
Learning the Fastest RNA Folding Path Based on Reinforcement Learning and Monte Carlo Tree Search


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Reinforcement Learning
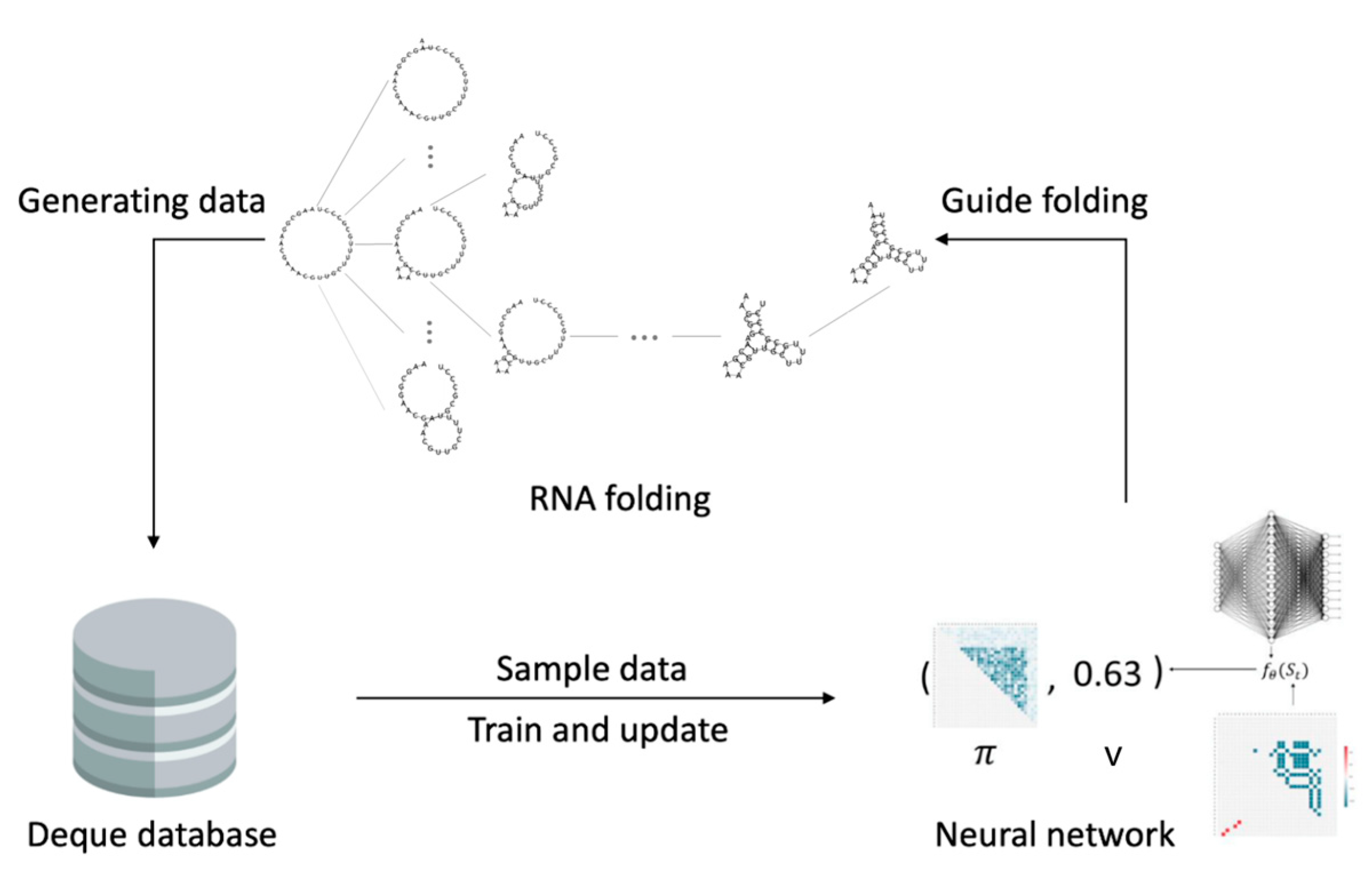
