Abstract
The activation reactions of methane mediated by metal carbide ions MC3+ (M = Ir and Pt) were comparatively studied at room temperature using the techniques of mass spectrometry in conjunction with theoretical calculations. MC3+ (M = Ir and Pt) ions reacted with CH4 at room temperature forming MC2H2+/C2H2 and MC4H2+/H2 as the major products for both systems. Besides that, PtC3+ could abstract a hydrogen atom from CH4 to generate PtC3H+/CH3, while IrC3+ could not. Quantum chemical calculations showed that the MC3+ (M = Ir and Pt) ions have a linear M-C-C-C structure. The first C–H activation took place on the Ir atom for IrC3+. The terminal carbon atom was the reactive site for the first C–H bond activation of PtC3+, which was beneficial to generate PtC3H+/CH3. The orbitals of the different metal influence the selection of the reactive sites for methane activation, which results in the different reaction channels. This study investigates the molecular-level mechanisms of the reactive sites of methane activation.
1. Introduction
Methane has attracted attention as the main component of natural gas and the conversion of methane into value-added chemicals is very important [1,2]. However, methane is extremely stable, with high C–H bond strengths (439 kJ/mol), negligible electron affinity and low polarizability [3]. At present, most of the catalytic conversion of methane needs to be carried out under high-temperature or high-pressure conditions [4]. Metal carbides are a kind of molecules with the potential to activate methane and have been studied by several groups [5]. Research on the mechanisms of the activation of methane by metal carbides is of great value and it may be helpful to find new catalysts of metal carbides [6,7]. At the same time, it is very difficult to study the activation mechanisms of methane. In recent years, people have found that the study of the gas-phase reaction of methane is an important means to study related reaction mechanisms [8,9].
Past studies have shown that gas-phase clusters are an ideal model for studying the reaction mechanisms in condensed-phase systems. The study of gas-phase clusters can reveal the specific reaction mechanisms, including the active sites, and provide references for condensed-phase catalytical processes [10]. At present, researchers are concerned about the reactions of methane with metal ions such as Os+ [11], Pt+ [12], Ta+ [13] and Rh(0) [14] and metal oxides such as MgO+ [15], PbO+ [16], V4O10+ [17] and Re2O7+ [18]. In addition to metal ions and their oxides, studies have shown that metal carbides can also activate methane, such as FeC6− [19], Mo2C2− [20], Ta2C4− [21], FeC3− [6], AuC+ [22], FeC4+ [23] and MC+ [7]. These studies have explored some possible mechanisms for the activation of methane by metal carbide clusters. For example, the study of AuC+ reveals a special hydride-transfer mechanism (HT) [22], while the study of FeC3- shows that methane and atomic clusters generate CC-coupling reaction products at high temperatures and explained the possible mechanism of non-oxidized methane aromatization at the molecular level [6]. The study of FeC4+ shows that the cluster can activate the C–H bonds of methane via the hydrogen-atom transfer (HAT) mechanism at ambient temperature; the study used the frontier orbital theory to explain the root cause of the HAT reaction [23]. Although there have been some studies on the mechanisms of metal carbides to activate methane in the past, there is still no clear investigation on the reactive sites of methane activation. Therefore, further research is needed to study the reactive sites for the activation of methane. Here, we reported the reactions of MC3+ (M = Pt and Ir) with methane.
2. Experimental and Computational Methods
The experiments were performed using an ion trap mass spectrometer equipped with a laser vaporization–supersonic expansion ion source that was reported previously [24,25]. The MC3+ (M = Pt and Ir) ions were generated by pulsed laser ablation of a rotating and translating metal/carbon (metal:carbon = 1:4) target. The nascent ablated plasma was entrained by a helium carrier gas with a backing pressure of about 0.5 MPa. The ions were mass-selected by a quadrupole and then were sent into a linear ion trap, where the ions were accumulated and cooled by helium gas. The MC3+ ions reacted with CH4, CD4 and 13CH4, introduced by a pulsed valve. After a 10 ms reaction, the trapped ions were ejected for mass detection.
