
Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-β1 Signaling Pathway

 , and
, and
Abstract
:1. Introduction
2. Results
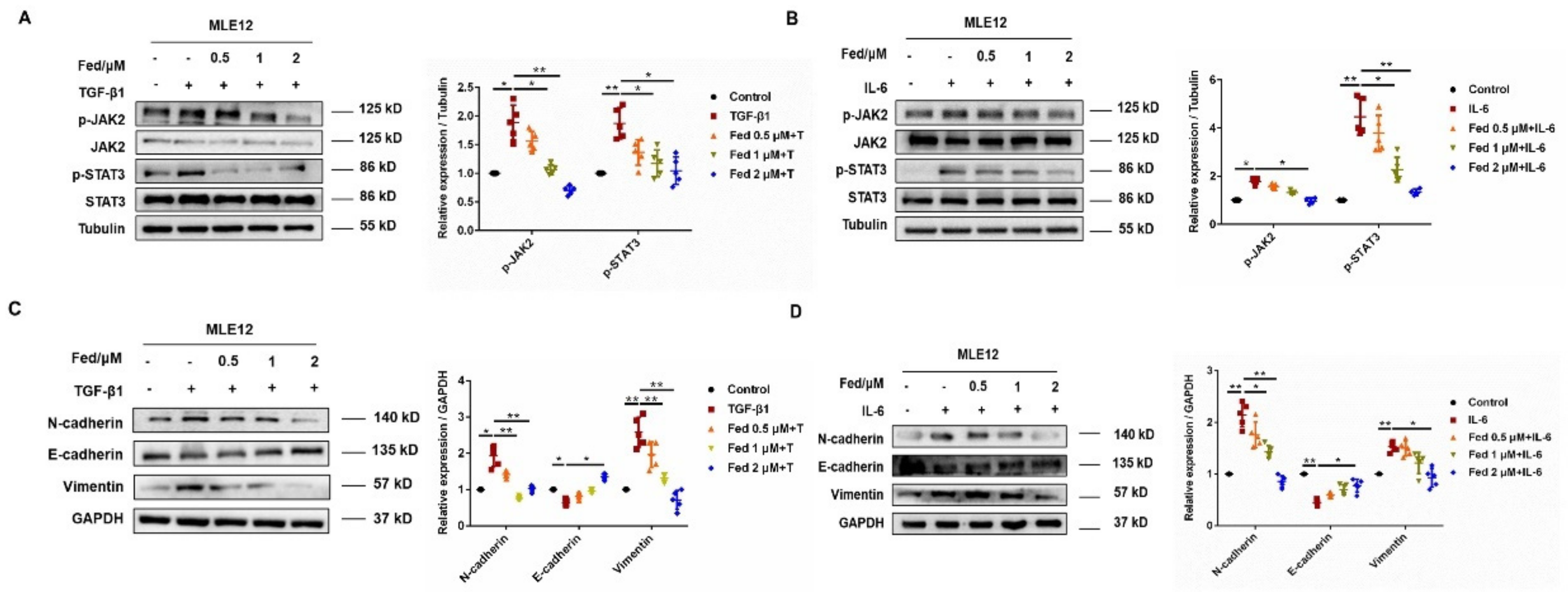
2.1. Fedratinib Inhibited Mesenchymal Marker Expression in Epithelial Cells and Fibroblast-to-Myofibroblast Transition by Suppressing JAK2/STAT3 Signaling
2.2. Fed Regulated the Production of Inflammatory Cytokines in Lung Epithelial Cells
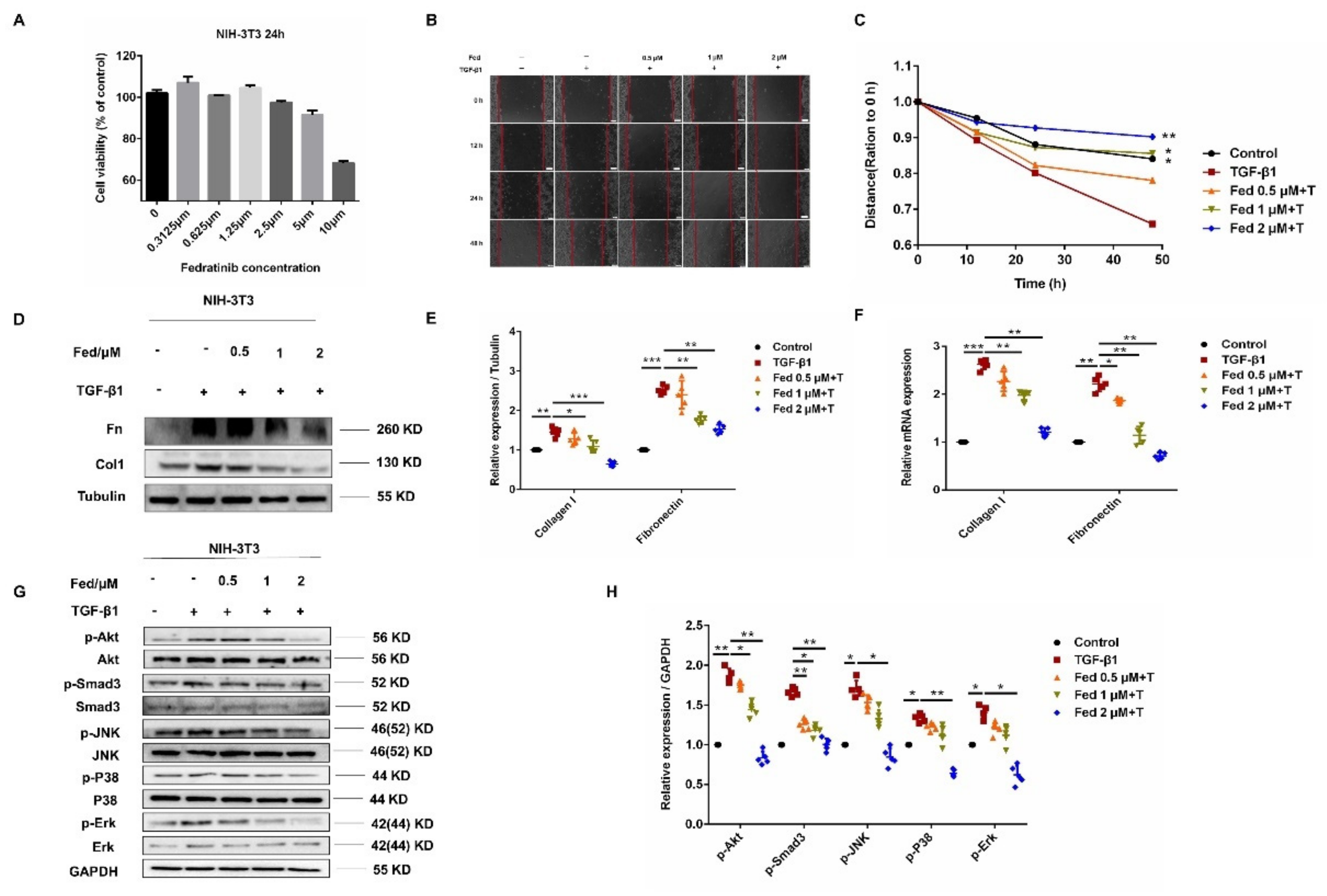
2.3. Fed Suppresses TGF-β1-Induced Myofibroblasts Proliferation, Migration, Activation and Apoptosis Resistance
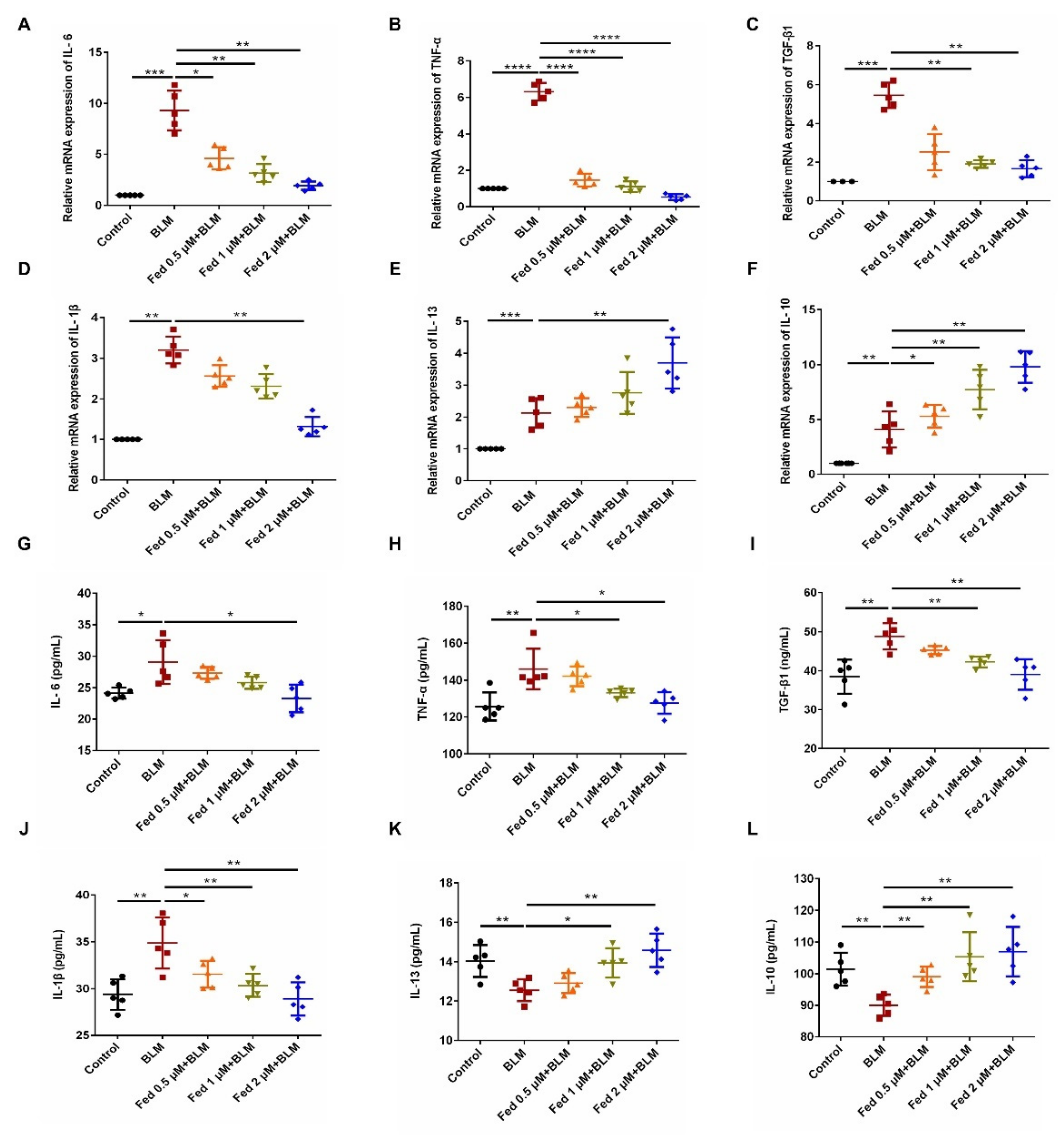
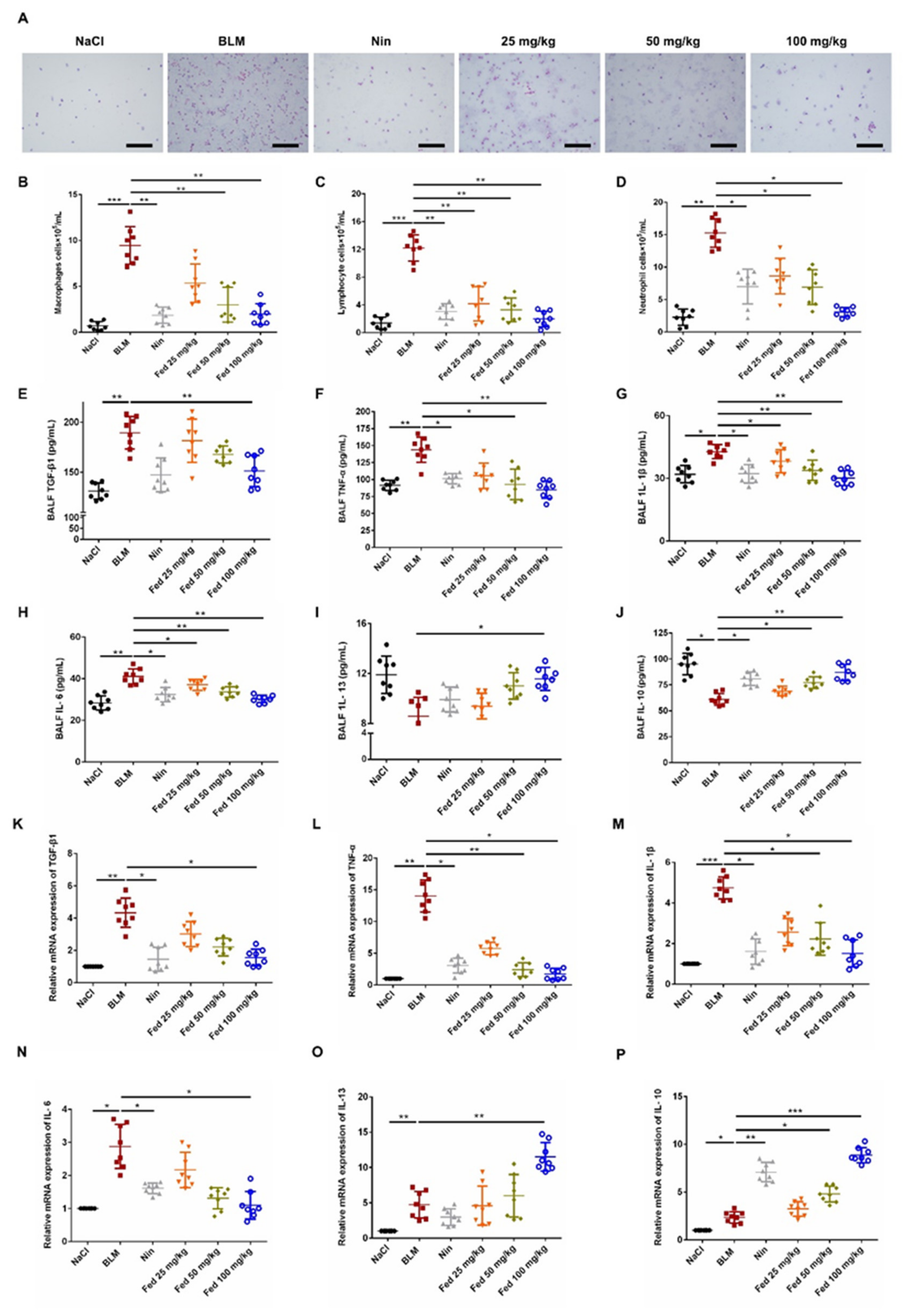
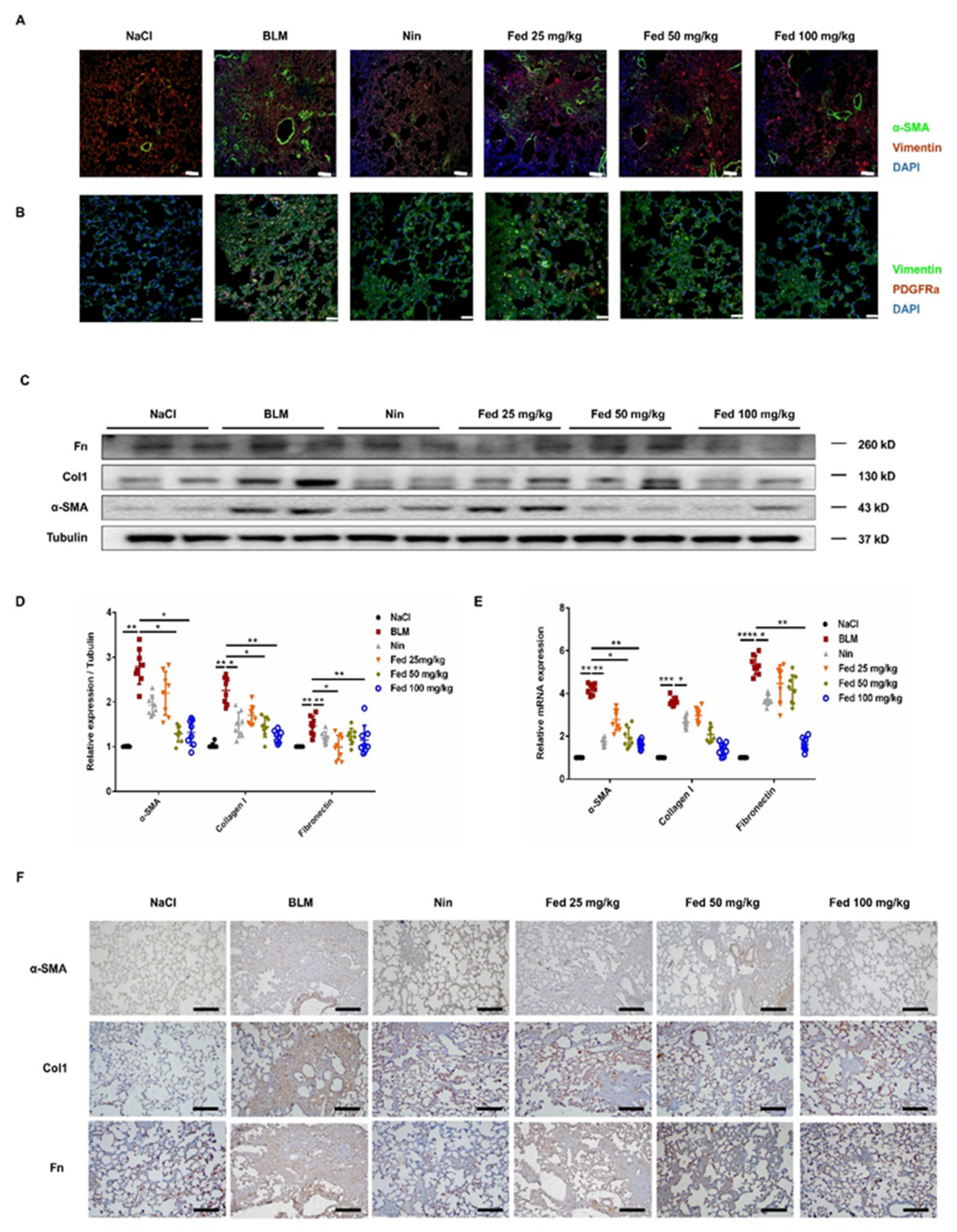
2.4. Influence of Fed on Bleomycin-Induced Lung Fibrosis and Inflammation
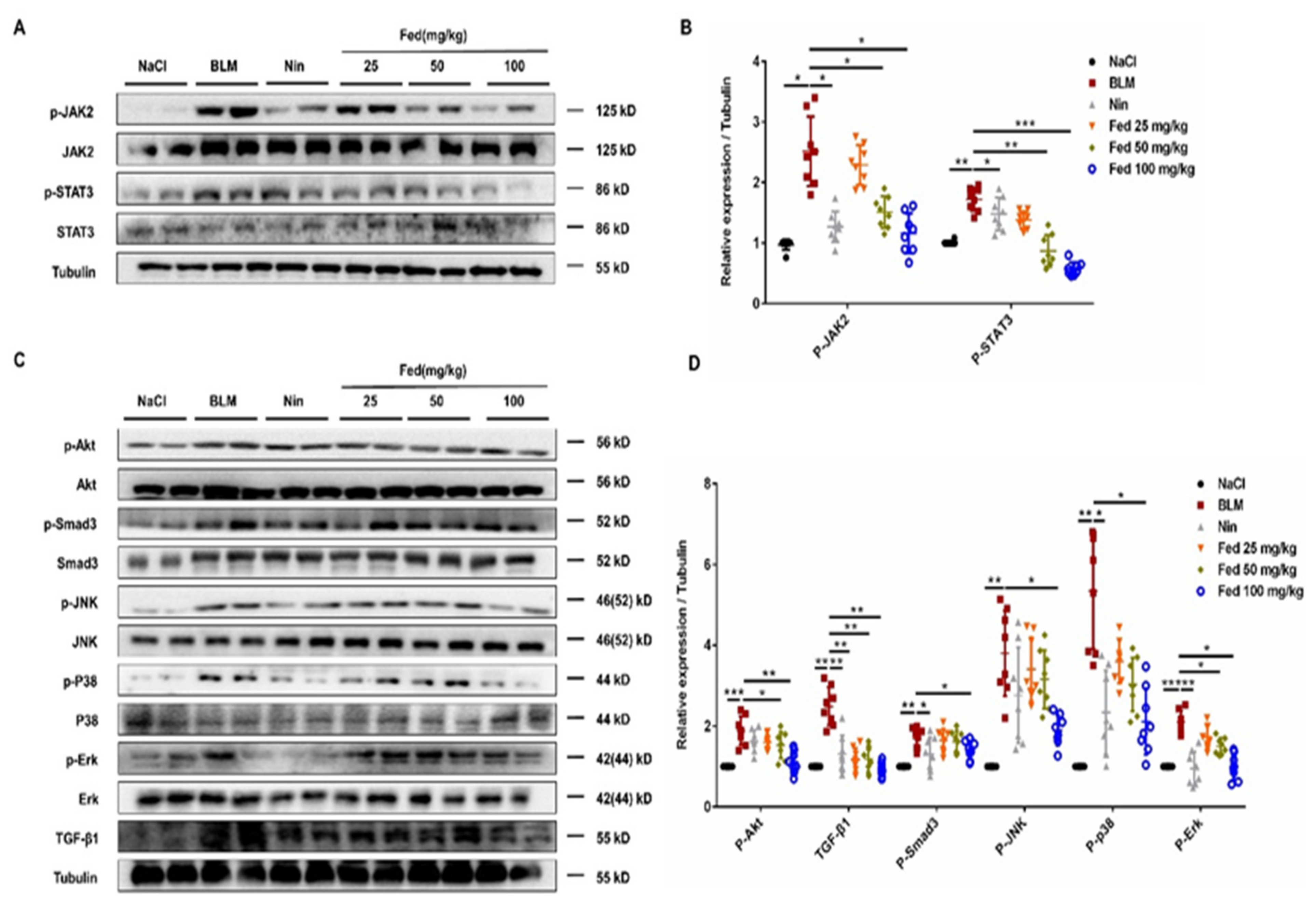
2.5. Fed Alleviates Myofibroblast Activation in Bleomycin-Induced Mice by Inhibiting Both JAK2/STAT3 and TGF-β1 Signaling
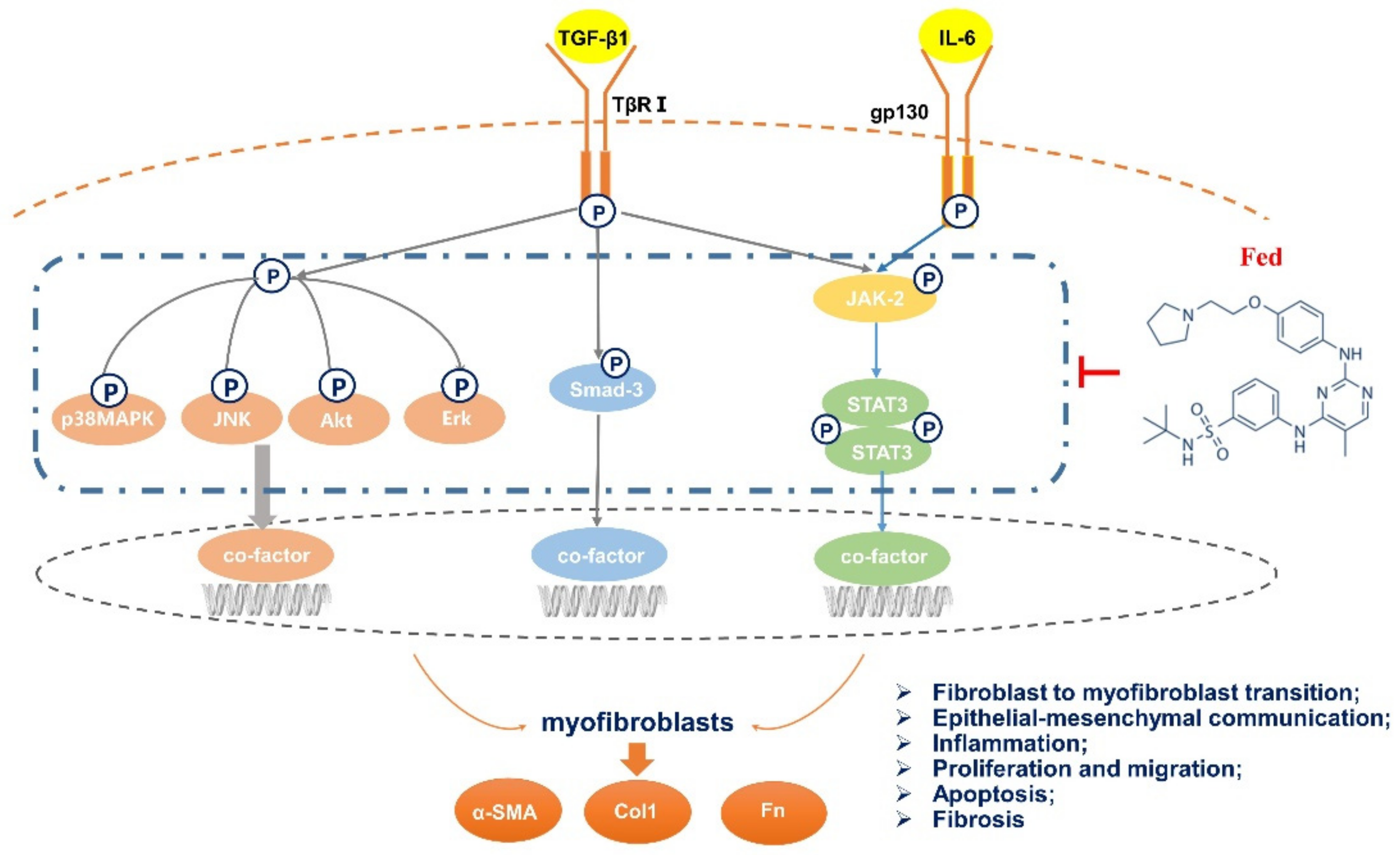
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Animals
4.4. Bleomycin Administration
