
Classification of So-Called Non-Covalent Interactions Based on VSEPR Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The σ-hole Bond and π-hole Bond Concept and the Classification of Interactions
3. The Electron Charge Shifts Accompanying Lewis Acid–Lewis Base Interactions
4. Changes of Structures of Interacting Centres
5. Changes of Structures of Interacting Centres Follow the VSEPR Model
6. The Hydrogen Bond and the Halogen Bond Related to VSEPR Model
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaplan, I.G. Intermolecular Interactions: Physical Picture, Computational Methods and Model Potentials; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed Comparison of the Pnicogen Bond with Chalcogen, Halogen, and Hydrogen Bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Forces, N. (Ed.) Steve Scheiner; Springer: Cham, Switzerland; Heidelberg, Germany, 2015. [Google Scholar]
- Novoa, J.J. (Ed.) Intermolecular Interactions in Crystals: Fundamentals of Crystal Engineering; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Dong, W.; Li, Q.; Scheiner, S. Comparative Strengths of Tetrel, Pnicogen, Chalcogen, and Halogen Bonds and Contributing Factors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunitz, J.D. Analysis of the Structure of Organic Molecules; Cornell University Press: New York, NY, USA, 1979. [Google Scholar]
- Bürgi, H.B. Stereochemistry and Reaction Paths as Determined from Crystal Structure Data–A Relationship between Structure and Energy. Angew. Chem. Internat. Ed. 1975, 14, 460–473. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D. From Crystal Statics to chemical dynamics. Acc. Chem. Res. 1983, 16, 153–161. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen Bond and Other Lewis Acid–Lewis Base Interactions as Preliminary Stages of Chemical Reactions. Molecules 2020, 25, 4668. [Google Scholar] [CrossRef] [PubMed]
- Cuma, M.; Scheiner, S.; Kar, T. Effect of adjoining aromatic ring upon excited state proton transfer, o-hydroxybenzaldehyde. J. Mol. Struct. 1999, 467, 37–49. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Krygowski, T.M. The proton transfer path for C=O…H-O systems modelled from crystal structure data. Chem. Phys. Lett. 1999, 305, 247–250. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding, A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Zundel, G. Series of Ten Lectures On: Proton Polarizability of Hydrogen Bonds and Proton Transfer Processes, Their Role in Electrochemistry and Biology; Institut für Physikalische Chemie der Universität München: München, Germany, 1997. [Google Scholar]
