
Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors

 ,
,  , , , , , , , ,
, , , , , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Construction of pET3aHisTEVP-zwf Vector and Purification of the Recombinant Protein HpG6PD
2.2. Selection of Inhibitors of HpG6PD of Helicobacter pylori
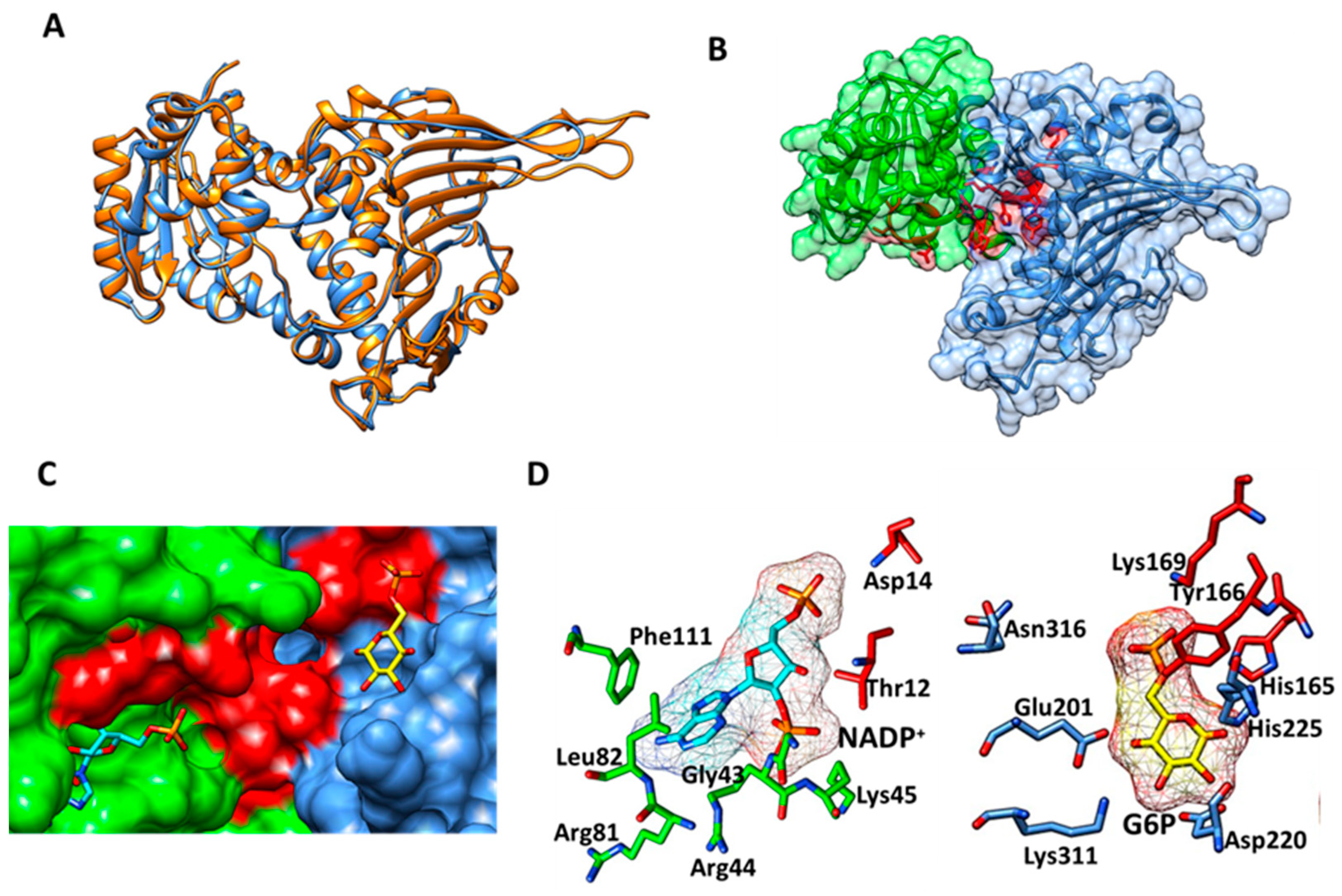
2.3. Homology Modeling of HpG6PD
2.4. Molecular Docking Studies with HpG6PD
2.5. MD Simulation Characterization of HpG6PD-Ligand Complexes
3. Materials and Methods
3.1. pET3aHisTEVP-zwf Vector Construction
3.2. Expression and Purification of the Recombinant G6PD of Helicobacter Pylori
3.3. Selection of Inhibitors of HpG6PD of Helicobacter pylori
3.4. Homology Modeling and Comparison of HpG6PD
3.5. Blind Molecular Docking
3.6. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- McGee, D.J.; May, C.A.; Garner, R.M.; Himpsl, J.M.; Mobley, H.L. Isolation of Helicobacter pylori genes that modulate urease activity. J. Bacteriol. 1999, 181, 2477–2484. [Google Scholar] [CrossRef]
- Sachs, G.; Scott, D.R.; Wen, Y. Gastric infection by Helicobacter pylori. Curr. Gastroenterol. Rep. 2011, 13, 540–546. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Cunha, E.S.; Chen, X.; Sanz-Gaitero, M.; Mills, D.J.; Luecke, H. Cryo-EM structure of Helicobacter pylori urease with an inhibitor in the active site at 2.0 Å resolution. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Treiber, G.; Ammon, S.; Schneider, E.; Klotz, U. Amoxicillin/metronidazole/omeprazole/clarithromycin: A new, short quadruple therapy for Helicobacter pylori eradication. Helicobacter 1998, 3, 54–58. [Google Scholar] [CrossRef]
- Langtry, H.D.; Wilde, M.I. Omeprazole. A review of its use in Helicobacter pylori infection, gastro-oesophageal reflux disease and peptic ulcers induced by nonsteroidal anti-inflammatory drugs. Drugs 1998, 56, 447–486. [Google Scholar] [CrossRef] [PubMed]
- Rimbara, E.; Fischbach, L.A.; Graham, D.Y. Optimal therapy for Helicobacter pylori infections. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 79–88. [Google Scholar] [CrossRef]
- Mucito-Varela, E.; Castillo-Rojas, G.; Cevallos, M.A.; Lozano, L.; Merino, E.; Lopez-Leal, G.; Lopez-Vidal, Y. Complete Genome Sequence of Helicobacter pylori Strain 29CaP Isolated from a Mexican Patient with Gastric Cancer. Genome Announc. 2016, 4, e01512–e01515. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.H.; Covert, M.W.; Famili, I.; Church, G.M.; Edwards, J.S.; Palsson, B.O. Genome-scale metabolic model of Helicobacter pylori 26695. Am. Soc. Microbiol. 2002, 16, 4582–4593. [Google Scholar] [CrossRef]
- Mendz, G.L.; Hazell, S.L. Evidence for a pentose phosphate pathway in Helicobacter pylori. Fems Microbiol. Lett. 1991, 84, 331–336. [Google Scholar] [CrossRef]