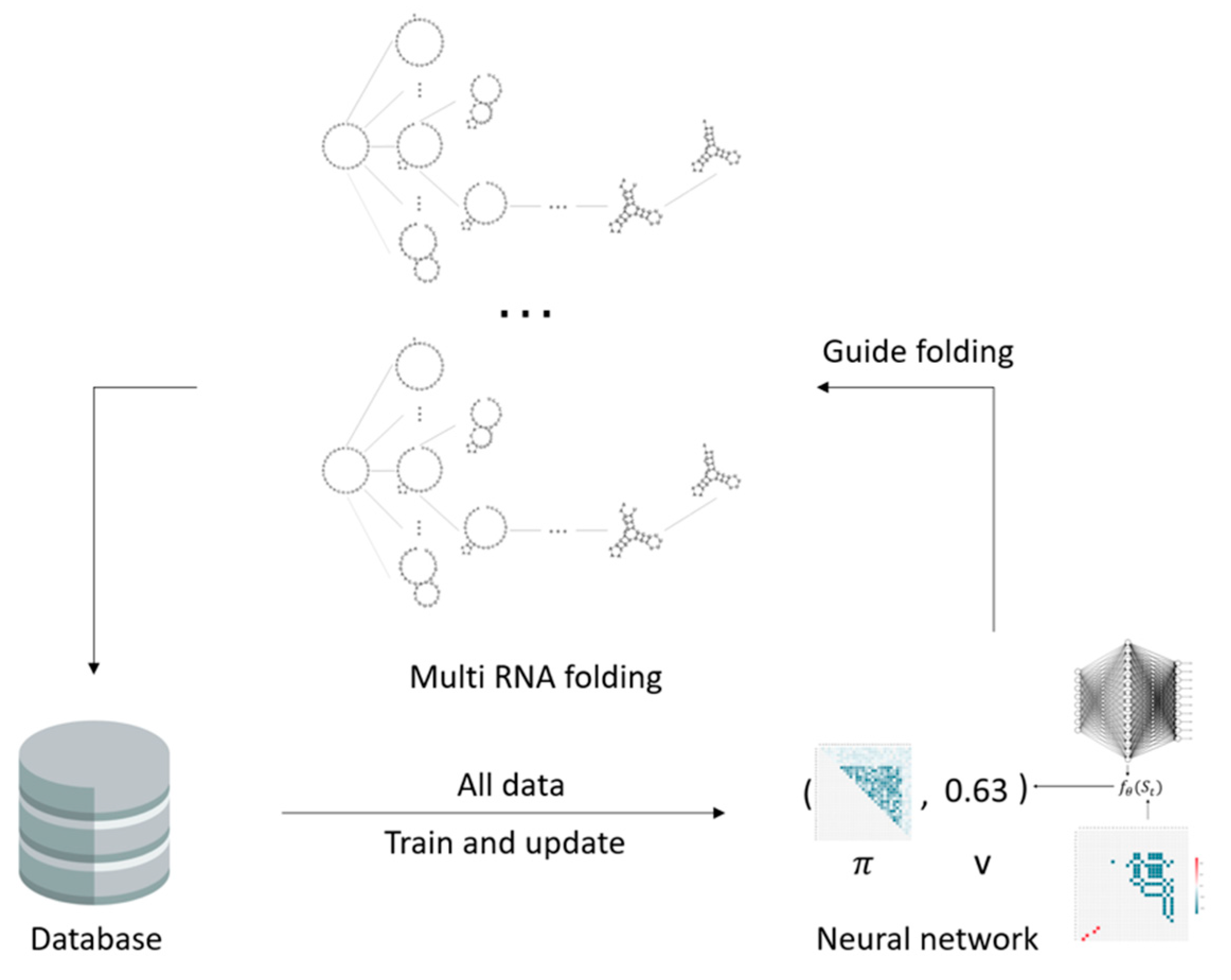
2.2. Monte Carlo Tree Search
- (1)
- Selection
