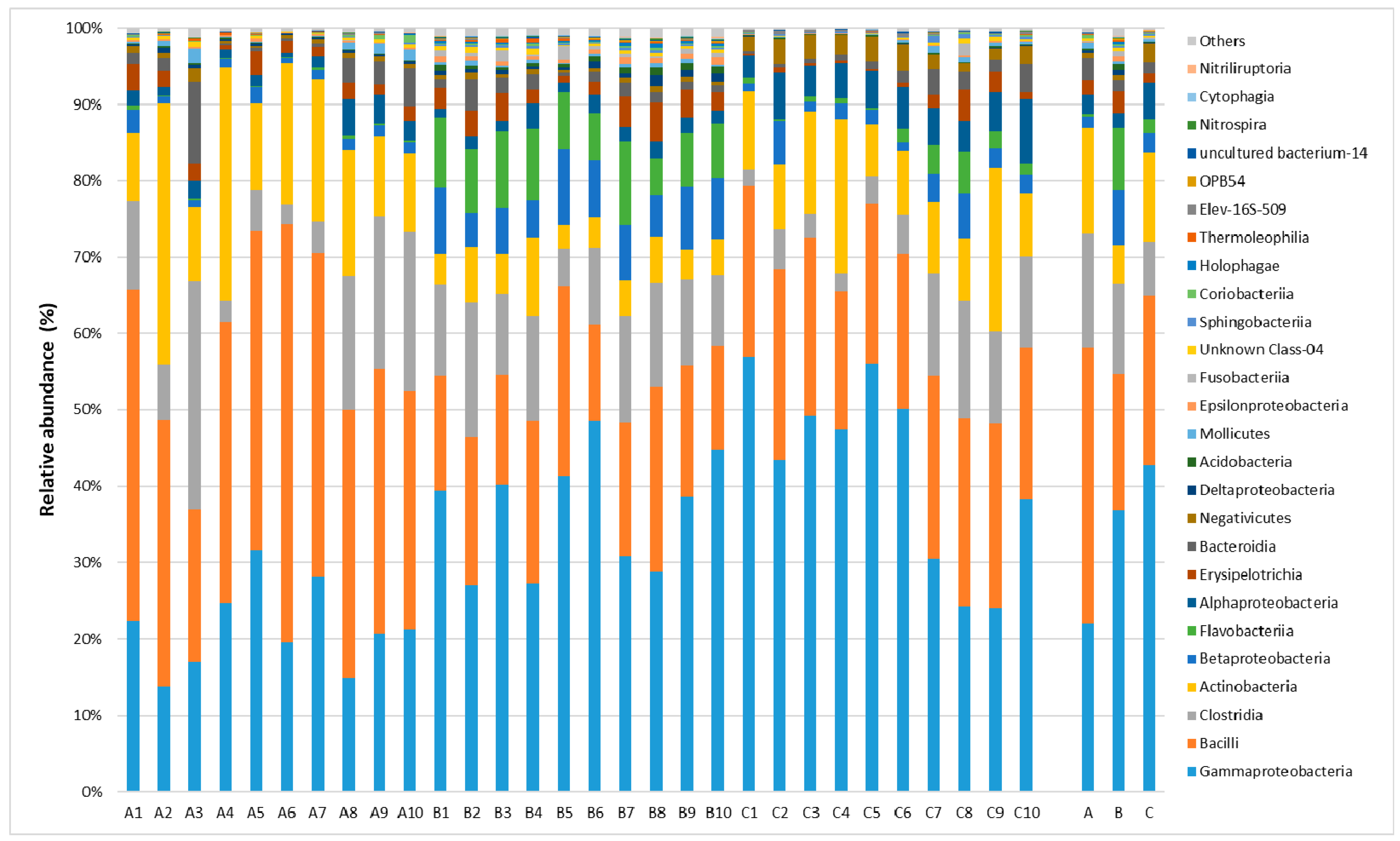
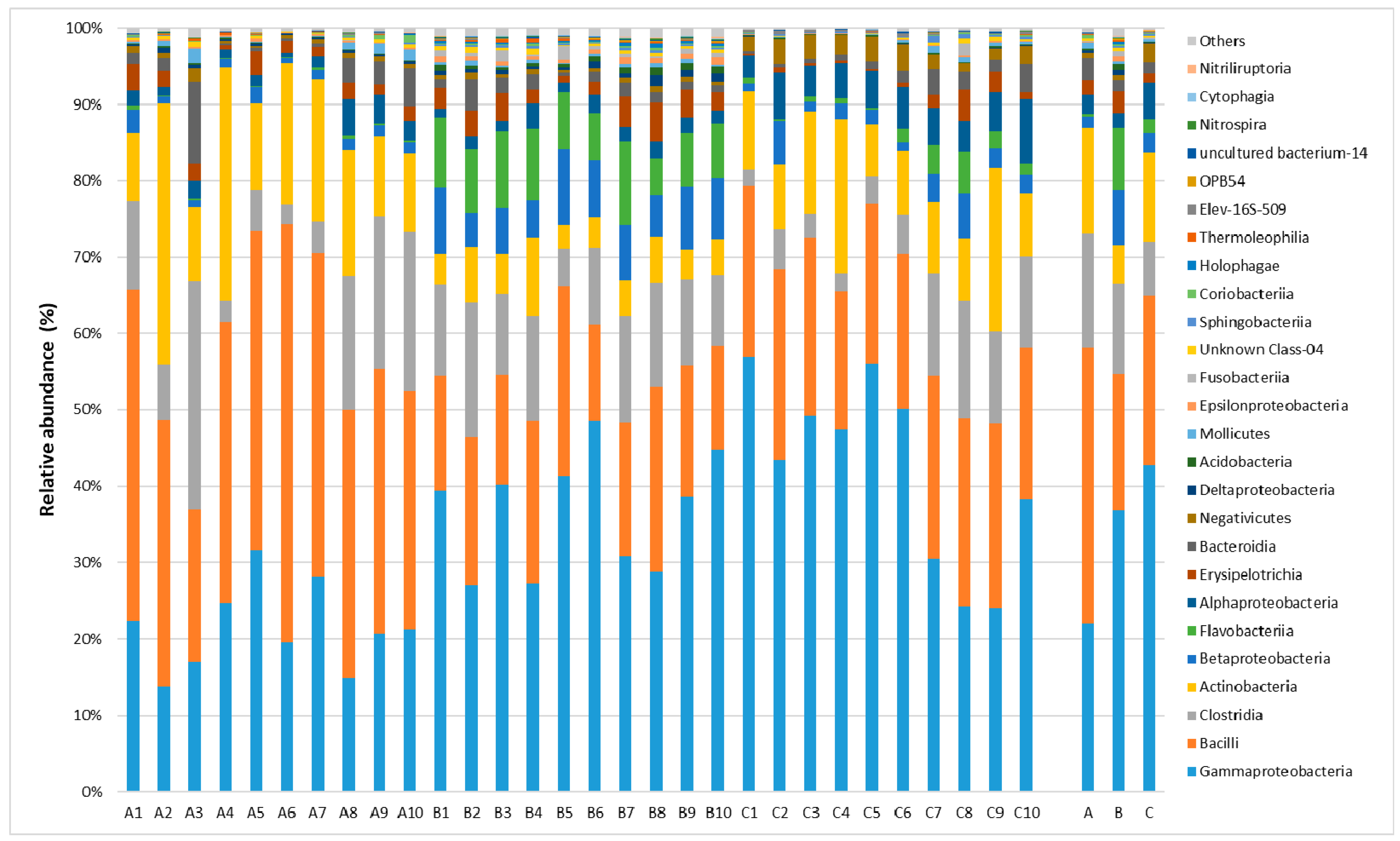
2.1. Taxonomic Analysis of the Milk Microbiome
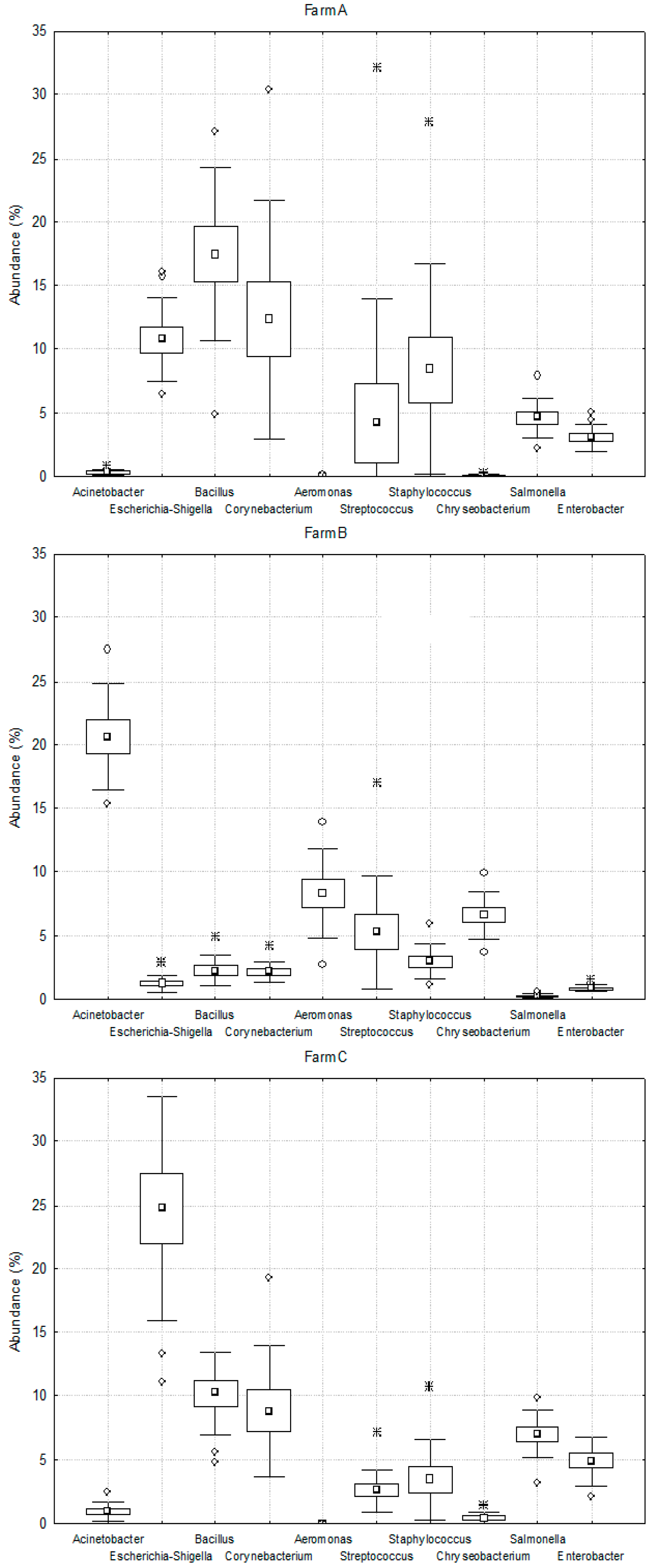
2.2. Lactic Fermentation Bacteria
2.3. Spoiled Milk Bacteria
2.4. Pathogenic Bacteria Causing Mastitis
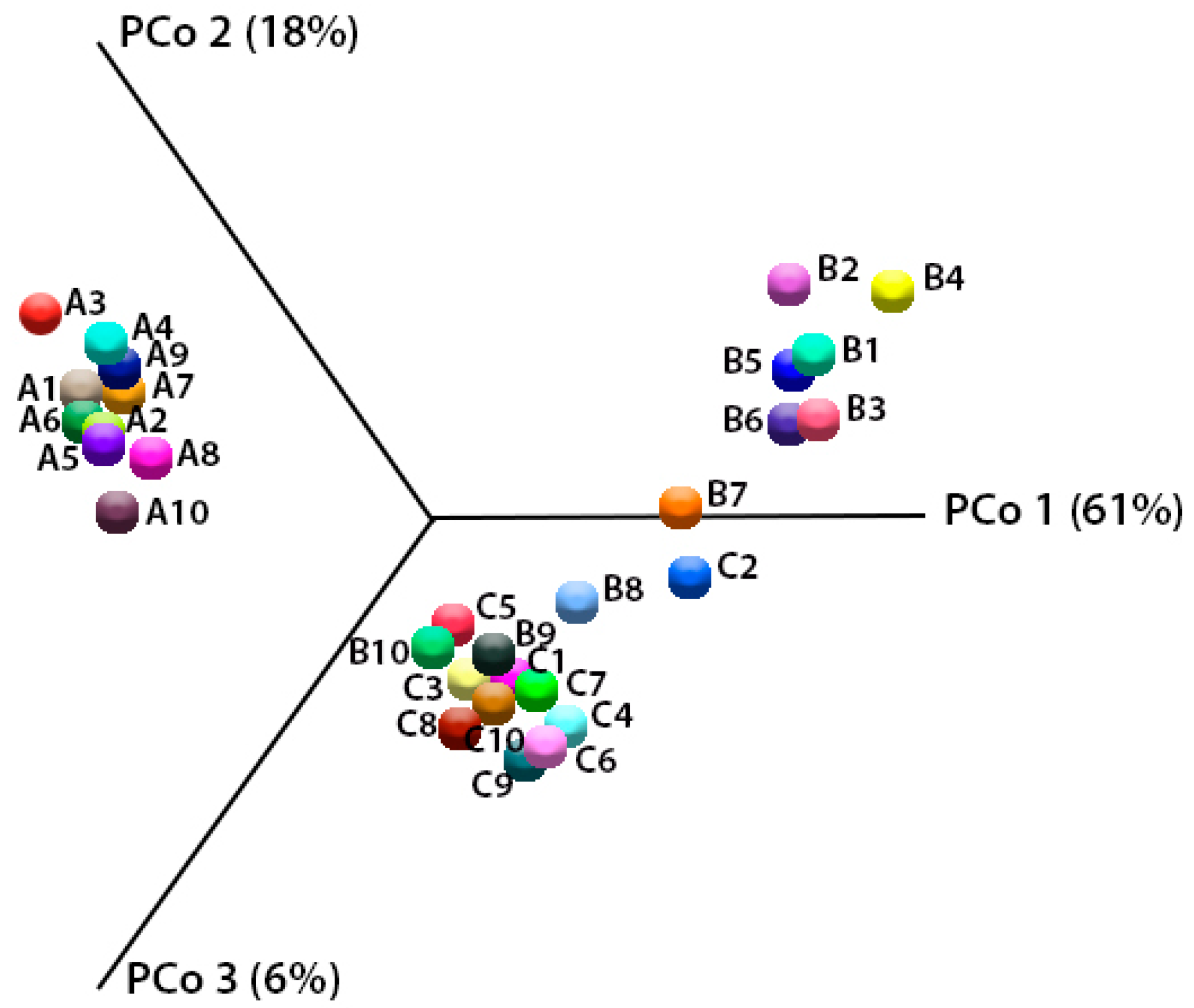
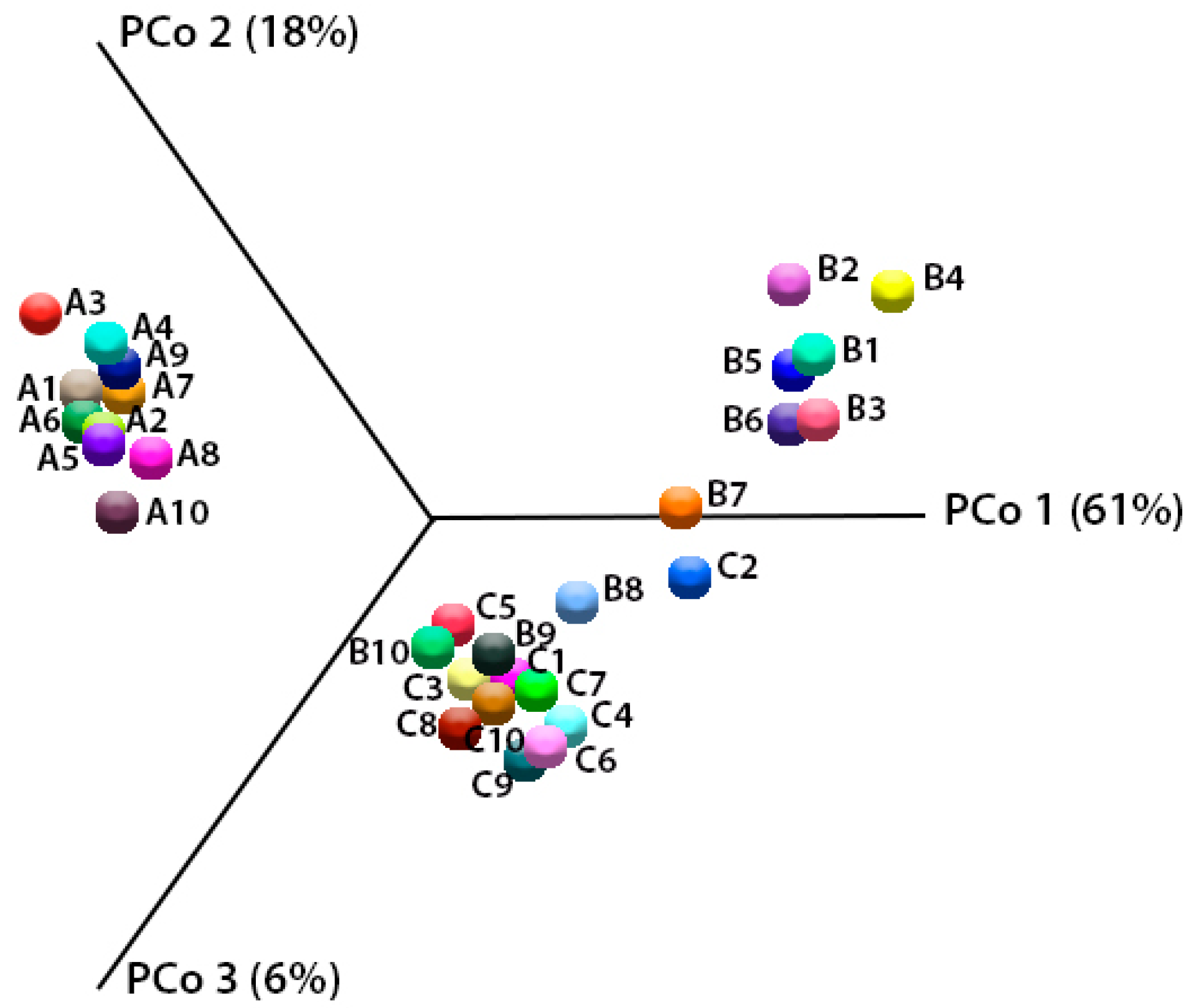
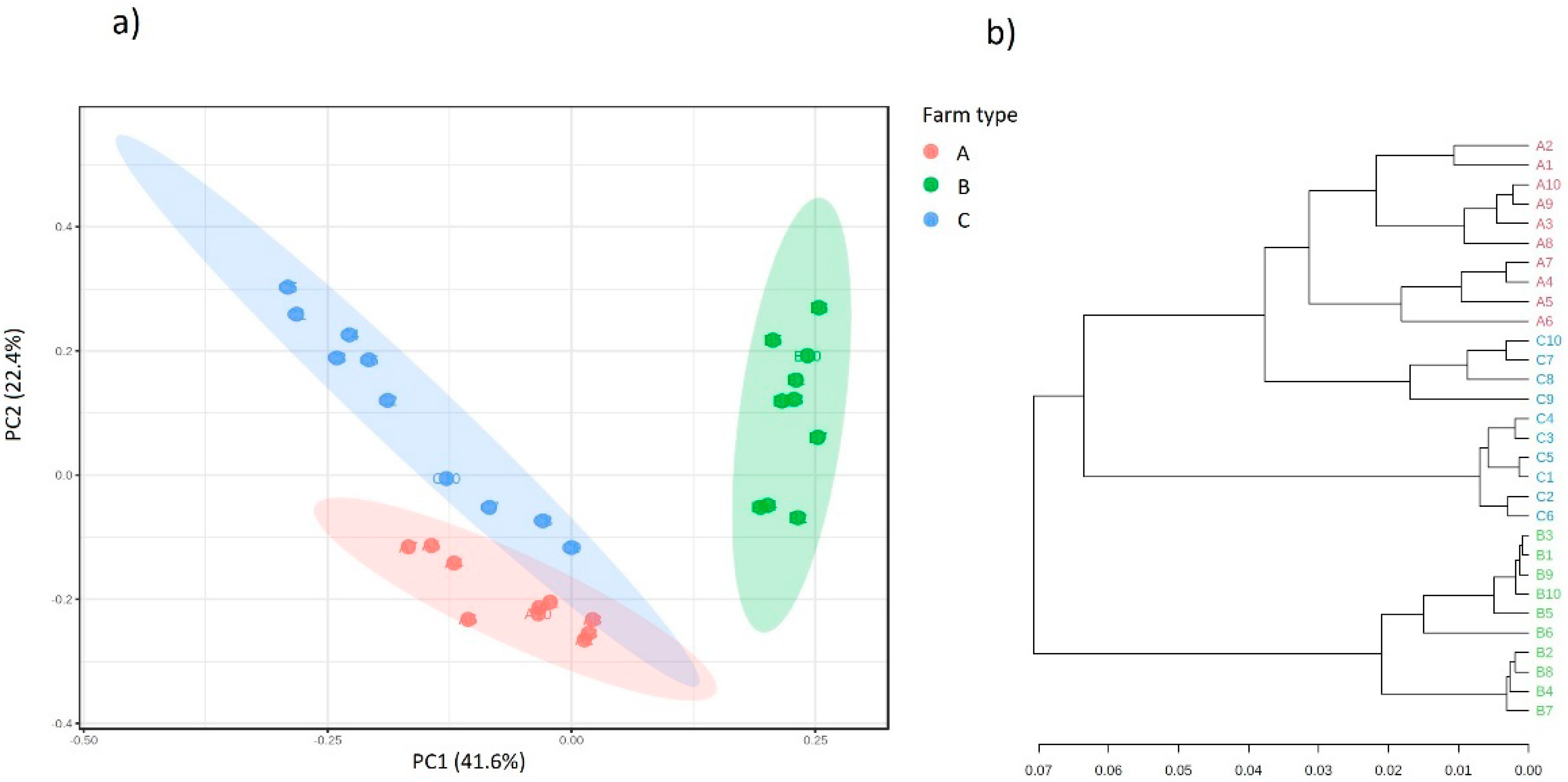
2.5. Analysis of Biodiversity
2.6. Analysis of Functional Potential and Antibiotic Resistance
3. Materials and Methods
3.1. Characteristics of Farms and Collection of Samples
3.2. Isolation of DNA
3.3. PCR Amplification and NGS Sequencing
3.4. Bioinformatic Analysis
3.5. Prediction the Functional Profile from Targeted Metagenomic Data
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Skeie, S.B.; Håland, M.; Thorsen, I.M.; Narvhus, J.; Porcellato, D. Bulk tank raw milk microbiota differs within and between farms: A moving goalpost challenging quality control. J. Dairy Sci. 2019, 102, 1959–1971. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.H.; Cho, Y.S.; Rackerby, B.; Goddik, L.; Park, S.H. Shifts of microbiota during cheese production: Impact on production and quality. Appl. Microbiol. Biotechnol. 2021, 105, 2307–2318. [Google Scholar] [CrossRef]