- Limbach, H.-H.; Tolstoy, P.M.; Pérez-Hernández, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO Hydrogen Bond Geometries and NMR Chemical Shifts: From Equilibrium Structures to Geometric H/D Isotope Effects, with Applications for Water, Protonated Water, and Compressed Ice. Israel J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Sigalov, M.; Shenderovich, I.G.; Mulloyarova, V.V.; Denisov, G.S.; Tolstoy, P.M. Correlations of NHN hydrogen bond energy with geometry and 1H NMR chemical shift difference of NH protons for aniline complexes. J. Chem. Phys. 2019, 150, 114305. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.S.J.; Colgan, A.C.; Phipps, R.J. Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates. Nat. Chem. 2020, 12, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis pairs: A new strategy to small molecule activation and hydrogenation catalysis. Dalton Trans. 2009, 17, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B-N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Keaton, R.J.; Blacquiere, J.M.; Baker, R.T. Base Metal Catalyzed Dehydrogenation of Ammonia−Borane for Chemical Hydrogen Storage. J. Am. Chem. Soc. 2007, 129, 1844–1845. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, A.; Besora, M.; Harvey, J.N.; Manners, I. Computational Analysis of Amine− Borane Adducts as Potential Hydrogen Storage Materials with Reversible Hydrogen Uptake. Inorg. Chem. 2008, 47, 5910–5918. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Hydrogen Bond and Other Lewis acid–Lewis Base Interactions–Mechanisms of Formation. In Practical Aspects of Computational Chemistry IV; Leszczynski, J., Shukla, M.K., Eds.; Springer Science: New York, NY, USA, 2016; Chapter 9; pp. 245–278. [Google Scholar]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds–mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef] [PubMed]
- Pendas, A.M.; Blanco, M.A.; Francisco, E. The nature of the hydrogen bond: A synthesis from the interacting quantum atoms picture. J. Chem. Phys. 2006, 125, 184112. [Google Scholar] [CrossRef] [PubMed]
- Alcock, N.W. Secondary bonding to nonmetallic elements. Adv. Inorg. Chem. Radiochem. 1972, 15, 1–58. [Google Scholar]
- Crabtree, R.H. Hypervalency, secondary bonding and hydrogen bonding: Siblings under the skin. Chem. Soc. Rev. 2017, 46, 1720–1729. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Hirota, E. Infrared diode laser study of the hydrogen bifluoride anion: FHF− and FDF−. J. Chem. Phys. 1986, 84, 2953–2960. [Google Scholar] [CrossRef]
