
Quantifying Conformational Isomerism in Chain Molecules by Linear Raman Spectroscopy: The Case of Methyl Esters



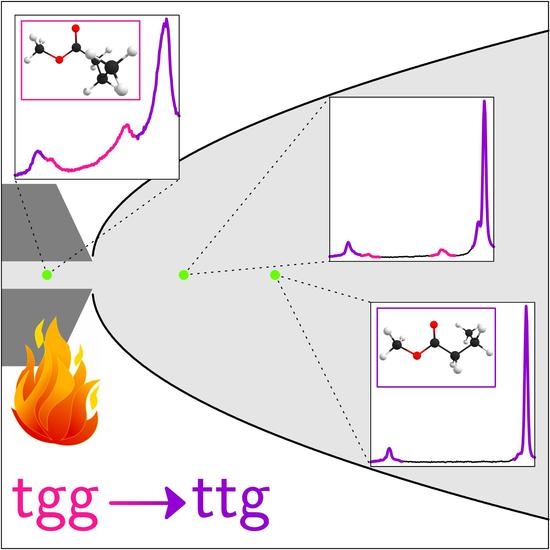
Abstract
:
1. Introduction
2. Results
2.1. Systematic Error Sources in Experimental Intensities
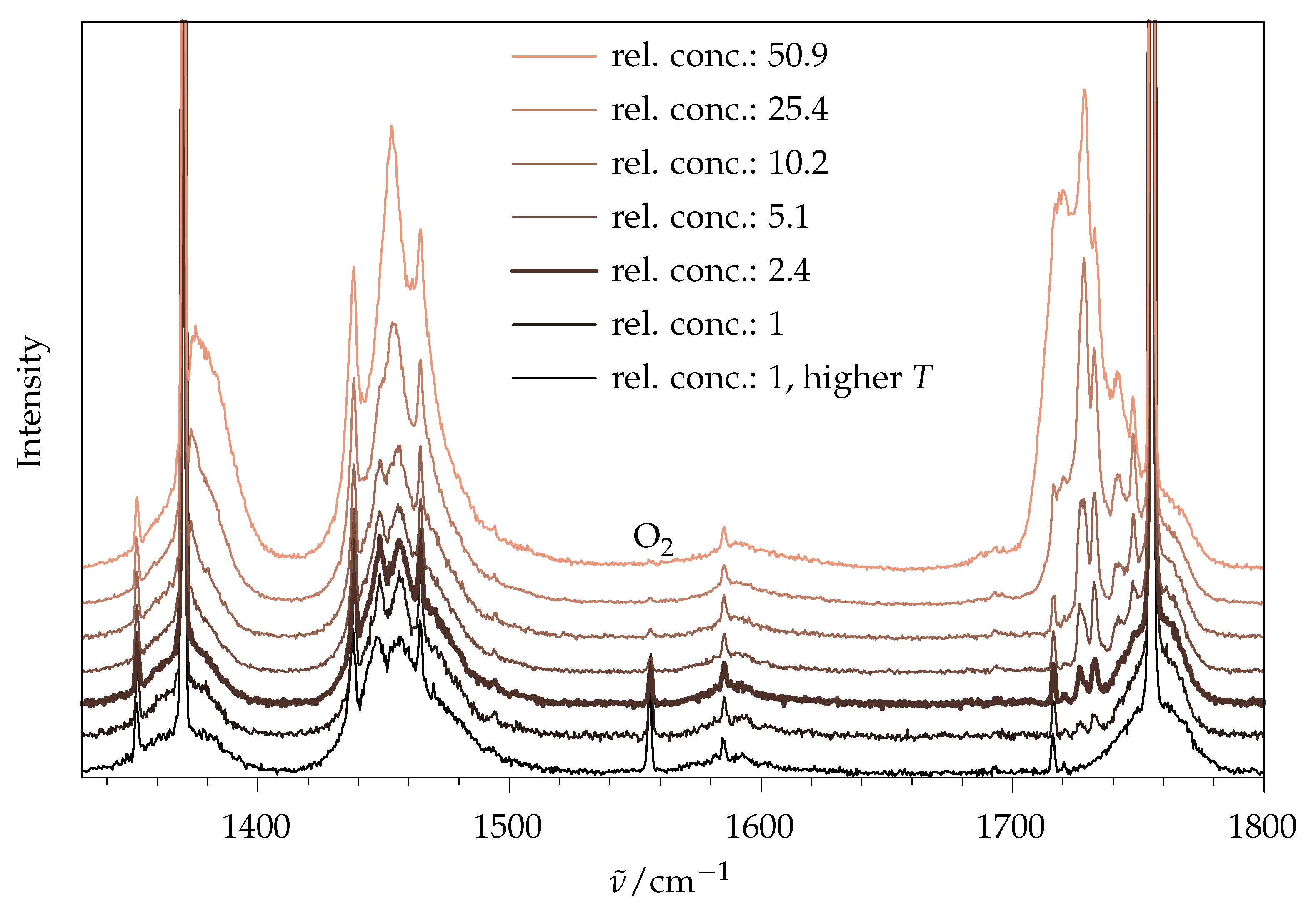
2.1.1. Aggregation Effects
2.1.2. Day-to-Day Reproducibility, Statistical Noise and Impurities
2.1.3. Error Due to Angle of Observation
2.1.4. Error Due to Transmittance of Monochromator
2.1.5. Error Due to Inhomogeneous Illumination
2.1.6. Error Due to Wavelength-Dependent Quantum Efficiency
2.1.7. Error Due to Vibrational Temperature