- Hoffman, P.S.; Goodwin, A.; Johnsen, J.; Magee, K.; Veldhuyzen van Zanten, S.J. Metabolic activities of metronidazole-sensitive and -resistant strains of Helicobacter pylori: Repression of pyruvate oxidoreductase and expression of isocitrate lyase activity correlate with resistance. J. Bacteriol. 1996, 178, 4822–4829. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morales-Luna, L.; Hernandez-Ochoa, B.; Ramirez-Nava, E.J.; Martinez-Rosas, V.; Ortiz-Ramirez, P.; Fernandez-Rosario, F.; Gonzalez-Valdez, A.; Cardenas-Rodriguez, N.; Serrano-Posada, H.; Centeno-Leija, S.; et al. Characterizing the Fused TvG6PD::6PGL Protein from the Protozoan Trichomonas vaginalis, and Effects of the NADP(+) Molecule on Enzyme Stability. Int. J. Mol. Sci. 2020, 21, 4831. [Google Scholar] [CrossRef]
- Ramirez-Nava, E.J.; Ortega-Cuellar, D.; Gonzalez-Valdez, A.; Castillo-Rodriguez, R.A.; Ponce-Soto, G.Y.; Hernandez-Ochoa, B.; Cardenas-Rodriguez, N.; Martinez-Rosas, V.; Morales-Luna, L.; Serrano-Posada, H.; et al. Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5. Int. J. Mol. Sci. 2019, 20, 5279. [Google Scholar] [CrossRef] [PubMed]
- Rowland, P.; Basak, A.K.; Gover, S.; Levy, H.R.; Adams, M.J. The three–dimensional structure of glucose 6–phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 Å resolution. Structure 1994, 2, 1073–1087. [Google Scholar] [CrossRef]
- Hansen, T.; Schlichting, B.; Schonheit, P. Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: Expression of the g6pd gene and characterization of an extremely thermophilic enzyme. Fems Microbiol. Lett. 2002, 216, 249–253. [Google Scholar] [CrossRef]
- Acero-Navarro, K.E.; Jimenez-Ramirez, M.; Villalobos, M.A.; Vargas-Martinez, R.; Perales-Vela, H.V.; Velasco-Garcia, R. Cloning overexpression and purification of glucose-6-phosphate dehydrogenase of Pseudomonas aeruginosa. Protein Expr. Purif. 2018, 142, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Purification and characterization of glucose-6-phosphate dehydrogenase from camel liver. Enzym. Res. 2014, 714054. [Google Scholar] [CrossRef]
- Adem, S.; Ciftci, M. Purification of rat kidney glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, and glutathione reductase enzymes using 2′,5′-ADP Sepharose 4B affinity in a single chromatography step. Protein Expr. Purif. 2012, 81, 1–4. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Biochemical characterization of buffalo liver glucose-6-phosphate dehydrogenase isoforms. Protein J. 2015, 34, 193–204. [Google Scholar] [CrossRef]
- Preuss, J.; Richardson, A.D.; Pinkerton, A.; Hedrick, M.; Sergienko, E.; Rahlfs, S.; Becker, K.; Bode, L. Identification and characterization of novel human glucose-6-phosphate dehydrogenase inhibitors. J. Biomol. Screen. 2013, 18, 286–297. [Google Scholar] [CrossRef]
- Ramirez-Nava, E.J.; Hernandez-Ochoa, B.; Navarrete-Vazquez, G.; Arreguin-Espinosa, R.; Ortega-Cuellar, D.; Gonzalez-Valdez, A.; Martinez-Rosas, V.; Morales-Luna, L.; Martinez-Miranda, J.; Sierra-Palacios, E.; et al. Novel inhibitors of human glucose-6-phosphate dehydrogenase (HsG6PD) affect the activity and stability of the protein. Biochim. Biophys. Acta Gen. Subj. 2021, 3, 129828. [Google Scholar] [CrossRef]
