
Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling



Abstract
:1. Introduction
2. Results and Discussion
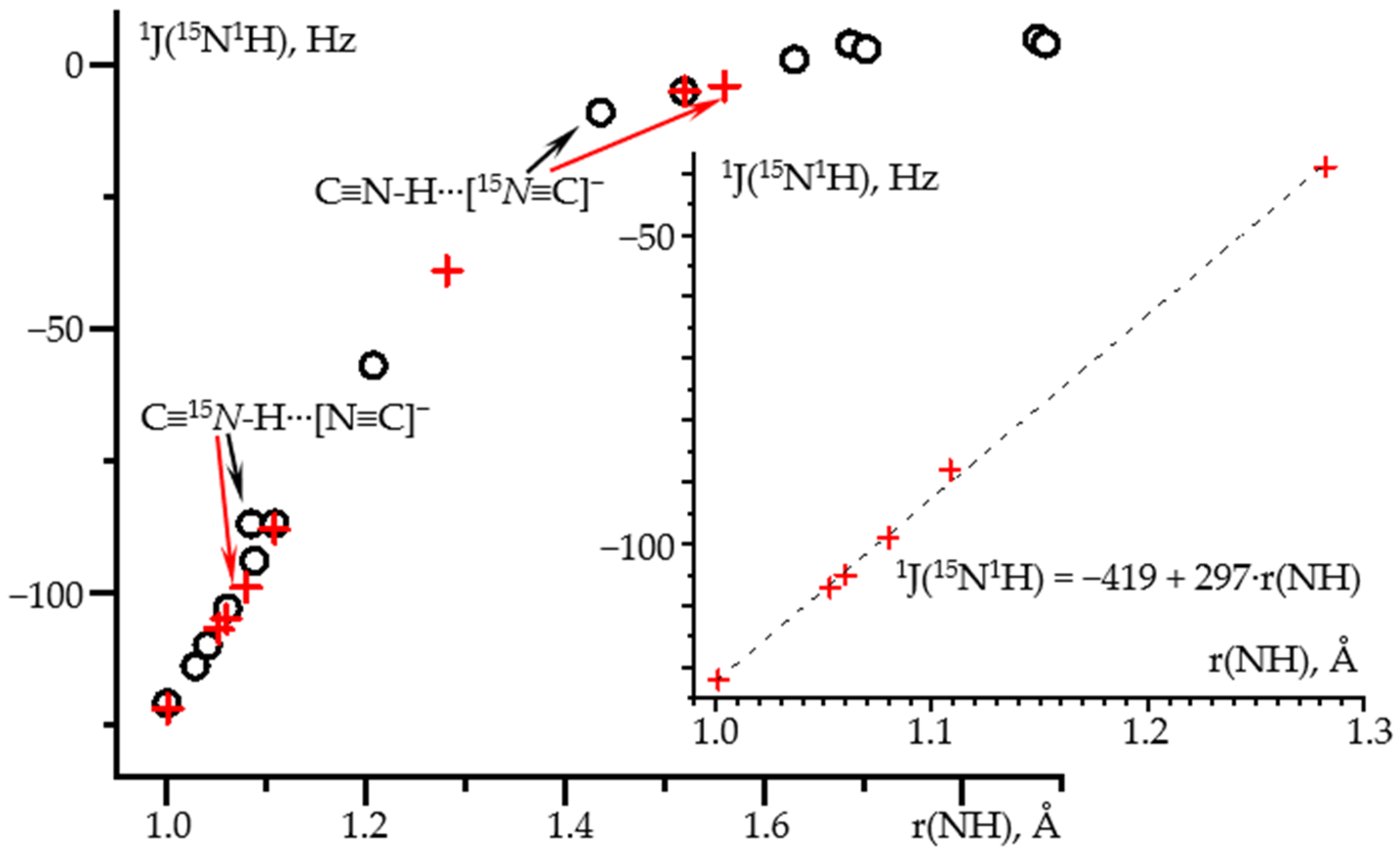
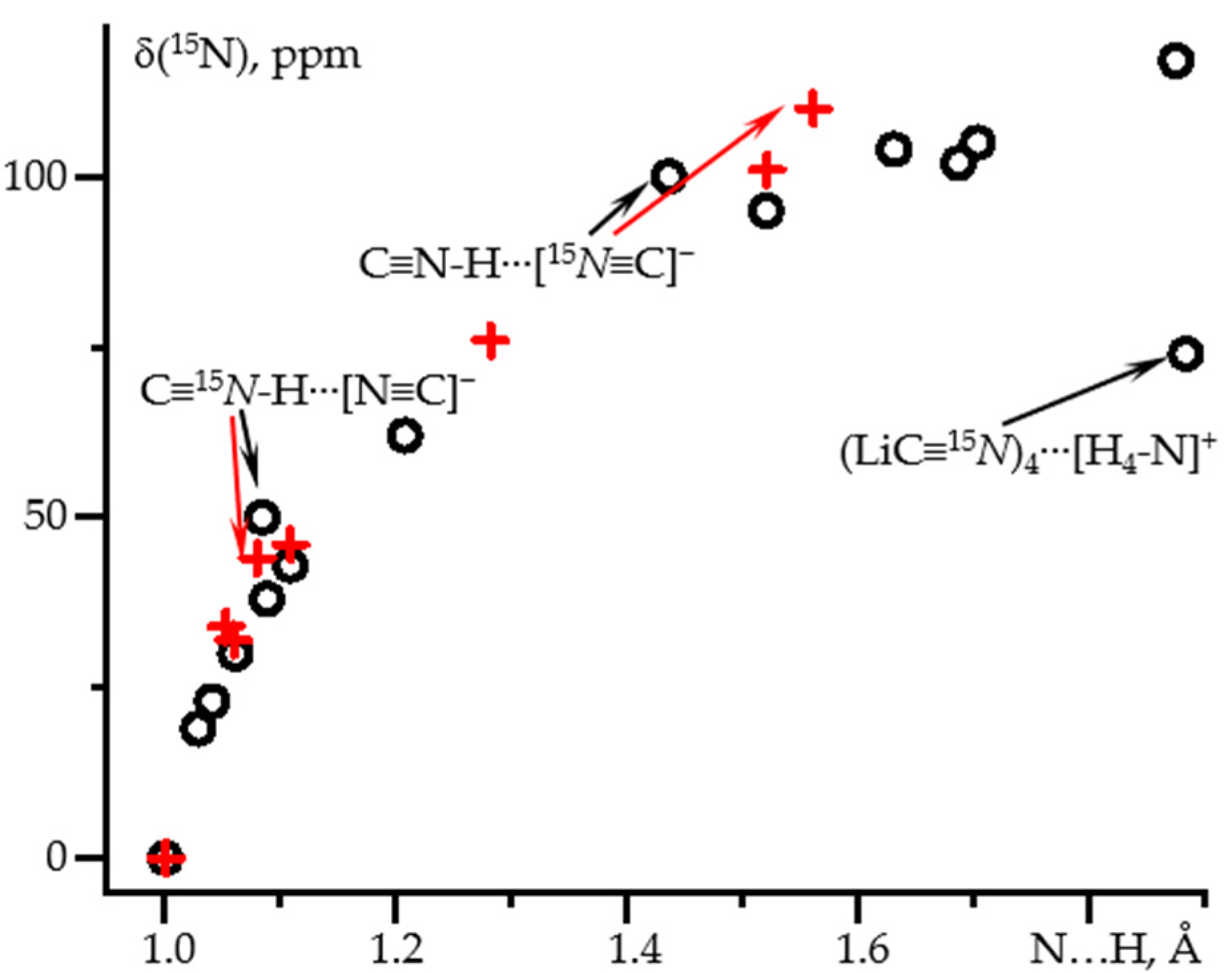
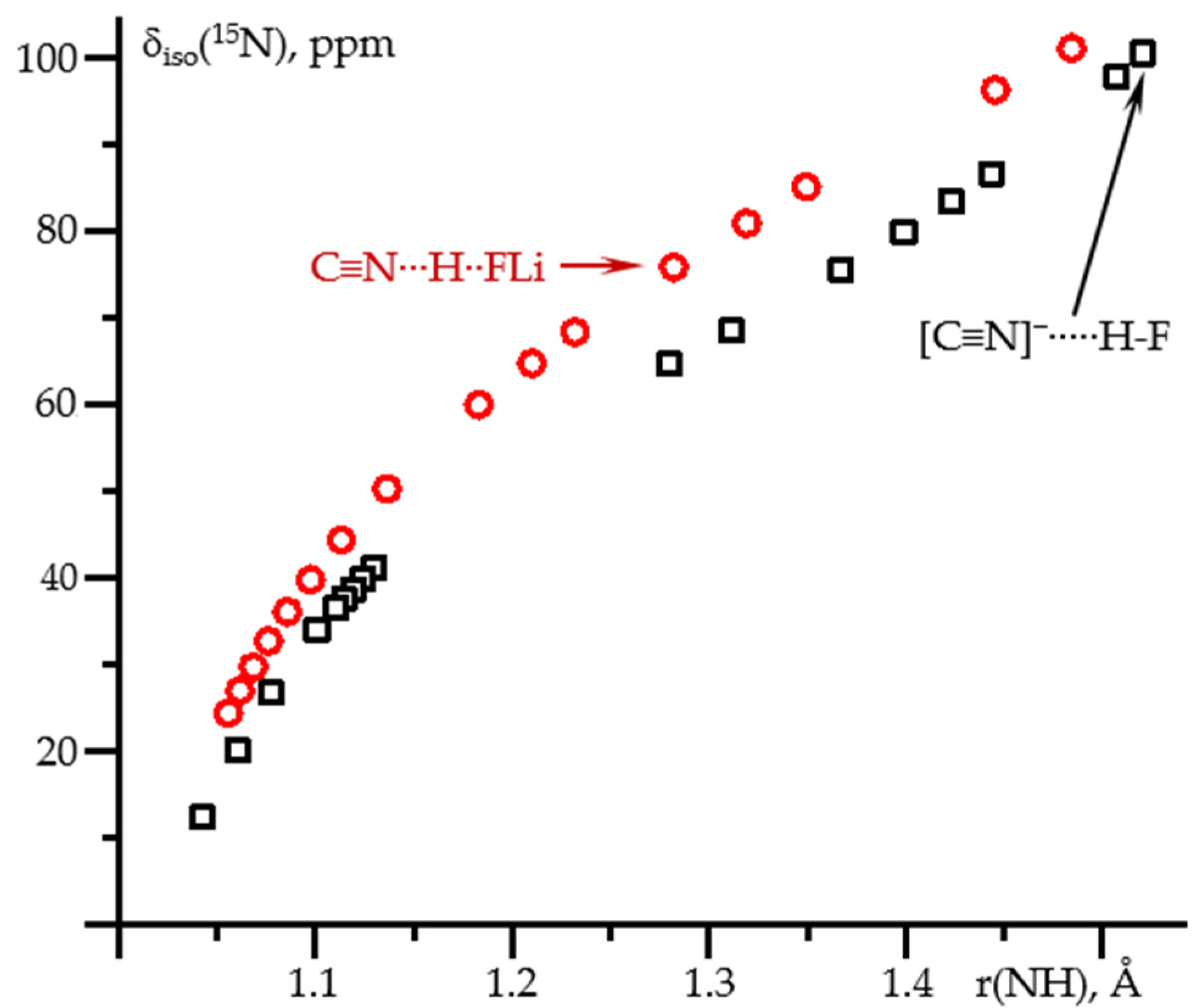
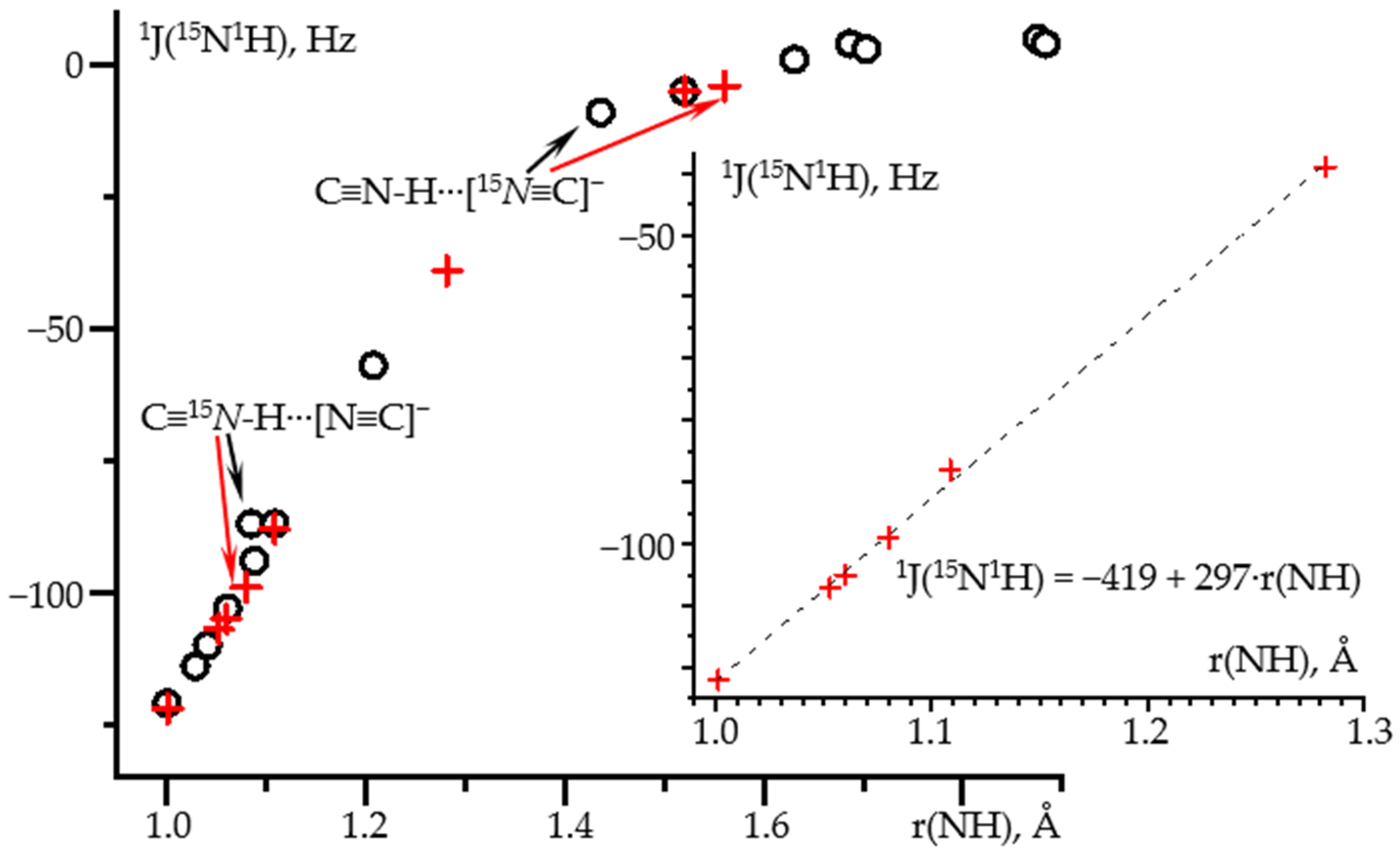
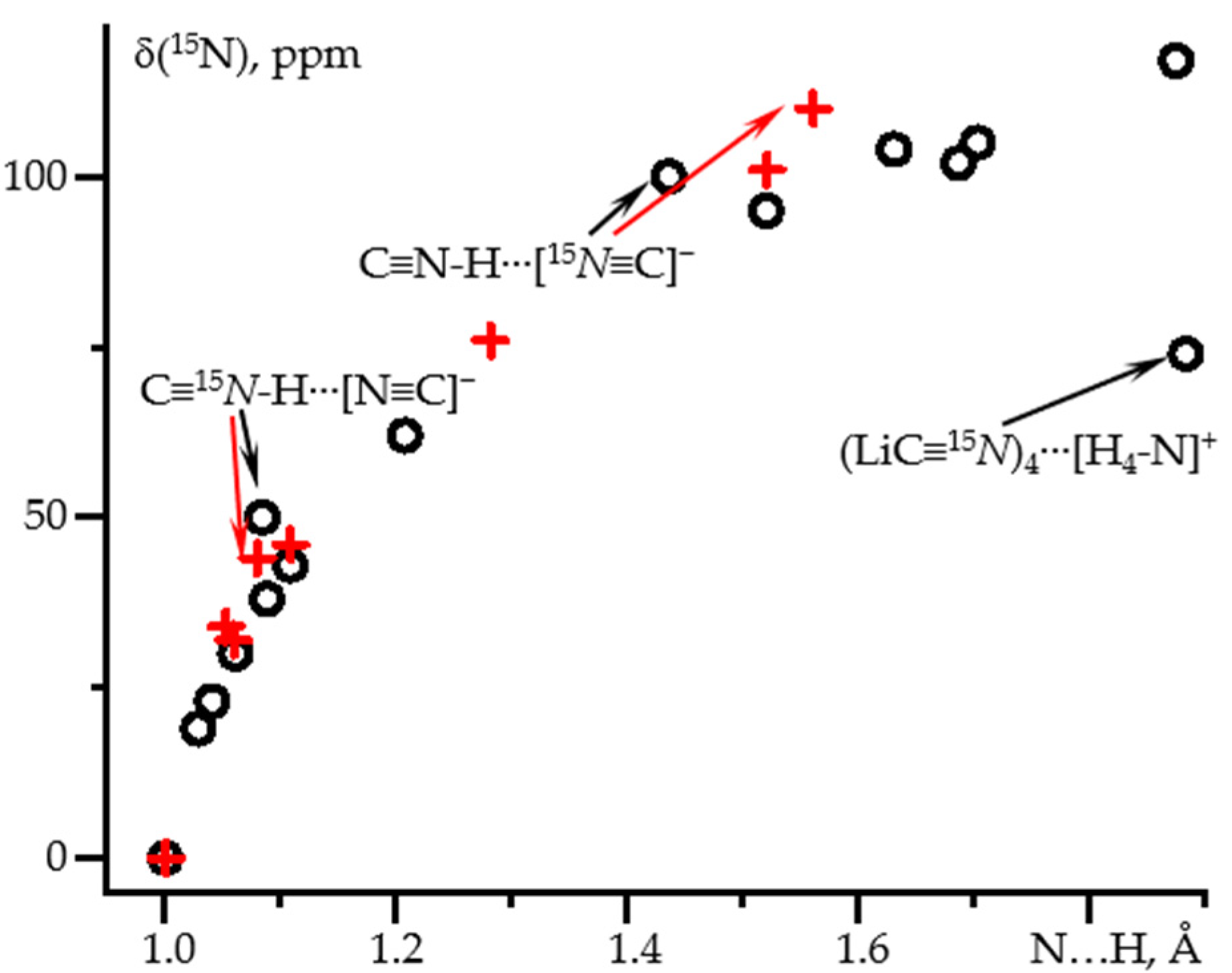
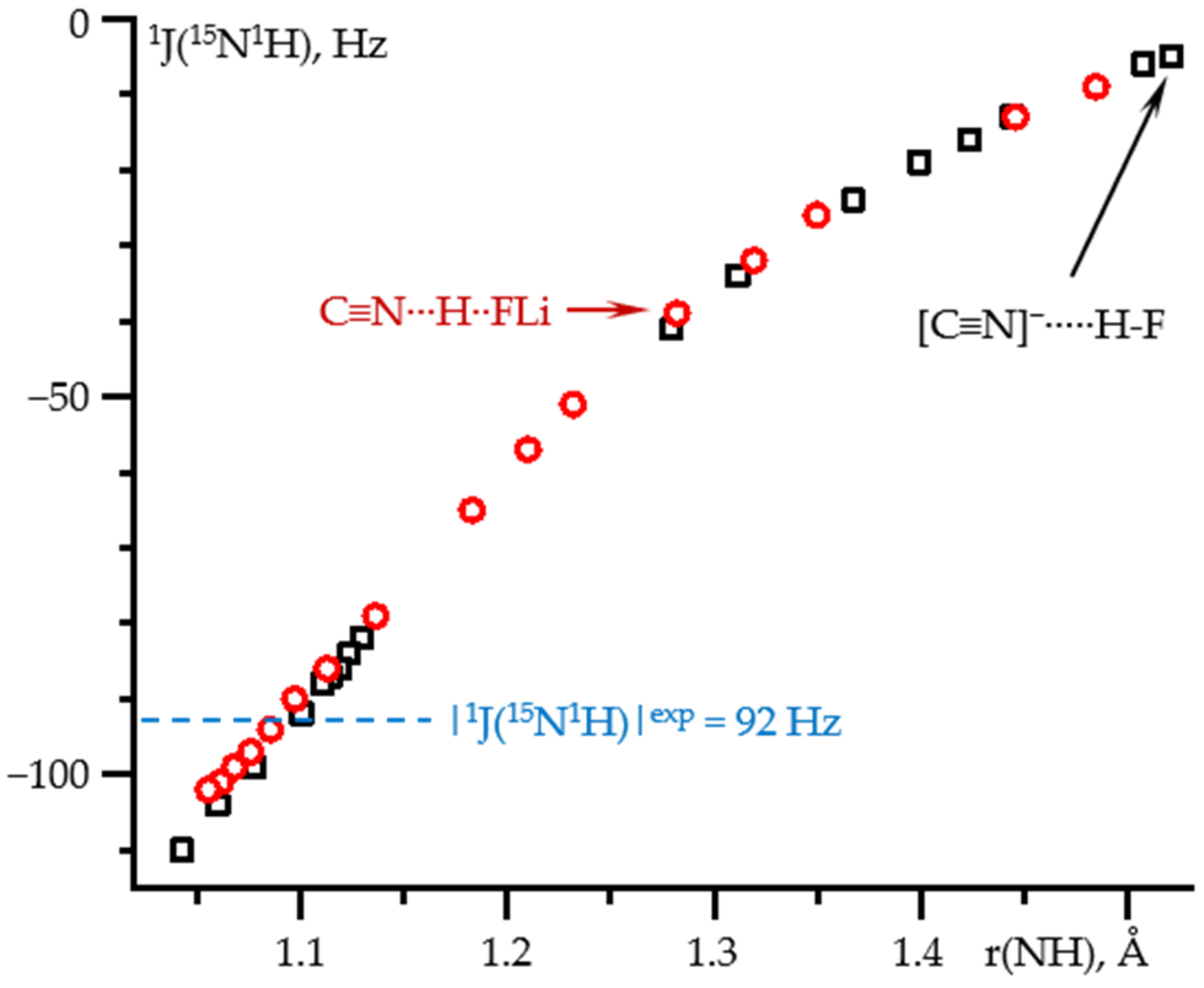
2.1. δiso(15N) and 1J(15N1H) as Functions of the N-H Distance in C≡N-H⋯F− and C≡N-H⋯FLi
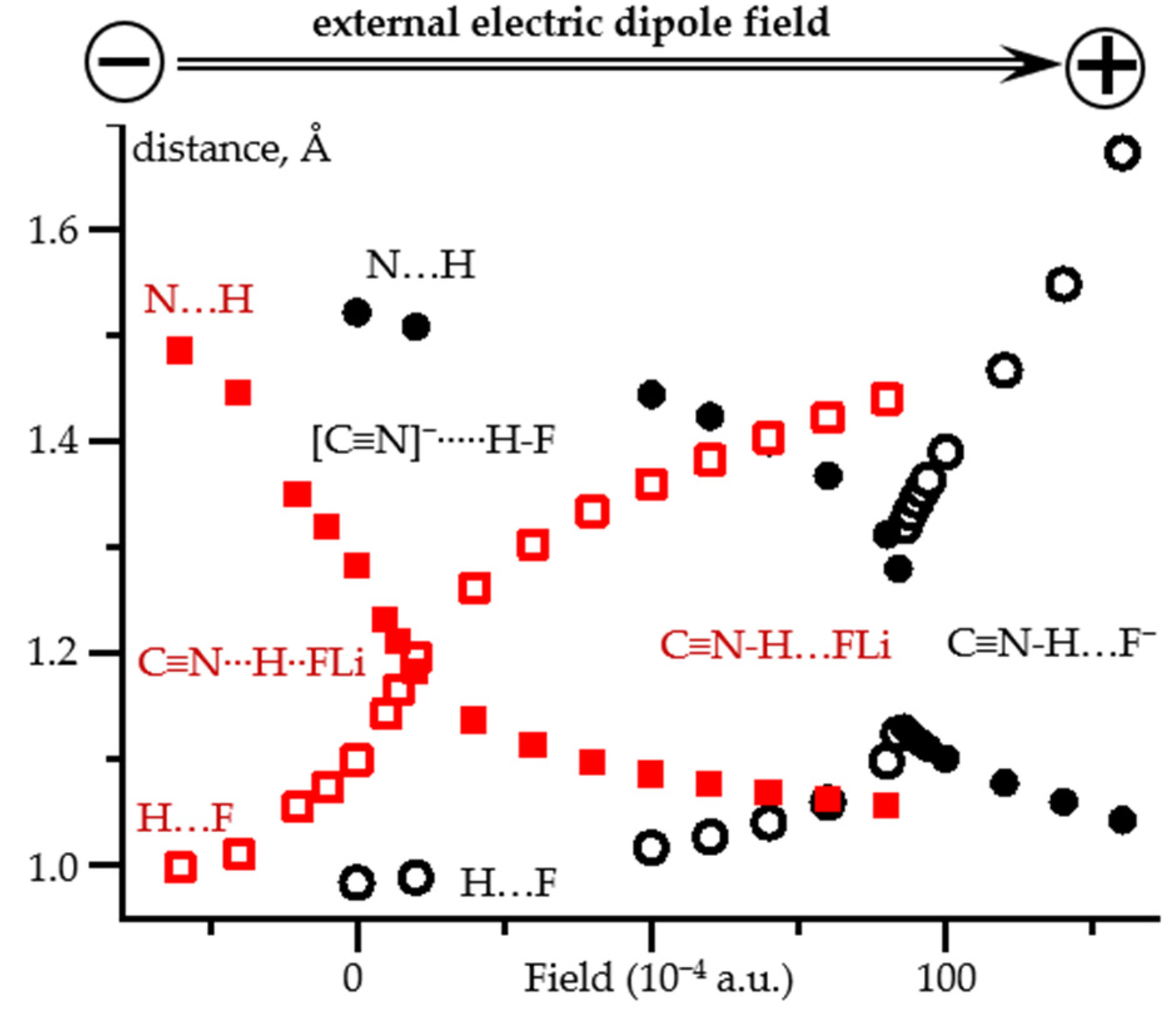
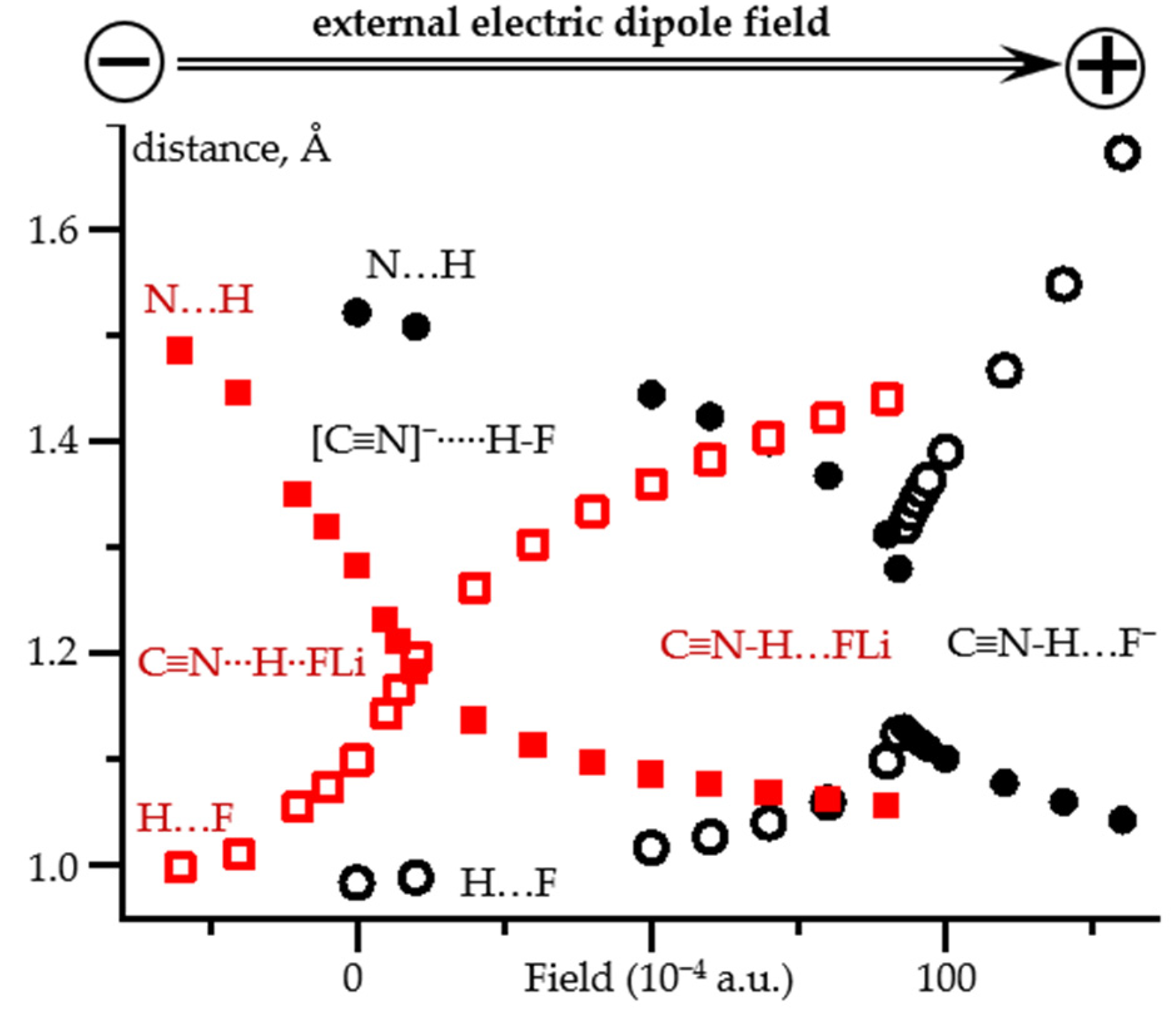
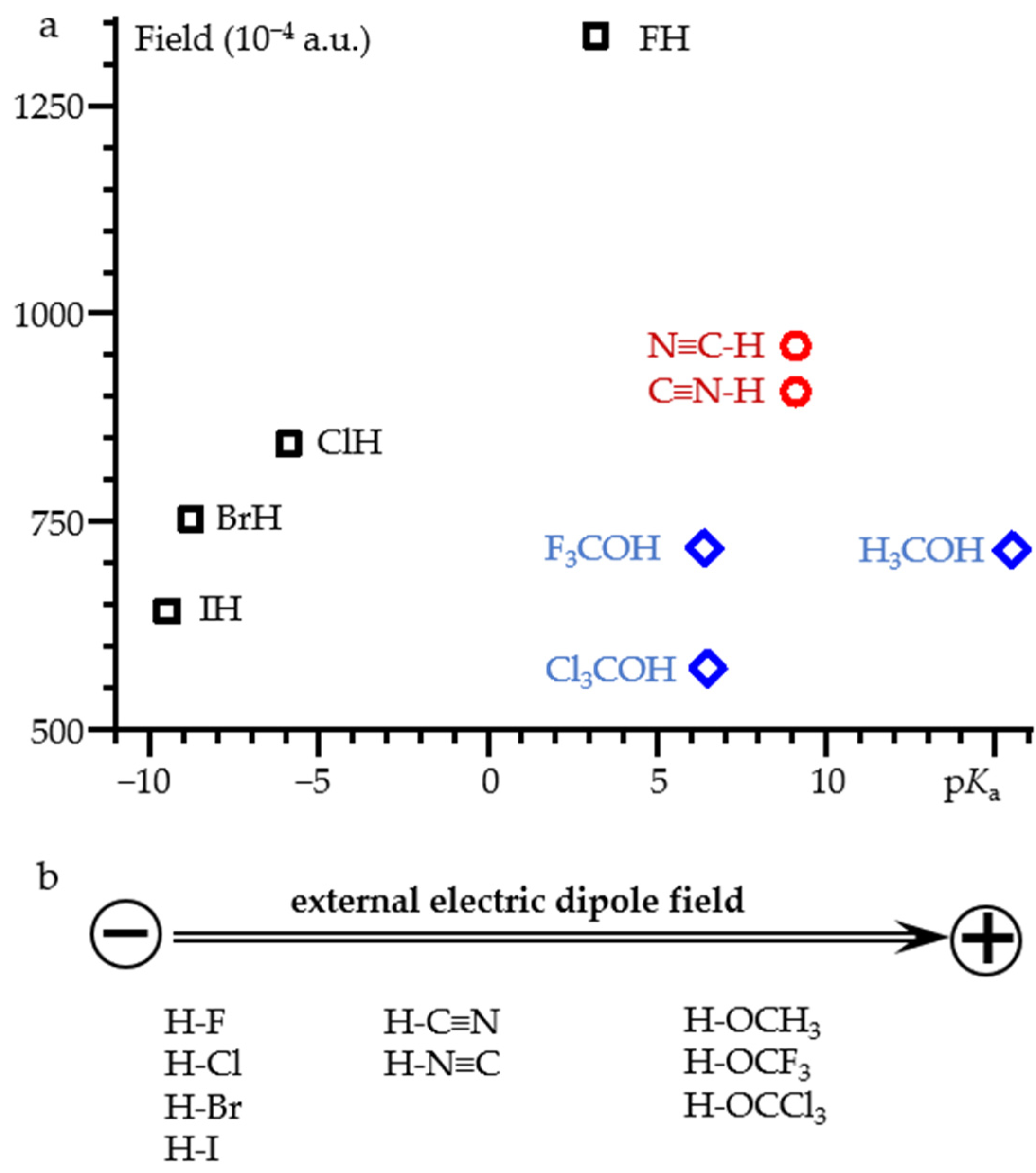
2.2. Field-Induced Proton Dissociation
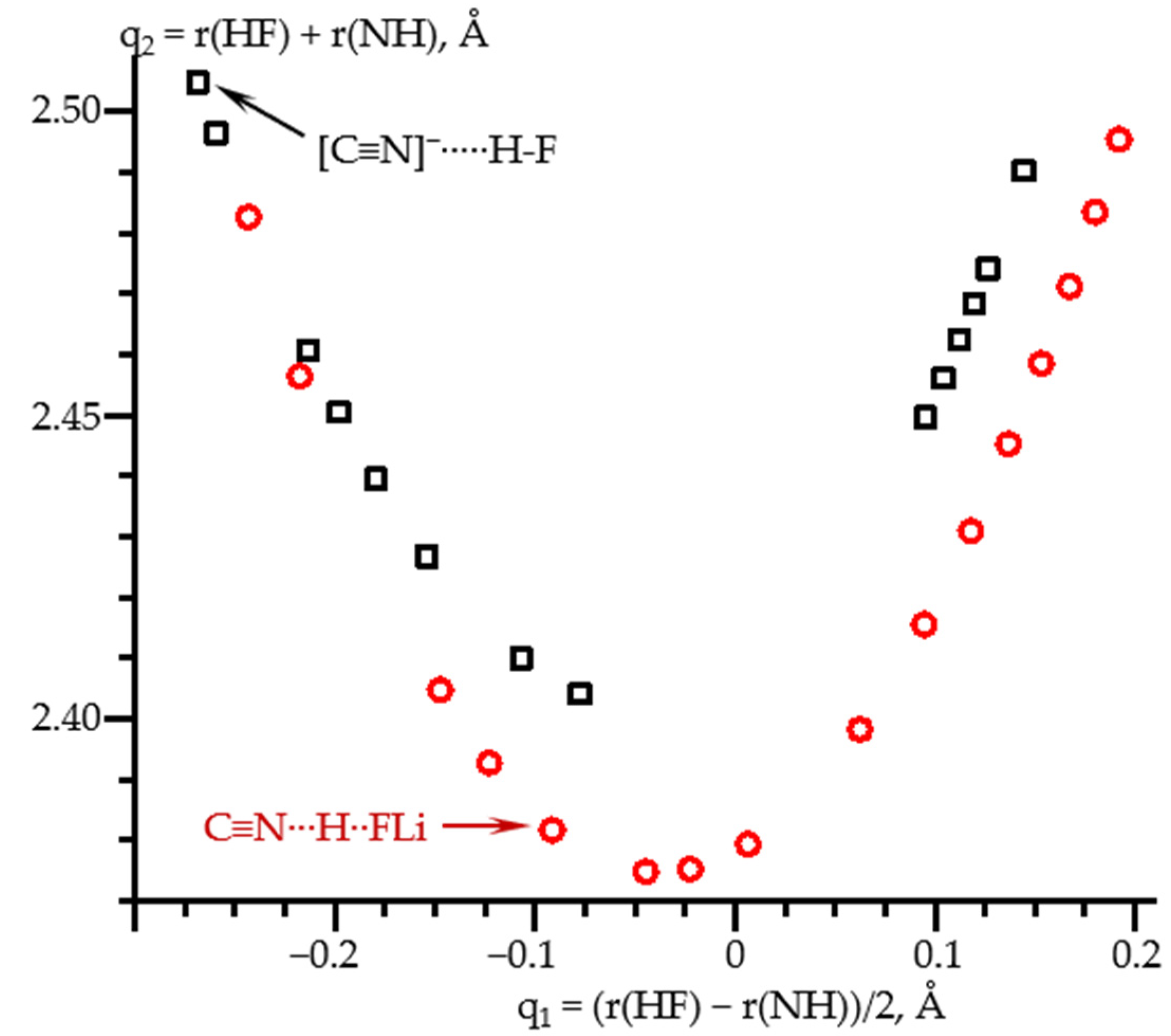
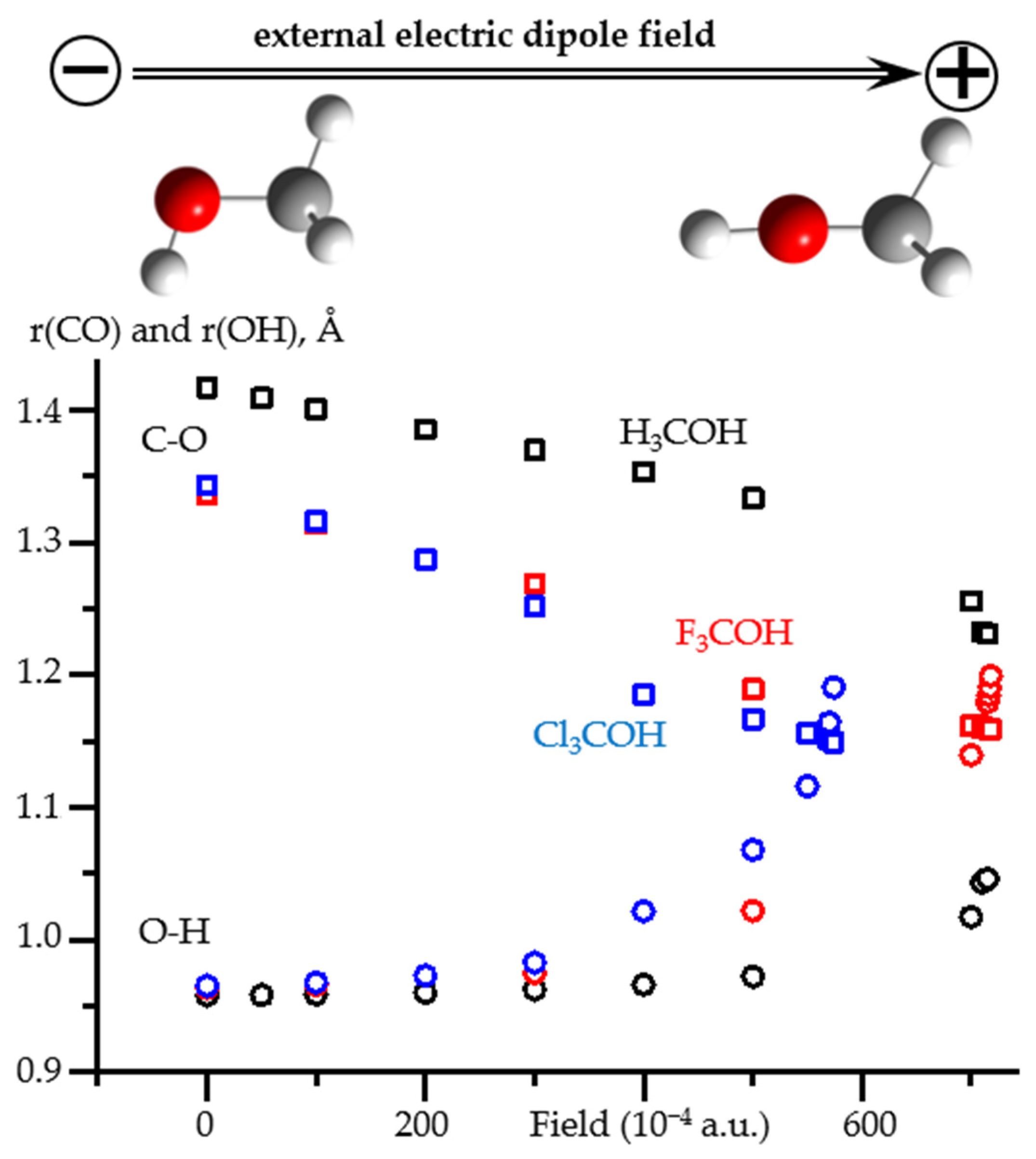
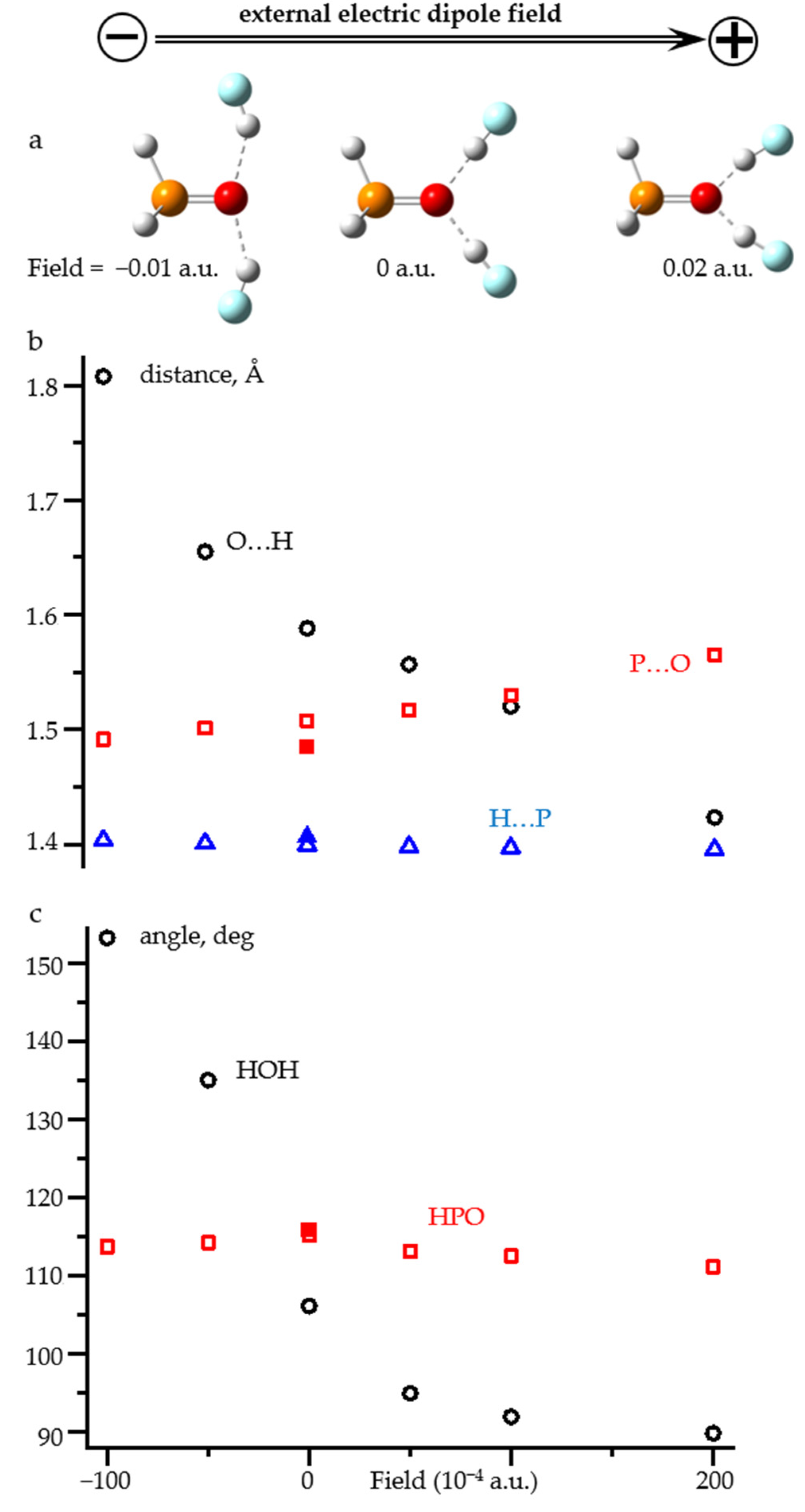
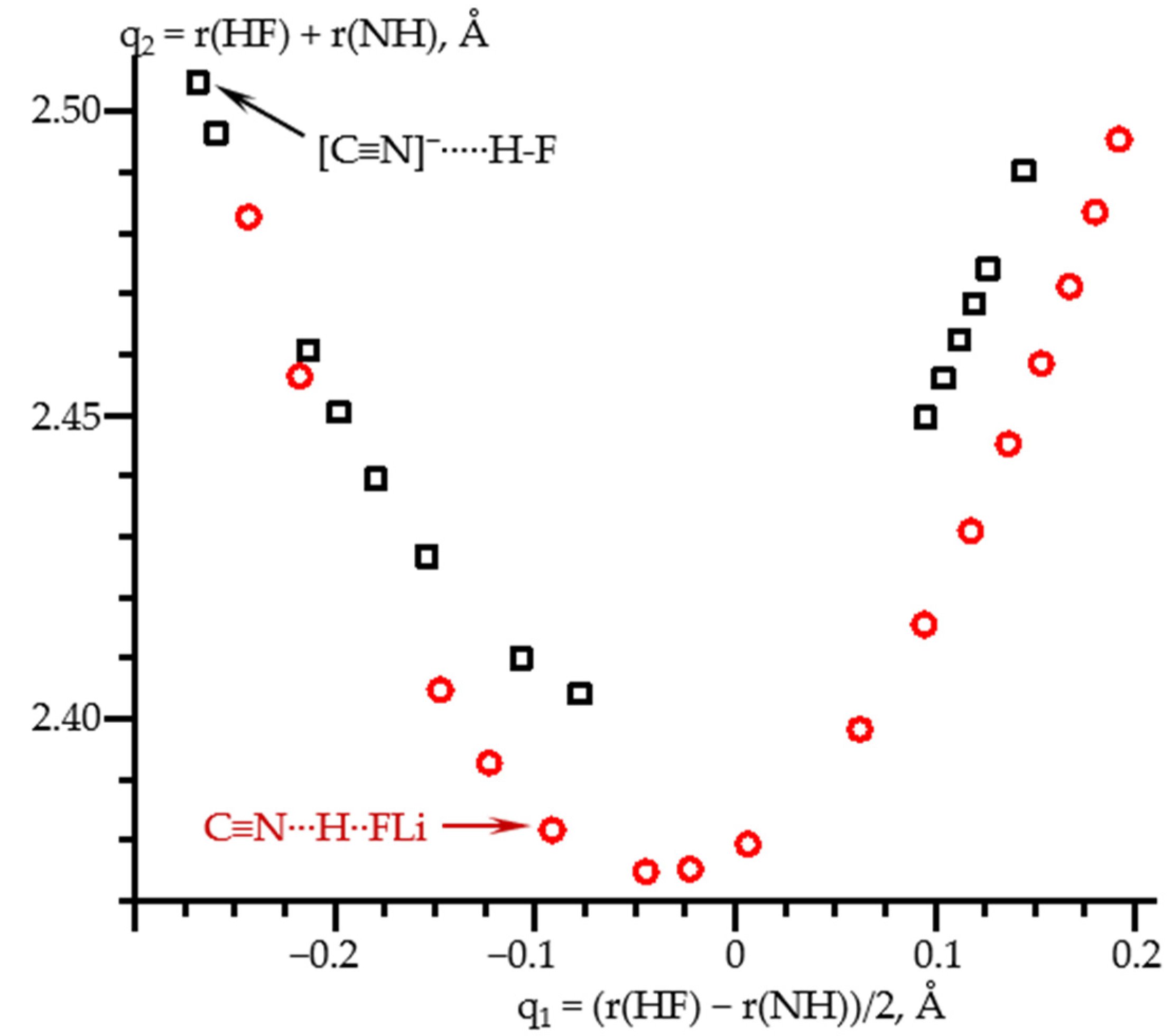
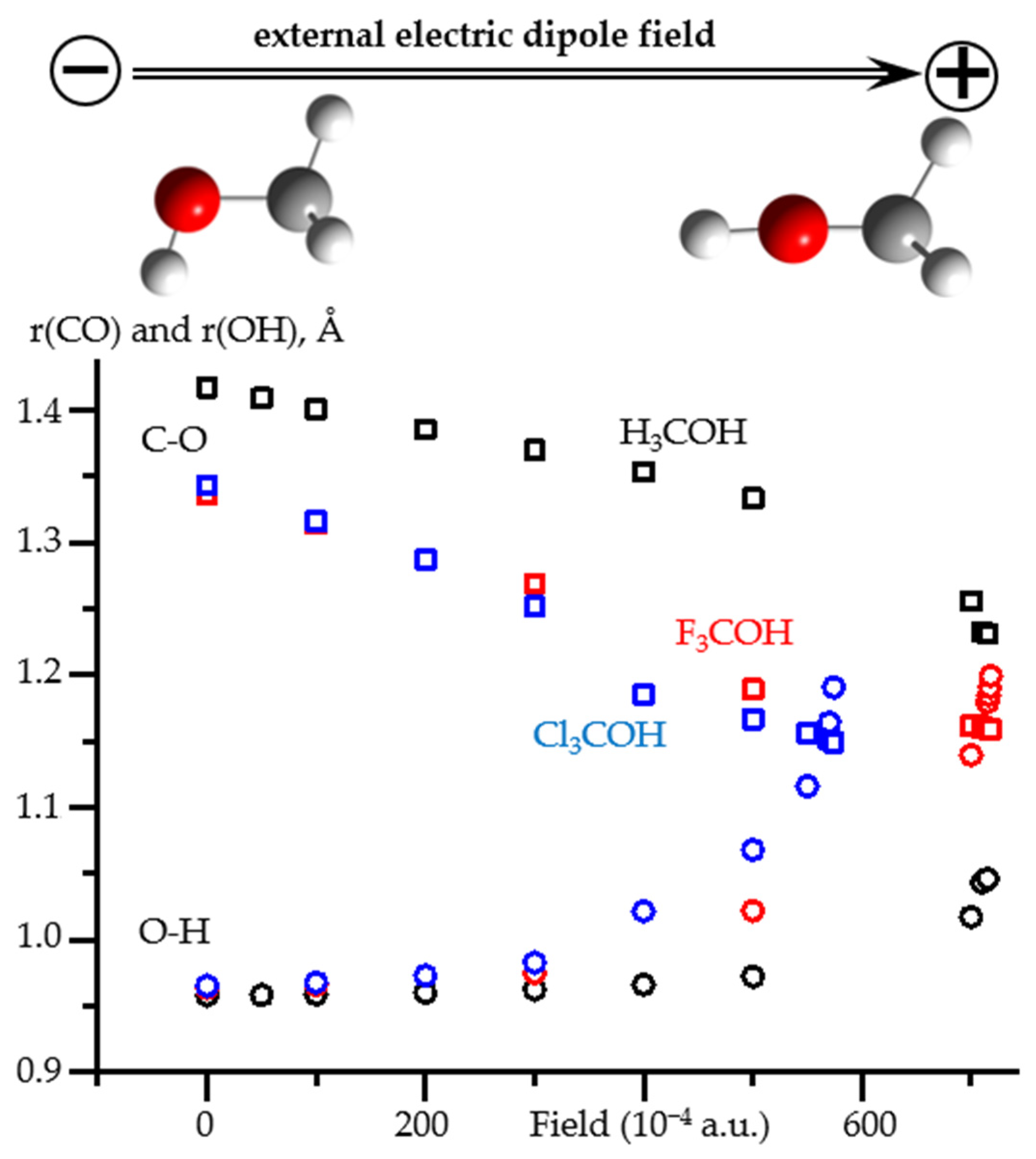
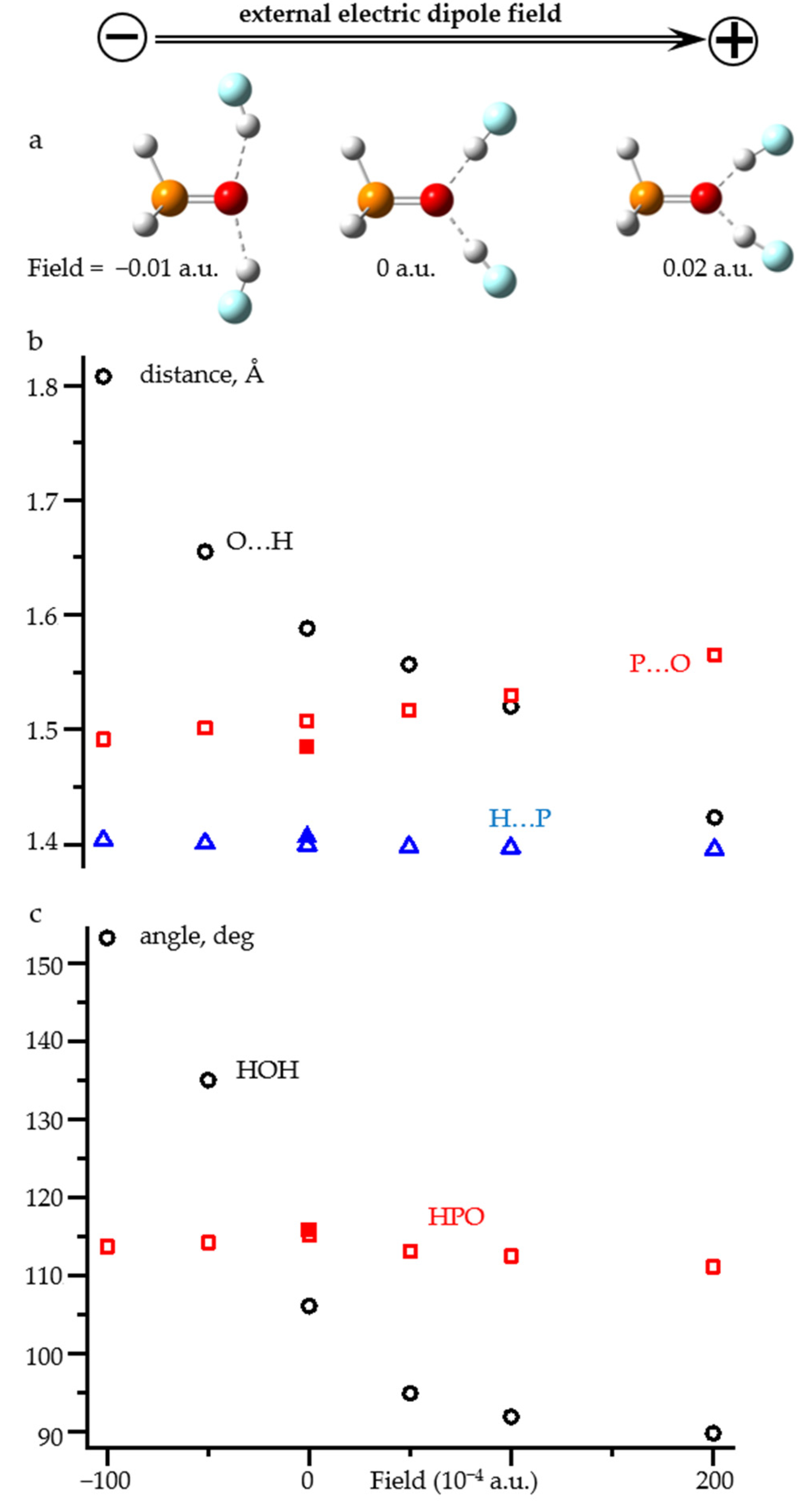
2.3. Field-Induced Structural Changes
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Novak, A. Hydrogen bonding in solids. Correlation of spectroscopic and crystallographic data. Struct. Bond. 1974, 18, 177–216. [Google Scholar] [CrossRef]
- Grech, E.; Malarski, Z.; Sobczyk, L. Isotope Effects in NH…N Hydrogen Bonds. Chem. Phys. Lett. 1986, 128, 259–263. [Google Scholar] [CrossRef]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and Spectral Properties of the Protonated Homodimer of Pyridine in the Liquid and Solid States. A Combined NMR, X-ray Diffraction and Inelastic Neutron Scattering Study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef] [PubMed]
- Iogansen, A.V. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Tolstoy, P.M.; Titova, A.A.; Kostin, M.A.; Denisov, G.S. Estimations of FH⋯ X hydrogen bond energies from IR intensities: Iogansen’s rule revisited. J. Comp. Chem. 2021, 42, 564–571. [Google Scholar] [CrossRef]
- Asfin, R.E.; Denisov, G.S.; Tokhadze, K.G. The infrared spectra and enthalpies of strongly bound dimers of phosphinic acids in the gas phase. (CH2Cl)2POOH and (C6H5)2POOH. J. Mol. Struct. 2002, 608, 161–168. [Google Scholar] [CrossRef]
- Lau, Y.K.; Ikuta, S.; Kebarle, P. Thermodynamics and kinetics of the gas-phase reactions: H3O+(H2O)n−1 + H2O = H3O+(H2O)n. J. Am. Chem. Soc. 1982, 104, 1462–1469. [Google Scholar] [CrossRef]
- Larson, J.W.; McMahon, T.B. Gas-phase bifluoride ion. An ion cyclotron resonance determination of the hydrogen bond energy in fluoride ion FHF- from gas-phase fluoride transfer equilibrium measurements. J. Am. Chem. Soc. 1982, 104, 5848–5849. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Genoni, A.; Jayatilaka, D.; Turner, M.J.; Sugimoto, K.; Nishibori, E.; Grabowsky, S. The advanced treatment of hydrogen bonding in quantum crystallography. J. Appl. Crystallogr. 2021, 54, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Woińska, M.; Grabowski, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [Green Version]