2.1.8. Combined Error Treatment
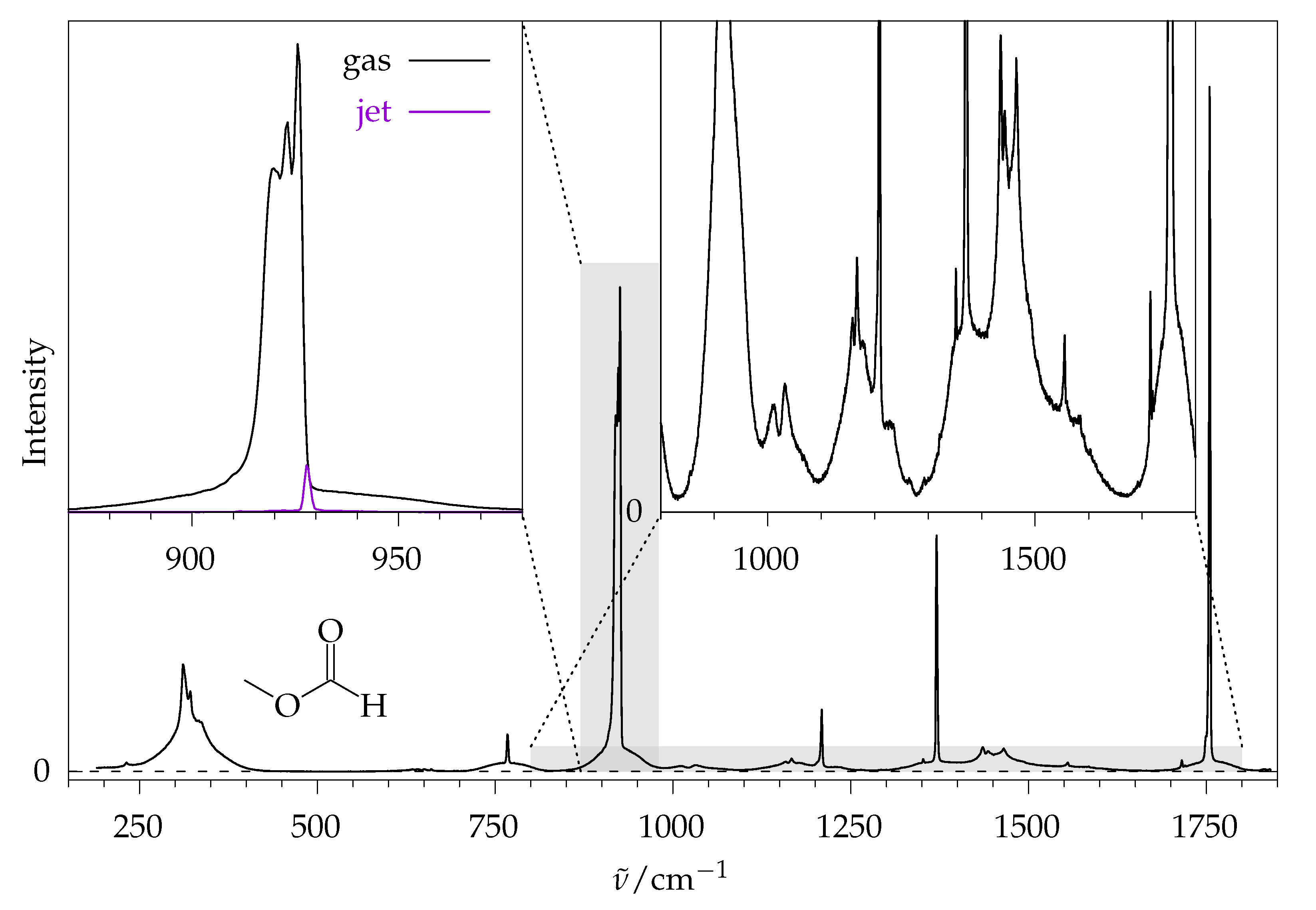
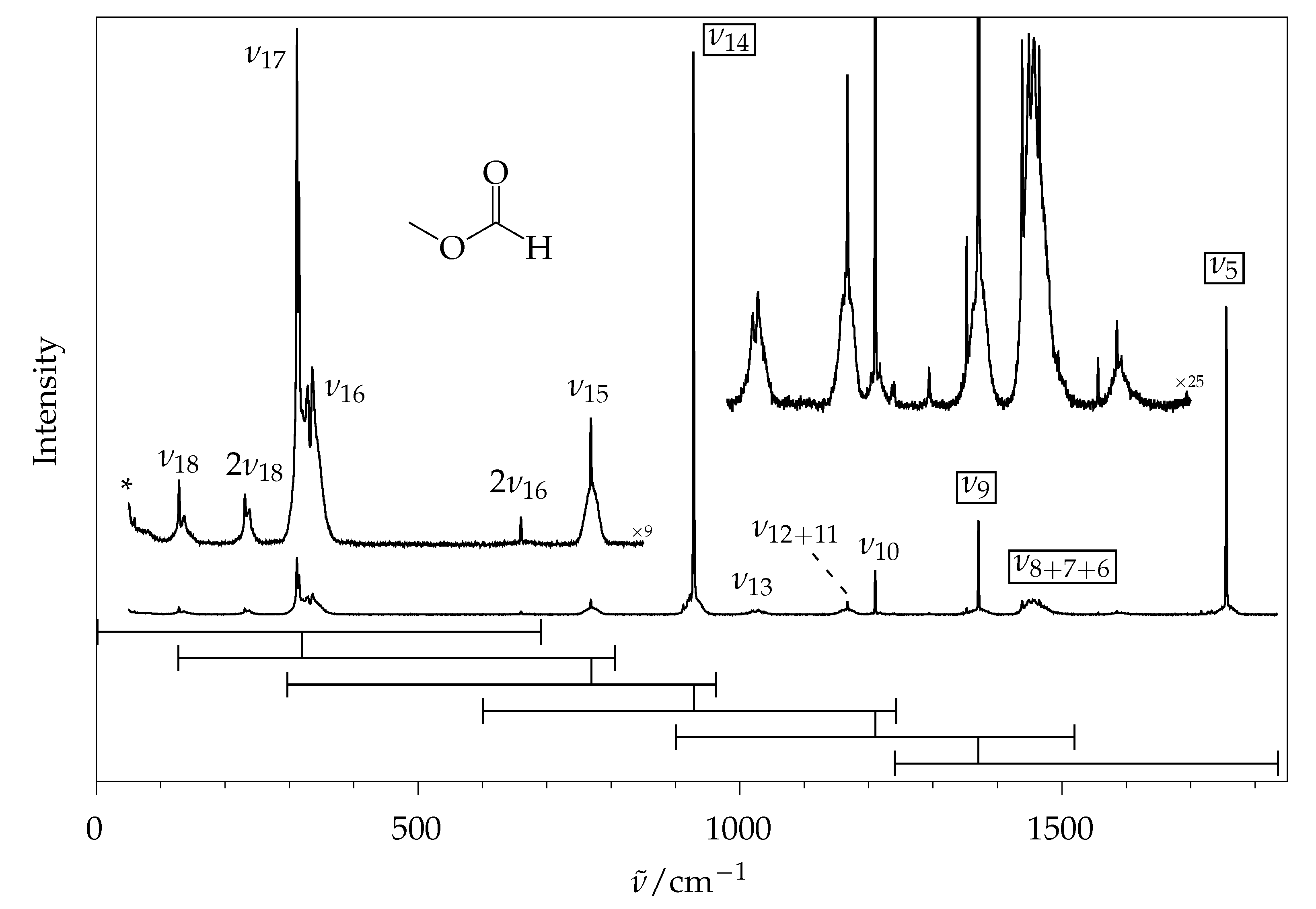
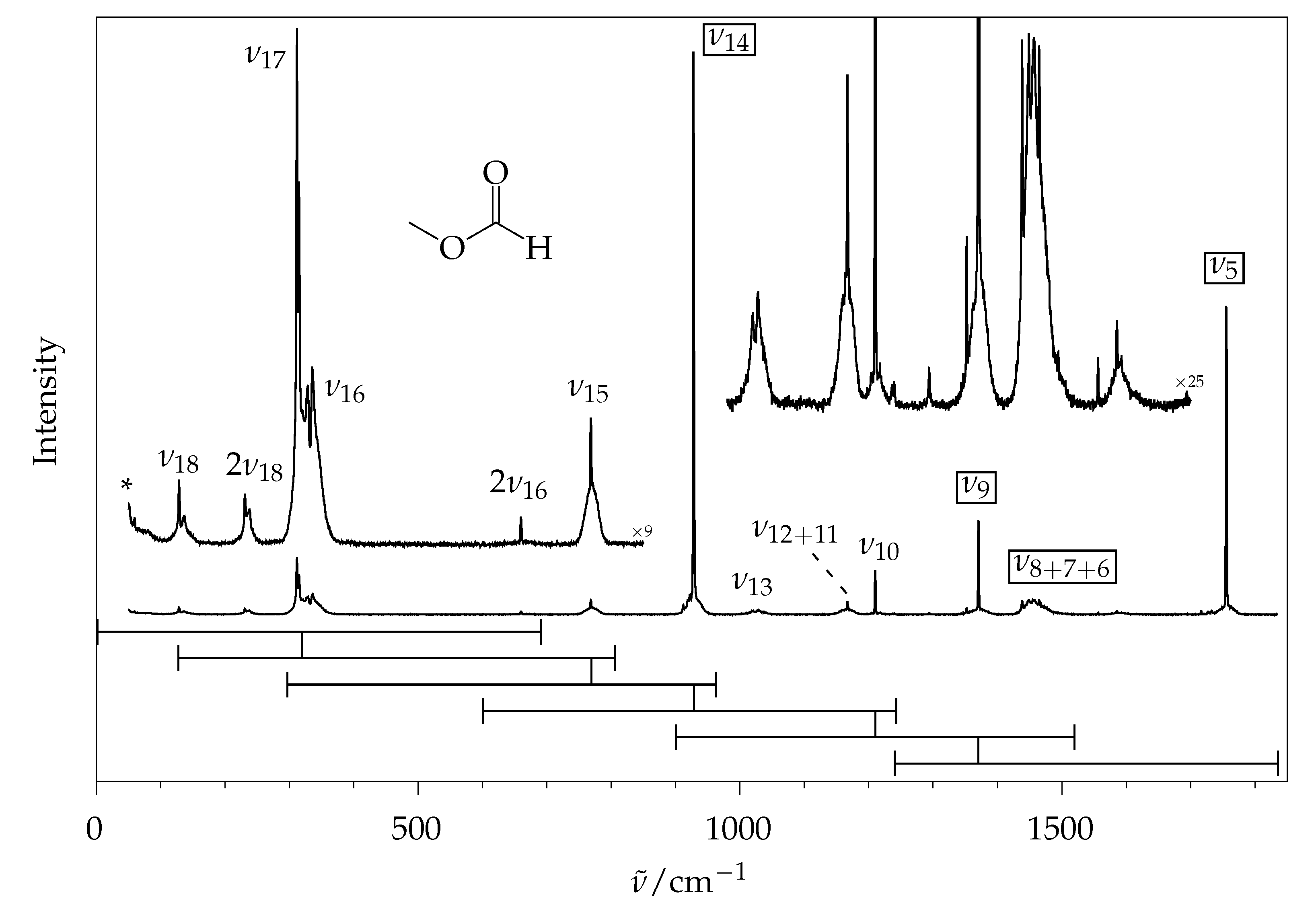
2.2. Methyl Methanoate
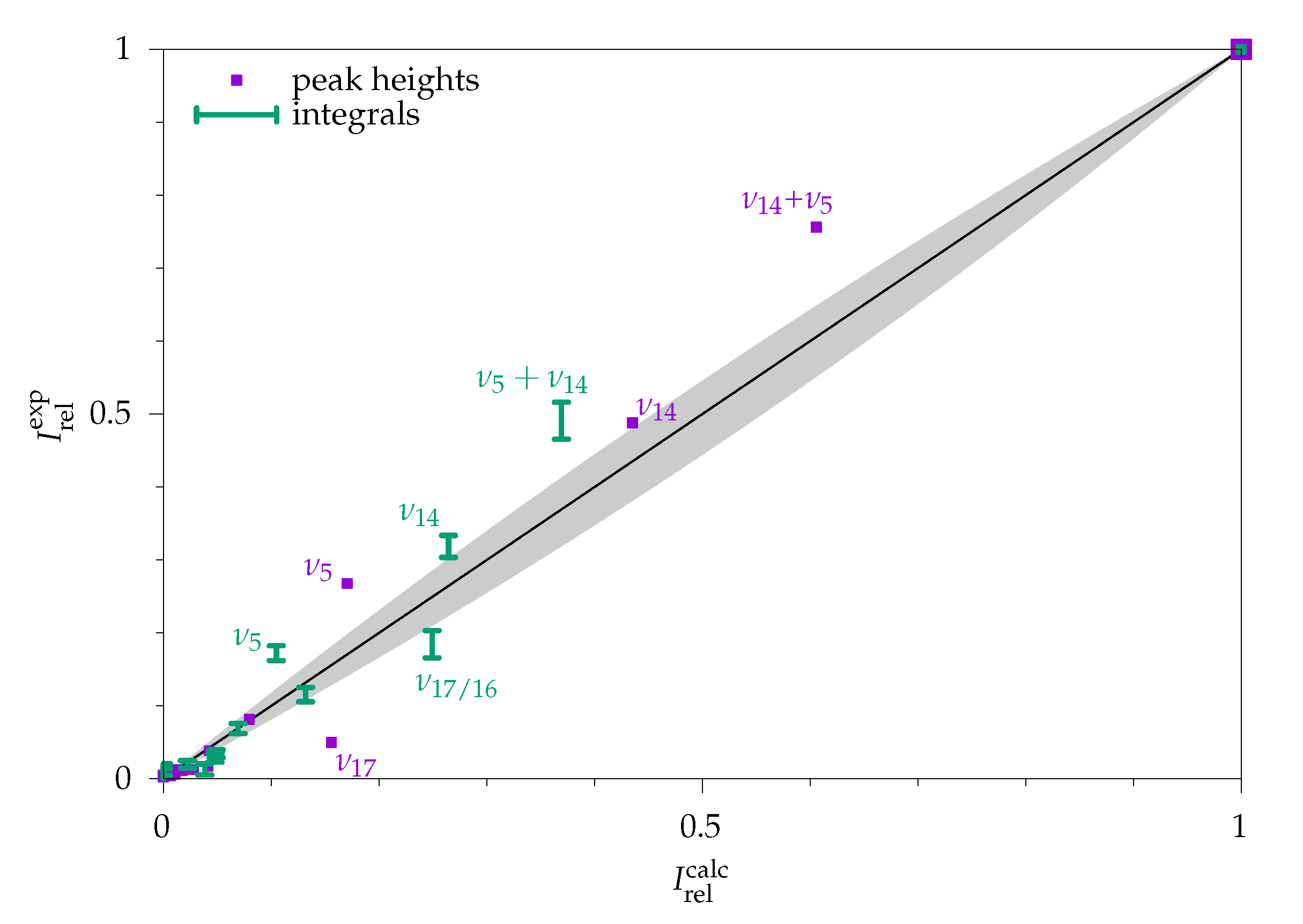
2.2.1. Comparison to Theory
2.2.2. Comparison to Gas Phase Reference Data
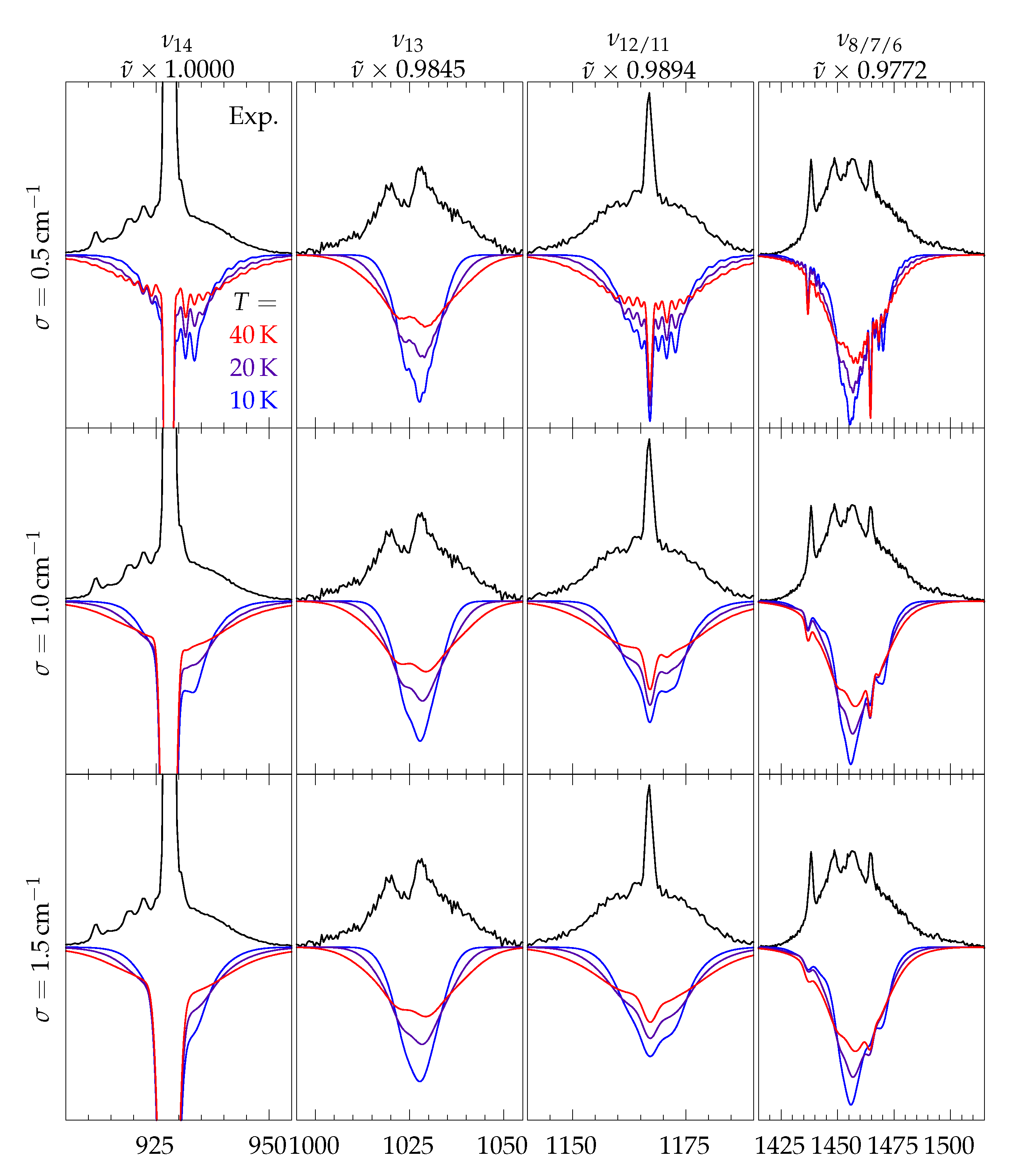
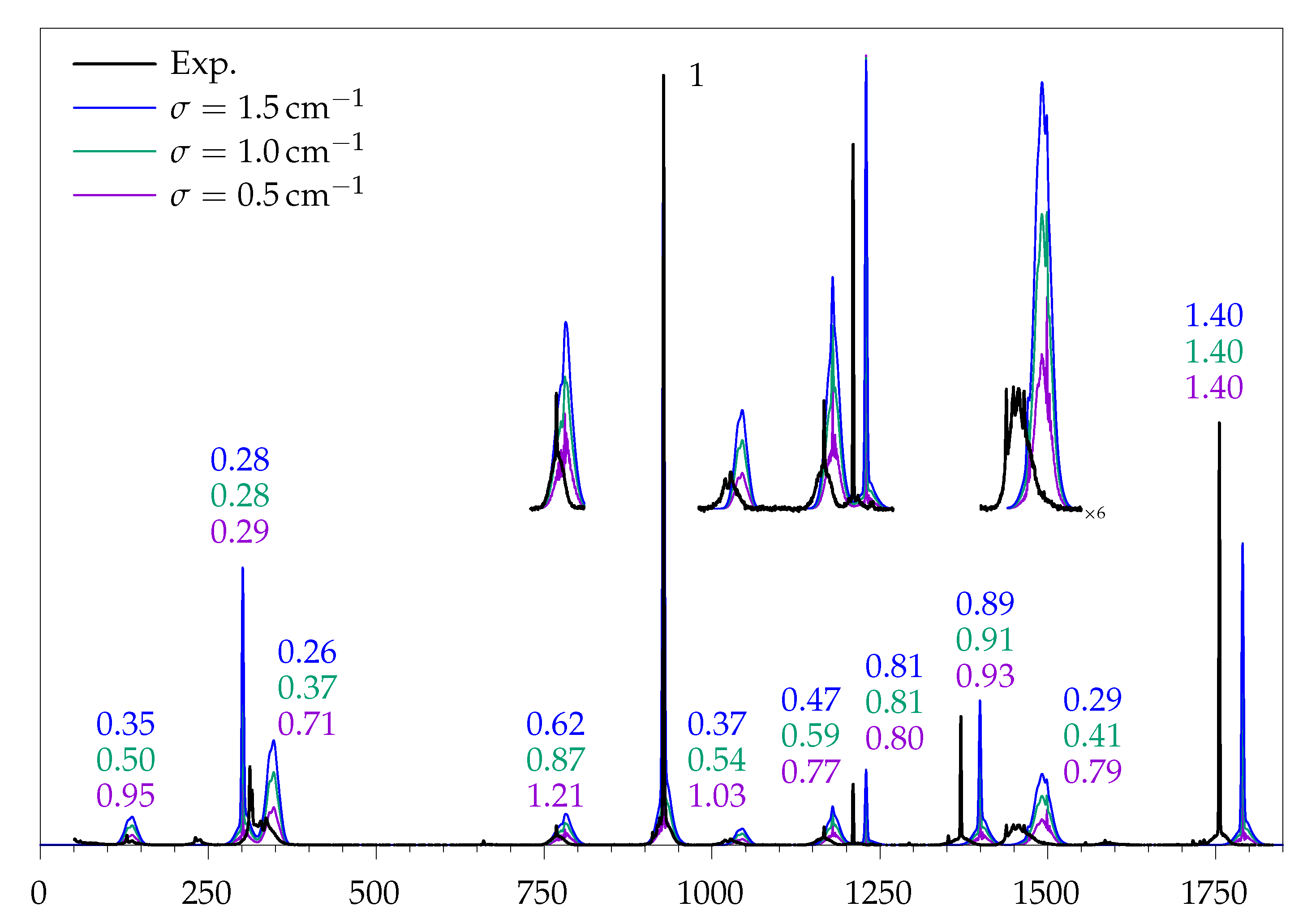
2.3. Simulation of Rotational Contours
2.4. Rovibrational Simulation of Methyl Methanoate
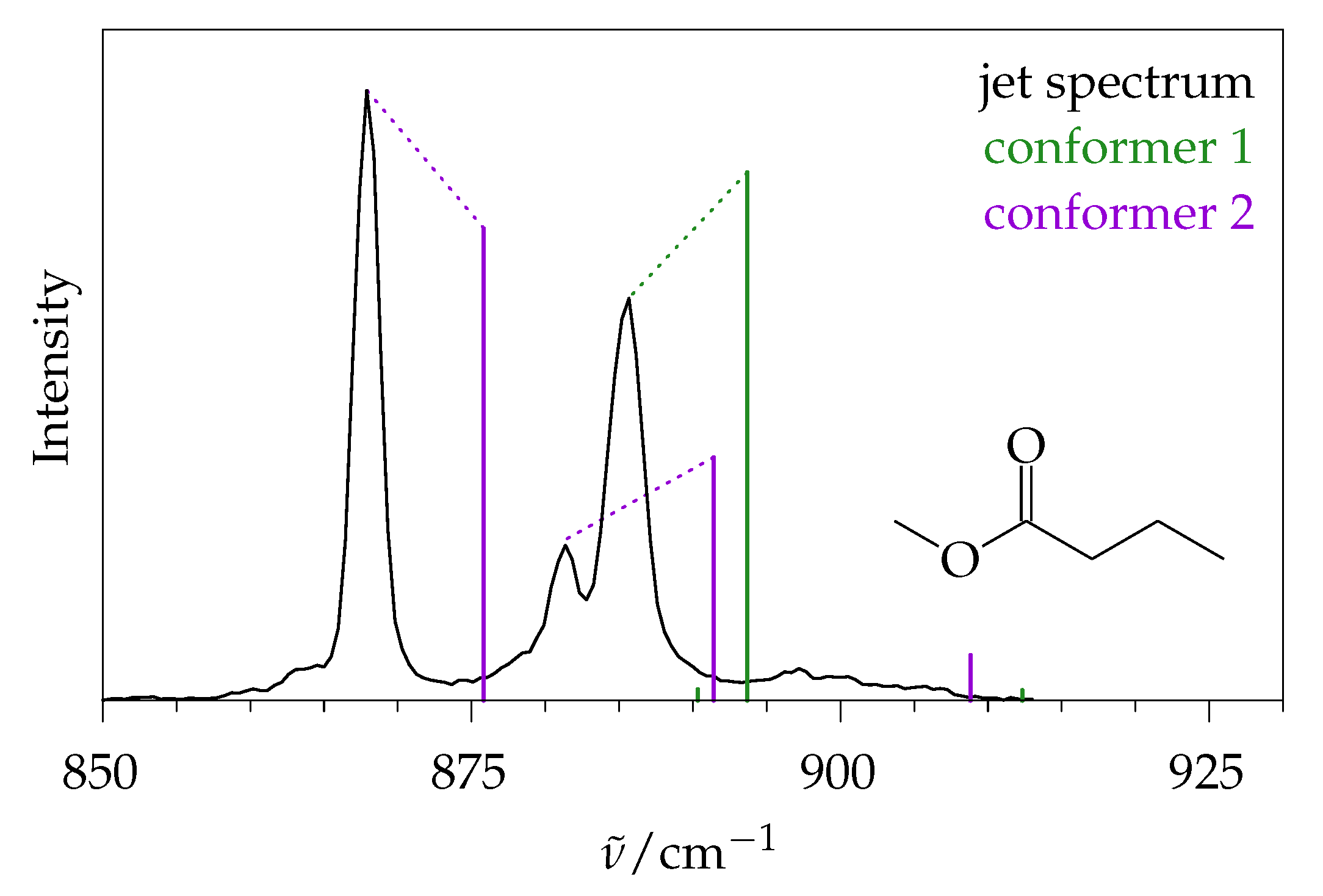
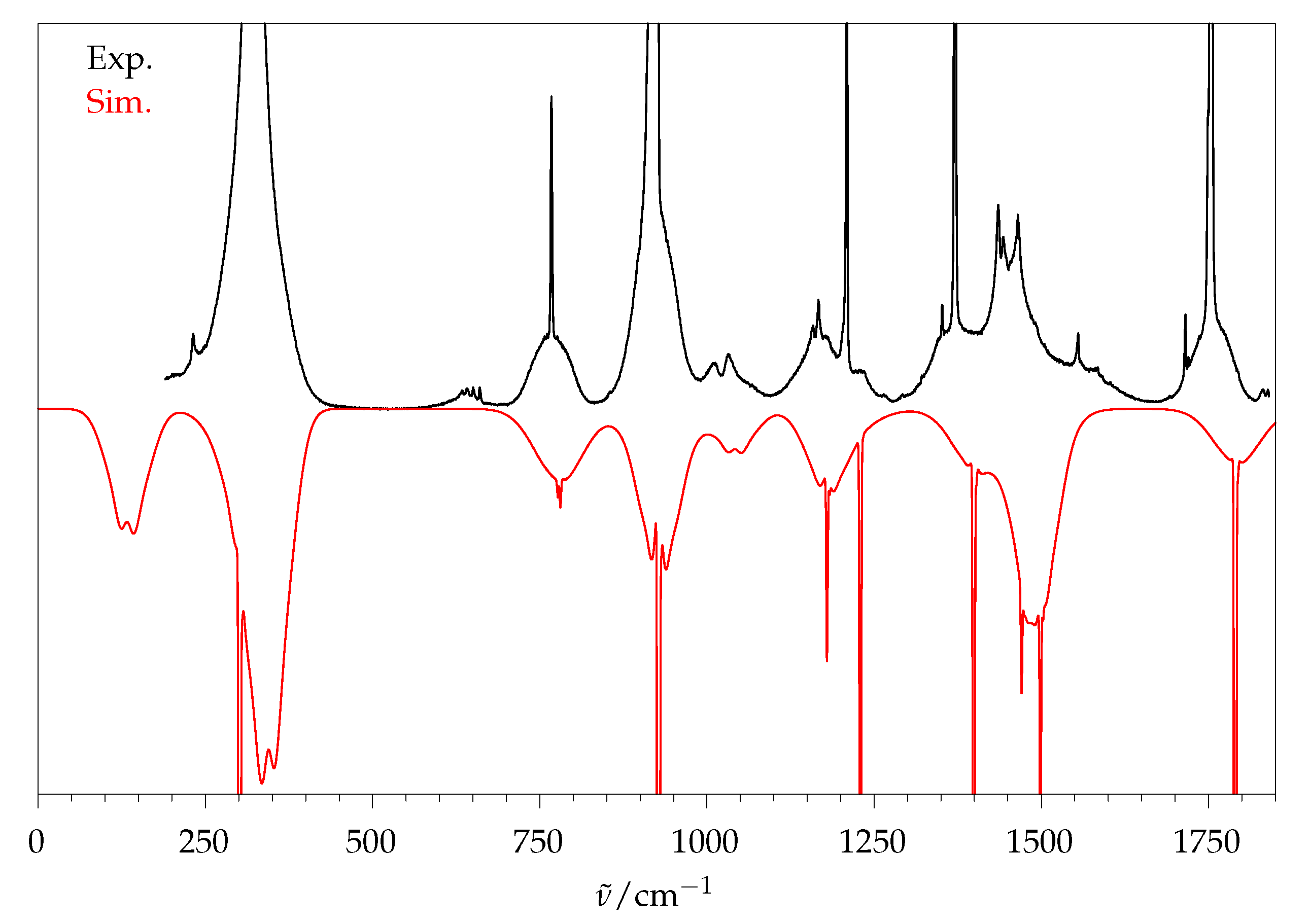
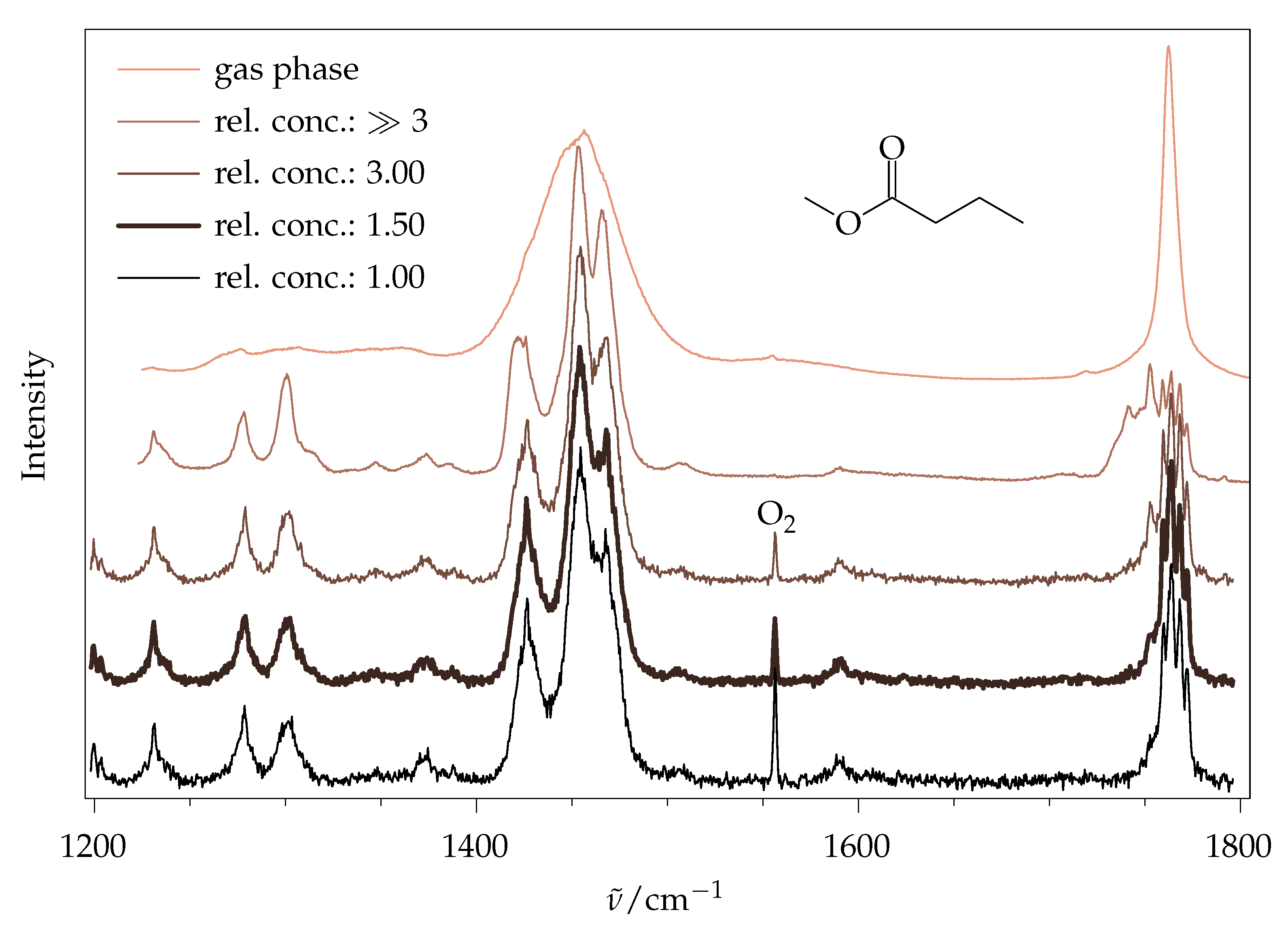
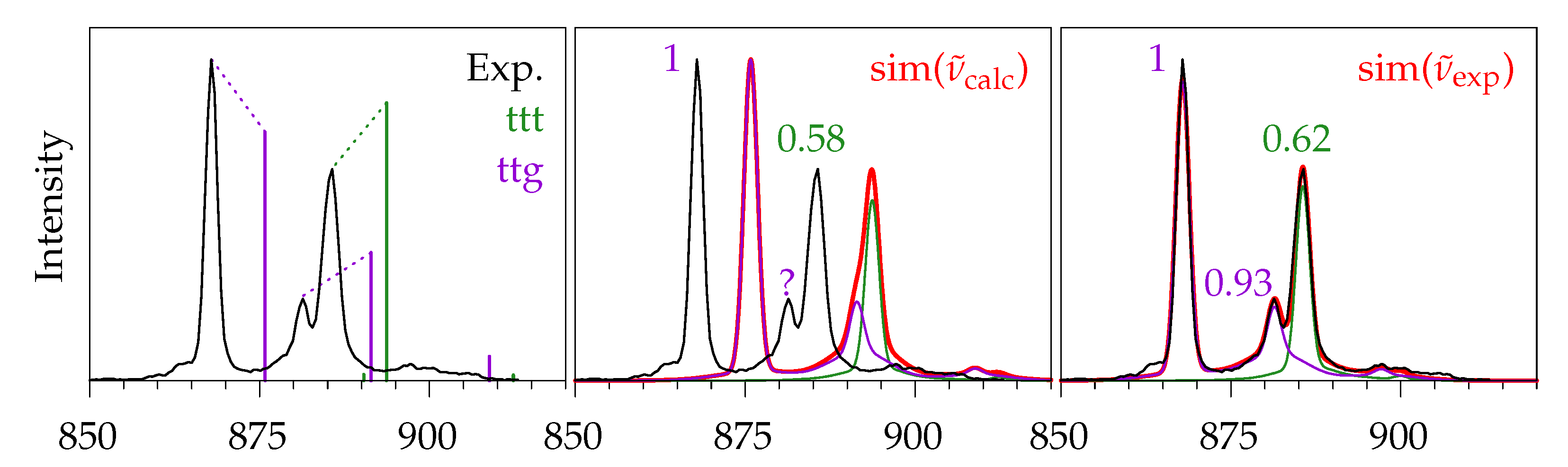