- Quigley, L.; O’Sullivan, O.; Stanton, C.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. The complex microbiota of raw milk. FEMS Microbiol. Rev. 2013, 37, 664–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from-omics studies. Mol. Biosyst. 2013, 12, 2359–2372. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nan, X.; Zhao, Y.; Jiang, L.; Wang, M.; Wang, H.; Zhang, F.; Xue, F.; Hua, D.; Liu, J.; et al. Rumen microbiome structure and metabolites activity in dairy cows with clinical and subclinical mastitis. J. Anim. Sci. Biotechnol. 2021, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Abebe, R.; Hatiya, H.; Abera, M.; Megersa, B.; Asmare, K. Bovine mastitis: Prevalence, risk factors and isolation of Staphylococcus aureus in dairy herds at Hawassa milk shed, South Ethiopia. BMC Vet. Res. 2016, 12, 270. [Google Scholar] [CrossRef] [Green Version]
- Bhakat, C.; Mohammad, A.; Mandal, D.K.; Mandal, A.; Rai, S.; Chatterjee, A.; Ghosh, M.K.; Dutta, T.K. Readily usable strategies to control mastitis for production augmentation in dairy cattle: A review. Vet. World 2020, 13, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- De Vliegher, S.; Fox, L.K.; Piepers, S.; McDougall, S.; Barkema, H.W. Invited review: Mastitis in dairy heifers: Nature of the disease, potential impact, prevention, and control. J. Dairy Sci. 2012, 95, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Fusco, V.; Chieffi, D.; Fanelli, F.; Logrieco, A.F.; Cho, G.-S.; Kabisch, J.; Böhnlein, C.; Franz, C.M.A.P. Microbial quality and safety of milk and milk products in the 21st century. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2013–2049. [Google Scholar] [CrossRef]