Theoretical calculations were performed using the Gaussian 09 package [26]. All of the calculations were performed using the BMK functional with the def2-TZVP basis sets [27,28]. Vibrational frequency calculations were employed to identify the nature of reaction intermediates, transition states (TSs) and products. The Molclus program [29] was used to search for the possible stable structures of the MC3+, MC2H2+, MC4H2+ and MC3H+ (M = Pt and Ir). The low-lying stable isomers were then re-optimized at the BMK/def2TZVP level to confirm the relative energy sequence. Transition-state optimizations were performed with the synchronous transit-guided quasi-Newton (STQN) method and were verified through intrinsic reaction coordinate (IRC) calculations [30,31]. Vibrational frequency calculations were performed to identify the nature of reaction intermediates, transition states (TSs) and products.
3. Results and Discussion
The mass spectra for the reactions of mass-selected MC3+ (M = Pt and Ir) ions with He (a1 and a2), CH4 (b1 and b2), CD4 (c1 and c2) and 13CH4 (d1 and d2) in the ion trap at room temperature are shown in Figure 1. No product ion was observed in the mass spectra when using pure He as reactant gas, while two main peaks, at m/z = 219 and 243 for the Ir-system, and m/z = 222 and 246 for the Pt-system for the reactions with CH4, which can be attributed to the product ions with chemical formulas MC2H2+ and MC4H2+ (M = 196Pt and 193Ir), were observed to be the major reaction products. A weak mass peak at m/z = 233 assigned to the ion with chemical formula PtC3H+ for PtC3+/CH4 was also observed, while no IrC3H+ was found. The mass spectra suggest that two main reaction channels for both systems and one hydrogen-atom abstraction channel for PtC3+ were observed. The first channel is the formation of the MC2H2+ cation with the release of a neutral C2H2 (reaction 1). The second channel is the generation of the MC4H2+ ion with concomitant elimination of a dihydrogen molecule (reaction 2). The third channel is the generation of the PtC3H+ ion with the release of CH3 as shown in reaction 3.
MC3+ + CH4 → MC2H2+ + C2H2
MC3+ + CH4 → MC4H2+ + H2
PtC3+ + CH4 → PtC3H+ + CH3
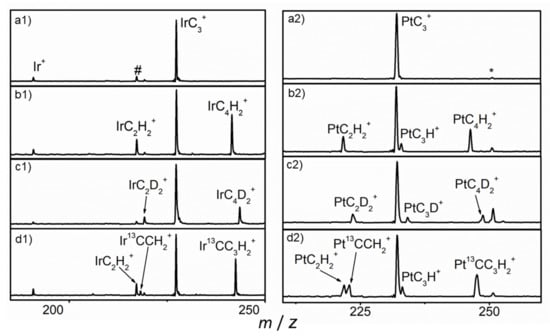
Figure 1.
Mass spectra from the reactions of MC3+ (M = Pt and Ir) with He (a1 and a2), CH4 (b1 and b2), CD4 (c1 and c2) and 13CH4 (d1 and d2). # and * denote IrCO+ and PtC3H2O+.
Impurity PtC3•H2O+ and IrCO+ ions were formed due to the small amount of contaminant in the chamber of the instrument. Isotopic-labeling experiments conducted using the CD4 sample showed that there were peaks of IrC2D2+, IrC4D2+, PtC2D2+, PtC4D2+ and PtC3D+, which demonstrates that the H atoms in the products were all from methane. Both the PtC2H2+ and Pt13CCH2+ product ions were observed to have approximately the same intensity, when using 13CH4, indicating that one or both carbon atoms of the eliminated C2H2 neutral molecule came from the PtC3+ ion. The intensity of the peak of Ir13CCH2+ is similar with that of IrC2H2+, which eliminates the background peak. The peaks of Ir13CC3H2+ and Pt13CC3H2+ were observed, which confirms the reaction channels.
In order to gain insight into the reaction mechanisms, the various possible structures of the products MC3+ (M = Pt and Ir) were obtained by calculations at the BMK/def2-TZVP level and are shown in Figure S1. The most stable structures of MC3+ (M = Pt and Ir) have a linear structure M-C-C-C, with the ground state 1Σ+ (IrC3+) and 2Σ− (PtC3+). For the products (Figure S2), the most stable structures of MC2H2+ and MC4H2+ (M = Pt and Ir) are metal cations with C2H2 and linear H-C-C-C-C-H, respectively. For MC3H+ (M = Pt and Ir), the most stable structures of MC3H+ (M = Pt and Ir) have a linear structure M-C-C-C-H, given in Figure S2.