- Capelli, S.C.; Bürgi, H.B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Lengthening of the covalent O–H bond in O–H⋯O hydrogen bonds re-examined from low-temperature neutron diffraction data of organic compounds. Acta Crystallogr. Sect. B Struct. Sci. 1994, 50, 348–357. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-Dependent Mechanisms of Intramolecular Proton Transfer in Aprotic Solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379. [Google Scholar] [CrossRef] [PubMed]
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. 1H/15N NMR Chemical Shielding, Dipolar 15N,2H Coupling and Hydrogen Bond Geometry Correlations in a Novel Series of Hydrogen-Bonded Acid-Base Complexes of Collidine with Carboxylic Acids. Magn. Reson. Chem. 2001, 39, S18–S29. [Google Scholar] [CrossRef]
- Limbach, H.H.; Pietrzak, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S. NMR parameters and geometries of OHN and ODN hydrogen bonds of pyridine–acid complexes. Chem. Eur. J. 2004, 10, 5195–5204. [Google Scholar] [CrossRef]
- Sharif, S.; Shenderovich, I.G.; González, L.; Denisov, G.S.; Silverman, D.N.; Limbach, H.-H. NMR and Ab initio Studies of Small Complexes Formed between Water and Pyridine Derivatives in Solid and Liquid Phase. J. Phys. Chem. A 2007, 111, 6084–6093. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.E. A Spectroscopic Overview of Intramolecular Hydrogen Bonds of NH…O,S,N Type. Molecules 2021, 26, 2409. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Sigalov, M.; Shenderovich, I.G.; Mulloyarova, V.V.; Denisov, G.S.; Tolstoy, P.M. Correlations of NHN hydrogen bond energy with geometry and 1H NMR chemical shift difference of NH protons for aniline complexes. J. Chem. Phys. 2019, 150, 114305. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Pavlov, D.V.; Albanov, A.I.; Tarasova, O.A.; Nedolya, N.A. Experimental and theoretical study of the intramolecular C–H⋯N and C–H⋯S hydrogen bonding effects in the 1H and 13C NMR spectra of the 2-(alkylsulfanyl)-5-amino-1-vinylpyrroles: A particular state of amine nitrogen. Magn. Res. Chem. 2013, 51, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Kozlecki, T.; Tolstoy, P.M.; Kwocz, A.; Vovk, M.A.; Kochel, A.; Polowczyk, I.; Tretyakov, P.Y.; Filarowski, A. Conformational state of β-hydroxynaphthylamides: Barriers for the rotation of the amide group around CN bond and dynamics of the morpholine ring. Spectrochim. Acta A 2015, 149, 254–262. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yermolayev, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry 2021, 13, 756. [Google Scholar] [CrossRef]