- Rocha, D.C.; Rocha, C.S.; Davi, S.T.; Calado, S.L.M.; Gomes, M.P. Veterinary antibiotics and plant physiology: An overview. Sci. Total Environ. 2021, 767, 144902. [Google Scholar] [CrossRef]
- Mohan, H.; Rajput, S.S.; Jadhav, E.B.; Sankhla, M.S.; Sonone, S.S.; Jadhav, S.; Kumar, R. Ecotoxicity, Occurrence, and Removal of Pharmaceuticals and Illicit Drugs from Aquatic Systems. Biointerface Res. Appl. Chem. 2021, 11, 12530–12546. [Google Scholar] [CrossRef]
- Ma, Z.; Lee, S.; Fan, P.; Zhai, Y.; Lim, J.; Galvão, K.N.; Nelson, C.; Jeong, K.C. Diverse β-lactam antibiotic-resistant bacteria and microbial community in milk from mastitic cows. Appl. Microbiol. Biotechnol. 2021, 105, 2109–2121. [Google Scholar] [CrossRef]
- Church, N.A.; McKillip, J.L. Antibiotic resistance crisis: Challenges and imperatives. Biologia 2021, 76, 1535–1550. [Google Scholar] [CrossRef]
- Altamirano, F.L.G.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [Green Version]
- van Teeseling, M.C.F.; de Pedro, M.A.; Cava, F. Determinants of Bacterial Morphology: From Fundamentals to Possibilities for Antimicrobial Targeting. Front. Microbiol. 2017, 8, 1264. [Google Scholar] [CrossRef] [Green Version]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [Green Version]