- Pylaeva, S.A.; Elgabarty, H.; Sebastiani, D.; Tolstoy, P.M. Symmetry and dynamics of FHF− anion in vacuum, in CD2Cl2 and in CCl4. Ab initio MD study of fluctuating solvent–solute hydrogen and halogen bonds. Phys. Chem. Chem. Phys. 2017, 19, 26107–26120. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. [FHF]−-The Strongest Hydrogen Bond under the Influence of External Interactions. Crystals 2016, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7758. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. Can Counter-Intuitive Halogen Bonding Be Coulombic? ChemPhysChem 2021, 22, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Grabowski, S.J. Understanding Hydrogen Bonds, Theoretical and Experimental Views; The Royal Society of Chemistry: Cambridge, UK, 2021. [Google Scholar]
- Kollman, P.A.; Liebman, J.F.; Allen, L.C. The Lithium Bond. J. Am. Chem. Soc. 1970, 92, 1142–1150. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Hill, J.A.S.S. A theoretical study of hydrogen- and lithium-bonded complexes of F–H/Li and Cl–H/Li with NF3, NH3, and NH2(CH3). J. Chem. Phys. 2011, 135, 164303. [Google Scholar] [CrossRef] [PubMed]
- Lipkowski, P.; Grabowski, S.J. Could the lithium bond be classified as the σ-hole bond?–QTAIM and NBO analysis. Chem. Phys. Lett. 2014, 591, 113–118. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zordan, F.; Brammer, L.; Sherwood, P. Supramolecular Chemistry of Halogens: Complementary Features of Inorganic (M-X) and Organic (C-X´) Halogens Applied to M-X···X´-C halogen Bond Formation. J. Am. Chem. Soc. 2005, 127, 5979–5989. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. The π-hole revisited. Phys. Chem. Chem. Phys. 2021, 23, 16458–16468. [Google Scholar] [CrossRef]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical Investigations on Heteronuclear Chalcogen—Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical investigations on chalcogen− chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef]
- Sanz, P.; Yañez, M.; Mó, O. Competition between X···H···Y Intramolecular Hydrogen Bonds and X····Y (X = O, S, and Y = Se, Te) Chalcogen−Chalcogen Interactions. J. Phys. Chem. A 2002, 106, 4661–4668. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Alikhani, E.; Fuster, F.; Madebene, B.; Grabowski, S.J. Topological reaction sites–very strong chalcogen bonds. Phys. Chem. Chem. Phys. 2014, 16, 2430–2442. