
Halogens in Acetophenones Direct the Hydrogen Bond Docking Preference of Phenol via Stacking Interactions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Methods
2.2. Computational Methods
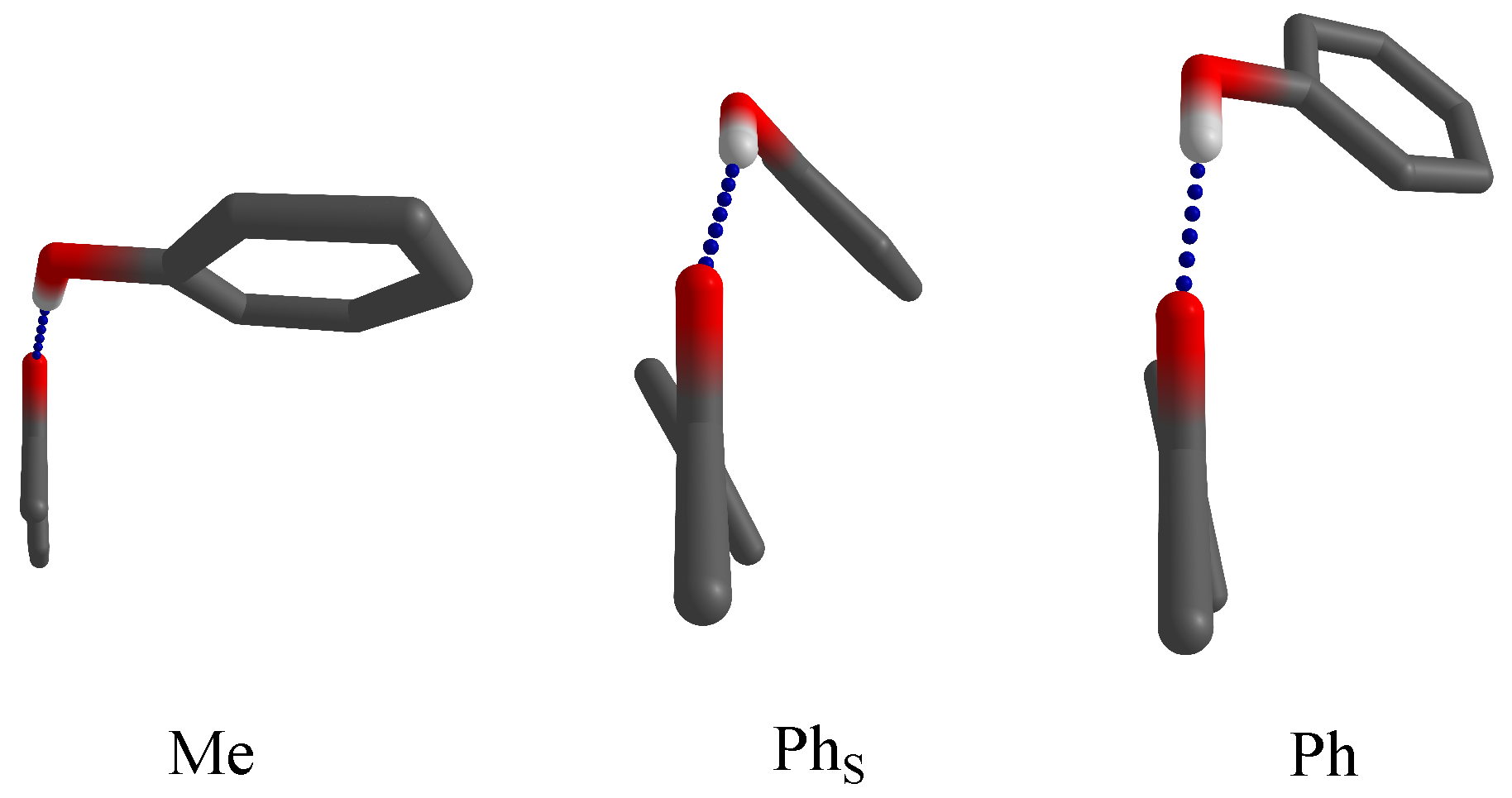
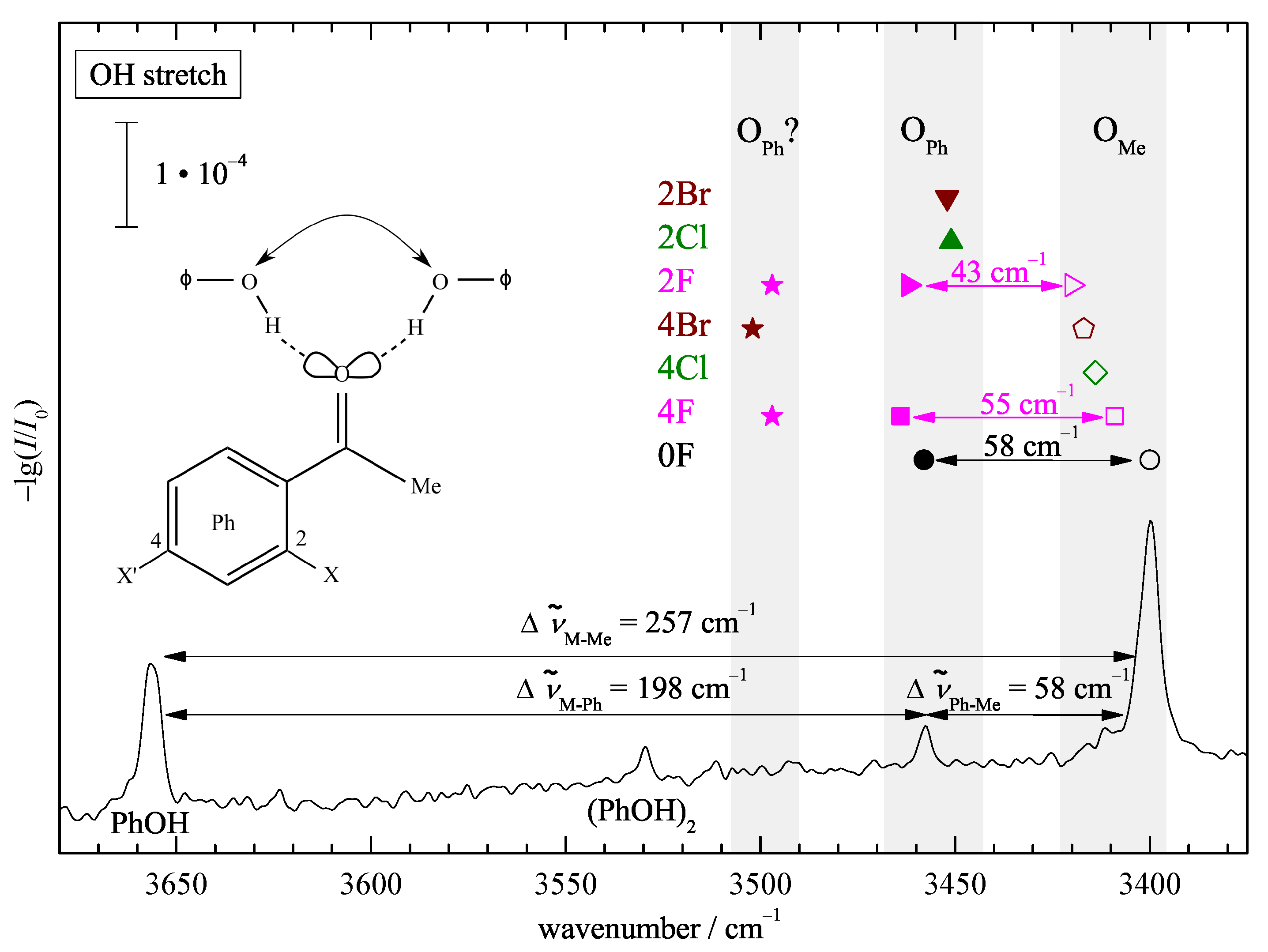
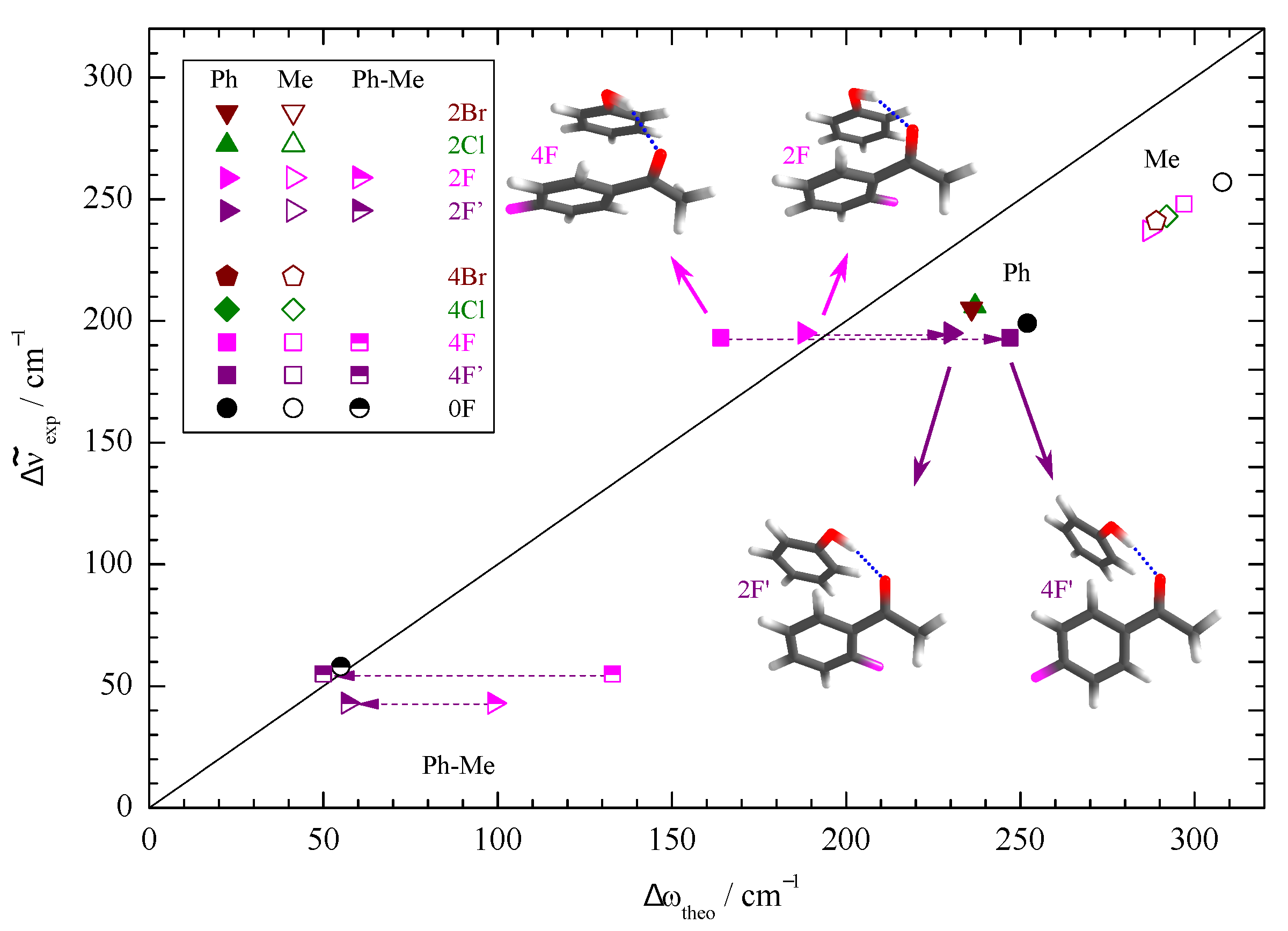
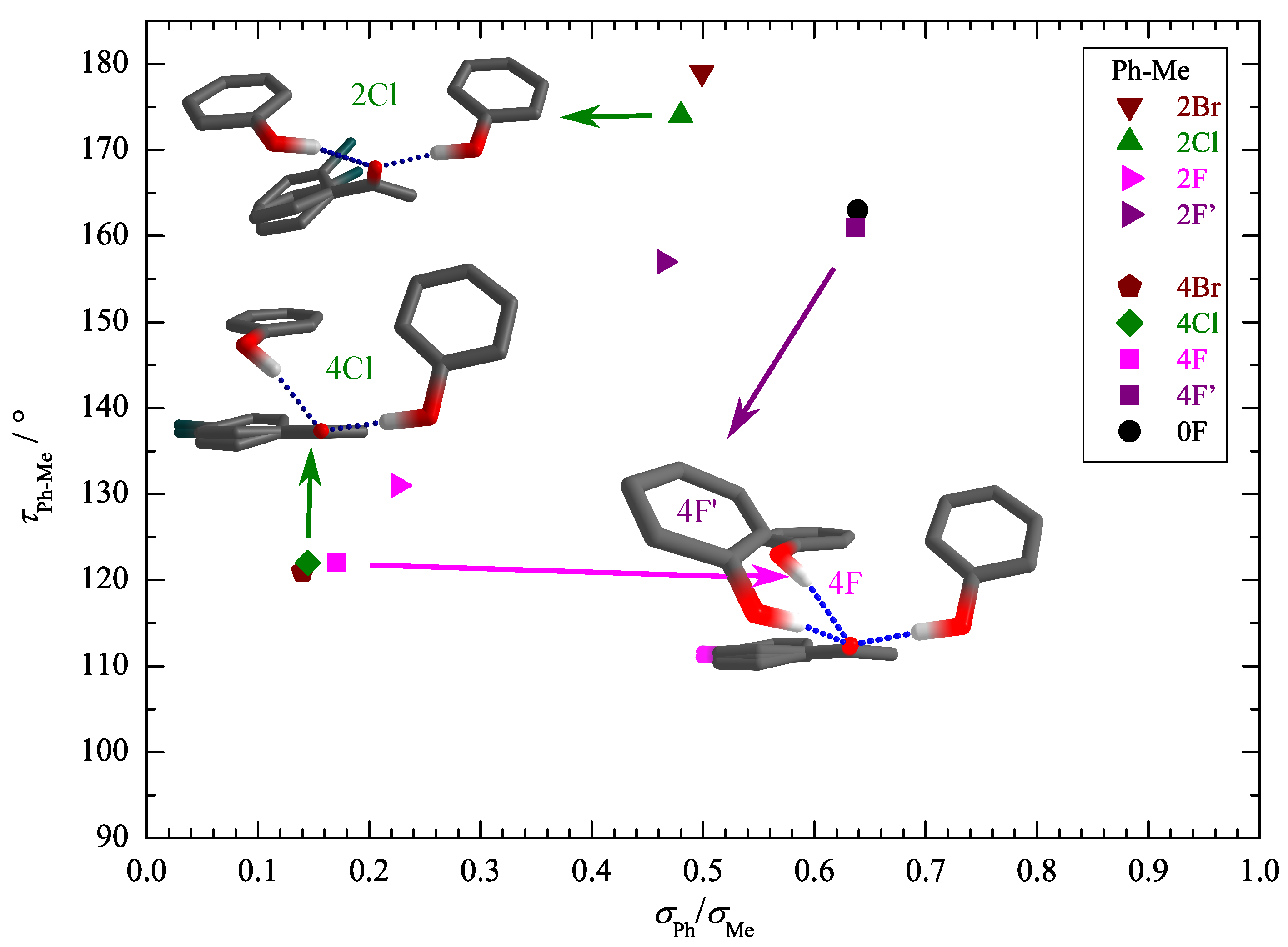
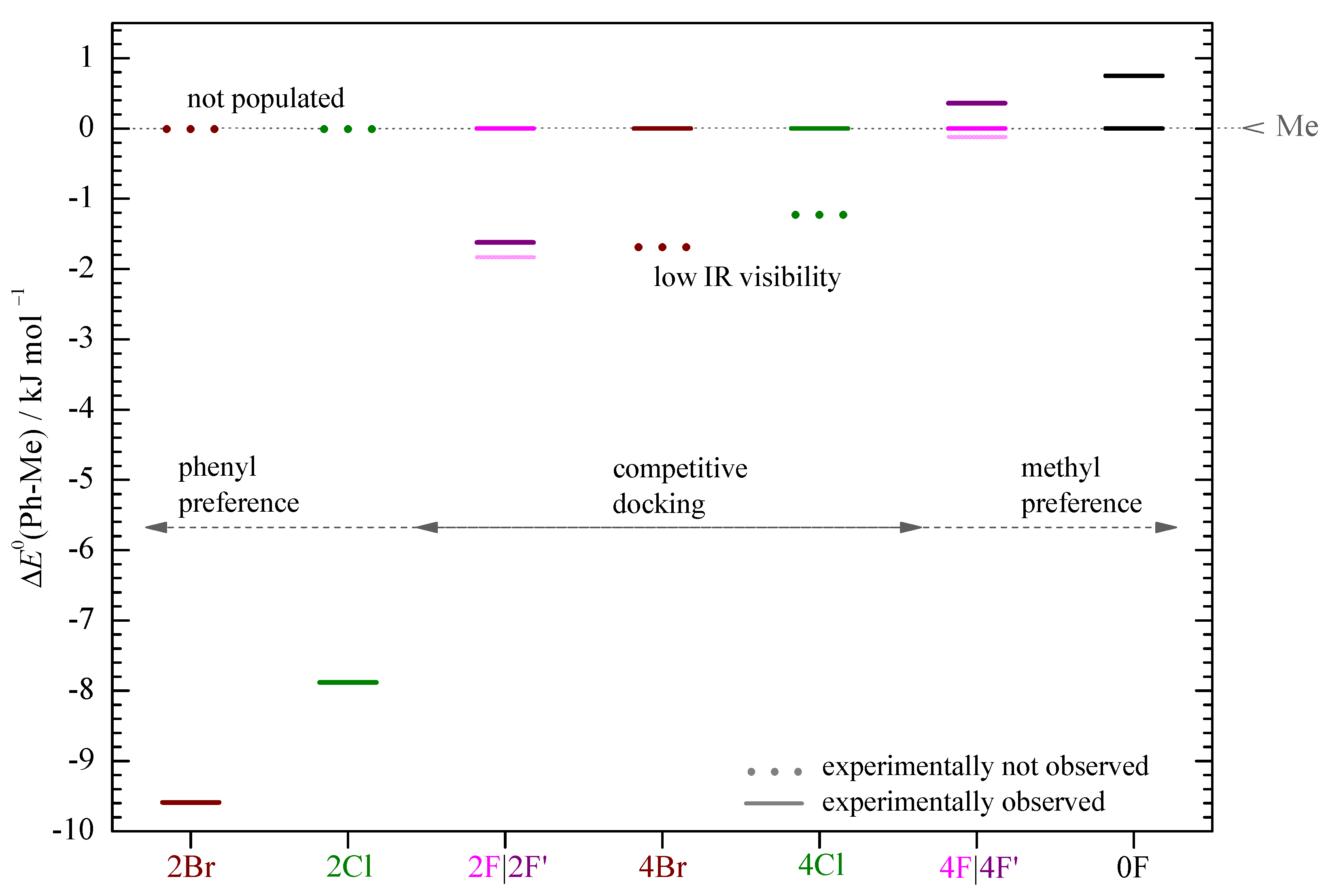
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| B3LYP | Becke 3-parameter Lee–Yang–Parr density functional |
| D3 | Third-generation Grimme parametrization of density functional dispersion correction |
| BJ | Becke and Johnson damping function for small interatomic distances |
| ZPVE | Harmonic zero-point vibrational energy |
| 2Br | 2-Bromo-acetophenone |
| 4Br | 4-Bromo-acetophenone |
| 2Cl | 2-Chloro-acetophenone |
| 4Cl | 4-Chloro-acetophenone |
| 0F | Acetophenone |
