
In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands


 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
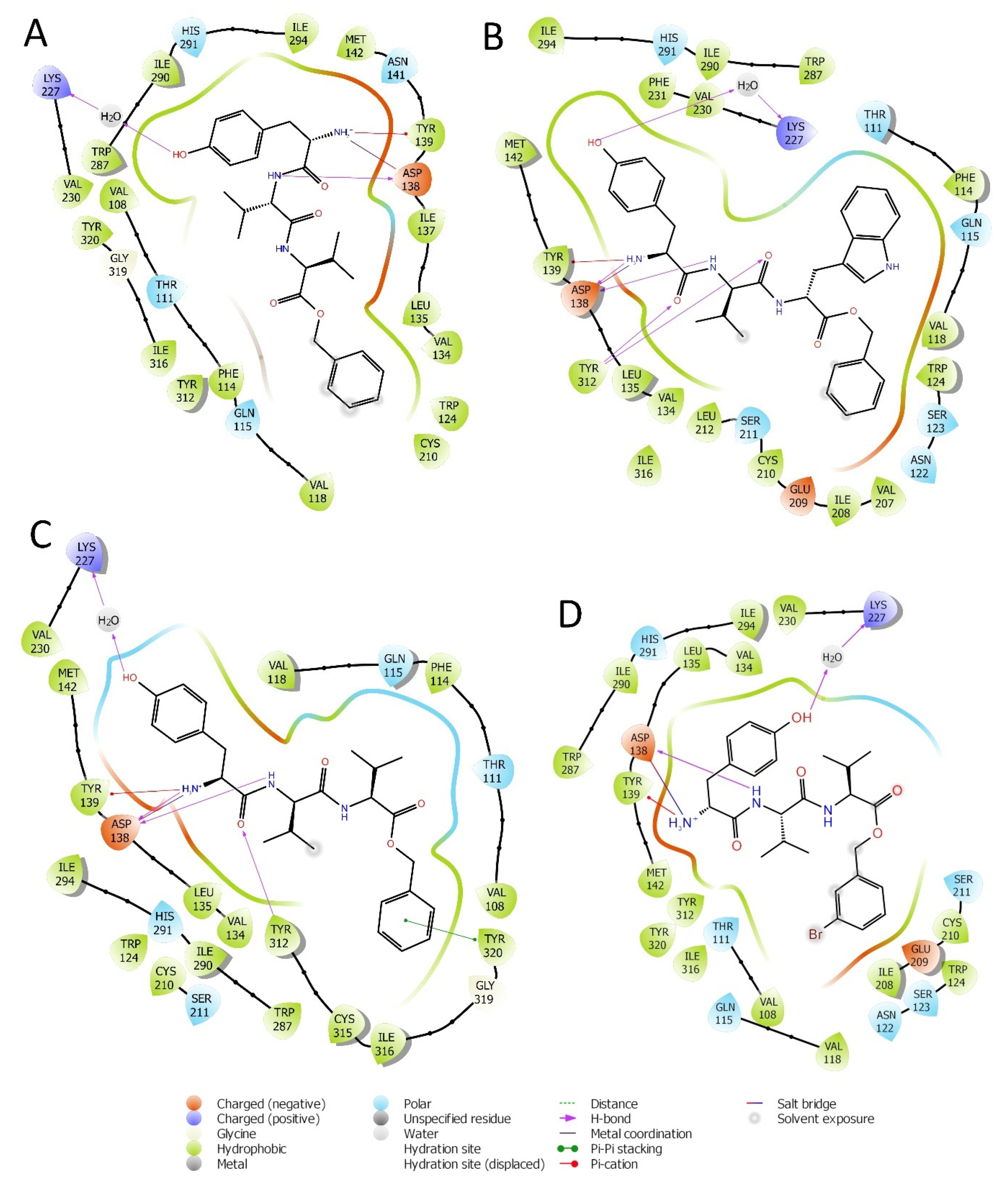
2.1. Structure Based Design
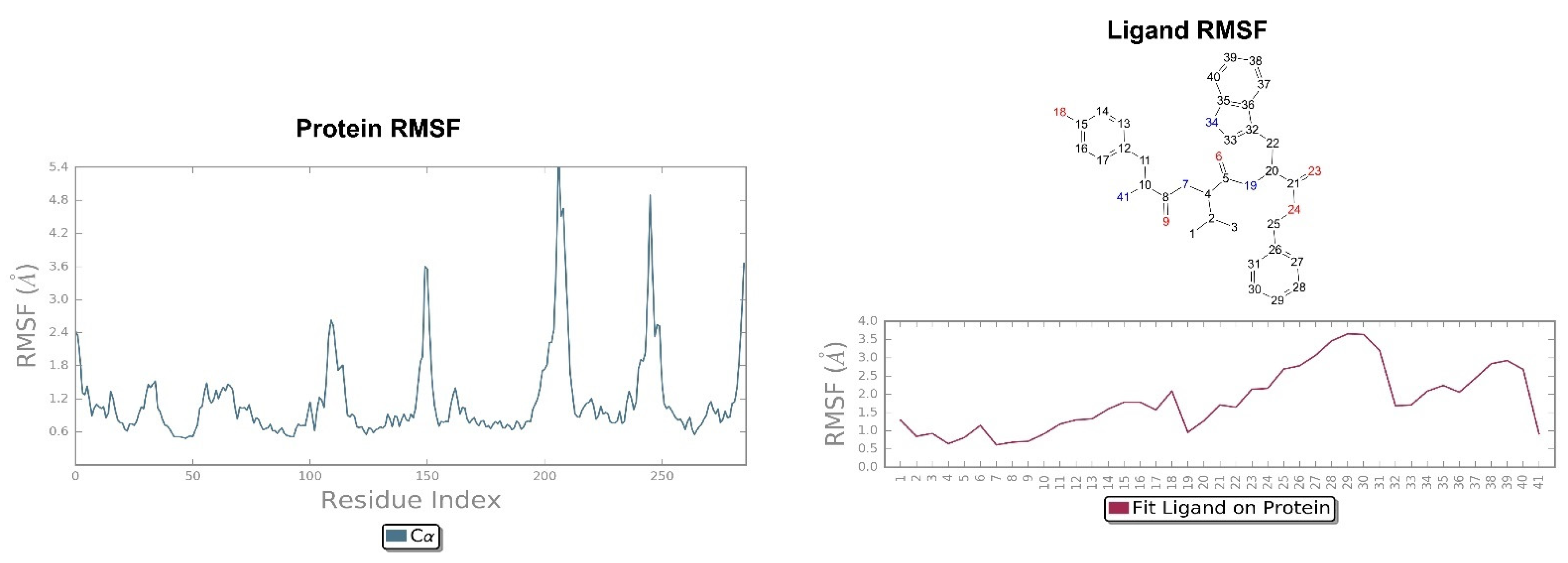
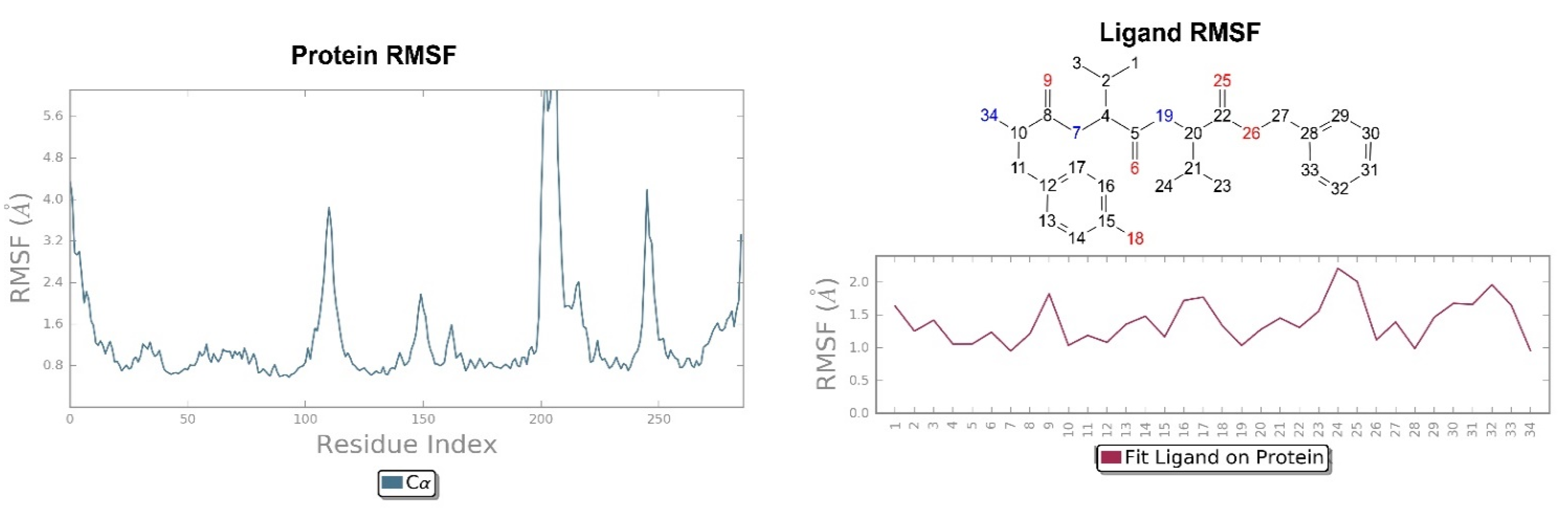
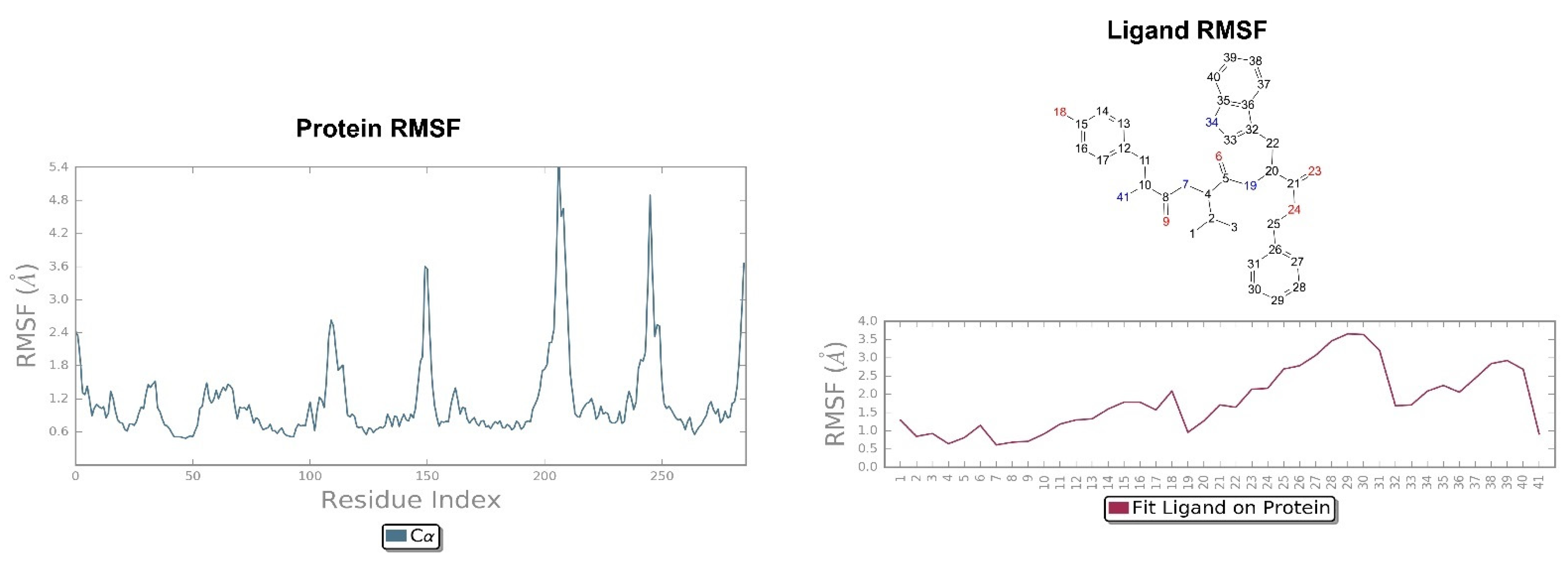
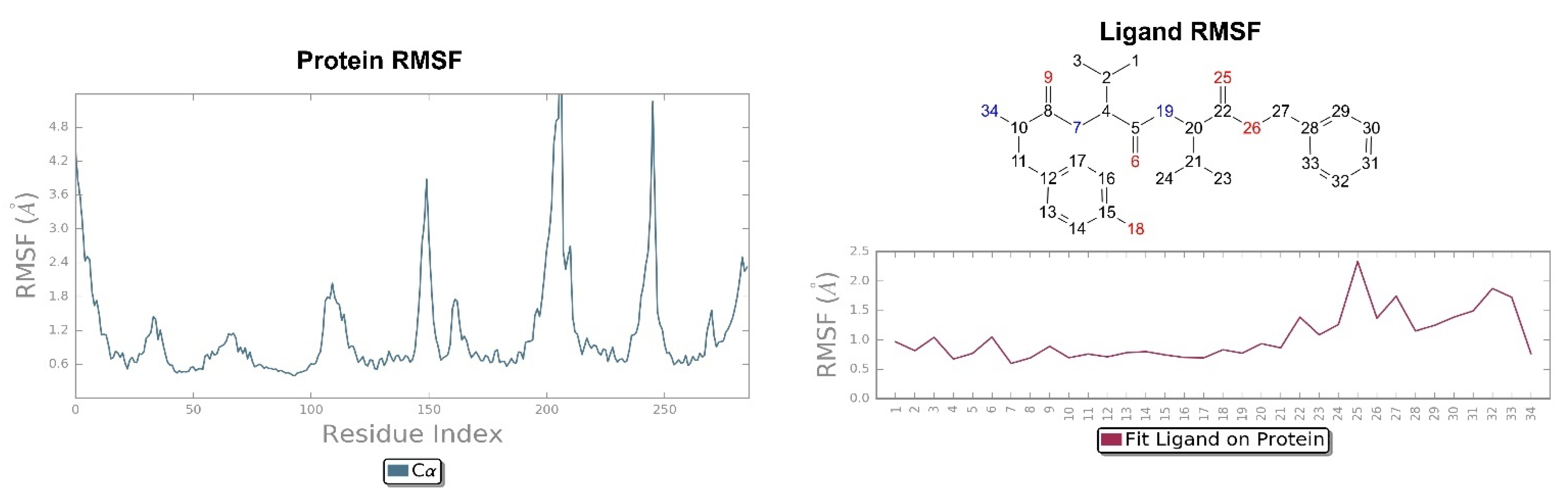
2.2. Molecular Dynamics Simulation
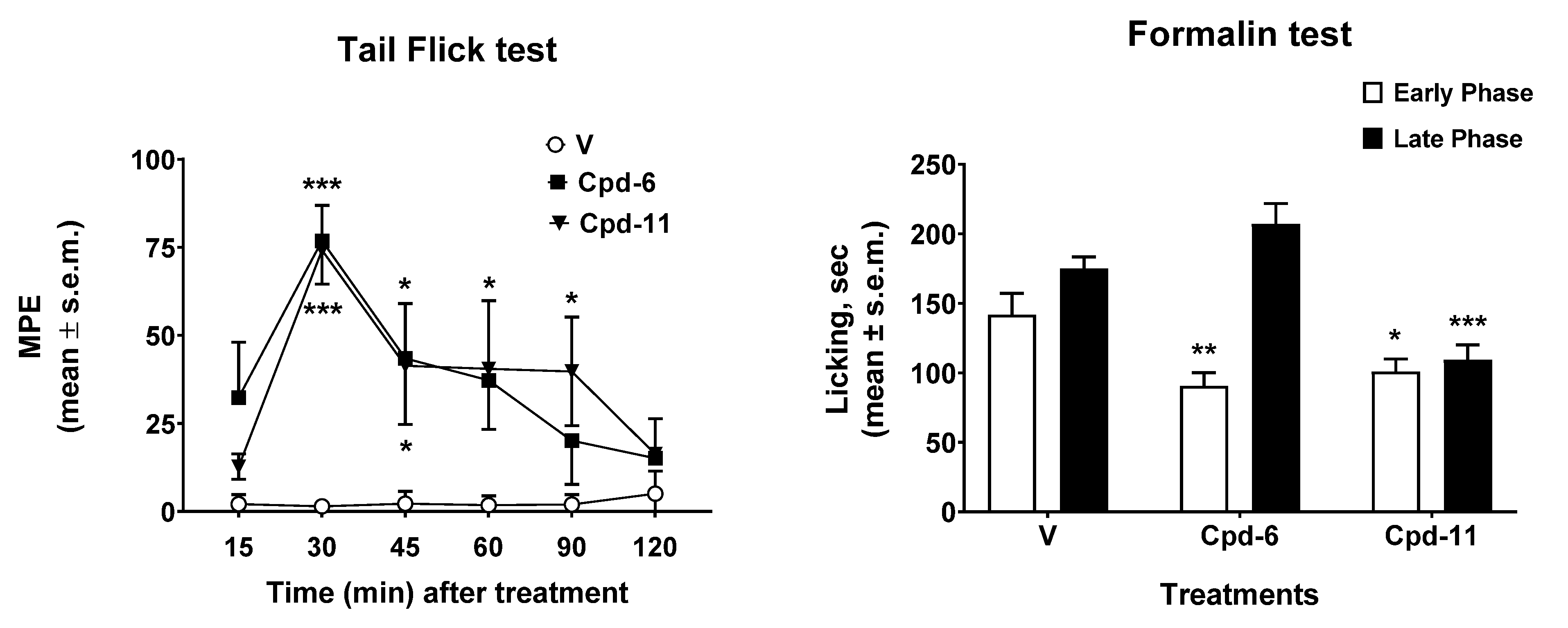
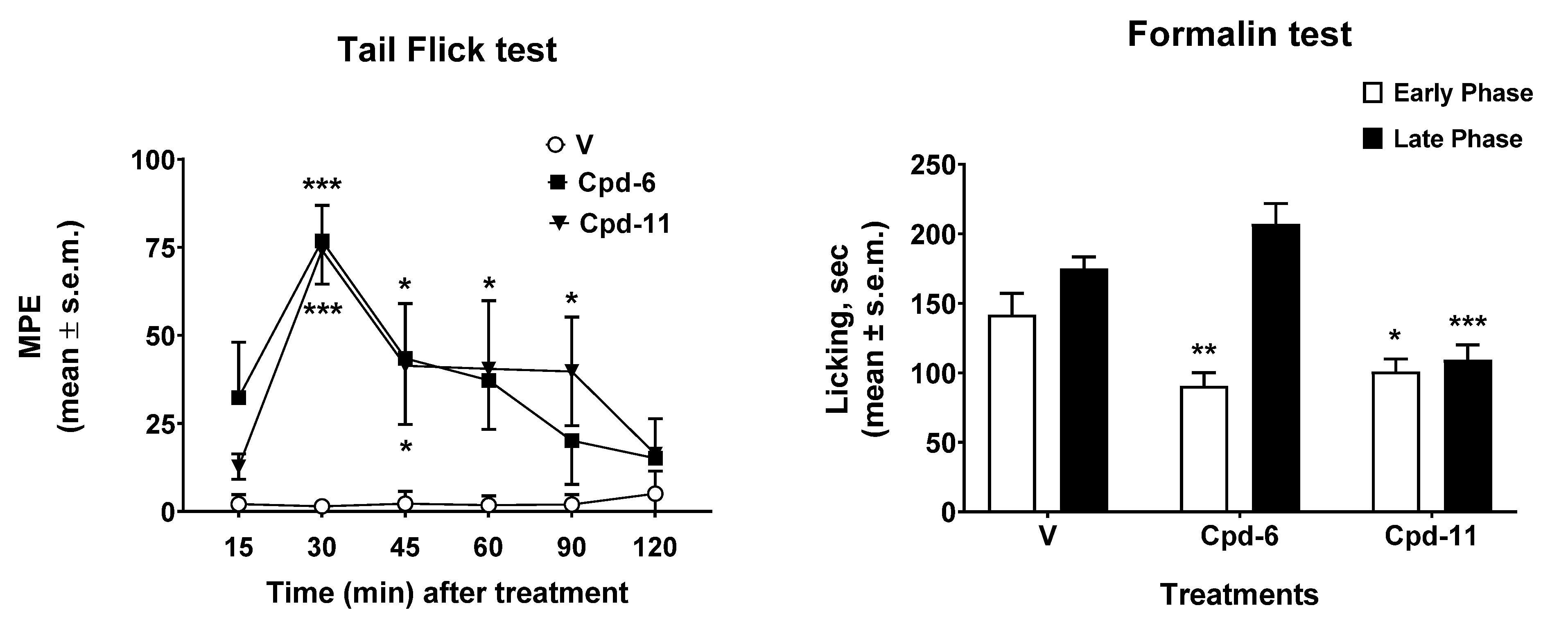
2.3. Antinociceptive Effect In Vivo
2.4. In Silico ADME and Drug-Likeness Evaluation
3. Methods and Materials
3.1. Library Preparation
3.2. Protein Preparation
3.3. Receptor Grid Generation
3.4. Glide Docking of the Co-Crystallized Ligand
3.5. Virtual Screening Workflow
3.6. Molecular Dynamics
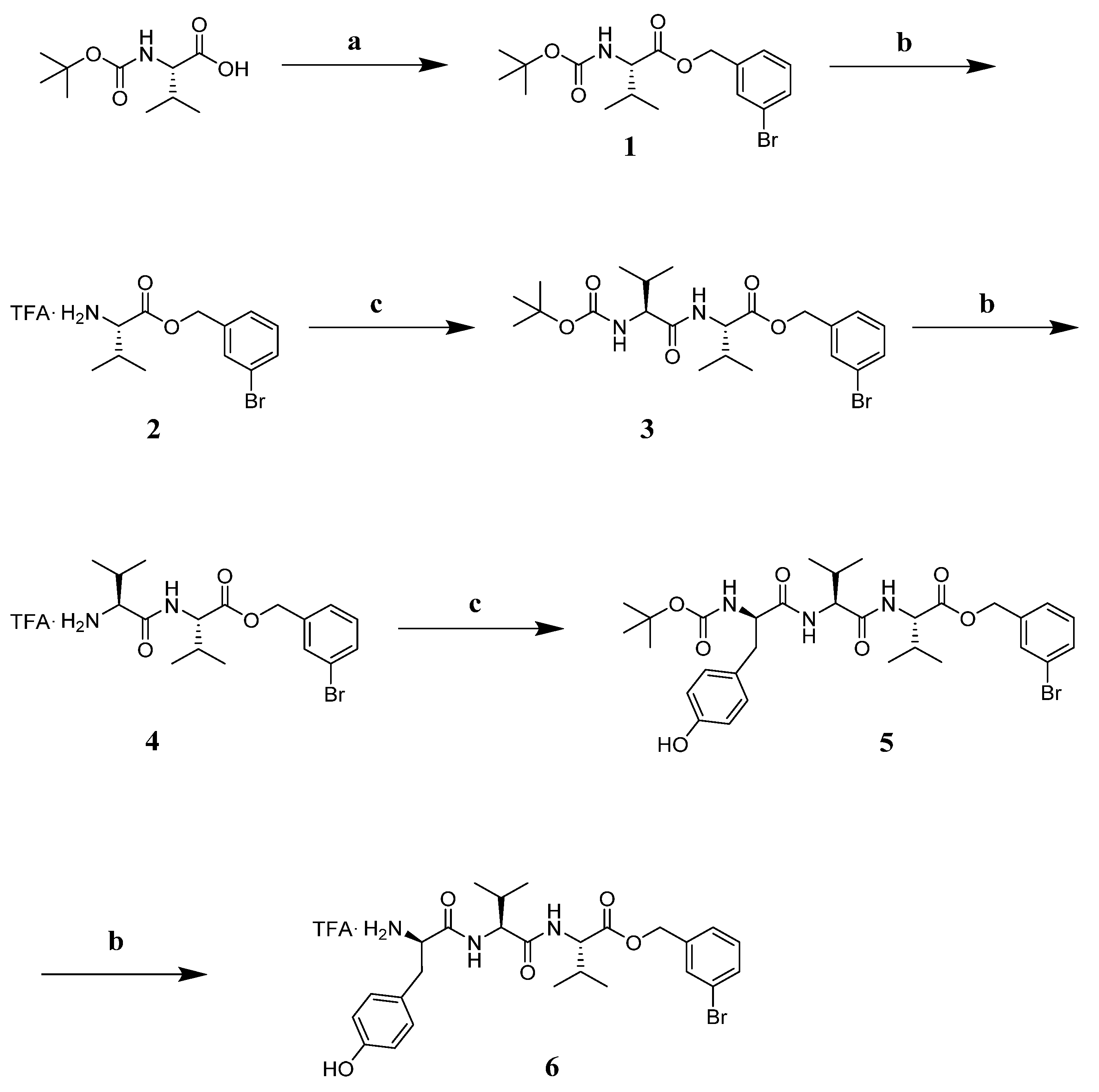
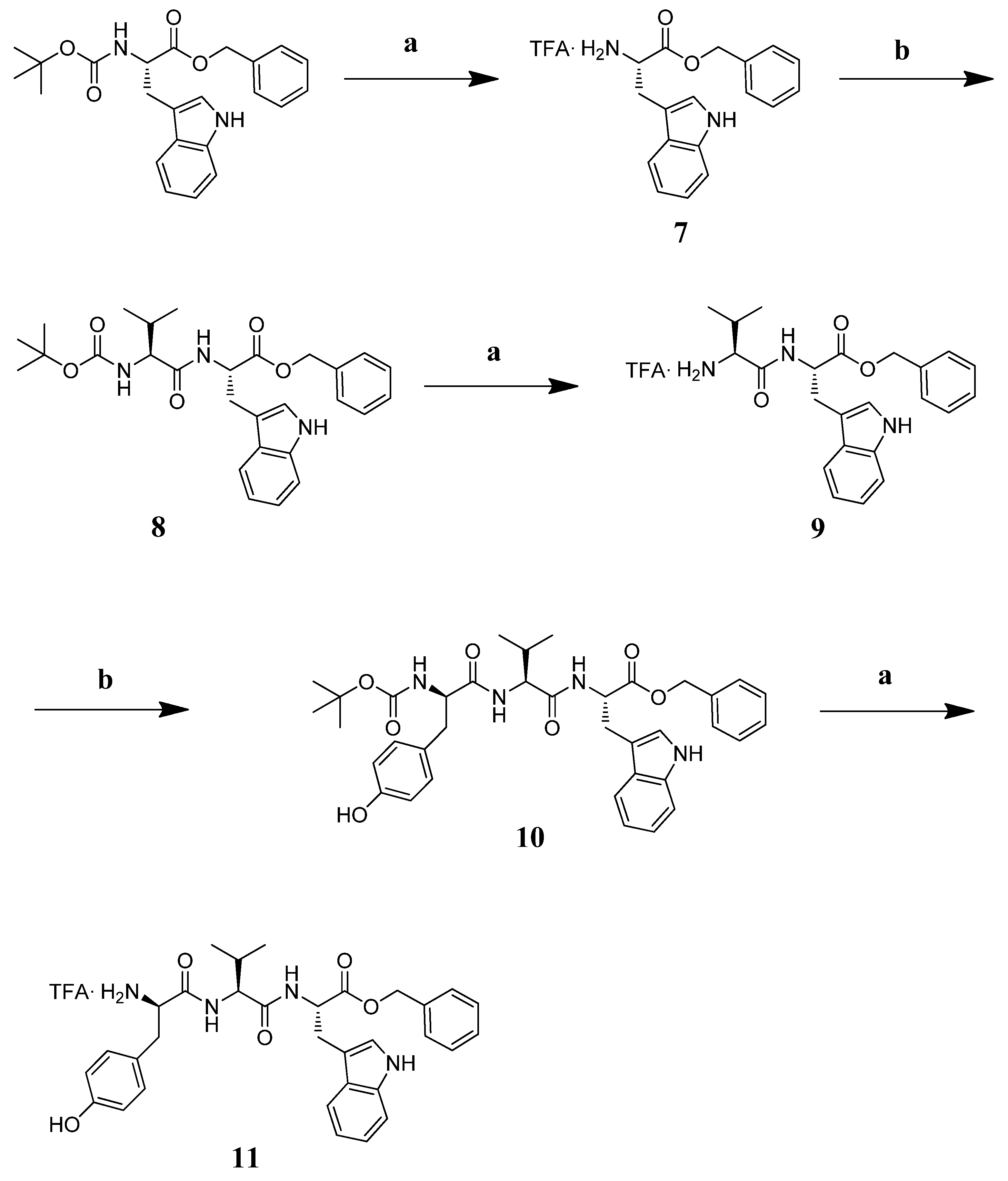
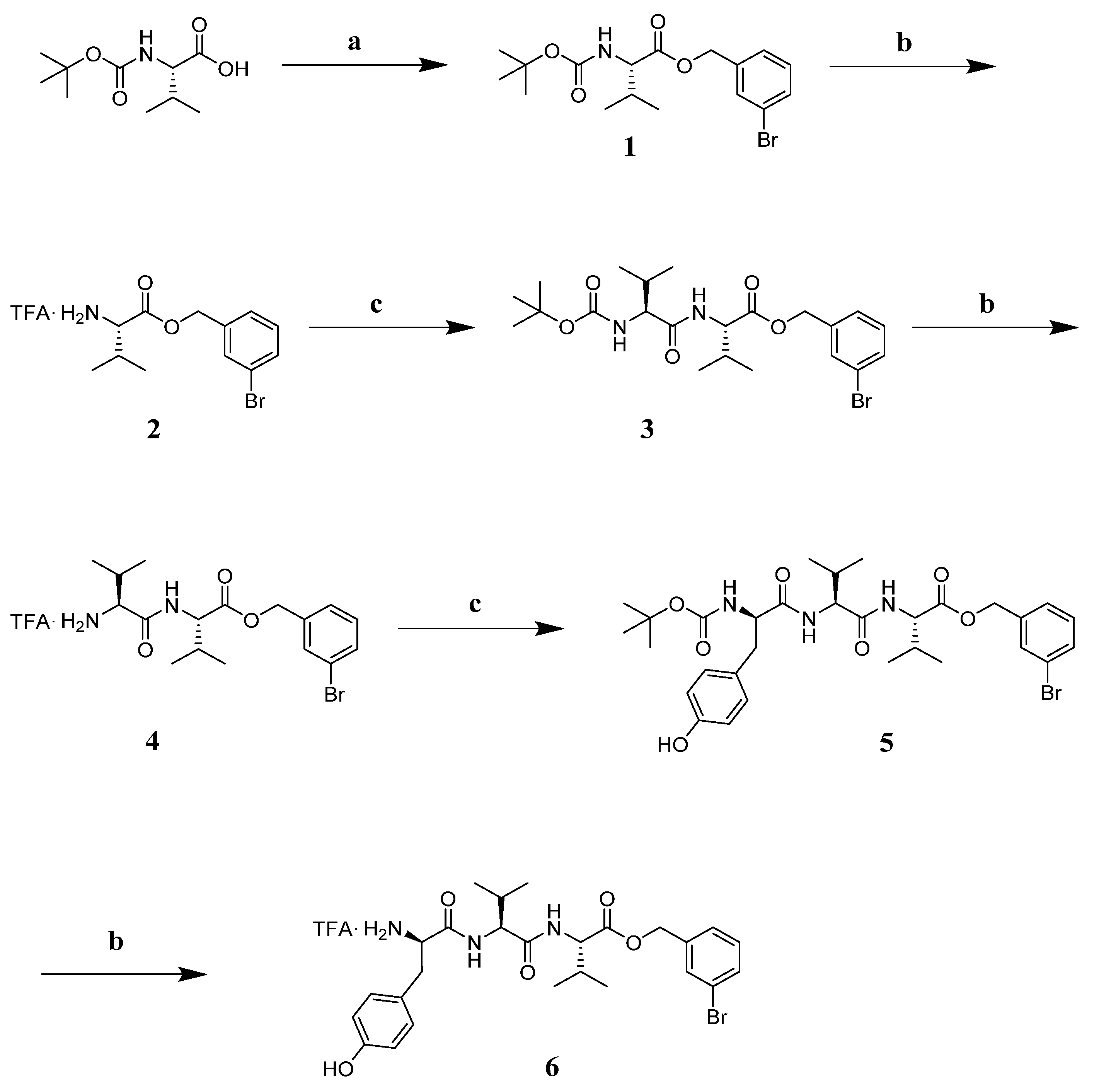
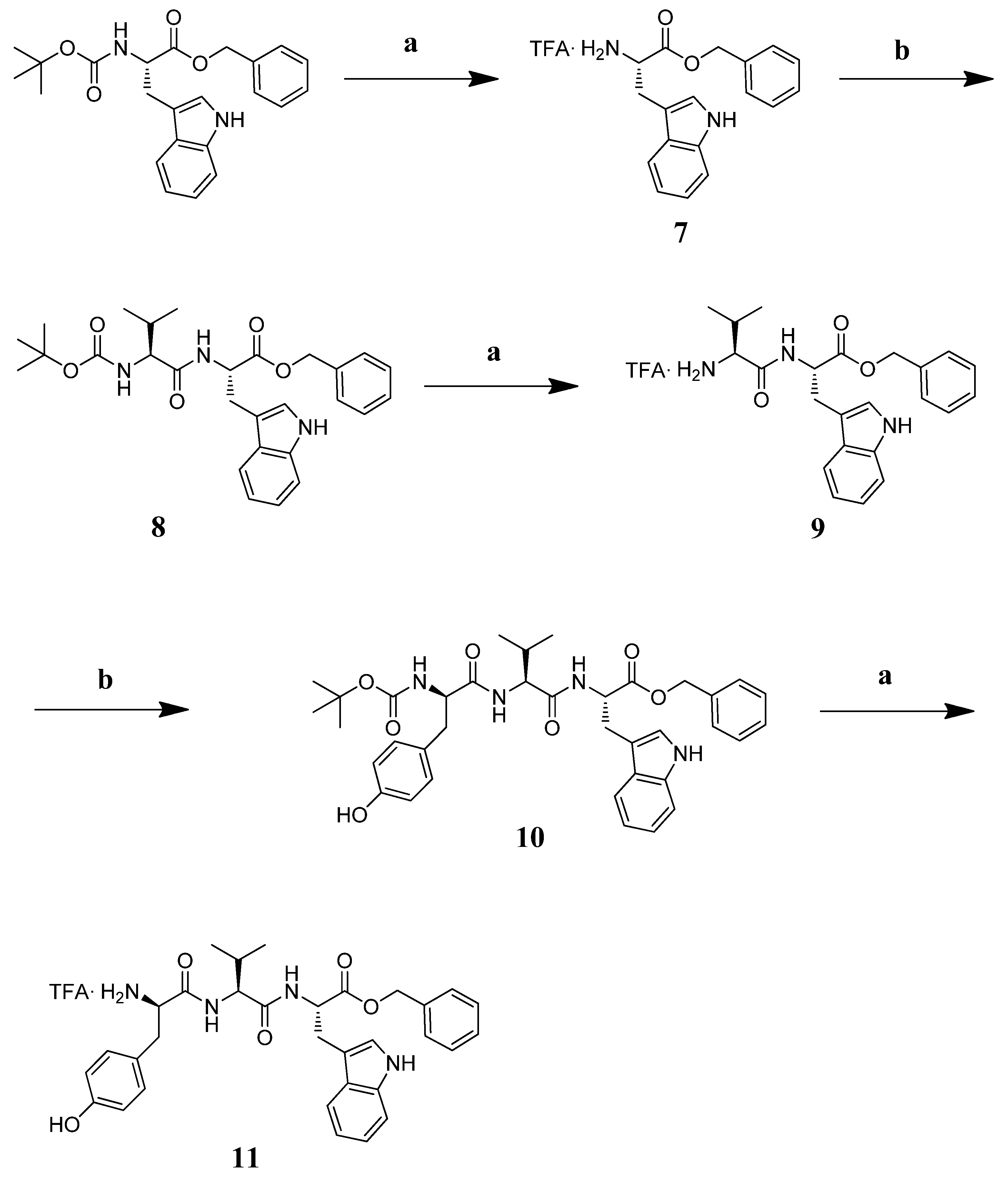
3.7. Chemistry
3.7.1. General
3.7.2. Synthesis
3.7.3. Characterization of Peptides and Intermediates
3.8. In Vivo Assays
3.8.1. Animals
3.8.2. Treatment Procedure
3.8.3. Surgery for Intracerebroventricular Injection
3.8.4. Tail Flick Test
3.8.5. Formalin Test
3.8.6. Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Sample Availability
References
- Brownstein, M.J. A brief Hystory of opiates, opioid peptides, and opioid receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 5391–5393. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; Von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J.; Dolphin, A.C. Regulation of µ-Opioid Receptors: Desensitization, Phosphorylation, Internalization, and Tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [Green Version]