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1, 2-dicarba-closo-dodecaboranes with short P⋯ P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Bauer, S.; Tschirschwitz, S.; Lönnecke, P.; Franck, R.; Kirchner, B.; Clark, M.L.; Hey-Hawkins, E. Enantiomerically pure bis (phosphanyl) carbaborane (12) compounds. Eur. J. Inorg. Chem. 2009, 2776–2788. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, Energies, Bonding, and NMR Properties of Pnicogen Complexes H2XP:NXH2 (X ═ H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Can two trivalent N atoms engage in a direct N⋯ N noncovalent interaction? Chem. Phys. Lett. 2011, 514, 32–35. [Google Scholar] [CrossRef]
- Scheiner, S.; Wysokiński, R.; Michalczyk, M.; Zierkiewicz, W. Pnicogen Bonds Pairing Anionic Lewis Acid With Neutral and Anionic Bases. J. Phys. Chem. A 2020, 124, 4998–5006. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Fleurat-Lessard, P.; Hristov, I.; Ziegler, T. Free Energy Profiles for the Identity SN2 Reactions Cl− + CH3Cl and NH3 + H3BNH3: A Constraint Ab Initio Molecular Dynamics Study. J. Phys. Chem. A 2004, 108, 9461–9468. [Google Scholar] [CrossRef]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole Strengths and Interactions of F3MX Molecules (M = C, Si, Ge and X = F., Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bond-σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Comparison between Tetrel Bonded Complexes Stabilized by σ and π Hole Interactions. Molecules 2018, 23, 1416. [Google Scholar] [CrossRef] [Green Version]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force. Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-Centered Halogen Bonding: A Factor in Recognition Phenomena and Reactivity. Cryst. Growth Des. 2011, 11, 4238–4246. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. A computational analysis of the bonding in boron trifluoride and boron trichloride and their complexes with ammonia. Inorg. Chem. 1993, 32, 2622–2625. [Google Scholar] [CrossRef]
- Bessac, F.; Frenking, G. Why Is BCl3 a Stronger Lewis Acid with Respect to String Bases than BF3? Inorg. Chem. 2003, 42, 7990–7994. [Google Scholar] [CrossRef] [PubMed]
- Jonas, V.; Frenking, G.; Reetz, M.T. Comparative Theoretical Study of Lewis Acid-Base Complexes of BH3, BF3, BCl3, AlCl3, and SO2. J. Am. Chem. Soc. 1994, 116, 8741–8753. [Google Scholar] [CrossRef]
- Van der Veken, B.J.; Sluyts, E.J. Reversed Lewis Acidity of Mixed Boron Halides: An Infrared Study of the Van der Waals Complexes of BFxClywith CH3F in Cryosolution. J. Am. Chem. Soc. 1997, 119, 11516–11522. [Google Scholar] [CrossRef]