- (2)
- Expansion
- (3)
- Simulation
- (4)
- Backpropagation
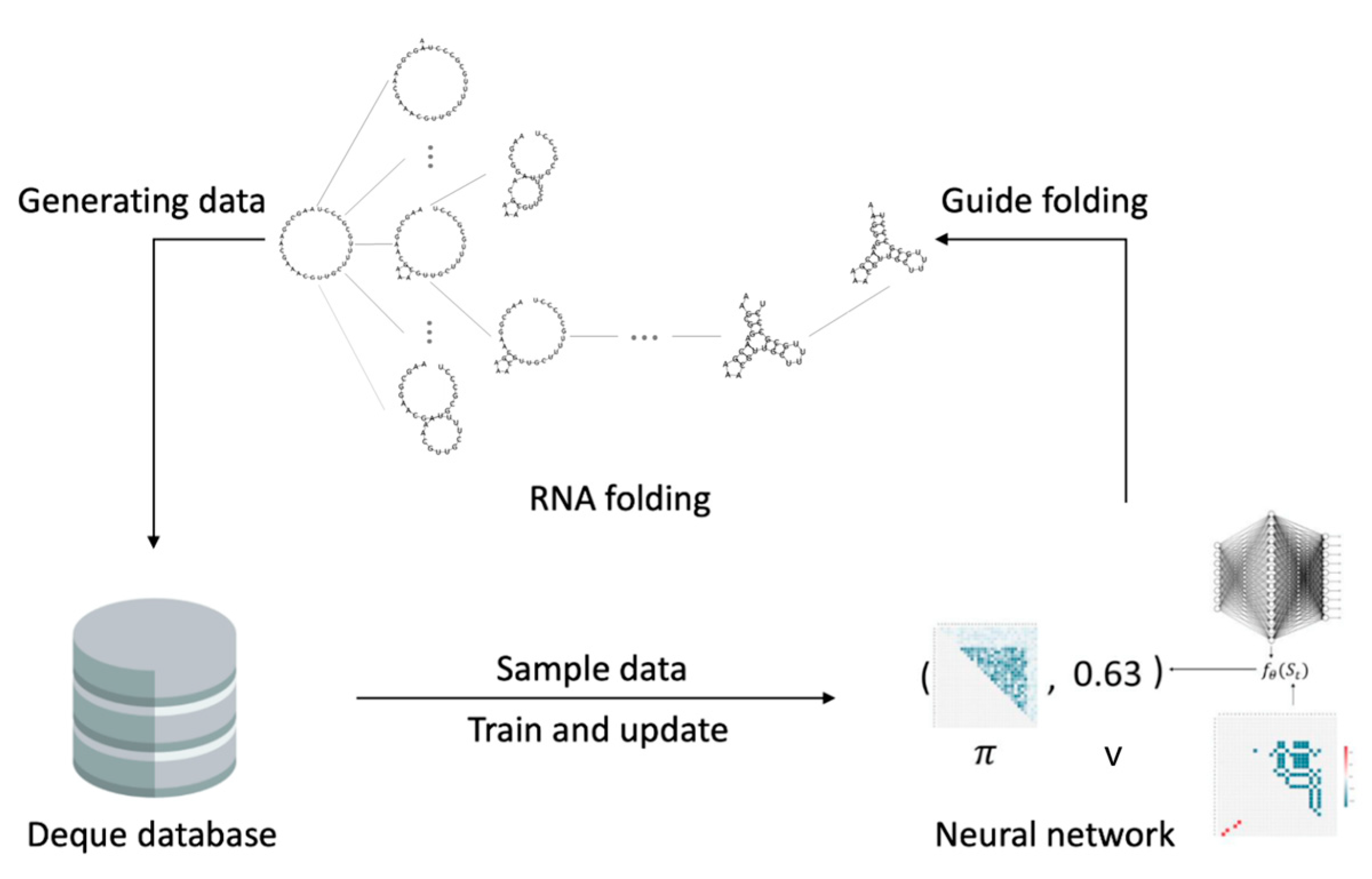