- Chavkin, C.; James, I.F.; Goldstein, A. Dynorphin is a specific endogenous ligand of the κ opioid receptor. Science 1982, 215, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Carlezon, W.A.; Thome, J.; Olson, V.G.; Lane-Ladd, S.B.; Brodkin, E.S.; Hiroi, N.; Duman, R.S.; Neve, R.L.; Nestler, E.J. Regulation of Cocaine Reward by CREB. Science 1998, 282, 2272–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, A.; Brantl, V.; Herz, A.; Emrich, H.M. Psychotomimesis mediated by kappa opiate receptors. Science 1986, 233, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinary potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Roth, B.L. New Technologies for Elucidating Opioid Receptor Function. Trends Pharmacol. Sci. 2016, 37, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.B.; Atkinson, R.N.; Rothman, R.B.; Fix, S.E.; Mascarella, S.W.; Vinson, N.A.; Dersch, C.M.; Cantrell, B.E.; Zimmerman, D.M.; Carroll, F.I. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J. Med. Chem. 2001, 44, 2687–2690. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Bruijnzeel, A.W. Kappa-Opioid receptor signaling and brain reward function. Brain Res. Rev. 2009, 62, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [Green Version]
- Urbano, M.; Guerrero, M.; Rosen, H.; Roberts, E. Antagonists of the kappa opioid receptor. Bioorg. Med. Chem. Lett. 2014, 24, 2021–2032. [Google Scholar] [CrossRef]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Meng, I.D.; Johansen, J.P.; Harasawa, I.; Fields, H.L. Kappa Opioids Inhibit Physiologically Identified Medullary Pain Modulating Neurons and Reduce Morphine Antinociception. J. Neurophysiol. 