- Fau, S.; Frenking, G. Theoretical investigation of the weakly bonded donor—acceptor complexes X3B—H2, X3B—C2H4, and X3B—C2H2 (X= H, F, Cl). Mol. Phys. 1999, 96, 519–527. [Google Scholar]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Grabowski, S.J. Boron and other triel Lewis acid centers: From hypovalency to hypervalency. Chem. Phys. Chem. 2014, 15, 2985–2993. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bond and coordination of triel centres–Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Montero-Campillo, M.M.; Sanz, P.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Alkaline-earth (Be, Mg and Ca) bonds at the origin of huge acidity enhancements. Phys. Chem. Chem. Phys. 2018, 20, 2413–2420. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Mani, D.; Arunan, E. The X–C⋯ Y (X= O/F, Y= O/S/F/Cl/Br/N/P)’carbon bond’and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. F-Halogen Bond: Conditions for Its Existence. J. Phys. Chem. A 2020, 124, 7290–7299. [Google Scholar] [CrossRef]
- Avramopoulos, A.; Papadopoulos, M.G.; Sadlej, A.J. Strong interactions through the X⋯ Au–Y bridge: The Au bond? Chem. Phys. Lett. 2003, 370, 765–769. [Google Scholar] [CrossRef]
- Li, H.; Li, Q.; Li, R.; Li, W.; Cheng, J. Prediction and characterization of HCCH⋅⋅⋅AuX (X = OH, F, Cl, Br, CH3, CCH, CN, and NC) complexes: A π Au-bond. J. Chem. Phys. 2011, 135, 074304. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Ugalde, J.M.; Andrada, D.M.; Frenking, G. Comparison of Hydrogen and Gold Bonding in [XHX]−, [XAuX]−, and Isoelectronic [NgHNg]+, [NgAuNg]+ (X = Halogen, Ng = Noble Gas). Chem. Eur. J. 2016, 22, 11317–11327. [Google Scholar] [CrossRef]
- Yanez, M.; Sanz, P.; Mo, O.; Alkorta, I.; Elguero, J. Beryllium Bonds, Do They Exist? J. Chem Theory Comput. 2009, 5, 2763–2771. [Google Scholar] [CrossRef]
- Eskandari, K. Characteristics of beryllium bonds; a QTAIM study. J. Mol. Model. 2012, 18, 3481–3487. [Google Scholar] [CrossRef]
- Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Modulating the Strength of Hydrogen Bonds through Beryllium Bonds. J. Chem. Theory Comput. 2012, 8, 2293–2300. [Google Scholar] [CrossRef]
- Yang, X.; Li, Q.; Cheng, J.; Li, W. A new interaction mechanism of LiNH2 with MgH2: Magnesium bond. J. Mol. Model. 2013, 19, 247–253. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Maynard, S.J. A computational study of model hydrogen-, halogen-, beryllium-and magnesium-bonded complexes of aziridine derivatives. Mol. Phys. 2016, 114, 1609–1618. [Google Scholar] [CrossRef]