2.3. The Fastest Folding Path Learning
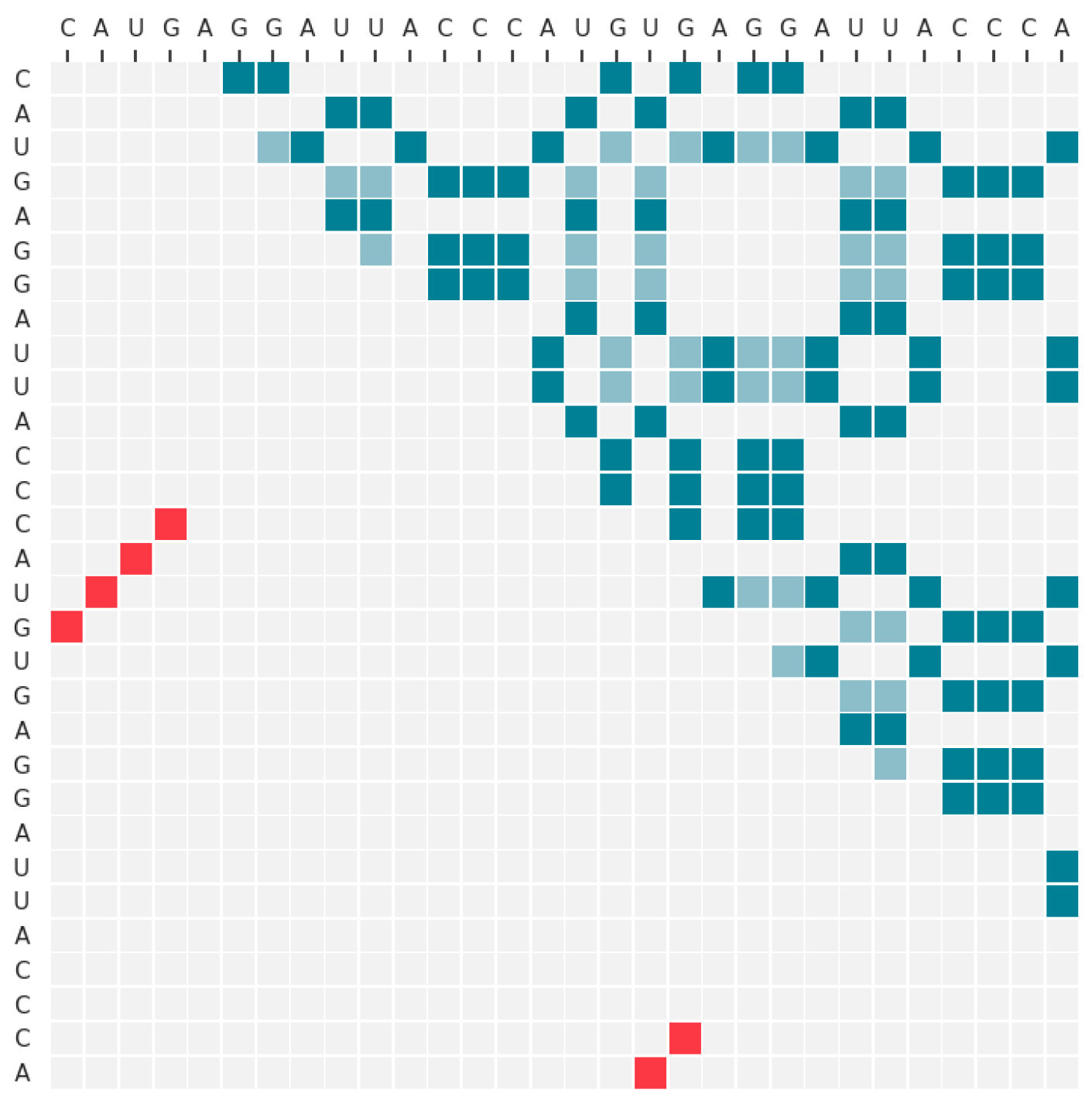
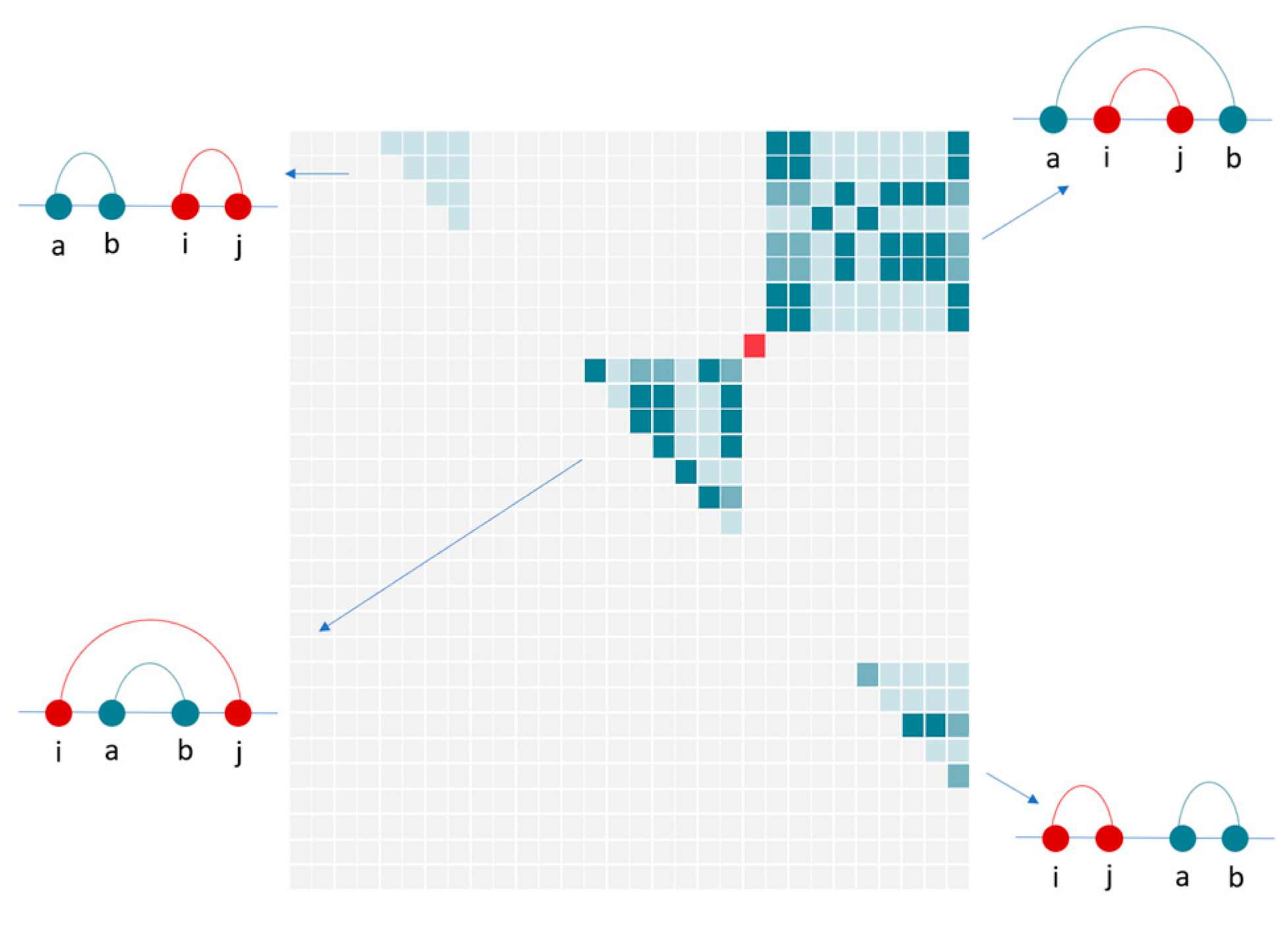