4.5. Pulmonary Function Testing
4.6. Hydroxyproline Measurement
4.7. Histological Examination
4.8. Cell Viability Analysis
4.9. Wound Healing Assay
4.10. Quantitative Real-Time PCR (qRT-PCR)
4.11. Western Blotting Analysis
4.12. Immunofluorescence Staining
4.13. Immunohistochemistry Staining
4.14. Bronchoalveolar Lavage
4.15. ELISA Detection
4.16. Apoptosis Analysis
4.17. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [Green Version]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [Green Version]
- Lyu, C.L.; Liu, J.Q.; Chen, M.; Chen, B.; Xiao, Z.J. The impact of meisoindigo on apoptosis and proliferation of SET2 cell line by JAK-STAT pathway. Zhonghua Xue Ye Xue Za Zhi 2019, 40, 29–34. [Google Scholar] [CrossRef]
- Shammo, J.M.; Stein, B.L. Mutations in MPNs: Prognostic implications, window to biology, and impact on treatment decisions. Hematol. Am. Soc. Hematol. Educ. Progr. 2016, 2016, 552–560. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of casein kinase II reduces TGFbeta induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 2015, 74, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prele, C.M.; Wong, J.; Hogaboam, C.M.; McAnulty, R.J.; Laurent, G.J.; Zhang, S.S.; Selman, M.; Mutsaers, S.E.; Knight, D.A. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am. J. Pathol. 2012, 180, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Pedroza, M.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escriva, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londono-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Allahverdian, S.; Saunders, A.D.R.; Liu, E.; Dorscheid, D.R. IL-13 signaling through IL-13 receptor alpha2 mediates airway epithelial wound repair. FASEB J. 2019, 33, 3746–3757. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Stubbe, J. Bleomycins: Towards better therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [Green Version]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef]