- Sobczyk, L.; Obrzud, M.; Filarowski, A. H/D Isotope Effects in Hydrogen Bonded Systems. Molecules 2013, 18, 4467–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisov, G.S.; Mavri, J.; Sobczyk, L. Potential Energy Shape for the Proton Motion in Hydrogen Bonds Reflected in Infrared and NMR Spectra. In Hydrogen Bonding—New Insights; Grabowski, S.J., Ed.; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar] [CrossRef]
- Denisov, G.S.; Bureiko, S.F.; Kucherov, S.Y.; Tolstoy, P.M. Correlation relationships between the energy and spectroscopic parameters of complexes with F⋯HF hydrogen bond. Dokl. Phys. Chem. 2017, 475, 115–118. [Google Scholar] [CrossRef]
- Grabowski, S.J. Intramolecular Hydrogen Bond Energy and Its Decomposition—O–H⋯O Interactions. Crystals 2021, 11, 5. [Google Scholar] [CrossRef]
- Iribarren, Í.; Montero-Campillo, M.M.; Alkorta, I.; Elguero, J.; Quiñonero, D. Cations brought together by hydrogen bonds: The protonated pyridine–boronic acid dimer explained. Phys. Chem. Chem. Phys. 2019, 21, 5796–5802. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Elguero, J. Theoretical studies of conformational analysis and intramolecular dynamic phenomena. Struct. Chem. 2019, 30, 2029–2055. [Google Scholar] [CrossRef]
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D Isotope Effects on NMR Chemical Shifts of [FHF]− Ion Based on Calculations of Nuclear Magnetic Shielding Tensor Surface. Z. Phys. Chem. 2003, 217, 1549–1563. [Google Scholar] [CrossRef]
- Grabowski, S.J. Study of correlations for dihydrogen bonds by quantum-chemical calculations. Chem. Phys. Lett. 1999, 312, 542–547. [Google Scholar] [CrossRef]
- Kizior, B.; Panek, J.J.; Szyja, B.M.; Jezierska, A. Structure-Property Relationship in Selected Naphtho- and Anthra-Quinone Derivatives on the Basis of Density Functional Theory and Car–Parrinello Molecular Dynamics. Symmetry 2021, 13, 564. [Google Scholar] [CrossRef]
- Alkorta, I.; Walker, N.R.; Legon, A.C. Non-Covalent Interactions of the Lewis Acids Cu–X, Ag–X, and Au–X (X = F and Cl) with Nine Simple Lewis Bases B: A Systematic Investigation of Coinage–Metal Bonds by Ab Initio Calculations. Inorganics 2021, 9, 13. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. Hydrogen Bonds with BF4− Anion as a Proton Acceptor. Crystals 2020, 10, 460. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Effect of Noncovalent Interactions on the 31P Chemical Shift Tensor of Phosphine Oxides, Phosphinic, Phosphonic, and Phosphoric Acids, and Their Complexes with Lead(II). J. Phys. Chem. C 2013, 117, 26689–26702. [Google Scholar] [CrossRef]