In order to gain insight into the reaction mechanism, the potential energy profiles (PESs) were calculated. All of the three Reactions (1)–(3) leading to the stable structures of the products were exothermic. The pathways for the IrC3+ + CH4 reaction leading to the IrC2H2+/C2H2 and IrC4H2+/H2 products are shown in Figure 2 and details are given in Figures S3–S4. An encounter complex (3I1), that is −1.51 eV lower in energy than the ground state reactants, is formed initially. The Ir atom serves as the active site of the reaction and intermediate 1I2 is formed via the first C–H bond activation. Then, the CH3 moiety is transferred to C3 to form a C–C bond; meanwhile, one H atom is transferred to the Ir atom (1I2 → 1TS2 → 1I3). Subsequently, two hydrogen atoms transfer from the metal Ir atom to the C atom to form two new C–H bonds (1I3 → 1TS3 → 1I4 → 1TS4 → 1I5). The H atom of the CH2 moiety is activated and transferred to the Ir metal (1I5 → 1TS5 → 1I6); then, the H atom migrates from the Ir metal to the C atom which is not coordinated to other H atoms (1I6 → 1TS6 → 1I7). After the CC bond is cleft, the intermediate 2I8 is formed, which involves two equivalent C2H2 moieties—either one can be liberated to form the final product P1 (3IrC2H2+/C2H2). For the pathway for the generation of IrC4H2+/H2, there is the rearrangement steps from 1I3 to form intermediate 1I10 (1I3 → 1TS8 → 1I9 → 1TS9 → 1I10). The final product 1IrC4H2+ is generated with the liberation of H2. Another possible pathway is given in Figure S4.
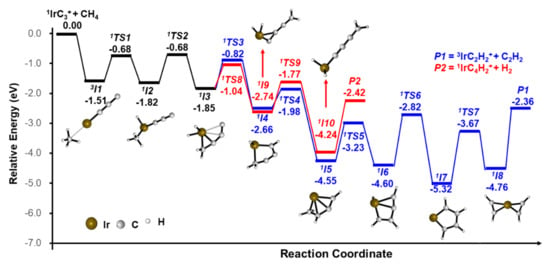
Figure 2.
Reaction pathways for generating IrC2H2+/C2H2 (blue trace) and IrC4H2+/H2 (red trace) from the reaction of IrC3+ and CH4, calculated at the BMK level. The zero-point vibrational energy-corrected energies (in eV) relative to the entrance channel are given.
The pathways for the PtC3+ + CH4 reaction are shown in Figure 3. Details and other possible pathways are given in Figures S5–S7. The reaction pathway starts from the encounter complex 2I1, followed by the formation of a stable intermediate 2I2 via the approaching of CH4 to the terminal carbon of PtC3+ (2I1 → 2TS1 → 2I2), where the first C–H activation takes place. The CH3 moiety of 2I2 is transferred to form 2I3, where CH3 is loosely coordinated to PtC3H+. The final product P1 (−0.46 eV) is generated with the liberation of CH3. The energy of P1 is higher than the transition states from 2I2 to P2/P3, which can explain the weak peak of PtCH3+, compared with PtC4H2+ and PtC2H2+. From 2I2, the H atom from the CH3 moiety is activated and transferred to the metal center to form intermediate 2I4 (2I2→ 2TS3 → 2I4). The H atom subsequently migrates from the Pt metal to the terminal C atom to form intermediate 2I5 (2I4 → 2TS4 → 2I5). After rearrangement (2I5 → 2TS5 → 2I6), the H atom of the CH2 moiety is activated and transferred to the Pt metal again (2I6 → 2TS6 → 2I7). Then, the H atom migrates from the Pt metal to C atom which is not coordinated to other H atoms (2I7 → 2TS7 → 2I8). After the C and Pt atoms are bonded, the stable intermediate 2I9 with a five-membered ring is generated. After the CC bond is cleft, the intermediate 2I10 is formed, which involves two equivalent C2H2 moieties—either one can be liberated to form the final product P2 (2PtC2H2+/C2H2). For another pathway to generate PtC4H2+/H2, after the rearrangement from 2I5 to form 2I11, the H atom of the CH2 moiety is activated and transferred to the Pt metal again (2I11 → 2TS11 → 2I12). The subsequent reaction pathway is the H–H bond formation; then, the final product PtC4H2+ is generated with the liberation of H2.