| 2F | 2-Fluoro-acetophenone |
| 4F | 4-Fluoro-acetophenone |
| PhOH | Phenol |
References
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-Stacking Interactions: Alive and Well in Proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141, 11027–11035. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, A.N.; Wheeler, S.E. Tuning Stacking Interactions between Asp–Arg Salt Bridges and Heterocyclic Drug Fragments. J. Chem. Inf. Model. 2019, 59, 149–158. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A.; Lawson, K.R.; Perkins, J.; Urch, C.J. Electrostatic Control of Aromatic Stacking Interactions. J. Am. Chem. Soc. 2005, 127, 8594–8595. [Google Scholar] [CrossRef]
- Šponer, J.; Riley, K.E.; Hobza, P. Nature and magnitude of aromatic stacking of nucleic acid bases. Phys. Chem. Chem. Phys. 2008, 10, 2595–2610. [Google Scholar] [CrossRef]
- Grimme, S. Do Special Noncovalent π–π Stacking Interactions Really Exist? Angew. Chem. Int. Ed. 2008, 47, 3430–3434. [Google Scholar] [CrossRef]
- Frey, J.A.; Holzer, C.; Klopper, W.; Leutwyler, S. Experimental and Theoretical Determination of Dissociation Energies of Dispersion-Dominated Aromatic Molecular Complexes. Chem. Rev. 2016, 116, 5614–5641. [Google Scholar] [CrossRef] [Green Version]
- Poblotzki, A.; Gottschalk, H.C.; Suhm, M.A. Tipping the Scales: Spectroscopic Tools for Intermolecular Energy Balances. J. Phys. Chem. Lett. 2017, 8, 5656–5665. [Google Scholar] [CrossRef]
- Zimmermann, C.; Gottschalk, H.C.; Suhm, M.A. Three-dimensional docking of alcohols to ketones: An experimental benchmark based on acetophenone solvation energy balances. Phys. Chem. Chem. Phys. 2020, 22, 2870–2877. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, C.; Fischer, T.L.; Suhm, M.A. Pinacolone-Alcohol Gas-Phase Solvation Balances as Experimental Dispersion Benchmarks. Molecules 2020, 25, 5095. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, G.C.; Charles, S.W. Infrared spectral perturbations in matrix experiments. Pure Appl. Chem. 1963, 7, 111–124. [Google Scholar] [CrossRef]
- Thijs, R.; Zeegers-Huyskens, T. Infrared and Raman studies of hydrogen bonded complexes involving acetone, acetophenone and benzophenone—I. Thermodynamic constants and frequency shifts of the ΔνOH and ΔνC=O stretching vibrations. Spectrochim. Acta Part A Mol. Spectrosc. 1984, 40, 307–313. [Google Scholar] [CrossRef]
- Gottschalk, H.C.; Altnöder, J.; Heger, M.; Suhm, M.A. Control over the Hydrogen-Bond Docking Site in Anisole by Ring Methylation. Angew. Chem. Int. Ed. 2016, 55, 1921–1924. [Google Scholar] [CrossRef]
- Lei, J.; Zhang, J.; Feng, G.; Grabow, J.U.; Gou, Q. Conformational preference determined by inequivalent n-pairs: Rotational studies on acetophenone and its monohydrate. Phys. Chem. Chem. Phys. 2019, 21, 22888–22894. [Google Scholar] [CrossRef]
- Fischer, T.L.; Wagner, T.; Gottschalk, H.C.; Nejad, A.; Suhm, M.A. A Rather Universal Vibrational Resonance in 1:1 Hydrates of Carbonyl Compounds. J. Phys. Chem. Lett. 2021, 12, 138–144. [Google Scholar] [CrossRef]
- Suhm, M.A.; Kollipost, F. Femtisecond single-mole infrared spectroscopy of molecular clusters. Phys. Chem. Chem. Phys. 2013, 15, 10702–10721. [Google Scholar] [CrossRef]
- Altnöder, J.; Lee, J.J.; Otto, K.E.; Suhm, M.A. Molecular Recognition in Glycolaldehyde, the Simplest Sugar: Two Isolated Hydrogen Bonds Win Over One Cooperative Pair. ChemistryOpen 2012, 1, 269–275. [Google Scholar] [CrossRef]
- Hartwig, B.; Lange, M.; Poblotzki, A.; Medel, R.; Zehnacker, A.; Suhm, M.A. The reduced cohesion of homoconfigurational 1,2-diols. Phys. Chem. Chem. Phys. 2020, 22, 1122–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karir, G.; Lüttschwager, N.O.B.; Suhm, M.A. Phenylacetylene as a gas phase sliding balance for solvating alcohols. Phys. Chem. Chem. Phys. 2019, 21, 7831–7840. [Google Scholar] [CrossRef] [Green Version]
- Lüttschwager, N.O.B. NoisySignalIntegration.jl: A Julia Package to Determine Uncertainty in Numeric Integrals of Noisy x-y Data. GitHub.com. Available online: https://github.com/nluetts/NoisySignalIntegration.jl (accessed on 8 August 2021).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree–Fock model of intermolecular interactions. J. Chem. Phys. 2005, 123, 024101. [Google Scholar] [CrossRef]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Accounts 2011, 128, 295–305. [Google Scholar] [CrossRef]
- TURBOMOLE V7.3 2018, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 8 August 2021).