- Giba, I.S.; Tolstoy, P.M. Self-Assembly of Hydrogen-Bonded Cage Tetramers of Phosphonic Acid. Symmetry 2021, 13, 258. [Google Scholar] [CrossRef]
- Bankiewicz, B.; Palusiak, M. Cooperation/Competition between Halogen Bonds and Hydrogen Bonds in Complexes of 2,6-Diaminopyridines and X-CY3 (X = Cl, Br; Y = H, F). Symmetry 2021, 13, 766. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Vener, M.V. Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds. Symmetry 2021, 13, 425. [Google Scholar] [CrossRef]
- Gholami, S.; Aarabi, M.; Grabowski, S.J. Coexistence of Intra- and Intermolecular Hydrogen Bonds: Salicylic Acid and Salicylamide and Their Thiol Counterparts. J. Phys. Chem. A 2021, 125, 1526–1539. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Simplified Calculation Approaches Designed to Reproduce the Geometry of Hydrogen Bonds in Molecular Complexes in Aprotic Solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Solimannejad, M.; Elguero, J. Competition of hydrogen bonds and halogen bonds in complexes of hypohalous acids with nitrogenated bases. J. Phys. Chem. A 2008, 112, 10856–10863. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. Experimentally Established Benchmark Calculations of 31P NMR Quantities. Chem. Methods 2021, 1, 61–70. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Vener, M.V.; Shenderovich, I.G. Local-structure effects on 31P NMR chemical shift tensors in solid state. J. Chem. Phys. 2019, 150, 144706. [Google Scholar] [CrossRef]
- Shenderovich, I.G. 1,3,5-Triaza-7-Phosphaadamantane (PTA) as a 31P NMR Probe for Organometallic Transition Metal Complexes in Solution. Molecules 2021, 26, 1390. [Google Scholar] [CrossRef] [PubMed]
- Battistin, F.; Balducci, G.; Milani, B.; Alessio, E. Water-Soluble Ruthenium(II) Carbonyls with 1,3,5-Triaza-7-phosphoadamantane. Inorg. Chem. 2018, 57, 6991–7005. [Google Scholar] [CrossRef] [PubMed]
- Battistin, F.; Balducci, G.; Iengo, E.; Demitri, N.; Alessio, E. Neutral 1,3,5-Triaza-7-phosphaadamantane-Ruthenium(II) Complexes as Precursors for the Preparation of Highly Water-Soluble Derivatives. Eur. J. Inorg. Chem. 2016, 2016, 2850–2860. [Google Scholar] [CrossRef] [Green Version]