- Baugh, L.; Phan, I.; Begley, D.W.; Clifton, M.C.; Armour, B.; Dranow, D.M.; Taylor, B.M.; Muruthi, M.M.; Abendroth, J.; Fairman, J.W.; et al. Increasing the structural coverage of tuberculosis drug targets. Tuberc. (Edinb.) 2015, 2, 142–148. [Google Scholar] [CrossRef]
- Ortiz, C.; Botti, H.; Buschiazzo, A.; Comini, M.A. Glucose-6-Phosphate Dehydrogenase from the Human Pathogen Trypanosoma cruzi Evolved Unique Structural Features to Support Efficient Product Formation. J. Mol. Biol. 2019, 11, 2143–2162. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 2.9.1–2.9.37. [Google Scholar] [CrossRef]
- Feig, M. Local Protein Structure Refinement via Molecular Dynamics Simulations with locPREFMD. J. Chem. Inf. Model. 2016, 56, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.; Lam, V.M.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Cryst. D. Biol. Cryst. 2005, 61, 495–504. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Gover, S.; Naylor, C.E.; Vandeputte-Rutten, L.; Adams, M.J.; Levy, H.R. An examination of the role of asp-177 in the His-Asp catalytic dyad of Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: X-ray structure and pH dependence of kinetic parameters of the D177N mutant enzyme. Biochemistry 2000, 39, 15002–15011. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; la Mora-De la Mora, D.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcántara, G.; Oria-Hernández, J. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–675. [Google Scholar] [CrossRef]
- Dominguez-Mendoza, E.A.; Galvan-Cipres, Y.; Martinez-Miranda, J.; Miranda-Gonzalez, C.; Colin-Lozano, B.; Hernandez-Nunez, E.; Hernandez-Bolio, G.I.; Palomino-Hernandez, O.; Navarrete-Vazquez, G. Design Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect. Molecules 2021, 26, 799. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2019. [Google Scholar]
- Williams, T.; Kelley, C.; Bersch, C.; Bröker, H.-B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C. Gnuplot 5.2. An interactive plotting program. Available online: http://www.gnuplot.info/docs_5.2/Gnuplot_5.2.pdf (accessed on 22 July 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Chemical Structure | HpG6PD Inhibition (%) at (400 μM) | HsG6PD Inhibition (%) at (400 μM) | HpG6PD IC50 |
|---|---|---|---|---|
| YGC-1 |  | 84 | 8 | 310 |
| YGC-3 |  | 50 | 0 | 383 |
| YGC-4 |  | 44 | 18 | 500 |
| MGD-1 |  | 50 | 4 | 465 |
| MGD-2 |  | 55 | 2 | 340 |
| TDA-1 |  | 91 | 10 | 204 |
| JMM-3 |  | 67 | 3 | 304 |
| Compound | Interactions | ΔG (kcal/mol) |
|---|---|---|
| G6P | His165, Tyr166, Lys169, Glu201, Asp220, His225, Lys311, Ans316 | |
| NADP+ | Thr12, Asp14, Gly43, Arg44, Lys45, Arg81, Leu82, Phe111 | |
| YGC-1 | Zone 1: Gly10, Thr12, Gly13, Asp14, Arg44, Leu82, Ser106, Ile107, Ser108, Phe111, Glu134. | −7.67 |
| Zone 2: Leu53, Thr54, Cys57, Glu58, Lys59, Leu62, Arg65, Arg69, Phe71, Leu72 | −8.05 | |