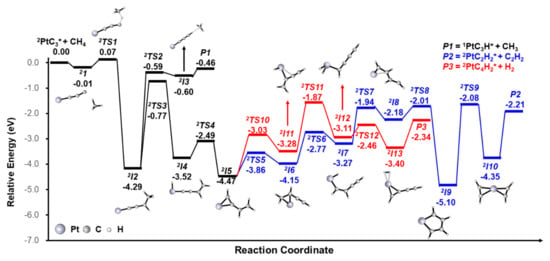
Figure 3.
Reaction pathways for generating PtC2H2+/C2H2 (blue trace), PtC4H2+/H2 (red trace) and PtC3H+/CH3 from the reaction of PtC3+ and CH4, calculated at the BMK level. The zero-point vibrational energy-corrected energies (in eV) relative to the entrance channel are given.
For the reactions of the IrC3+ and PtC3+ with CH4, we could find the hydrogen abstraction reaction products PtC3H+ and no IrC3H+ was observed. Based on our calculations, the reactive sites play a key role. There are two possible sites, the metal and terminal carbon atom (the other two carbon atoms are fully bonded). For IrC3+/CH4, the barrier to the first C–H activation on the terminal C atom (+0.04 eV; see Figure S4) is higher in energy than 1TS1 (−0.68 eV). Based on the lack of IrC3H+ observed, we confirm that the reactive site of the first C–H activation is the Ir atom. For PtC3+/CH4, the energy required for the first C–H bond activation on Pt and C atoms is +0.01 (2TSX3) and +0.07 eV (2TS1), given in Figure 3 and Figure S6, respectively. Though the Pt atom, as a reactive site, is favorable, the transition state of H atom transfer as the next step (+0.19 eV, 2TSX4; Figure S4) is higher in energy than 2TS1. Based on the observation of PtC3H+ in the experiment, we confirm that the terminal carbon atom can be a reactive site for PtC3+/CH4.
As we all know, metals have a stronger adsorption capacity for methane than non-metals. The different reactive sites can be explained by the analysis of the orbital. In the PtC3+ species, platinum uses two of the six valence orbitals (s and d) to form a σ- and π-bond with the adjacent carbon atom. This leaves seven electrons occupying the four remaining non-bonding orbitals on platinum, such that there are no empty orbitals on the metal. Therefore, it is hard for the C−H bond to donate electron density to the metal and the energy of the transition state 2TSX3 is a little higher than the 2TS1 in Figure 3. For the IrC3+ species, Ir uses two of the six valence orbitals (s and d) to form a bond with the adjacent carbon atom, which leaves six electrons occupying the four remaining non-bonding orbitals on the Ir atom, such that there are enough empty orbitals on the metal, which is beneficial for the C–H bond activation of CH4. This can explain why the barrier to the first C–H activation on the terminal C atom (+0.04 eV) is higher than that of 1TS1 (−0.68 eV). Our work demonstrates that the orbitals of the metal influence the selection of the reactive sites for the activation of methane. The different reactive sites result in different reaction channels.
In conclusion, the activation of methane mediated by MC3+ (M = Ir and Pt) were comparatively studied at room temperature by gas-phase experiments with theoretical calculations. Mass spectrometric studies on the reactions of the MC3+ (M = Ir and Pt) ions with CH4 show that two main reaction channels were observed. The first channel is the formation of the MC2H2+ cation with the release of neutral C2H2. The second channel is the generation of the MC4H2+ ion with concomitant elimination of a dihydrogen molecule. Besides that, PtC3H+ could be found in the experiments, while IrC3H+ could not. Quantum chemical calculations suggest that the MC3+ (M = Ir and Pt) ions have a linear M-C-C-C+ structure. The Ir atom is the reactive site for the reaction of IrC3+/CH4. The generation of the PtC3H+ and PESs can confirm that the terminal carbon atom is the reactive site for PtC3+/CH4. The orbitals of the metal influence the selection of the reactive sites for methane activation, which results in the different reaction channels. Our work is helpful for understanding reactive sites of methane activation.