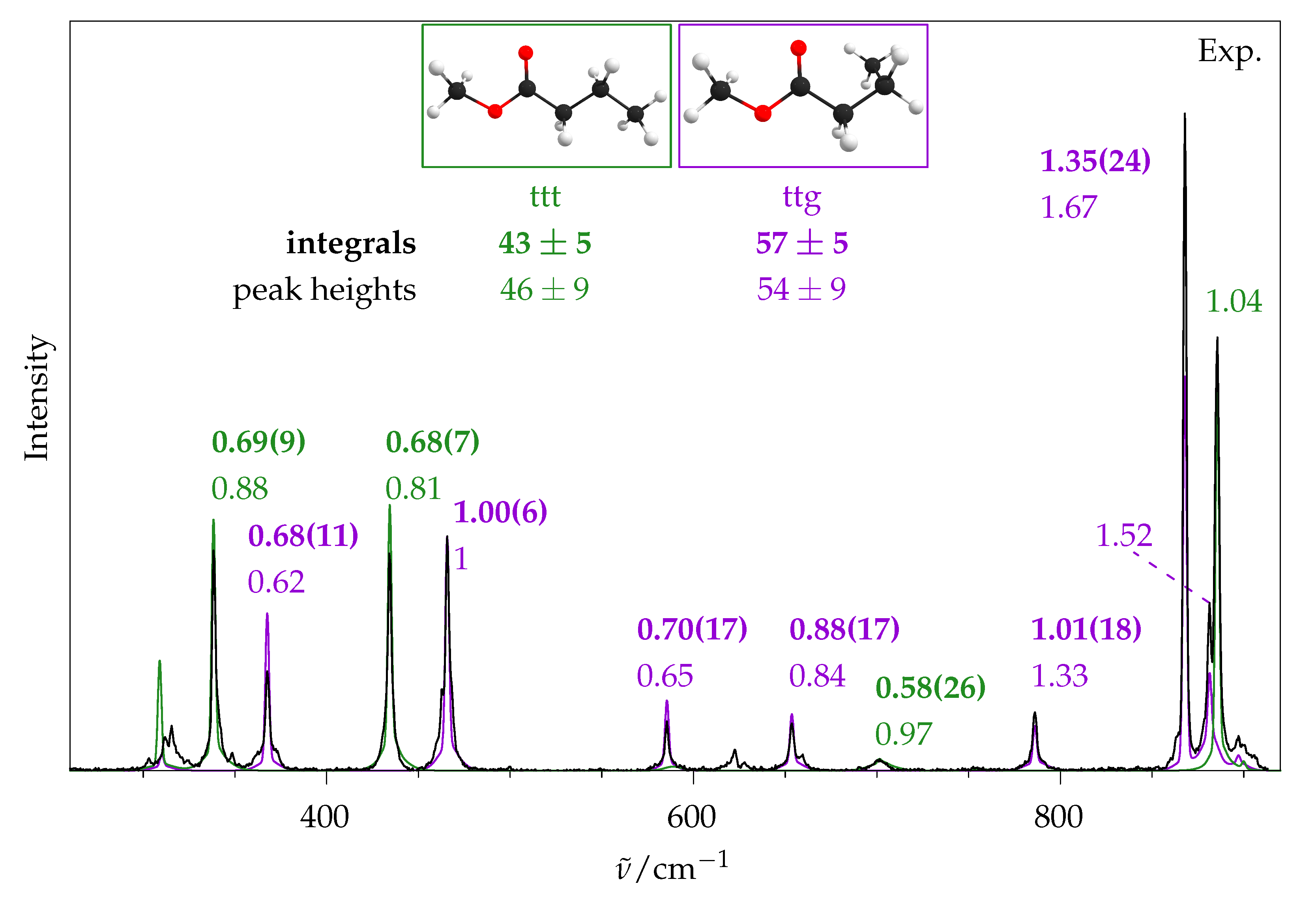
2.5. Methyl Butanoate
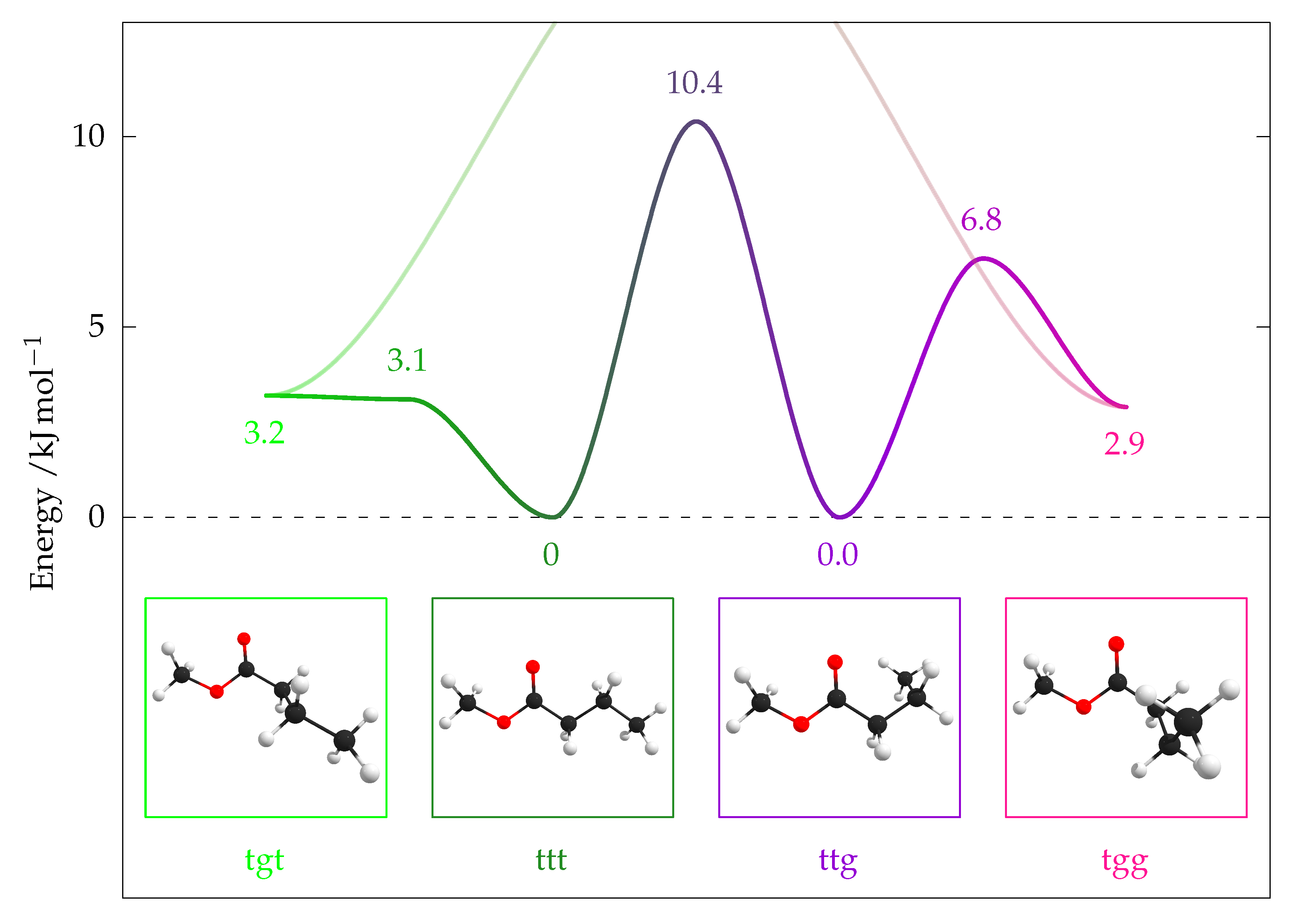
2.5.1. Quantum Chemical Predictions
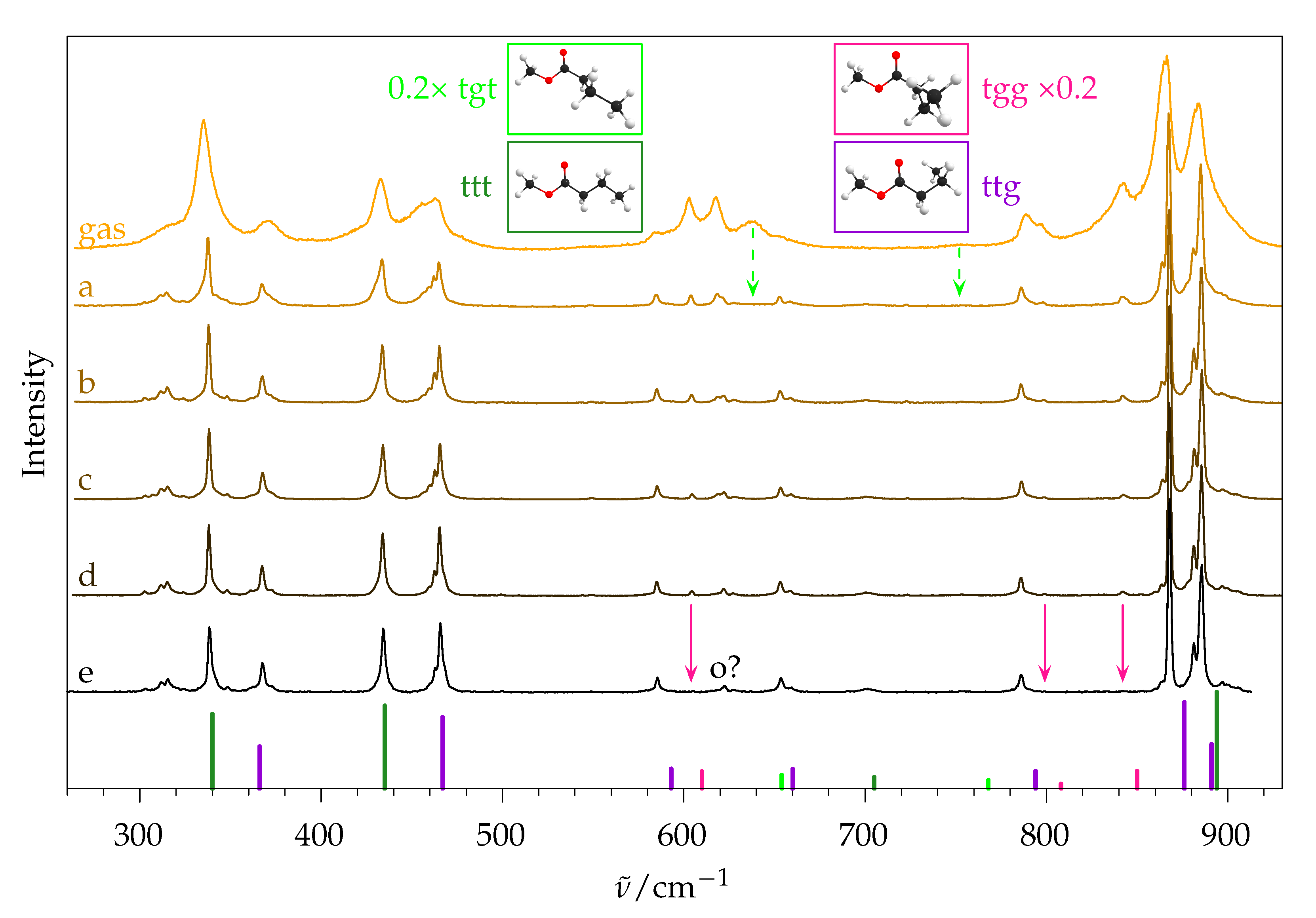
2.5.2. Experimental Spectra
3. Materials and Methods
3.1. Quantum Chemical Calculations
3.2. Rovibrational Simulation
3.3. Experimental Setup
3.4. Integration of Signals
3.5. Chemicals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| ESI | Electronic Supplementary Information |
| CCD | Charge Coupled Device |
| ZPE | Zero Point Energy |
| DFT | Density Functional Theory |
| PES | Potential Energy Surface |
| t | trans |
| c | cis |
| g | gauche |