- Biasin, V.; Crnkovic, S.; Sahu-Osen, A.; Birnhuber, A.; El Agha, E.; Sinn, K.; Klepetko, W.; Olschewski, A.; Bellusci, S.; Marsh, L.M.; et al. PDGFRalpha and alphaSMA mark two distinct mesenchymal cell populations involved in parenchymal and vascular remodeling in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L684–L697. [Google Scholar] [CrossRef]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Xu, S.; Mao, Y.; Wu, J.; Feng, J.; Li, J.; Wu, L.; Yu, Q.; Zhou, Y.; Zhang, J.; Chen, J.; et al. TGF-beta/Smad and JAK/STAT pathways are involved in the anti-fibrotic effects of propylene glycol alginate sodium sulphate on hepatic fibrosis. J. Cell. Mol. Med. 2020, 24, 5224–5237. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Silva, T.E.; Soares, E.S.P.E.; Colombo, B.S.; Silva, M.C.; Wildner, L.M.; Bazzo, M.L.; Rateke, E.C.; Frode, T.S.; Mello, S.V.; et al. From stable disease to acute-on-chronic liver failure: Circulating cytokines are related to prognosis in different stages of cirrhosis. Cytokine 2017, 91, 162–169. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-beta pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Chen, W.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.M.; Sun, W.; Ning, B.F.; Zhou, T.F.; Li, X.F.; Zhong, W.; Cheng, Z.; Xia, M.Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.T. JAK/STAT3 signaling is required for TGF-beta-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Akcora, B.O.; Dathathri, E.; Ortiz-Perez, A.; Gabriel, A.V.; Storm, G.; Prakash, J.; Bansal, R. TG101348, a selective JAK2 antagonist, ameliorates hepatic fibrogenesis in vivo. FASEB J. 2019, 33, 9466–9475. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [Green Version]
- Elias, J.A.; Lentz, V.; Cummings, P.J. Transforming growth factor-beta regulation of IL-6 production by unstimulated and IL-1-stimulated human fibroblasts. J. Immunol. 1991, 146, 3437–3443. [Google Scholar] [PubMed]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.S.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef]
- Pardanani, A.; Tefferi, A.; Jamieson, C.; Gabrail, N.Y.; Lebedinsky, C.; Gao, G.; Liu, F.; Xu, C.; Cao, H.; Talpaz, M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015, 