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. WIREs Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Ebata, T.; Watanabe, T.; Mikami, N. Evidence for the Cyclic Form of Phenol Trimer: Vibrational Spectroscopy of the OH Stretching Vibrations of Jet-Cooled Phenol Dimer and Trimer. J. Phys. Chem. 1995, 99, 5761–5764. [Google Scholar] [CrossRef]
- Ebata, T.; Fujii, A.; Mikami, N. Vibrational spectroscopy of small-sized hydrogen-bonded clusters and their ions. Int. Rev. Phys. Chem. 1998, 17, 331–361. [Google Scholar] [CrossRef]
- Gottschalk, H.C.; Fischer, T.L.; Meyer, V.; Hildebrandt, R.; Schmitt, U.; Suhm, M.A. A Sustainable Slit Jet FTIR Spectrometer for Hydrate Complexes and Beyond. Instruments 2021, 5, 12. [Google Scholar] [CrossRef]
- Heger, M.; Suhm, M.A.; Mata, R.A. Communication: Towards the binding energy and vibrational red shift of the simplest organic hydrogen bond: Harmonic constraints for methanol dimer. J. Chem. Phys. 2014, 141, 101105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medel, R.; Suhm, M.A. Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: Different correction patterns for density functional and wave-function-based methods. Phys. Chem. Chem. Phys. 2021, 23, 5629–5643. [Google Scholar] [CrossRef]
- Jedwabny, W.; Dyguda-Kazimierowicz, E.; Pernal, K.; Szalewicz, K.; Patkowski, K. Extension of an Atom–Atom Dispersion Function to Halogen Bonds and Its Use for Rational Design of Drugs and Biocatalysts. J. Phys. Chem. 2021, 125, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.B.; Bistoni, G.; Sparta, M.; Saitow, M.; Riplinger, C.; Auer, A.A.; Neese, F. Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework. J. Chem. Theory Comput. 2016, 12, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Dishman, A.F.; Volkman, B.F. Unfolding the Mysteries of Protein Metamorphosis. ACS Chem. Biol. 2018, 13, 1438–1446. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ketone | Me | Ph | Ph | Phenol Preference |
|---|---|---|---|---|
| 0F | 81–89 | 19–11 | Me | |
| 2F | 29–52 | <59 | (<50) | Me, Ph |
| 2Cl | (<11) | >89 | Ph | |
| 2Br | (<12) | >88 | Ph | |
| 4F | 40–81 | <30 | (<52) | Me, Ph |
| 4Cl | >38 | (<62) | Me, Ph | |
| 4Br | >28 | (<72) | Me, Ph |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, C.; Lange, M.; Suhm, M.A. Halogens in Acetophenones Direct the Hydrogen Bond Docking Preference of Phenol via Stacking Interactions. Molecules 2021, 26, 4883. https://doi.org/10.3390/molecules26164883
Zimmermann C, Lange M, Suhm MA. Halogens in Acetophenones Direct the Hydrogen Bond Docking Preference of Phenol via Stacking Interactions. Molecules. 2021; 26(16):4883. https://doi.org/10.3390/molecules26164883
Chicago/Turabian StyleZimmermann, Charlotte, Manuel Lange, and Martin A. Suhm. 2021. "Halogens in Acetophenones Direct the Hydrogen Bond Docking Preference of Phenol via Stacking Interactions" Molecules 26, no. 16: 4883. https://doi.org/10.3390/molecules26164883
APA StyleZimmermann, C., Lange, M., & Suhm, M. A. (2021). Halogens in Acetophenones Direct the Hydrogen Bond Docking Preference of Phenol via Stacking Interactions. Molecules, 26(16), 4883. https://doi.org/10.3390/molecules26164883