2005, 93, 1138–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Chiara, G.; Imperato, A. Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J. Pharmacol. Exp. Ther. 1988, 244, 1067–1080. [Google Scholar]
- Narita, M.; Funada, M.; Suzuki, T. Regulations of opioid dependence by opioid receptor types. Pharmacol. Ther. 2001, 89, 1–15. [Google Scholar] [CrossRef]
- Spanagel, R.; Herz, A.; Shippenberg, T.S. The Effects of opioid peptides on dopamine release in the nucleus accumbens: An in vivo microdialysis study. J. Neurochem. 1990, 55, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Dortch-Carnes, J.; Potter, D.E. Bremazocine: A κ-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005, 11, 195–212. [Google Scholar] [CrossRef]
- Land, B.B.; Bruchas, M.R.; Schattauer, S.; Giardino, W.J.; Aita, M.; Messinger, D.; Hnasko, T.S.; Palmiter, R.D.; Chavkin, C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc. Natl. Acad. Sci. USA 2009, 106, 19168–19173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, C.F.; McMackin, M.Z.; Campi, K.L.; Doig, I.E.; Takahashi, E.Y.; Pride, M.C.; Trainor, B.C. Effects of kappa opioid receptors on conditioned place aversion and social interaction in males and females. Behav. Brain Res. 2014, 262, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Baner, K.; Westkaemper, R.; Siebert, D.; Rice, K.C.; Steinberg, S.; Ernsberger, P.; Rothman, R.B. Salvinorin A: A potent naturally occurring nonnitrogenous k opioid selective agonist. Proc. Natl. Acad. Sci. USA 2002, 99, 11934–11939. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Land, B.B.; Chavkin, C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010, 1314, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchas, M.R.; Macey, T.A.; Lowe, J.D.; Chavkin, C. Kappa Opioid Receptor Activation of p38 MAPK Is GRK3- and Arrestin-dependent in Neurons and Astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, J.P.; Xu, M.; Mackie, K.; Chavkin, C. Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J. Biol. Chem. 2003, 278, 34631–34640. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Land, B.B.; Aita, M.; Xu, M.; Barot, S.K.; Li, S.; Chavkin, C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa opioid-dependent dysphoria. J. Neurosci. 2007, 27, 11614–11623. [Google Scholar] [CrossRef] [Green Version]
- El Rawas, R.; Amaral, I.M.; Hofer, A. Is p38 MAPK Associated to Drugs of Abuse-Induced Abnormal Behaviors? Int. J. Mol. Sci. 2020, 21, 4833. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Schindler, A.G.; Shankar, H.; Messinger, D.I.; Miyatake, M.; Land, B.B.; Lemos, J.C.; Hagan, C.E.; Neumaier, J.F.; Quintana, A. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 2011, 71, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.A.; Smith, M.L.; Kieffer, B.L.; Evans, C.J. Ligand-directed signalling within the opioid receptor family. Br. J. Pharmacol. 