Supplementary Materials
The following are available online, Figure S1: The optimized geometries of the [IrC3]+ and [PtC3]+ isomers at the BMK/def2-TZVP level. The relative energies relative to the global minimum structure (in eV), symmetry and electronic states are shown; Figure S2: The optimized geometries of the [IrC2H2]+/[PtC2H2]+ (top), [IrC4H2]+/[PtC4H2]+ (middle) and [IrC3H]+/[PtC3H]+ (bottom) isomers at the BMK/def2-TZVP level. The relative energies relative to the global minimum structure (in eV), symmetry and electronic states are shown; Figure S3: The detailed potential energy profiles of the reaction of IrC3+ and CH4 in Figure 2. The energies are given in eV; Figure S4: The other possible potential energy profiles of the reaction of IrC3+ and CH4. The energies are given in eV; Figure S5. The detailed potential energy profiles of the reaction of PtC3+ and CH4 in Figure 3. The energies are given in eV; Calculated geometries and potential energy profiles. This material is available free of charge via the Internet.
Author Contributions
Conceptualization, X.W. and H.W.; methodology, X.W. and H.W.; formal analysis, W.L.; data curation, W.L.; writing—original draft preparation, Z.L. and X.W.; writing—review and editing, X.W.; supervision, X.W. All authors have read and agreed to the published version of the manuscript.
Funding
Support from the National Natural Science Foundation of China (Grant No. 21603037, 21973016).
Data Availability Statement
Not available.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 21603037, 21973016).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- Choudhary, T.; Aksoylu, E.; Goodman, D.W. Nonoxidative Activation of Methane. Catal. Rev. 2003, 45, 151–203. [Google Scholar] [CrossRef]
- Peter, M.; Marks, T.J. Platinum Metal-Free Catalysts for Selective Soft Oxidative Methane Ethylene Coupling. Scope and Mechanistic Observations. J. Am. Chem. Soc. 2015, 137, 15234–15240. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1594. [Google Scholar] [CrossRef]
- Schwarz, H.; Shaik, S.; Li, J. Electronic Effects on Room-Temperature, Gas-Phase C-H Bond Activations by Cluster Oxides and Metal Carbides: The Methane Challenge. J. Am. Chem. Soc. 2017, 139, 17201–17212. [Google Scholar] [CrossRef]
- Li, H.-F.; Jiang, L.-X.; Zhao, Y.-X.; Liu, Q.-Y.; Zhang, T.; He, S.-G. Formation of Acetylene in the Reaction of Methane with Iron Carbide Cluster Anions FeC3—Under High-Temperature Conditions. Angew. Chem. Int. Ed. 2018, 57, 2662–2666. [Google Scholar] [CrossRef]
- Geng, C.; Weiske, T.; Li, J.; Shaik, S.; Schwarz, H. Intrinsic Reactivity of Diatomic 3d Transition-Metal Carbides in the Thermal Activation of Methane: Striking Electronic Structure Effects. J. Am. Chem. Soc. 2018, 141, 599–610. [Google Scholar] [CrossRef]
- Ding, X.-L.; Wu, X.-N.; Zhao, Y.-X.; He, S.-G. C–H Bond Activation by Oxygen-Centered Radicals over Atomic Clusters. Accounts Chem. Res. 2011, 45, 382–390. [Google Scholar] [CrossRef]
- Gong, Y.; Zhou, M.; Andrews, L. Spectroscopic and Theoretical Studies of Transition Metal Oxides and Dioxygen Complexes. Chem. Rev. 2009, 109, 6765–6808. [Google Scholar] [CrossRef]
- Böhme, D.K.; Schwarz, H. Gas-Phase Catalysis by Atomic and Cluster Metal Ions: The Ultimate Single-Site Catalysts. Angew. Chem. Int. Ed. 2005, 44, 2336–2354. [Google Scholar] [CrossRef]
- Zhang, G.; Li, S.; Jiang, Y. Dehydrogenation of Methane by Gas-Phase Os+: A Density Functional Study. Organometallics 2003, 22, 3820–3830. [Google Scholar] [CrossRef]