- Shenderovich, I.G.; Buntkowsky, G.; Schreiber, A.; Gedat, E.; Sharif, S.; Albrecht, J.; Golubev, N.S.; Findenegg, G.H.; Limbach, H.-H. Pyridine-15N—a Mobile NMR Sensor for Surface Acidity and Surface Defects of Mesoporous Silica. J. Phys. Chem. B 2003, 107, 11924–11939. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Mauder, D.; Akcakayiran, D.; Findenegg, G.H.; Shenderovich, I.G. Does Water Affect the Acidity of Surfaces? The Proton-Donating Ability of Silanol and Carboxylic Acid Groups at Mesoporous Silica. ChemPhysChem 2012, 13, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominikowska, J.; Palusiak, M. Tuning Aromaticity of para-Substituted Benzene Derivatives with an External Electric Field. ChemPhysChem 2018, 19, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Effect of an external electric field on the dissociation energy and the electron density properties: The case of the hydrogen bonded dimer HF⋯HF. J. Chem. Phys. 2009, 130, 044104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bene, J.E.; Jordan, M.J.T. To what extent do external fields and vibrational and isotopic effects influence NMR coupling constants across hydrogen bonds? Two-bond Cl-N spin-spin coupling constants (2hJCl-N) in model ClH:NH3 complexes. J. Phys. Chem. A 2002, 106, 5385–5392. [Google Scholar] [CrossRef]
- Bevitt, J.; Chapman, K.; Crittenden, D.; Jordan, M.J.T.; Del Bene, J.E. An ab initio study of anharmonicity and field effects in hydrogen-bonded complexes of the deuterated analogues of HCl and HBr with NH3 and N(CH3)3. J. Phys. Chem. A 2001, 105, 3371–3378. [Google Scholar] [CrossRef]
- Ramos, M.; Alkorta, I.; Elguero, J.; Golubev, N.S.; Denisov, G.S.; Benedict, H.; Limbach, H.-H. Theoretical study of the influence of electric fields on hydrogen-bonded acid−base complexes. J. Phys. Chem. A 1997, 101, 9791–9800. [Google Scholar] [CrossRef]
- Suydam, I.T.; Snow, C.D.; Pande, V.S.; Boxer, S.G. Electric fields at the active site of an enzyme: Direct comparison of experiment with theory. Science 2006, 313, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Sellner, B.; Valiev, M.; Kathmann, S.M. Charge and electric field fluctuations in aqueous NaCl electrolytes. J. Phys. Chem. B 2013, 117, 10869–10882. [Google Scholar] [CrossRef] [PubMed]
- Torii, H. Theoretical analysis and modeling of the electrostatic responses of the vibrational and NMR spectroscopic properties of the cyanide anion. J. Mol. Liq. 2019, 284, 773–779. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501. [Google Scholar] [CrossRef]
- Nardo, V.M.; Cassone, G.; Ponterio, R.C.; Saija, F.; Sponer, J.; Tommasini, M.; Trusso, S. Electric-field-induced effects on the dipole moment and vibrational modes of the centrosymmetric indigo molecule. J. Phys. Chem. A 2020, 124, 10856–10869. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G.; Sponer, J.; Trusso, S.; Saija, F. Ab initio spectroscopy of water under electric fields. Phys. Chem. Chem. Phys. 2019, 21, 21205–21212. [Google Scholar] [CrossRef]