2.3.1. Input Data
2.3.2. Neural Network
2.3.3. Action Space
2.3.4. Reinforcement Learning
2.3.5. Gym Environment
3. Results
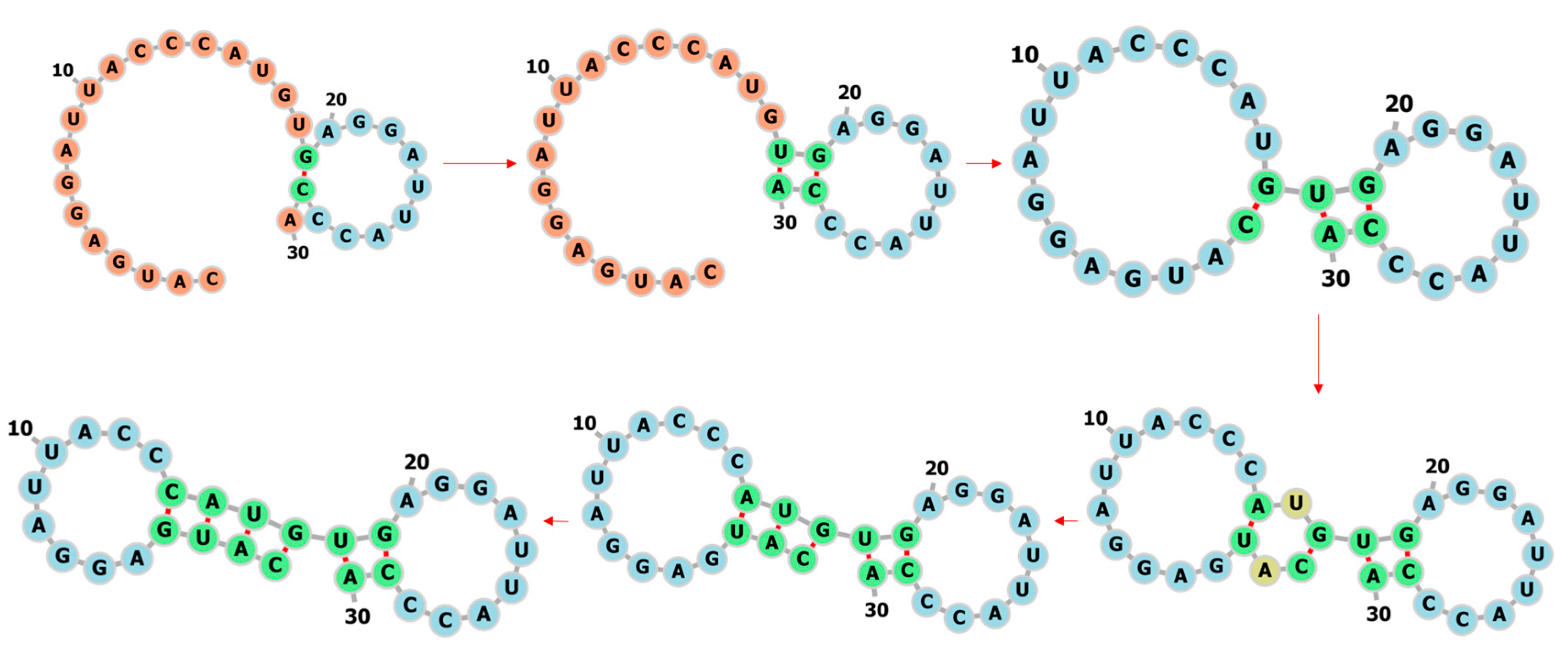
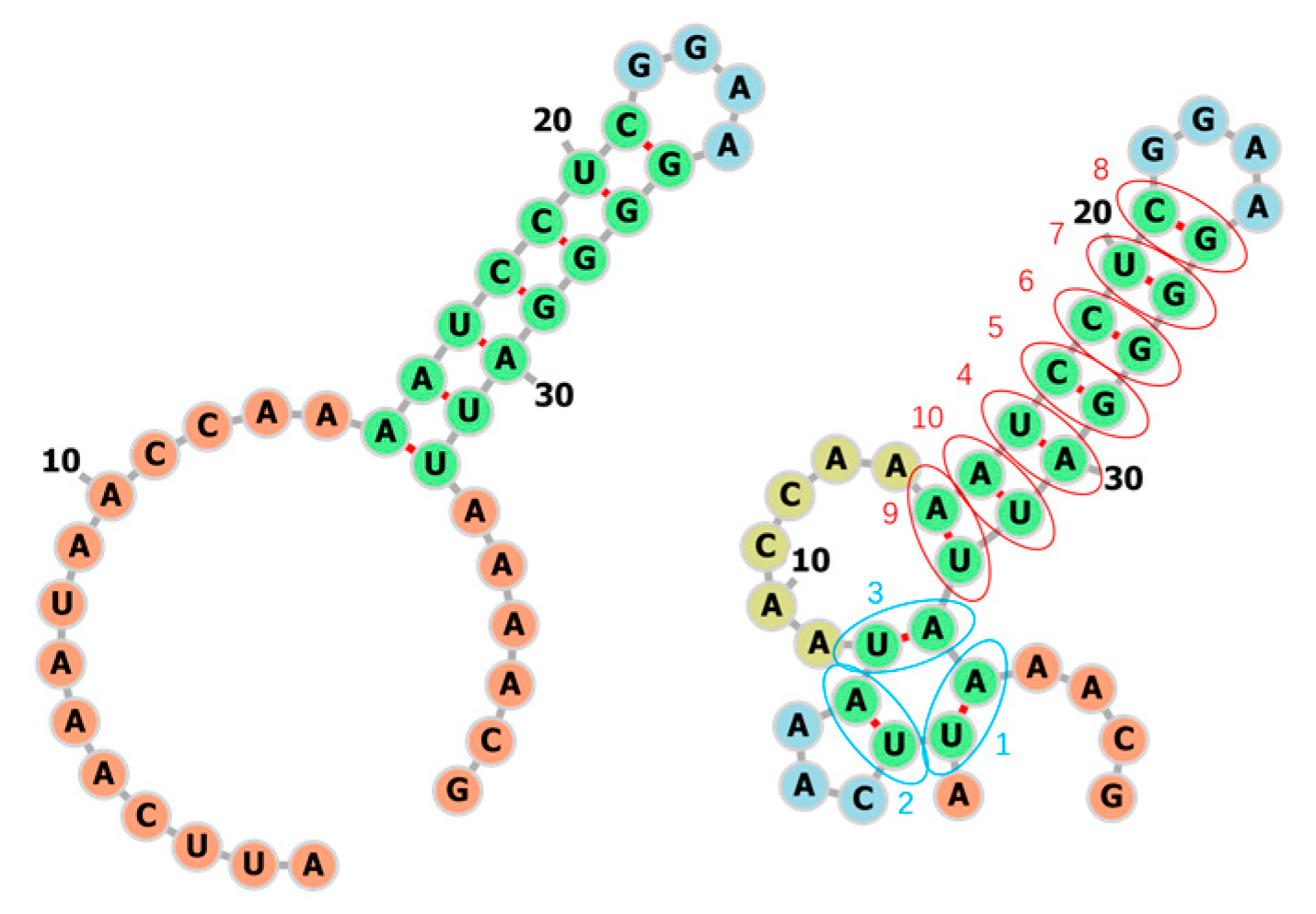