2012, 167, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Schattauer, S.S.; Miyatake, M.; Shankar, H.; Zietz, C.; Levin, J.R.; Liu-Chen, L.Y.; Gurevich, V.V.; Rieder, M.J.; Chavkin, C. Ligand directed signaling differences between rodent and human κ-opioid receptors. J. Biol. Chem. 2012, 287, 41595–41607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Bruchas, M.R.; Ippolito, D.L.; Gendron, L.; Chavkin, C. Sciatic Nerve ligation-induced proliferation of spinal cord astrocytes is mediated by k opioid activation of p38 mitogen-activated protein kinase. J. Neurosci. 2007, 27, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.L.; Scopton, A.P.; Rives, M.L.; Bikbulatov, R.V.; Polepally, P.R.; Brown, P.J.; Kenakin, T.; Javitch, J.A.; Zjawiony, J.K.; Roth, B.L. Identification of novel functionally selective kappa-opioid receptor scaffolds. Mol. Pharmacol. 2014, 85, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Bikbulatov, R.V.; Mocanu, V.; Dicheva, N.; Parker, C.E.; Wetsel, W.C.; Mosier, P.D.; Westkaemper, R.B.; Allen, J.A.; Zjawiony, J.K.; et al. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel Salvinorin A analogues as active state probes of the κ-opioid receptor. Biochemistry 2009, 48, 6898–6908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yongye, A.B.; Pinilla, C.; Medina-Franco, J.L.; Giulianotti, M.A.; Dooley, C.T.; Appel, J.R.; Nefzi, A.; Scior, T.; Houghten, R.A.; Martinez-Mayorga, K. Integrating computational and mixture-based screening of combinatorial libraries. J. Mol. Model. 2011, 17, 1473–1482. [Google Scholar] [CrossRef]
- Lopez-Vallejo, F.; Giulianotti, M.A.; Houghten, R.A.; Medina-Franco, J.L. Expanding the medicinally relevant chemical space with compound libraries. Drug Discov. Today 2012, 17, 718–726. [Google Scholar] [CrossRef]
- Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of novel µ-opioid receptor inverse agonist from a combinatorial library of tetrapeptides through structure-based virtual screening. Molecules 2019, 24, 3872. [Google Scholar] [CrossRef] [Green Version]
- Torino, D.; Mollica, A.; Pinnen, F.; Feliciani, F.; Lucente, G.; Fabrizi, G.; Portalone, G.; Davis, P.; Lai, J.; Ma, S.W.; et al. Synthesis and evaluation of new endomorphin-2 analogues containing (Z)-alpha,beta-didehydrophenylalanine (Delta(Z)Phe) residues. J. Med. Chem. 2010, 53, 4550–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; Sansone, A.; Masi, A.; Lucente, G.; Punzi, P.; Mollica, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Davis, P.; et al. Synthesis and activity of endomorphin-2 and morphiceptin analogues with proline surrogates in position 2. Eur. J. Med. Chem. 2010, 45, 4594–4600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardy, E.; Robinson, J.E.; Li, C.; Olsen, R.H.J.; DiBerto, J.F.; Giguere, P.M.; Sassano, F.M.; Huang, X.P.; Zhu, H.; Urban, D.J. A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. Neuron 2015, 86, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenalti, G.; Zatsepin, N.A.; Betti, C.; Giguere, P.; Han, G.W.; Ishchenko, A.; Liu, W.; Guillemyn, K.; Zhang, H.; James, D. Structural basis for bifunctional peptide recognition at human δ-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanucci, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Lucente, G.; Mollica, A. Conformationally constrained histidines in the design of peptidomimetics: Strategies for the χ-Space control. Int. J. Mol. Sci. 2011, 