- Li, S.-Y.; Wu, D.; Li, Y.; Yu, D.; Liu, J.-Y.; Li, Z.-R. Insight into structural and π–magnesium bonding characteristics of the X2Mg⋯Y (X = H, F.; Y = C2H2, C2H4 and C6H6) complexes. RSC Advances 2016, 6, 102754–102761. [Google Scholar] [CrossRef]
- Tama, R.; Mó, O.; Yáñez, M.; Montero-Campillo, M.M. Characterizing magnesium bonds: Main features of a non-covalent interaction. Theor. Chem. Acc. 2017, 136, 36. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A.; Leszczynski, J. Hydride bonding–Ab initio studies of BeH2…Li+, BeH2…Na+ and BeH2…Mg2+ model systems. Chem. Phys. Lett. 2006, 422, 334–339. [Google Scholar] [CrossRef]
- Richardson, T.B.; de Gala, S.; Crabtree, R.H. Unconventional Hydrogen Bonds: Intermolecular B-H…H-N Interactions. J. Am. Chem. Soc. 1995, 117, 12875–12876. [Google Scholar] [CrossRef]
- Klooster, W.T.; Koetzle, T.F.; Siegbahn, P.E.M.; Richardson, T.B.; Crabtree, R.H. Study of the N−H···H−B Dihydrogen Bond Including the Crystal Structure of BH3NH3 by Neutron Diffraction. J. Am. Chem. Soc. 1999, 121, 6337–6343. [Google Scholar] [CrossRef]
- Epstein, L.M.; Shubina, E.S. New types of hydrogen bonding in organometallic chemistry. Coord. Chem. Rev. 2002, 231, 165–181. [Google Scholar] [CrossRef]
- Bakhmutov, V.I. Dihydrogen Bonds, Principles, Experiments, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Custelcean, R.; Jackson, J.E. Topochemical Control of Covalent Bond Formation by Dihydrogen Bonding. J. Am. Chem. Soc. 1998, 120, 12935–12941. [Google Scholar] [CrossRef]
- Custelcean, R.; Jackson, J.E. Topochemical Dihydrogen to Covalent Bonding Transformation in LiBH4·TEA: A Mechanistic Study. J. Am. Chem. Soc. 2000, 122, 5251–5257. [Google Scholar] [CrossRef]
- Marincean, S.; Jackson, J.E. Quest for IR-pumped reactions in dihydrogen-bonded complexes. J. Phys. Chem. A 2004, 108, 5521–5526. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ruipérez, F. Dihydrogen bond interactions as a result of H2 cleavage at Cu, Ag and Au centres. Phys. Chem. Chem. Phys. 2016, 18, 12810–12818. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The Nature of Halogen∙∙∙Halogen Interactions: Are Short Halogen Contacts Due to Specific Attractive Forces or Due to Close Packing of Nonspherical Atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Formigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef]