References
- Chalmers, J.M.; Griffiths, P.R. (Eds.) Handbook of Vibrational Spectroscopy; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Klaeboe, P. Conformational studies by vibrational spectroscopy: A review of various methods. Vib. Spectrosc. 1995, 9, 3–17. [Google Scholar] [CrossRef]
- Zimmermann, C.; Gottschalk, H.C.; Suhm, M.A. Three-dimensional docking of alcohols to ketones: An experimental benchmark based on acetophenone solvation energy balances. Phys. Chem. Chem. Phys. 2020, 22, 2870–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüttschwager, N.O.B.; Suhm, M.A. Stretching and folding of 2-nanometer hydrocarbon rods. Soft Matter 2014, 10, 4885–4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef] [PubMed]
- Oakes, R.E.; Beattie, J.R.; Moss, B.W.; Bell, S.E. Conformations, vibrational frequencies and Raman intensities of short-chain fatty acid methyl esters using DFT with 6-31G(d) and Sadlej pVTZ basis sets. J. Mol. Struct. Theochem 2002, 586, 91–110. [Google Scholar] [CrossRef]
- Fernández-Sánchez, J.M.; Montero, S. Gas phase Raman scattering cross sections of benzene and perdeuterated benzene. J. Chem. Phys. 1989, 90, 2909–2914. [Google Scholar] [CrossRef]
- Hewett, D.M.; Bocklitz, S.; Tabor, D.P.; Sibert, E.L., III; Suhm, M.A.; Zwier, T.S. Identifying the first folded alkylbenzene via ultraviolet, infrared, and Raman spectroscopy of pentylbenzene through decylbenzene. Chem. Sci. 2017, 8, 5305–5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Castillo, A.O.; Abeysekera, C.; Hays, B.M.; Kleiner, I.; Nguyen, H.V.L.; Zwier, T.S. Conformational preferences and internal rotation of methyl butyrate by microwave spectroscopy. J. Mol. Spectrosc. 2017, 337, 51–58. [Google Scholar] [CrossRef]
- Forsting, T.; Suhm, M. Curry-Jet SETUP. 2019. Available online: https://figshare.com/articles/dataset/Curry-Jet_SETUP/6395840/1 (accessed on 23 July 2021).