5, e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, C.; Hasserjian, R.; Gotlib, J.; Cortes, J.; Stone, R.; Talpaz, M.; Thiele, J.; Rodig, S.; Pozdnyakova, O. Effect of treatment with a JAK2-selective inhibitor, fedratinib, on bone marrow fibrosis in patients with myelofibrosis. J. Transl. Med. 2015, 13, 294. [Google Scholar] [CrossRef]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Suzukawa, M.; Nagase, H.; Koizumi, Y.; Ro, S.; Kobayashi, K.; Yoshihara, H.; Kojima, Y.; Kamiyama-Hara, A.; Hebisawa, A.; et al. IL-9 Blockade Suppresses Silica-induced Lung Inflammation and Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bi, Z.; Liu, S.; Gao, S.; Cui, Y.; Huang, K.; Huang, M.; Mao, J.; Li, L.; Gao, J.; et al. Antifibrotic Mechanism of Cinobufagin in Bleomycin-Induced Pulmonary Fibrosis in Mice. Front. Pharmacol. 2019, 10, 1021. [Google Scholar] [CrossRef]
- Martinotti, S.; Ranzato, E. Scratch Wound Healing Assay. Methods Mol. Biol. 2020, 2109, 225–229. [Google Scholar] [CrossRef]
- Gong, L.; Zhu, L.; Yang, T. Fendrr involves in the pathogenesis of cardiac fibrosis via regulating miR-106b/SMAD3 axis. Biochem. Biophys. Res. Commun. 2020, 524, 169–177. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Wollin, L.; Shitrit, D. Fibroblast-matrix interplay: Nintedanib and pirfenidone modulate the effect of IPF fibroblast-conditioned matrix on normal fibroblast phenotype. Respirology 2018, 23, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (3′–5′) |
|---|---|---|
| Mouse GAPDH | AGGTCGGTGTGAACGGATTTG | GGGGTCGTTGATGGCAACA |
| Mouse α-SMA | GTCCCAGACATCAGGGAGTAA | GTCCCAGACATCAGGGAGTAA |
| Mouse Fibronectin | TCGGATACTTCAGCGTCAGGA | TCGGATACTTCAGCGTCAGGA |
| Mouse Collagen I | ATGTGGACCCCTCCTGATAGT | ATGTGGACCCCTCCTGATAGT |
| Mouse IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Mouse IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| Mouse IL-1β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Mouse TNF-α | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Mouse IL-13 | CCTGGCTCTTGCTTGCCTT | GGTCTTGTGTGATGTTGCTCA |