- Reed, E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C. Valency and Bonding, a Natural Bond Orbital Donor—Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Gillespie, R.J.; Hargittai, I. The VSEPR Model of Molecular Geometry; Allyn & Bacon: Boston, MA, USA, 1991. reprinted: Gillespie, R.J.; Hargittai, I. The VSEPR Model of Molecular Geometry; Dover Publications, Inc.: Mineola, NY, USA, 2012. [Google Scholar]
- Chen, M.M.L.; Hoffmann, R. Sulfuranes. Theoretical aspects of bonding, substituent site preferences, and geometrical distortions. J. Am. Chem. Soc. 1976, 98, 1647–1653. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Silvi, B. The octet rule and hypervalence: Two misunderstood concepts. Coord. Chem. Rev. 2002, 233-234, 53–62. [Google Scholar] [CrossRef]
- Noury, S.; Silvi, B.; Gillespie, R.J. Chemical Bonding in Hypervalent Molecules: Is the Octet Rule Relevant? Inorg. Chem. 2002, 41, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic Basis of Improper Hydrogen Bonding: A Subtle Balance of Hyperconjugation and Rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef]
- Grabowski, S.J. Halogen Bond and Its Counterparts: Bent’s Rule Explains the Formation of Nonbonding Interactions. J. Phys. Chem. A 2011, 115, 12340–12347. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3, and CNCH3 as σ Donors and π Acceptors. A Theoretical Study by the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Velde, G.T.E.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds and other interactions as a response to protect doublet/octet electron structure. J. Mol. Model. 2018, 24, 38. [Google Scholar] [CrossRef]
- Grabowski, S.J. σ-Hole Bond Versus Hydrogen Bond: From Tetravalent to Pentavalent, N., P and As Atoms. Chem. Eur. J. 2013, 19, 14600–14611. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, J.J.; Grabowski, S.J.; Roszak, S.; Leszczynski, J. H…σ interactions–an ab initio and ´atoms in molecules´ study. Chem. Phys. Lett. 2004, 393, 81–86. [Google Scholar] [CrossRef]
- Grabowski, S.J. Clusters of Ammonium Cation—Hydrogen Bond versus σ-Hole Bond. ChemPhysChem 2014, 15, 876–884. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Matta, C.; Boyd, R.J. (Eds.) Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Arnold, W.D.; Oldfield, E. The Chemical Nature of Hydrogen Bonding in Proteins via NMR: J-Couplings, Chemical Shifts, and AIM Theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 1154–11161. [Google Scholar] [CrossRef]
- Martin, J.C. “Frozen” Transition States: Pentavalent Carbon et al. Science 1983, 221, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Coordination of Be and Mg Centres by HCN Ligands–Be…N and Mg…N Interactions. ChemPhysChem 2018, 19, 1830–1840. [Google Scholar]