- Chranina, O.V.; Czerniakowski, F.P.; Denisov, G.S. UV-vis electrochromism due to proton transfer. J. Mol. Struct. 1988, 177, 309–315. [Google Scholar] [CrossRef]
- Wang, Z.; Danovich, D.; Ramanan, R.; Shaik, S. Oriented-external electric fields create absolute enantioselectivity in Diels–Alder reactions: Importance of the molecular dipole moment. J. Am. Chem. Soc. 2018, 140, 13350–13359. [Google Scholar] [CrossRef]
- Cassone, G. Nuclear quantum effects largely influence molecular dissociation and proton transfer in liquid water under an electric field. J. Phys. Chem. Lett. 2020, 11, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G.; Sofia, A.; Rinaldi, G.; Sponer, J. Catalyst-free hydrogen synthesis from liquid ethanol: An ab initio molecular dynamics study. J. Phys. Chem. C 2019, 123, 9202–9208. [Google Scholar] [CrossRef] [Green Version]
- Shenderovich, I.G.; Denisov, G.S. Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules 2021, 26, 1283. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. NMR properties of the cyanide anion, a quasisymmetric two-faced hydrogen bonding acceptor. Symmetry 2021, 13, 1298. [Google Scholar] [CrossRef]
- Golubev, N.S.; Detering, C.; Smirnov, S.N.; Shenderovich, I.G.; Denisov, G.S.; Limbach, H.-H.; Tolstoy, P.M. H/D Isotope Effects on NMR Chemical Shifts of Nuclei Involved in a Hydrogen Bridge of Hydrogen Isocyanide Complexes with Fluoride Anion. Phys. Chem. Chem. Phys. 2009, 11, 5154–5159. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Denisov, G.S. A review with comprehensive data on experimental indirect scalar NMR spin–spin coupling constants across hydrogen bonds. Magn. Res. Chem. 2008, 46, 599–624. [Google Scholar] [CrossRef]
- Golubev, N.S.; Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.-H. Nuclear Scalar Spin-Spin Coupling Reveals Novel Properties of Low-Barrier Hydrogen Bonds in a Polar Environment. Chem. Eur. J. 1999, 5, 492–497. [Google Scholar] [CrossRef]
- Dingley, A.J.; Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Gindin, V.A.; Golubev, N.S.; Dunger, A.; Reibke, R.; Kirpekar, S.; Malkina, O.L.; Limbach, H.-H. Nuclear Magnetic Resonance of Hydrogen Bonded Clusters between F⁻ and (HF)n: Experiment and Theory. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 422–428. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Lesnichin, S.B.; Limbach, H.-H.; Shenderovich, I.G. How Short is the Strongest Hydrogen Bond in the Proton-Bound Homodimers of Pyridine Derivatives? J. Phys. Chem. A 2014, 118, 10804–10812. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G.; Limbach, H.-H. Geometrical Features of Hydrogen Bonded Complexes Involving Sterically Hindered Pyridines. J. Phys. Chem. A 2006, 110, 10872–10879. [Google Scholar] [CrossRef] [PubMed]