- Wesendrup, R.; Schröder, D.; Schwarz, H. Catalytic Pt+-Mediated Oxidation of Methane by Molecular Oxygen in the Gas Phase. Angew. Chem. Int. Ed. 1994, 33, 1174–1176. [Google Scholar] [CrossRef]
- Irikura, K.K.; Beauchamp, J.L. Methane oligomerization in the gas phase by third-row transition-metal ions. J. Am. Chem. Soc. 1991, 113, 2769–2770. [Google Scholar] [CrossRef]
- Wang, G.; Chen, M.; Zhou, M. Activation of methane by Rh(0): Evidence for direct insertion of rhodium into the C–H bond at cryogenic temperatures. Chem. Phys. Lett. 2005, 412, 46–49. [Google Scholar] [CrossRef]
- Schröder, D.; Roithová, J. Low-Temperature Activation of Methane: It also Works Without a Transition Metal. Angew. Chem. Int. Ed. 2006, 45, 5705–5708. [Google Scholar] [CrossRef]
- Zhang, X.; Schwarz, H. Thermal Activation of Methane by Diatomic Metal Oxide Radical Cations: PbO+center dot as One of the Missing Pieces. Chemcatchem 2010, 2, 1391–1394. [Google Scholar] [CrossRef]
- Feyel, S.; Döbler, J.; Schröder, D.; Sauer, J.; Schwarz, H. Thermal Activation of Methane by Tetranuclear [V4O10]+. Angew. Chem. Int. Ed. 2006, 45, 4681–4685. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Wu, X.-N.; Wang, Z.-C.; He, S.-G.; Ding, X.-L. Hydrogen-atom abstraction from methane by stoichiometric early transition metal oxide cluster cations. Chem. Commun. 2010, 46, 1736–1738. [Google Scholar] [CrossRef]
- Li, H.-F.; Li, Z.-Y.; Liu, Q.-Y.; Li, X.-N.; Zhao, Y.-X.; He, S.-G. Methane Activation by Iron-Carbide Cluster Anions FeC6−. J. Phys. Chem. Lett. 2015, 6, 2287–2291. [Google Scholar] [CrossRef]
- Liu, Q.-Y.; Ma, J.-B.; Li, Z.-Y.; Zhao, C.; Ning, C.; Chen, H.; He, S. Activation of Methane Promoted by Adsorption of CO on Mo2C2−Cluster Anions. Angew. Chem. Int. Ed. 2016, 55, 5760–5764. [Google Scholar] [CrossRef]
- Li, H.-F.; Zhao, Y.-X.; Yuan, Z.; Liu, Q.-Y.; Li, Z.-Y.; Li, X.-N.; Ning, C.-G.; He, S.-G. Methane Activation by Tantalum Carbide Cluster Anions Ta2C4−. J. Phys. Chem. Lett. 2017, 8, 605–610. [Google Scholar] [CrossRef]
- Li, J.; Zhou, S.; Schlangen, M.; Weiske, T.; Schwarz, H. Hidden Hydride Transfer as a Decisive Mechanistic Step in the Reactions of the Unligated Gold Carbide [AuC]+ with Methane under Ambient Conditions. Angew. Chem. Int. Ed. 2016, 55, 13072–13075. [Google Scholar] [CrossRef]
- Geng, C.; Li, J.; Weiske, T.; Schwarz, H. A Reaction-Induced Localization of Spin Density Enables Thermal C–H Bond Activation of Methane by Pristine FeC4+. Chem.-A Eur. J. 2019, 25, 12940–12945. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-N.; Liu, Z.; Wu, H.; Zhang, D.; Li, W.; Huang, Z.; Wang, G.; Xu, F.; Ding, C.-F.; Zhou, M. Reactions of Transition-Metal Carbyne Cations with Ethylene in the Gas Phase. J. Phys. Chem. A 2020, 124, 2628–2633. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.-N.; Liu, Z.; Wu, H.; Zhang, D.; Ding, X.-L. The C/C Exchange in Activation/Coupling Reaction of Acetylene and Methane Mediated by Os+: A Comparison with Ir+, Pt+ and Au+. J. Phys. Chem. Lett. 2020, 11. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, G.; Petersson, A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef] [Green Version]
- Lu, L. Molclus program, Beijing Kein Research Center for Natural Science, China. 2016. Available online: http://www.keinsci.com/research/molclus.html (accessed on 1 September 2021).
- Peng, C.Y.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical-Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).