- Grabowski, S.J. Halogen bond with the multivalent halogen acting as the Lewis acid center. Chem. Phys. Lett. 2014, 605–606, 131–136. [Google Scholar] [CrossRef]
- Clauss, A.D.; Nelsen, S.F.; Ayoub, M.; Moore, J.W.; Landis, C.R.; Weinhold, F. Rabbit-ears hybrids, VSEPR sterics, and other orbital anachronisms. Chem. Educ. Res. Pract. 2014, 15, 417–434. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ugalde, J.M. High-level ab initio calculations on low barrier hydrogen bonds and proton bound homodimers. Chem. Phys. Lett. 2010, 493, 37–44. [Google Scholar] [CrossRef]
- Parthasarathy, R. Crystal structure of glycylglycine hydrochloride. Acta Cryst. 1969, B25, 509–518. [Google Scholar] [CrossRef]
- Taylor, R.; Kennard, O.; Versichel, W. Geometry of the N-H∙∙∙O=C Hydrogen Bond. 2. Three-Center (“Bifurcated”) and Four-Center (“Trifurcated”) Bonds. J. Am. Chem. Soc. 1984, 106, 244–248. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Hoffmann, R. Stabilizing H3−: Or Are We Stabilizing a Proton? ChemPhysChem 2012, 13, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Schelemper, E.O.; Britton, D. The Crystal Structure of p-Iodobenzonitrile. Acta Cryst. 1965, 18, 419–424. [Google Scholar] [CrossRef]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. IUCrJ 2017, 4, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J.; Casanova, D.; Formoso, E.; Ugalde, J.M. Tetravalent Oxygen and Sulphur Centres Mediated by Carborane Superacid: Theoretical Analysis. ChemPhysChem 2019, 20, 2443–2450. [Google Scholar] [CrossRef]


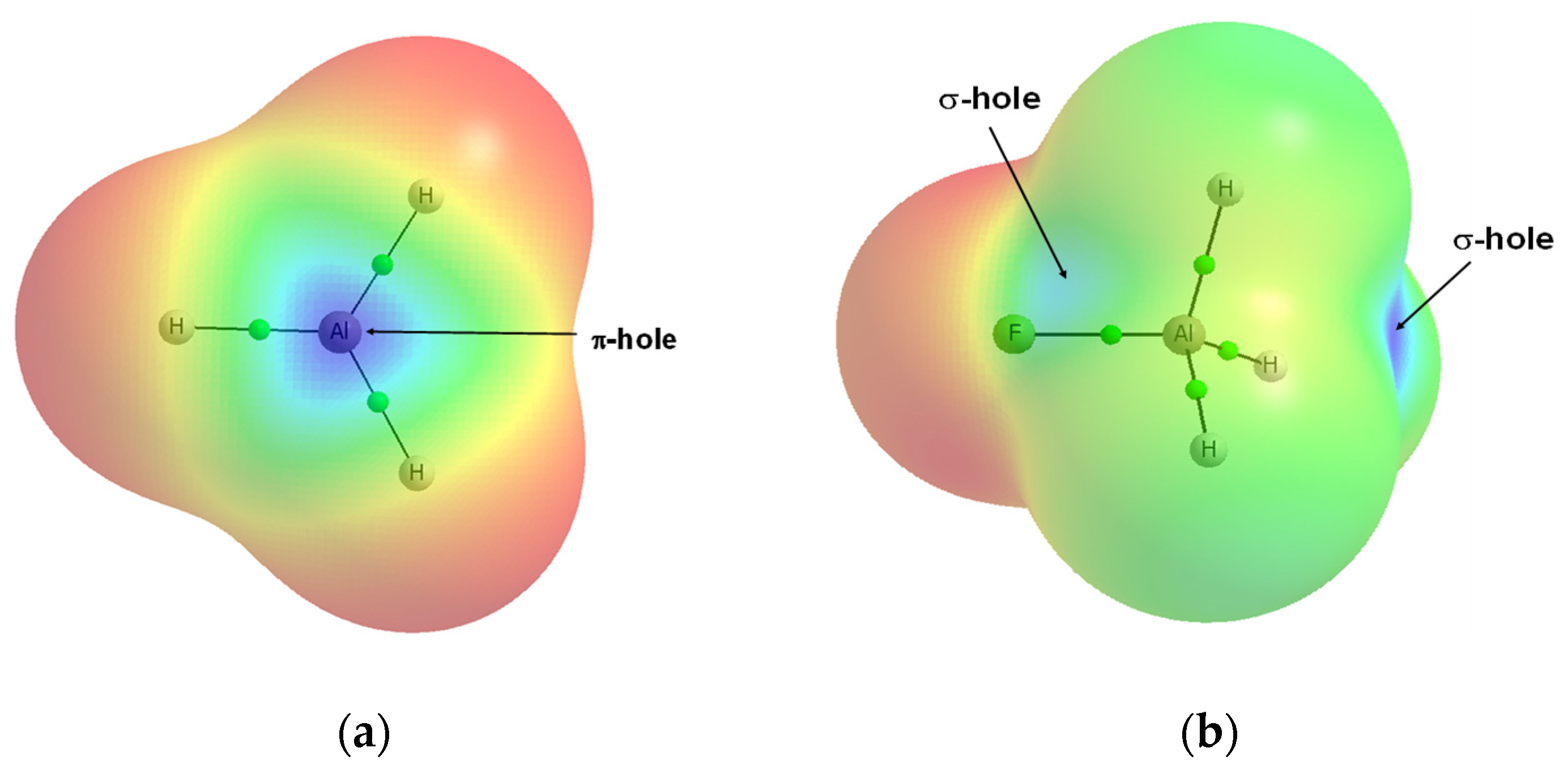
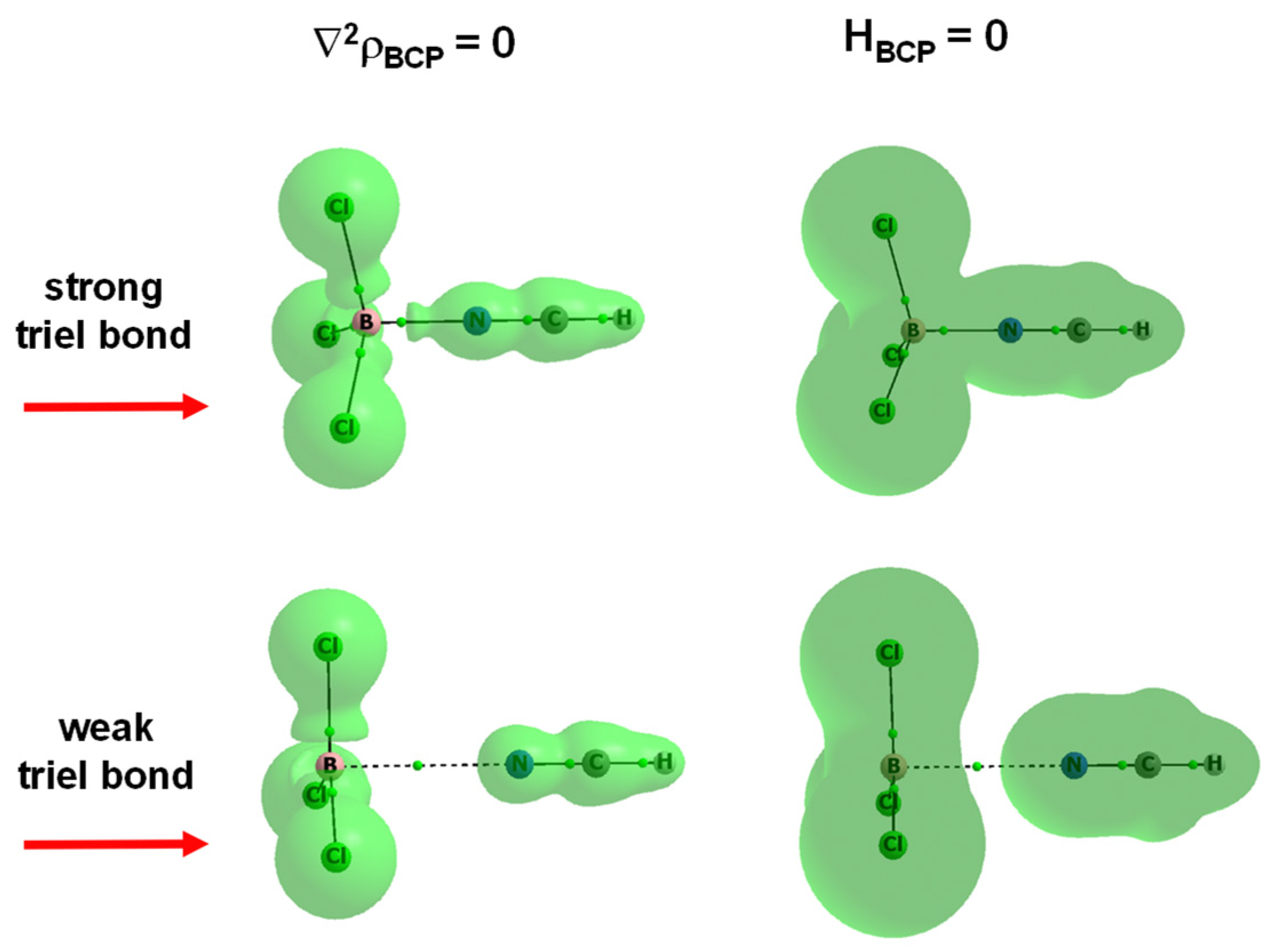
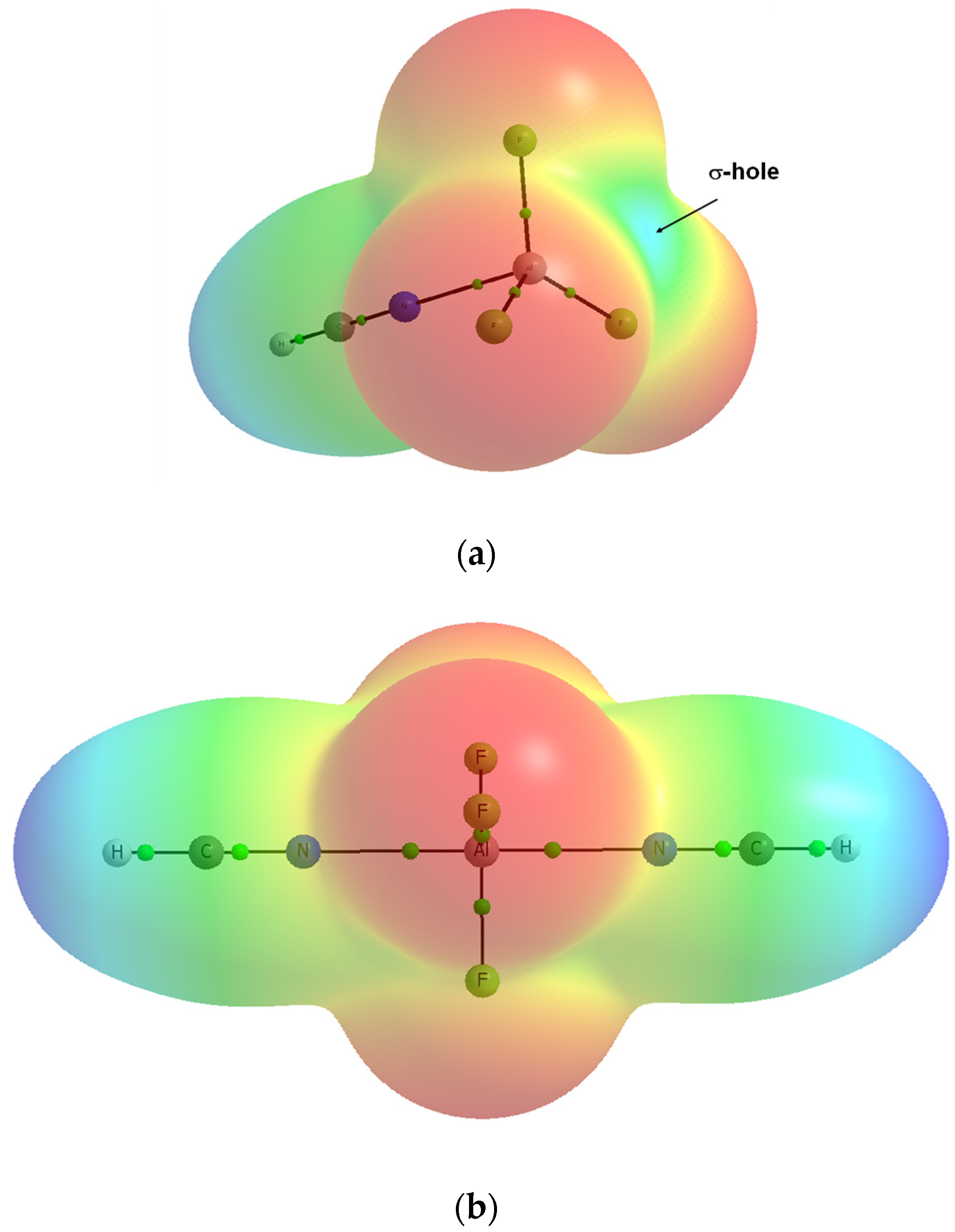
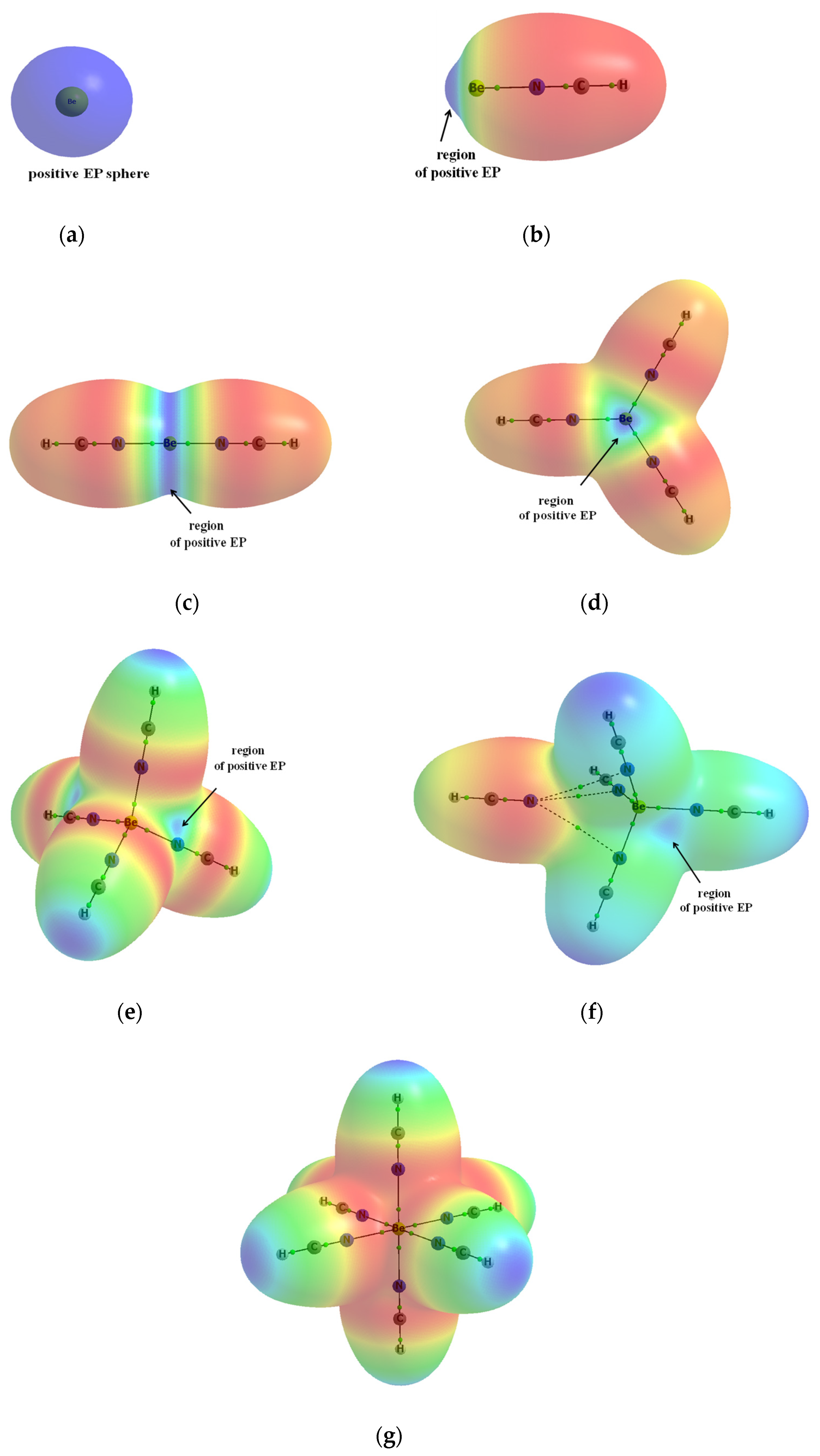


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, S.J. Classification of So-Called Non-Covalent Interactions Based on VSEPR Model. Molecules 2021, 26, 4939. https://doi.org/10.3390/molecules26164939
Grabowski SJ. Classification of So-Called Non-Covalent Interactions Based on VSEPR Model. Molecules. 2021; 26(16):4939. https://doi.org/10.3390/molecules26164939
Chicago/Turabian StyleGrabowski, Sławomir J. 2021. "Classification of So-Called Non-Covalent Interactions Based on VSEPR Model" Molecules 26, no. 16: 4939. https://doi.org/10.3390/molecules26164939
APA StyleGrabowski, S. J. (2021). Classification of So-Called Non-Covalent Interactions Based on VSEPR Model. Molecules, 26(16), 4939. https://doi.org/10.3390/molecules26164939