- Limbach, H.-H.; Tolstoy, P.M.; Pérez-Hernández, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO Hydrogen Bond Geometries and NMR Chemical Shifts: From Equilibrium Structures to Geometric H/D Isotope Effects, with Applications for Water, Protonated Water, and Compressed Ice. Isr. J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Autschbacha, J. Perspective: Relativistic effects. J. Chem. Phys. 2012, 136, 150902. [Google Scholar] [CrossRef] [Green Version]
- Ramabhadran, R.O.; Hua, Y.; Flood, A.H.; Raghavachari, K. C vs. N: Which end of the cyanide anion is a better hydrogen bond acceptor? J. Phys. Chem. A 2014, 118, 7418–7423. [Google Scholar] [CrossRef] [PubMed]
- Millar, L.J.; Ford, T.A. Ab initio investigations of some molecular complexes containing hydrogen cyanide: Hydrogen-bonded or donor–acceptor? J. Mol. Struct. 2005, 744, 195–205. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P=O Moiety as an Ambidextrous Hydrogen Bond Acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide adducts of triarylphosphine oxides. Dalton Trans. 2019, 48, 14312–14325. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Lindhardt, D.; Bhuvanesh, N.; Blümel, J. Di(hydroperoxy)cycloalkanes Stabilized via Hydrogen Bonding by Phosphine Oxides: Safe and Efficient Baeyer−Villiger Oxidants. ACS Sustain. Chem. Eng. 2018, 6, 6829–6840. [Google Scholar] [CrossRef]
- Szatyłowicz, H.; Krygowski, T.M.; Panek, J.J.; Jezierska, A. H-bonded complexes of aniline with HF/F− and anilide with HF in terms of symmetry-adapted perturbation, atoms in molecules, and natural bond orbitals theories. J. Phys. Chem. A 2008, 112, 9895–9905. [Google Scholar] [CrossRef]
- Szatyłowicz, H. Structural aspects of the intermolecular hydrogen bond strength: H-bonded complexes of aniline, phenol and pyridine derivatives. J. Phys. Org. Chem. 2008, 21, 897–914. [Google Scholar] [CrossRef]
- Borisenko, V.E.; Filarovski, A.I. The electrooptical parameters of aniline and its halogen derivatives in hydrogen bonded complexes. J. Mol. Struct. 1989, 196, 353–370. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Deng, W.; Cheeseman, J.R.; Frisch, M.J. Calculation of Nuclear Spin-Spin Coupling Constants of Molecules with First and Second Row Atoms in Study of Basis Set Dependence. J. Chem. Theory Comput. 2006, 2, 1028–1037. [Google Scholar] [CrossRef]