12, 2853–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017, 168, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Feliciani, F.; Pinnen, F.; Stefanucci, A.; Costante, R.; Cacciatore, I.; Lucente, G.; Mollica, A. Structure-activity relationships of biphalin analogs and their biological evaluation on opioid receptors. Mini Rev Med Chem. 2013, 13, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic efficiency as an important metric in drug design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef]
- Saha, S.; Das, R.; Divyanshi, D.; Tiwari, N.; Tiwari, A.; Dalai, R.; Kumar, A.; Goswami, C. Ratio of hydrophobic-hydrophilic and positive-negative residues at lipid-water-interface influences surface expression and channel-gating of TRPV1. BioRxiv 2020, 4, 272484. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of genetic polymorphisms of ABCB1 (MDR1, P-glycoprotein) on drug disposition and potential clinical implications: Update of the literature. Clin. Pharmacokinet. 2015, 54, 709–735. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Mbarik, M.; Poirier, S.J.; Doiron, J.; Selka, A.; Barnett, D.A.; Cormier, M.; Touaibia, M.; Surette, M.E. Phenolic acid phenetylestersand their corresponding ketones: Inhibition of 5-lypoxygenases and stability in human blood and HepaRG cells. Pharmacol. Res. Pers. 2019, 7, e00524. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2017-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-1: Epik; Schrödinger, LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-1: Protein Preparation Wizard; Epik; Schrödinger, LLC: New York, NY, USA, 2017.