- McInnes, R.S.; McCallum, G.E.; Lamberte, L.; van Schaik, W. Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 2020, 53, 35–43. [Google Scholar] [CrossRef]
- Tilocca, B.; Costanzo, N.; Morittu, V.M.; Spina, A.A.; Soggiu, A.; Britti, D.; Roncada, P.; Piras, C. Milk microbiota: Characterization methods and role in cheese production. J. Proteom. 2020, 210, 103534. [Google Scholar] [CrossRef] [PubMed]
- Ganda, E.K.; Bisinotto, R.S.; Lima, S.F.; Kronauer, K.; Decter, D.H.; Oikonomou, G.; Bicalho, R.C. Longitudinal metagenomics profiling of bovine milk to assess the impact of intramammary treatment using a third-generation cephalosporin. Sci. Rep. 2016, 6, 37565. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, G.; Machado, V.S.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16s rDNA. PLoS ONE 2012, 7, e47671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehn, J.S.; Gorden, P.J.; Munro, D.; Rong, R.; Dong, Q.; Plummer, P.J.; Phillips, G.J. Bacterial community profiling of milk samples as a means to understand culture-negative bovine clinical mastitis. PLoS ONE 2013, 8, e61959. [Google Scholar] [CrossRef] [Green Version]
- Wouters, J.T.M.; Ayad, E.A.E.; Hugenholtz, J.; Smit, G. Microbes from raw milk for fermented dairy products. Int. Dairy J. 2012, 12, 91–109. [Google Scholar] [CrossRef]