| Mouse TGF-β1 | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, H.; Luan, J.; Gao, S.; Li, S.; Jiang, Q.; Liu, R.; Liang, Q.; Zhang, R.; Zhang, F.; Li, X.; et al. Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-β1 Signaling Pathway. Molecules 2021, 26, 4491. https://doi.org/10.3390/molecules26154491
Ruan H, Luan J, Gao S, Li S, Jiang Q, Liu R, Liang Q, Zhang R, Zhang F, Li X, et al. Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-β1 Signaling Pathway. Molecules. 2021; 26(15):4491. https://doi.org/10.3390/molecules26154491
Chicago/Turabian StyleRuan, Hao, Jiaoyan Luan, Shaoyan Gao, Shuangling Li, Qiuyan Jiang, Rui Liu, Qing Liang, Ruiqin Zhang, Fangxia Zhang, Xiaohe Li, and et al. 2021. "Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-β1 Signaling Pathway" Molecules 26, no. 15: 4491. https://doi.org/10.3390/molecules26154491
APA StyleRuan, H., Luan, J., Gao, S., Li, S., Jiang, Q., Liu, R., Liang, Q., Zhang, R., Zhang, F., Li, X., Zhou, H., & Yang, C. (2021). Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-β1 Signaling Pathway. Molecules, 26(15), 4491. https://doi.org/10.3390/molecules26154491