- Harris, W.; Coe, D.; George, W. Vibrational spectra and structure of esters—II. Raman spectra and potential function calculations for HCOOCH3, DCOOCH3 and HCOOCD3. Spectrochim. Acta Part A 1976, 32, 1–10. [Google Scholar] [CrossRef]
- Smalley, R.E.; Wharton, L.; Levy, D.H. Molecular optical spectroscopy with supersonic beams and jets. Acc. Chem. Res. 1977, 10, 139–145. [Google Scholar] [CrossRef]
- Gawrilow, M.; Suhm, M.A. 2-Methoxyethanol: Harmonic tricks, anharmonic challenges and chirality-sensitive chain aggregation. Phys. Chem. Chem. Phys. 2020, 22, 15303–15311. [Google Scholar] [CrossRef]
- Bloino, J.; Biczysko, M.; Barone, V. Anharmonic Effects on Vibrational Spectra Intensities: Infrared, Raman, Vibrational Circular Dichroism, and Raman Optical Activity. J. Phys. Chem. A 2015, 119, 11862–11874. [Google Scholar] [CrossRef] [Green Version]
- Montero, S. Anharmonic Raman intensities of overtones, combination and difference bands. J. Chem. Phys. 1982, 77, 23–29. [Google Scholar] [CrossRef]
- Larsen, R.W.; Zielke, P.; Suhm, M.A. Hydrogen-bonded OH stretching modes of methanol clusters: A combined IR and Raman isotopomer study. J. Chem. Phys. 2007, 126, 194307. [Google Scholar] [CrossRef] [PubMed]
- Shimanouchi, T. Molecular Vibrational Frequencies. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1997; Available online: https://doi.org/10.18434/T4D303 (accessed on 23 July 2021).
- Long, D.A. The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; Wiley: Chichester, UK; New York, NY, USA, 2002. [Google Scholar]
- Luijks, G.; Timmerman, J.; Stolte, S.; Reuss, J. Raman analysis of SF6 molecular beams excited by a cw CO2 laser. Chem. Phys. 1983, 77, 169–184. [Google Scholar] [CrossRef]
- Amrein, A.; Quack, M.; Schmitt, U. High-resolution interferometric Fourier transform infrared absorption spectroscopy in supersonic free jet expansions: Carbon monoxide, nitric oxide, methane, ethyne, propyne, and trifluoromethane. J. Phys. Chem. US 1988, 92, 5455–5466. [Google Scholar] [CrossRef]
- Blom, C.; Günthard, H. Rotational isomerism in methyl formate and methyl acetate; a low-temperature matrix infrared study using thermal molecular beams. Chem. Phys. Lett. 1981, 84, 267–271. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Lagrangian approach to molecular vibrational Raman intensities using time-dependent hybrid density functional theory. J. Chem. Phys. 2007, 126, 201104. [Google Scholar] [CrossRef] [Green Version]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Zuber, G.; Hug, W. Rarefied Basis Sets for the Calculation of Optical Tensors. 1. The Importance of Gradients on Hydrogen Atoms for the Raman Scattering Tensor. J. Phys. Chem. A 2004, 108, 2108–2118. [Google Scholar] [CrossRef]
- Curl, R.F. Microwave Spectrum, Barrier to Internal Rotation, and Structure of Methyl Formate. J. Chem. Phys. 1959, 30, 1529–1536. [Google Scholar] [CrossRef]
- Fateley, W.; Miller, F.A. Torsional frequencies in the far infrared—I. Spectrochim. Acta 1961, 17, 857–868. [Google Scholar] [CrossRef]
- Gilson, T.R.; Hendra, P.J. Laser Raman Spectroscopy: A Survey of Interest Primarily to Chemists, and Containing a Comprehensive Discussion of Experiments on Crystals; Wiley-Interscience: Chichester, UK, 1970. [Google Scholar]
- Watson, J.K.G. Asymptotic energy levels of a rigid asymmetric top. Mol. Phys. 2007, 105, 679–688. [Google Scholar] [CrossRef]
- PGOPHER. A Program for Simulating Rotational, Vibrational and Electronic Spectra, C. M. Western, University of Bristol. Available online: http://pgopher.chm.bris.ac.uk (accessed on 23 July 2021).
- Avila, G.; Fernández, J.; Maté, B.; Tejeda, G.; Montero, S. Ro-vibrational Raman Cross Sections of Water Vapor in the OH Stretching Region. J. Mol. Spectrosc. 1999, 196, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 014108. [Google Scholar] [CrossRef]
- Puzzarini, C.; Stanton, J.F.; Gauss, J. Quantum-chemical calculation of spectroscopic parameters for rotational spectroscopy. Int. Rev. Phys. Chem. 2010, 29, 273–367. [Google Scholar] [CrossRef]
- Goubet, M.; Pirali, O. The far-infrared spectrum of azulene and isoquinoline and supporting anharmonic density functional theory calculations to high resolution spectroscopy of polycyclic aromatic hydrocarbons and derivatives. J. Chem. Phys. 2014, 140, 044322. [Google Scholar] [CrossRef]
- Licari, D.; Tasinato, N.; Spada, L.; Puzzarini, C.; Barone, V. VMS-ROT: A New Module of the Virtual Multifrequency Spectrometer for Simulation, Interpretation, and Fitting of Rotational Spectra. J. Chem. Theory Comput. 2017, 13, 4382–4396. [Google Scholar] [CrossRef]
- Hills, G.; Foster, R.; Jones, W. Raman spectra of asymmetric top molecules. Mol. Phys. 1977, 33, 1571–1588. [Google Scholar] [CrossRef]
- Jones, G.; Owen, N. Molecular structure and conformation of carboxylic esters. J. Mol. Struct. 1973, 18, 1–32. [Google Scholar] [CrossRef]
- Nejad, A.; Suhm, M.A.; Meyer, K.A.E. Increasing the weights in the molecular work-out of cis- and trans-formic acid: Extension of the vibrational database via deuteration. Phys. Chem. Chem. Phys. 2020, 22, 25492–25501. [Google Scholar] [CrossRef]
- Lüttschwager, N.O.B. Raman Spectroscopy of Conformational Rearrangements at Low Temperatures; Springer International Publishing: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Dahmani, R.; Sun, H.; Mouhib, H. Quantifying soft degrees of freedom in volatile organic compounds: Insight from quantum chemistry and focused single molecule experiments. Phys. Chem. Chem. Phys. 2020, 22, 27850–27860. [Google Scholar] [CrossRef]
- Beattie, J.R.; Bell, S.E.J.; Moss, B.W. A critical evaluation of Raman spectroscopy for the analysis of lipids: Fatty acid methyl esters. Lipids 2004, 39, 407–419. [Google Scholar] [CrossRef]
- Erlekam, U.; Frankowski, M.; von Helden, G.; Meijer, G. Cold collisions catalyse conformational conversion. Phys. Chem. Chem. Phys. 2007, 9, 3786. [Google Scholar] [CrossRef] [Green Version]
- Sadlej, A.J. Medium-size polarized basis sets for high-level correlated calculations of molecular electric properties. Collect. Czech. Chem. Commun. 1988, 53, 1995–2016. [Google Scholar] [CrossRef]
- Zwier, T.S.; (Department of Chemistry, Purdue University, West Lafayette, IN, USA). Personal communication, 2021.