- Young, W.; Hine, B.C.; Wallace, O.A.M.; Callaghan, M.; Bibiloni, R. Transfer of intestinal bacterial components to mammary secret ions in the cow. PeerJ 2015, 3, e888. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Sadiq, F.A.; Liu, T.J.; Li, Y.; Gu, J.S.; Yang, H.Y.; He, G.Q. Spoilage potential of psychrotrophic bacteria isolated from raw milk and the thermo-stability of their enzymes. J. Zhejiang Univ. Sci. B 2018, 19, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Oultram, J.W.H.; Ganda, E.K.; Boulding, S.C.; Bicalho, R.C.; Oikonomou, G. A Metataxonomic Approach Could Be Considered for Cattle Clinical Mastitis Diagnostics. Front. Vet. Sci. 2017, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, W.; Vogensen, F.K.; Lillevang, S.; Abu Al-Soud, W.; Sørensen, S.J.; Jakobsen, M. The fate of indigenous microbiota, starter cultures, Escherichia coli, Listeria innocua and Staphylococcus aureus in Danish raw milk and cheeses determined by pyrosequencing and quantitative real time (qRT)-PCR. Int. J. Food Microbiol. 2012, 153, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, V.D.; Ahir, V.B.; Koringa, P.G.; Jakhesara, S.J.; Rank, D.N.; Nauriyal, D.S.; Kunjadia, A.P.; Joshi, C.G. Milk microbiome signatures of subclinical mastitis-affected cattle analysed by shotgun sequencing. J. Appl. Microbiol. 2012, 112, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.N.; Istiaq, A.; Clement, R.A.; Sultana, M.; Crandall, K.A.; Siddiki, A.Z.; Hossain, M.A. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 2019, 9, 13536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, S.F.; Bicalho, M.L.S.; Bicalho, R.C. Evaluation of milk sample fractions for characterization of milk microbiota from healthy and clinical mastitis cows. PLoS ONE 2018, 13, e0193671. [Google Scholar] [CrossRef] [Green Version]
- Pyatov, V.; Vrtková, I.; Knoll, A. Detection of selected antibiotic resistance genes using multiplex PCR assay in mastitis pathogens in the Czech Republic. Acta Vet. Brno 2017, 86, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Molineri, A.I.; Camussone, C.; Zbrun, M.V.; Suárez Archilla, G.; Cristiani, M.; Neder, V.; Calvinho, L.; Signorini, M. Antimicrobial resistance of Staphylococcus aureus isolated from bovine mastitis: Systematic review and meta-analysis. Prev. Vet. Med. 2021, 188, 105261. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.P.; Murinda, S.E. Antimicrobial resistance of mastitis pathogens. Vet. Clin. N. Am. Food Anim. Pract. 2012, 28, 165–185. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, J. Beta-lactamase induction and cell wall metabolism in Gram-negative bacteria. Front. Microbiol. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web–based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega, T.R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst-a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 180–188. [Google Scholar] [CrossRef]
 —mean,
—mean, 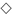 —outlers point, ∗—extreme point.