- Impact, Schrödinger, LLC, New York, NY; Prime; Schrödinger, LLC: New York, NY, USA, 2017.
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-1: Glide; Schrödinger, LLC: New York, NY, USA, 2017.
- Balboni, G.; Salvadori, S.; Guerrini, R.; Bianchi, C.; Santagada, V.; Calliendo, G.; Bryant, S.D.; Lazarus, L.H. Opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei. Peptides 2000, 21, 1663–1671. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, X.P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Steven, R.C.; Katritch, V. Structure-based discovery of new antagonist and biased agonist chemotypes for the kappa opioid receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wand, G.S.; Ney, R.L.; Baylin, S.; Eipper, B.; Mains, R.E. Characterization of a peptide alpha-amidation activity in human plasma and tissues. Metabolism 1985, 34, 1044–1052. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-1: Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, 2017. Maestro-Desmond Interoperability Tools; Schrödinger, LLC: New York, NY, USA, 2017.
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Stefanucci, A.; Pinnen, F.; Lucente, G.; Fidanza, S.; Pieretti, S. Antinociceptive profile of potent opioid peptide AM94, a fluorinated analogue of biphalin with non-hydrazine linker. J. Pept. Sci. 2013, 19, 233–239. [Google Scholar] [CrossRef]
- Mollica, A.; Carotenuto, A.; Novellino, E.; Limatola, A.; Costante, R.; Pinnen, F.; Stefanucci, A.; Pieretti, S.; Borsodi, A.; Samavati, R.; et al. Novel Cyclic Biphalin Analogue with Improved Antinociceptive Properties. ACS Med. Chem. Lett. 2014, 5, 1032–1036. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides Sequences | Docking Score |
|---|---|
| D-Tyr-L-Val-L-Val-OBz | −11.789 |
| D-Tyr-D-Val-L-Val-OBz | −11.467 |
| L-Tyr-L-Val-D-Val-OBz | −11.189 |
| D-Tyr-D-Val-D-Val-OBz | −11.154 |
| D-Tyr-L-Val-D-Val-OBz | −9.975 |
| L-Tyr-D-Val-D-Val-OBz | −9.598 |
| L-Tyr-L-Val-L-Val-OBz | −8.510 |
| L-Tyr-D-Val-L-Val-OBz | −8.188 |
| Peptides Sequences | Docking Score |
|---|---|
| D-Tyr-L-Val-L-Trp-OBz | −11.582 |
| L-Tyr-D-Val-L-Trp-OBz | −11.075 |
| D-Tyr-D-Val-L-Trp-OBz | −8.174 |
| L-Tyr-L-Val-L-Val-OBz | −7.523 |
| Peptides Sequences | Docking Score |
|---|---|
| D-Tyr-L-Val-L-Val-O-(3-Br)-OBz | −11.288 |
| D-Tyr-D-Val-L-Val-(3-Br)-OBz | −10.728 |
| D-Tyr-L-Val-D-Val-O-(3-Br)-OBz | −9.849 |
| L-Tyr-L-Val-D-Val-O-(3-Br)-OBz | −9.451 |
| D-Tyr-D-Val-D-Val-O-(3-Br)-OBz | −9.150 |
| L-Tyr-L-Val-L-Val-O-(3-Br)-OBz | −9.087 |
| L-Tyr-D-Val-D-Val-O-(3-Br)-OBz | −8.792 |
| L-Tyr-D-Val-L-Val-O-(3-Br)-OBz | −7.774 |
| Peptides | Lipophilicity | Drug-Likeness | Pharmacokinetics | ||||
|---|---|---|---|---|---|---|---|
| TPSA (Å) | CLogP (o/w) | Bioavailability Score | Lipinski Filter | GIA | P-gp Substrate | CYP3A4 Inhibitor | |
| 6 | 132.37 | 2.59 | 0.55 | Yes (1) | high | yes | no |
| 11 | 148.16 | 2.46 | 0.55 | Yes (1) | low | yes | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanucci, A.; Iobbi, V.; Della Valle, A.; Scioli, G.; Pieretti, S.; Minosi, P.; Mirzaie, S.; Novellino, E.; Mollica, A. In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands. Molecules 2021, 26, 4767. https://doi.org/10.3390/molecules26164767
Stefanucci A, Iobbi V, Della Valle A, Scioli G, Pieretti S, Minosi P, Mirzaie S, Novellino E, Mollica A. In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands. Molecules. 2021; 26(16):4767. https://doi.org/10.3390/molecules26164767
Chicago/Turabian StyleStefanucci, Azzurra, Valeria Iobbi, Alice Della Valle, Giuseppe Scioli, Stefano Pieretti, Paola Minosi, Sako Mirzaie, Ettore Novellino, and Adriano Mollica. 2021. "In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands" Molecules 26, no. 16: 4767. https://doi.org/10.3390/molecules26164767
APA StyleStefanucci, A., Iobbi, V., Della Valle, A., Scioli, G., Pieretti, S., Minosi, P., Mirzaie, S., Novellino, E., & Mollica, A. (2021). In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands. Molecules, 26(16), 4767. https://doi.org/10.3390/molecules26164767