| Zone 3: Ile184, Leu185, Leu188, Phe324, Val327, Asn328, Leu341, Leu343, Lys344 | −7.16 | |
| YGC-3 | Zone1: Gly10, Thr12, Gly13, Asp14, Arg44, Leu82, Ser106, Ser108, Pro109, Phe111, Glu134, Lys135, Pro136. | −8.18 |
| Zone 2: Glu51, Leu53, Thr54, Leu55, Leu56, Cys57, Lys59, Leu62, Gly68, Arg69. | −7.87 | |
| Zone 3: Ile184, Ile192, Phe324, Asn325, Val327, Asn328, Leu341, Lys344. | −7.81 | |
| MGD-1 | Zone 1: Gly10, Thr12, Gly13, Asp14, Leu15, Arg44, Val80, Arg81, Leu82, Asp83, Ile107, Ser108, Phe111 | −8.13 |
| Zone 2: Thr54, Cys57, Glu58, Lys59, Leu62, Arg65, Gly68, Arg69, Phe71, Leu72. | −8.20 | |
| Zone 3: Lys170, Asn174, Glu177, Leu178, Asn181, Asn182, Phe332, Thr354, His355, Asn356, Lys357. | ||
| MGD-2 | Zone 1: Gly10, Ala11, Thr12, Gly43, Arg44, Val80, Arg81, Leu82, Ile107, Phe111. | −9.15 |
| Zone 2: Leu53, Thr54, Leu56, Glu58, Lys59, Arg65, Leu72. | −9.07 | |
| Zone 3: Leu178, Leu185, Leu188, Phe324, Val327, Asn328, Leu330, Leu341, Leu343, Lys344 | −8.16 | |
| TDA-1 | Zone 1: Gly10, Thr12, Gly13, Gly43, Arg44, Arg81, Ile107, Ser108, Phe111 | −6.21 |
| Zone 2: Val22, Ser23, Thr25, Glu26, Thr29, Gln61, His63, Gln358, Phe360, Leu361, Arg362. | −7.21 | |
| Zone 3: Leu178, Asn182, Ile184, Ile185, Leu188, Val327, Asn328, Leu341, Leu343. | −6.25 | |
| JMM-3 | Zone 1: Gly10, Thr12, Gly13, Asp14, Leu15, Lys45, Arg81, Leu82, Asp83, Ser106, Ser108, Phe111. | −8.55 |
| Zone 2: Leu53, Thr54, Leu55, Cys57, Glu58, Lys59, Leu62, Arg65, Gly68, Arg69, Leu72 | −8.28 | |
| Zone 3: Asn182, Ile184, Leu188, Val327, Asn328, Leu341, Leu343. Lys344. | −9.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Aguayo-Ortiz, R.; Ortiz-Ramírez, P.; Morales-Luna, L.; Martínez-Rosas, V.; González-Valdez, A.; Gómez-Chávez, F.; Enríquez-Flores, S.; Wong-Baeza, C.; et al. Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors. Molecules 2021, 26, 4955. https://doi.org/10.3390/molecules26164955
Hernández-Ochoa B, Navarrete-Vázquez G, Aguayo-Ortiz R, Ortiz-Ramírez P, Morales-Luna L, Martínez-Rosas V, González-Valdez A, Gómez-Chávez F, Enríquez-Flores S, Wong-Baeza C, et al. Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors. Molecules. 2021; 26(16):4955. https://doi.org/10.3390/molecules26164955
Chicago/Turabian StyleHernández-Ochoa, Beatriz, Gabriel Navarrete-Vázquez, Rodrigo Aguayo-Ortiz, Paulina Ortiz-Ramírez, Laura Morales-Luna, Víctor Martínez-Rosas, Abigail González-Valdez, Fernando Gómez-Chávez, Sergio Enríquez-Flores, Carlos Wong-Baeza, and et al. 2021. "Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors" Molecules 26, no. 16: 4955. https://doi.org/10.3390/molecules26164955
APA StyleHernández-Ochoa, B., Navarrete-Vázquez, G., Aguayo-Ortiz, R., Ortiz-Ramírez, P., Morales-Luna, L., Martínez-Rosas, V., González-Valdez, A., Gómez-Chávez, F., Enríquez-Flores, S., Wong-Baeza, C., Baeza-Ramírez, I., Pérez de la Cruz, V., & Gómez-Manzo, S. (2021). Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors. Molecules, 26(16), 4955. https://doi.org/10.3390/molecules26164955