—mean, —outlers point, ∗—extreme point.
—outlers point, ∗—extreme point.
—mean, —outlers point, ∗—extreme point.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
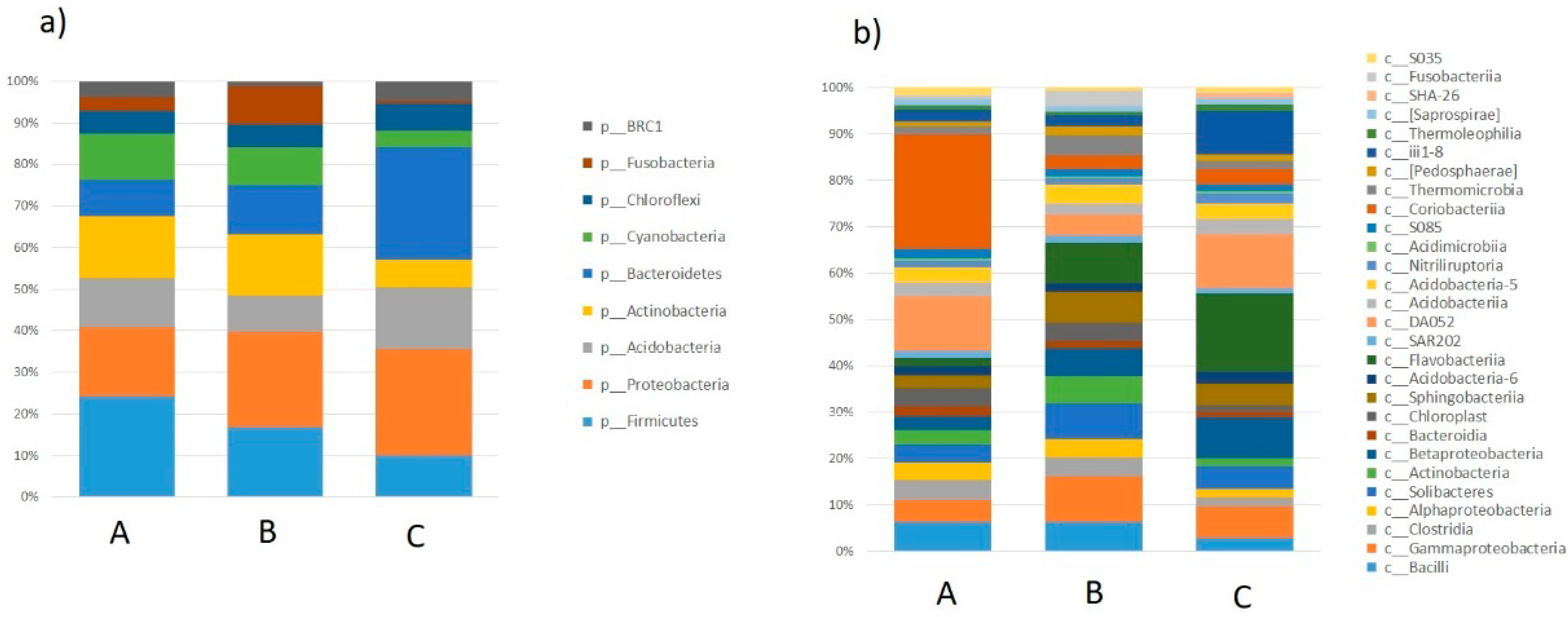{kind=link}
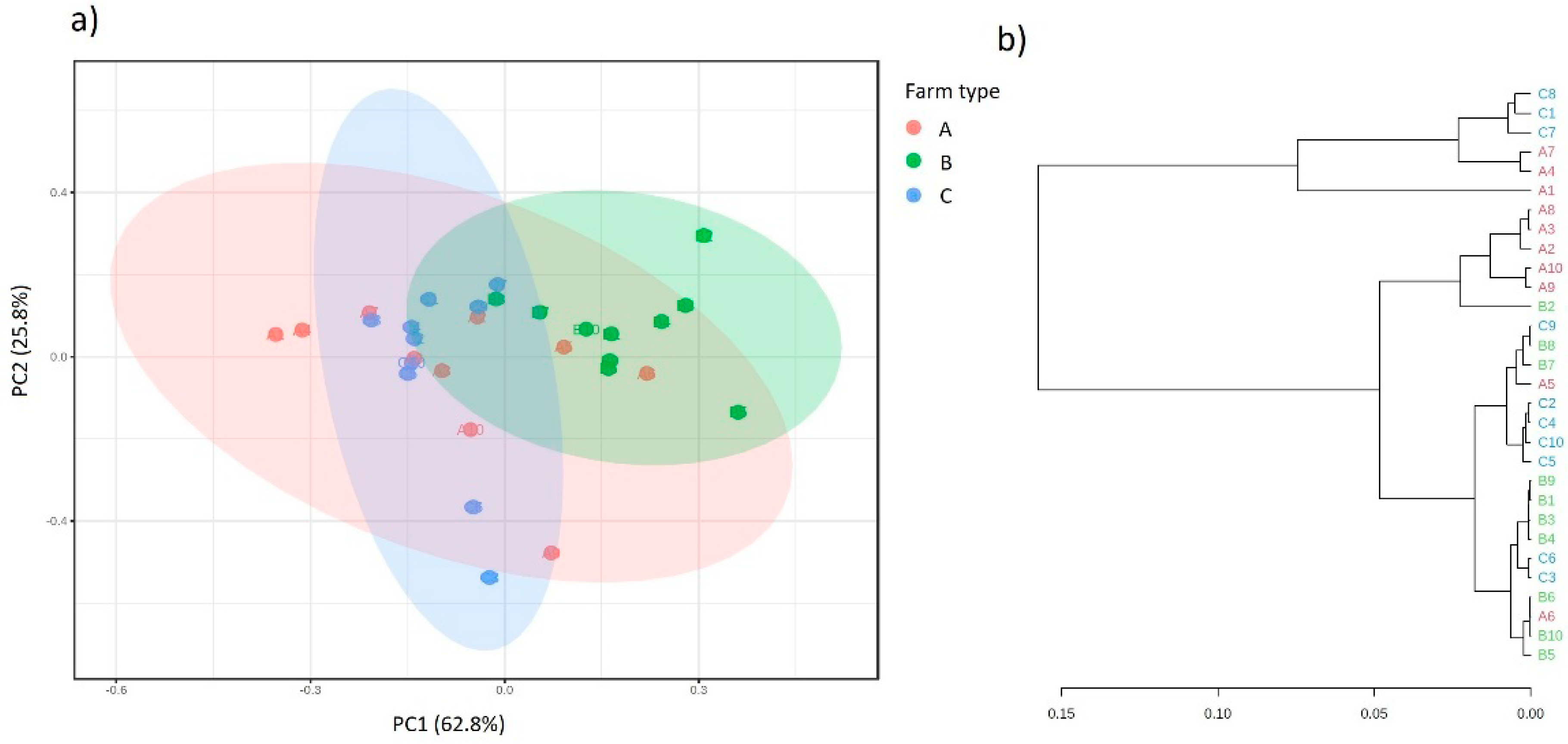{kind=link}
| Milk from Farm A | Milk from Farm B | Milk from Farm C | |
|---|---|---|---|
| Number of OTUs | 5490 ± 78 | 4486 ± 94 | 3488 ± 58 |
| Chao-1 bias-corrected | 5503 ± 14 | 4597 ± 18 | 3508 ± 14 |
| Shannon entropy | 8.59 ± 0.45 | 7.02 ± 0.36 | 6.33 ± 0.39 |
| Phylogenetic diversity | 10.23 ± 0.61 | 8.98 ± 0.49 | 7.12 ± 0.44 |
| KO Identifier | Gene Name |
|---|---|
| K01467 | beta-lactamase class C (ampC) |
| K02171 | BlaI family transcriptional regulator, penicillinase repressor (blaI) |
| K02172 | bla regulator protein blaR1 (blaR1) |
| K02545 | penicillin-binding protein 2 prime (mecA) |
| K02546 | BlaI family transcriptional regulator, methicillin resistance regulatory protein (mecI) |