- Jensen, F. The optimum contraction of basis sets for calculating spin–spin coupling constants. Theor. Chem. Acc. 2010, 126, 371–382. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Limbach, H.-H. Solid State NMR for Nonexperts: An Overview of Simple but General Practical Methods. Solids 2021, 2, 9. [Google Scholar] [CrossRef]
- Bryce, D.L.; Bernard, G.M.; Gee, M.; Lumsden, M.D.; Eichele, K.; Wasylishen, R.E. Practical Aspects of Modern Routine Solid-State Multinuclear Magnetic Resonance Spectroscopy: One-Dimensional Experiments. Can. J. Anal. Sci. Spectrosc. 2001, 46, 46–82. [Google Scholar] [CrossRef]
- Duer, M.J. (Ed.) Solid-State NMR Spectroscopy: Principles and Applications; Blackwell Science Ltd.: Oxford, UK, 2002. [Google Scholar]
- Manzoni, V.; Coutinho, K.; Canuto, S. An insightful approach for understanding solvatochromic reversal. Chem. Phys. Lett. 2016, 655–656, 30–34. [Google Scholar] [CrossRef]
- Vidal, Á.V.; Vicente Poutás, L.C.; Faza, O.N.; López, C.S. On the Use of Popular Basis Sets: Impact of the Intramolecular Basis Set Superposition Error. Molecules 2019, 24, 3810. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adduct | 1Structure: wB97XD/def2tzvp NMR: wB97XD/pcJ-2 | Structure: MP2/def2qzvpp NMR: wB97XD/pcJ-3 | ||||
|---|---|---|---|---|---|---|
| r(NH), Å | δiso(15N), ppm | 1J(15N1H), Hz | r(NH), Å | δiso(15N), ppm | 1J(15N1H), Hz | |
| C≡15N-1H | 1.0007 | 0 (277.6) | −121 | 1.0010 | 0 (274.3) | −122 |
| C≡15N-1H⋯Cl− | - | - | - | 1.0528 | 34 | −107 |
| (C≡15N-1H)3⋯F− | 1.0612 | 30 | −103 | 1.0599 | 32 | −105 |
| C≡15N-1H⋯[15N≡C]− | 1.0847 | 50 | −87 | 1.0800 | 44 | −99 |
| (C≡15N-1H)2⋯F− | 1.1086 | 43 | −87 | 1.1086 | 46 | −88 |
| C≡15N-1H⋯FLi | 1.2079 | 62 | −57 | 1.2821 | 76 | −39 |
| C≡15N-1H⋯F− | 1.5202 | 95 | −5 | 1.5208 | 101 | −5 |
| C≡15N-1H⋯[15N≡C]− | 1.4362 | 100 | −9 | 1.5609 | 110 | −4 |
| [15N≡C]− | - | 124 | - | - | 129 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shenderovich, I.G.; Denisov, G.S. Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling. Molecules 2021, 26, 4967. https://doi.org/10.3390/molecules26164967
Shenderovich IG, Denisov GS. Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling. Molecules. 2021; 26(16):4967. https://doi.org/10.3390/molecules26164967
Chicago/Turabian StyleShenderovich, Ilya G., and Gleb S. Denisov. 2021. "Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling" Molecules 26, no. 16: 4967. https://doi.org/10.3390/molecules26164967
APA StyleShenderovich, I. G., & Denisov, G. S. (2021). Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling. Molecules, 26(16), 4967. https://doi.org/10.3390/molecules26164967