- TURBOMOLE V7.4.1 2019, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 23 July 2021).
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Weigend, F. A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. [Google Scholar] [CrossRef]
- Bachorz, R.A.; Bischoff, F.A.; Glöß, A.; Hättig, C.; Höfener, S.; Klopper, W.; Tew, D.P. The MP2-F12 method in the TURBOMOLE program package. J. Comput. Chem. 2011, 32, 2492–2513. [Google Scholar] [CrossRef]
- Hättig, C.; Tew, D.P.; Köhn, A. Communications: Accurate and efficient approximations to explicitly correlated coupled-cluster singles and doubles, CCSD-F12. J. Chem. Phys. 2010, 132, 231102. [Google Scholar] [CrossRef]
- Marchetti, O.; Werner, H.J. Accurate calculations of intermolecular interaction energies using explicitly correlated wave functions. Phys. Chem. Chem. Phys. 2008, 10, 3400. [Google Scholar] [CrossRef]
- Eaton, J.W.; Bateman, D.; Hauberg, S.; Wehbring, R. GNU Octave Version 6.1.0 Manual: A High-Level Interactive Language for Numerical Computations. 2020. Available online: https://www.gnu.org/software/octave/doc/v6.1.0/ (accessed on 23 July 2021).
- Kramida, A.; Ralchenko, Y.; Reader, J.; NIST ASD Team. NIST Atomic Spectra Database; (ver. 5.8); National Institute of Standards and Technology: Gaithersburg, MD, USA, 2020. Available online: https://physics.nist.gov/asd (accessed on 23 July 2021).
- Karir, G.; Lüttschwager, N.O.B.; Suhm, M.A. Phenylacetylene as a gas phase sliding balance for solvating alcohols. Phys. Chem. Chem. Phys. 2019, 21, 7831–7840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüttschwager, N.O.B. NoisySignalIntegration.jl; A Julia Package to Determine Uncertainty in Numeric Integrals of Noisy x-y Data. Available online: https://github.com/nluetts/NoisySignalIntegration.jl (accessed on 23 July 2021).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | /cm | B/d | B/dD | B/a | P/d | P/a | Γ | Mode Description | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 132 ± 4 | 1.9 ± 1.2 | 6.7 | 5.7 | 5.1 | 7.3 | 5.6 | (CH) | |||
| 2 | 234 ± 4 | 1.9 ± 1.2 | ||||||||
| 312; 315 a |  | 27.3 ± 2.9 | 43.7 | 42.1 | 42.5 | 44.6 | 43.7 | () | ||
| 332 ± 4 | () | |||||||||
| 2 | 660 | 0.2 ± 0.6 | ||||||||
| 769 | 5.1 ± 0.9 | 8.6 | 6.3 | 6.3 | 8.4 | 6.3 | () −() | |||
| 928 | 47.2 ± 2.3 | 46.4 | 47.9 | 47.9 | 43.2 | 44.9 | () +() | |||
| 1024 ± 4 | 2.9 ± 0.8 | 3.9 | 3.8 | 3.8 | 4.0 | 3.9 | () | |||
| 1167 | | 4.3 ± 1 | 8.4 | 6.4 | 6.4 | 7.0 | 4.7 | (CH) | ||
| 1167 | (C–O) −() −(CH) | |||||||||
| 1210 | 3 ± 0.8 | 3.9 | 3.8 | 3.9 | 5.1 | 5.7 | () −() +(CH) | |||
| 1370 | 10.2 ± 1.1 | 12.2 | 9.6 | 9.6 | 12.7 | 10.4 | () | |||
| 1438 |  | 17.1 ± 1.5 | 23.1 | 18.5 | 18.5 | 25.2 | 20.5 | umbrella (CH) | ||
| 1452 ± 4 | (CH) | |||||||||
| 1465 | (CH) | |||||||||
| — | 1585 | 1.3 ± 0.8 | ||||||||
| 1755 | 25.5 ± 1.5 | 18.4 | 24.0 | 24.0 | 18.9 | 24.2 | (C=O) |
| Gas Reference [11] | This Work | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mode | ||||||||||
| 313 | 13 ± 3 | 311 | 32.6 ± 4.6 | 312 | 28 ± 4.5 | 301 | 40 | 302 ± 2 | 38.7 ± 3.2 | |
| 919 | 38 ± 3 | 926 | 38.4 ± 4.3 | 928 | 39.7 ± 1.8 | 928 | 34.6 | 948 ± 21 | 33.8 ± 3.1 | |
| 1207 | 6 ± 3 | 1209 | 3.1 ± 2.6 | 1210 | 2.5 ± 0.6 | 1229 | 2.9 | 1242 ± 14 | 3.5 ± 0.6 | |
| 1368 | 10 ± 3 | 1371 | 8.7 ± 4.5 | 1370 | 8.5 ± 0.8 | 1399 | 9 | 1398 ± 1 | 8.2 ± 1.1 | |
| 1751 | 34 ± 3 a | 1755 | 17.1 ± 2.3 | 1755 | 21.2 ± 1.2 | 1790 | 13.5 | 1809 ± 21 | 15.8 ± 2.6 | |
| Mode | /cm | |||||
|---|---|---|---|---|---|---|
| 132 | 0.28 ± 0.17 | 0.95 | 0.50 | 0.35 | 0.14 | |
| 312; 332 | 0.62 ± 0.07 | 0.50 | 0.33 | 0.27 | 0.12 | |
| 769 | 0.59 ± 0.10 | 1.21 | 0.87 | 0.62 | 0.22 | |
| 928 | 1 | 1 | 1 | 1 | 1 | |
| 1024 | 0.73 ± 0.20 | 1.03 | 0.54 | 0.37 | 0.15 | |
| 1167 | 0.51 ± 0.11 | 0.77 | 0.59 | 0.47 | 0.21 | |
| 1210 | 0.75 ± 0.19 | 0.80 | 0.81 | 0.81 | 1.5 | |
| 1370 | 0.83 ± 0.09 | 0.93 | 0.91 | 0.89 | 1.02 | |
| 1452 | 0.73 ± 0.07 | 0.79 | 0.41 | 0.29 | 0.08 | |
| 1755 | 1.37 ± 0.10 | 1.40 | 1.40 | 1.40 | 2.22 |
| Conformer | [6] | [9] | ||||
|---|---|---|---|---|---|---|
| tgt | 2.2 | (1.8) | 3.2 | (2.8) | 3.1 | 2.6 |
| tgt–ttt ‡ | 2.3 | (1.9) | 3.1 | (2.7) | – | 4.4 |
| ttt | 0 | (0) | 0 | (0) | 0 | 0.1 |
| ttt–ttg ‡ | 10.5 | (9.9) | 10.4 | (9.8) | 11.3 | 11.1 |
| ttg | −0.6 | (−0.6) | 0.0 | (0.1) | 0.2 | 0 |
| ttg–tgg ‡ | 5.9 | (5.4) | 6.8 | (6.4) | – | 7.2 |
| tgg | 1.8 | (1.8) | 2.9 | (2.9) | 3.9 | 2.5 |
| tgg–tgt ‡ | 15.5 | (14.7) | 16.3 | (15.5) | – | – |
| Method | ttt | ttg | |
|---|---|---|---|
| This | Raman Integrals | 43 ± 8 | 57 ± 8 |
| Work | Raman Peak Heights | 46 ± 9 | 54 ± 9 |
| CCSD(T)//B3LYP | 51 | 49 | |
| CCSD(T)//B3LYP | 47 | 53 | |
| CCSD(T)//B3LYP | 40 | 60 | |
| CCSD(T)//B3LYP | 37 | 63 | |
| Ref. [9] | Microwave | 41 ± 4 | 59 ± 6 |
| B2PLYP | 51 | 49 | |
| B2PLYP | 38 | 62 | |
| MP2 | 34 | 66 | |
| MP2 | 30 | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawrilow, M.; Suhm, M.A. Quantifying Conformational Isomerism in Chain Molecules by Linear Raman Spectroscopy: The Case of Methyl Esters. Molecules 2021, 26, 4523. https://doi.org/10.3390/molecules26154523
Gawrilow M, Suhm MA. Quantifying Conformational Isomerism in Chain Molecules by Linear Raman Spectroscopy: The Case of Methyl Esters. Molecules. 2021; 26(15):4523. https://doi.org/10.3390/molecules26154523
Chicago/Turabian StyleGawrilow, Maxim, and Martin A. Suhm. 2021. "Quantifying Conformational Isomerism in Chain Molecules by Linear Raman Spectroscopy: The Case of Methyl Esters" Molecules 26, no. 15: 4523. https://doi.org/10.3390/molecules26154523
APA StyleGawrilow, M., & Suhm, M. A. (2021). Quantifying Conformational Isomerism in Chain Molecules by Linear Raman Spectroscopy: The Case of Methyl Esters. Molecules, 26(15), 4523. https://doi.org/10.3390/molecules26154523