| K02547 | methicillin resistance protein (mecR1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hornik, B.; Czarny, J.; Staninska-Pięta, J.; Wolko, Ł.; Cyplik, P.; Piotrowska-Cyplik, A. The Raw Milk Microbiota from Semi-Subsistence Farms Characteristics by NGS Analysis Method. Molecules 2021, 26, 5029. https://doi.org/10.3390/molecules26165029
Hornik B, Czarny J, Staninska-Pięta J, Wolko Ł, Cyplik P, Piotrowska-Cyplik A. The Raw Milk Microbiota from Semi-Subsistence Farms Characteristics by NGS Analysis Method. Molecules. 2021; 26(16):5029. https://doi.org/10.3390/molecules26165029
Chicago/Turabian StyleHornik, Bartosz, Jakub Czarny, Justyna Staninska-Pięta, Łukasz Wolko, Paweł Cyplik, and Agnieszka Piotrowska-Cyplik. 2021. "The Raw Milk Microbiota from Semi-Subsistence Farms Characteristics by NGS Analysis Method" Molecules 26, no. 16: 5029. https://doi.org/10.3390/molecules26165029
APA StyleHornik, B., Czarny, J., Staninska-Pięta, J., Wolko, Ł., Cyplik, P., & Piotrowska-Cyplik, A. (2021). The Raw Milk Microbiota from Semi-Subsistence Farms Characteristics by NGS Analysis Method. Molecules, 26(16), 5029. https://doi.org/10.3390/molecules26165029