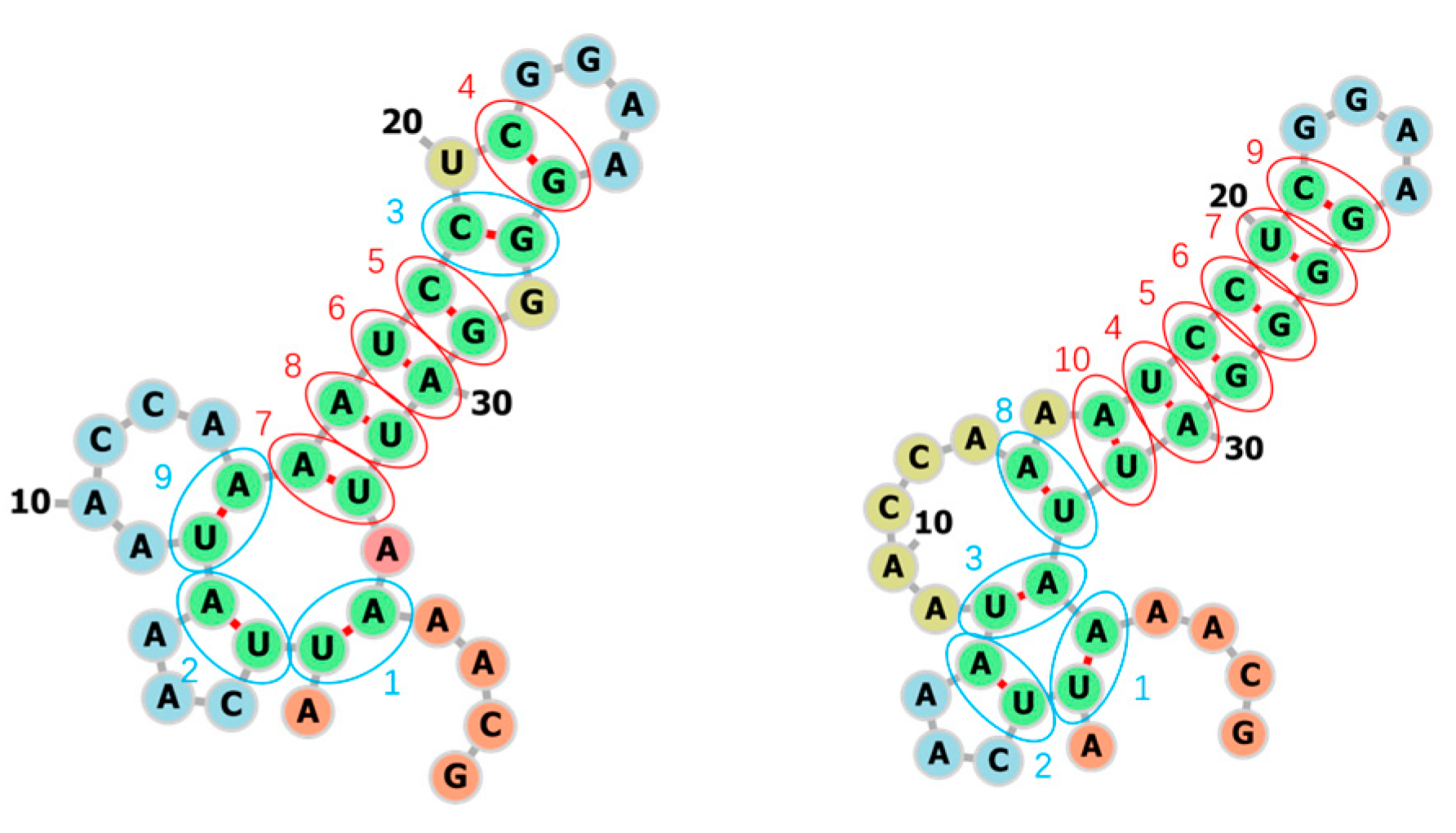
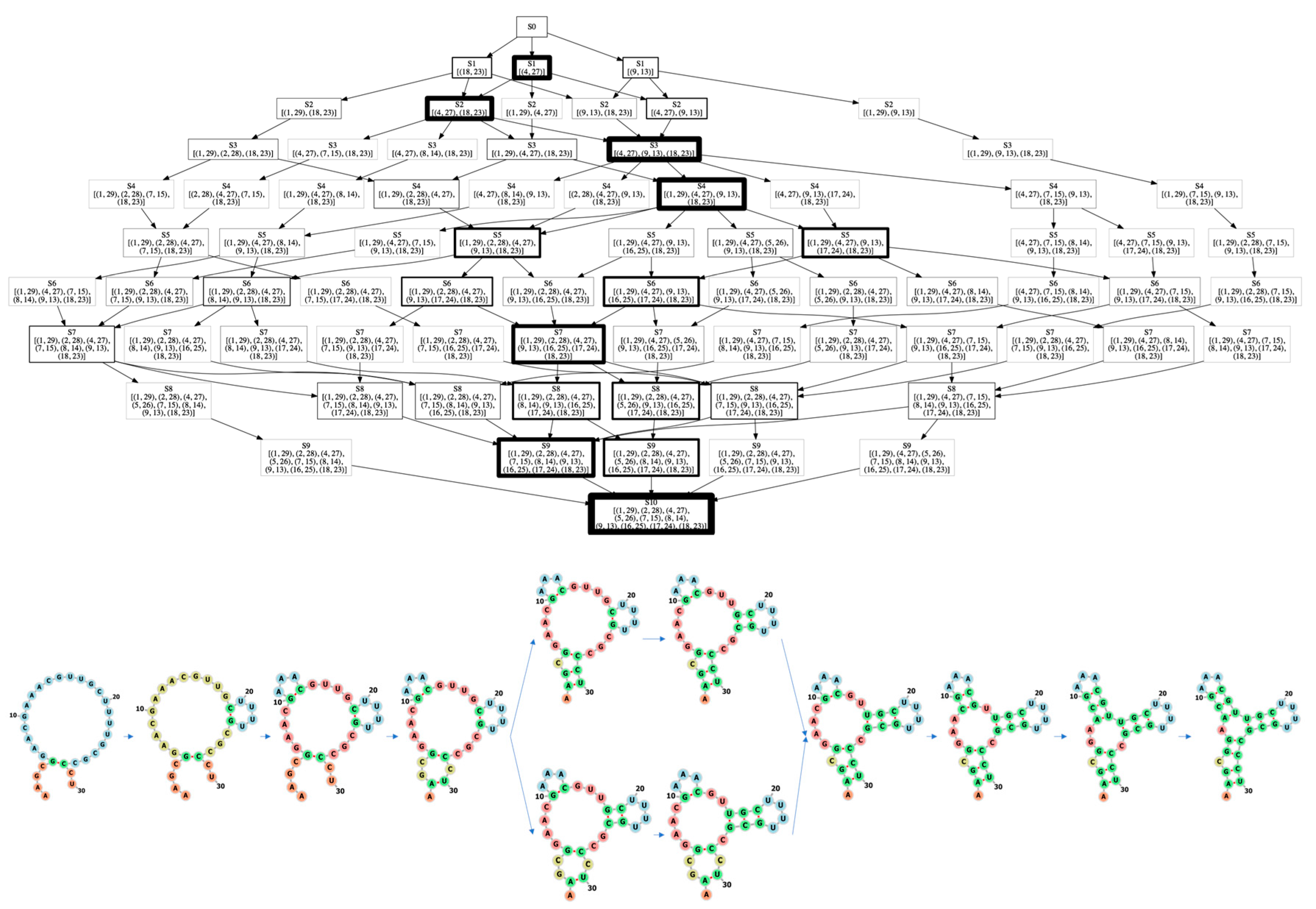
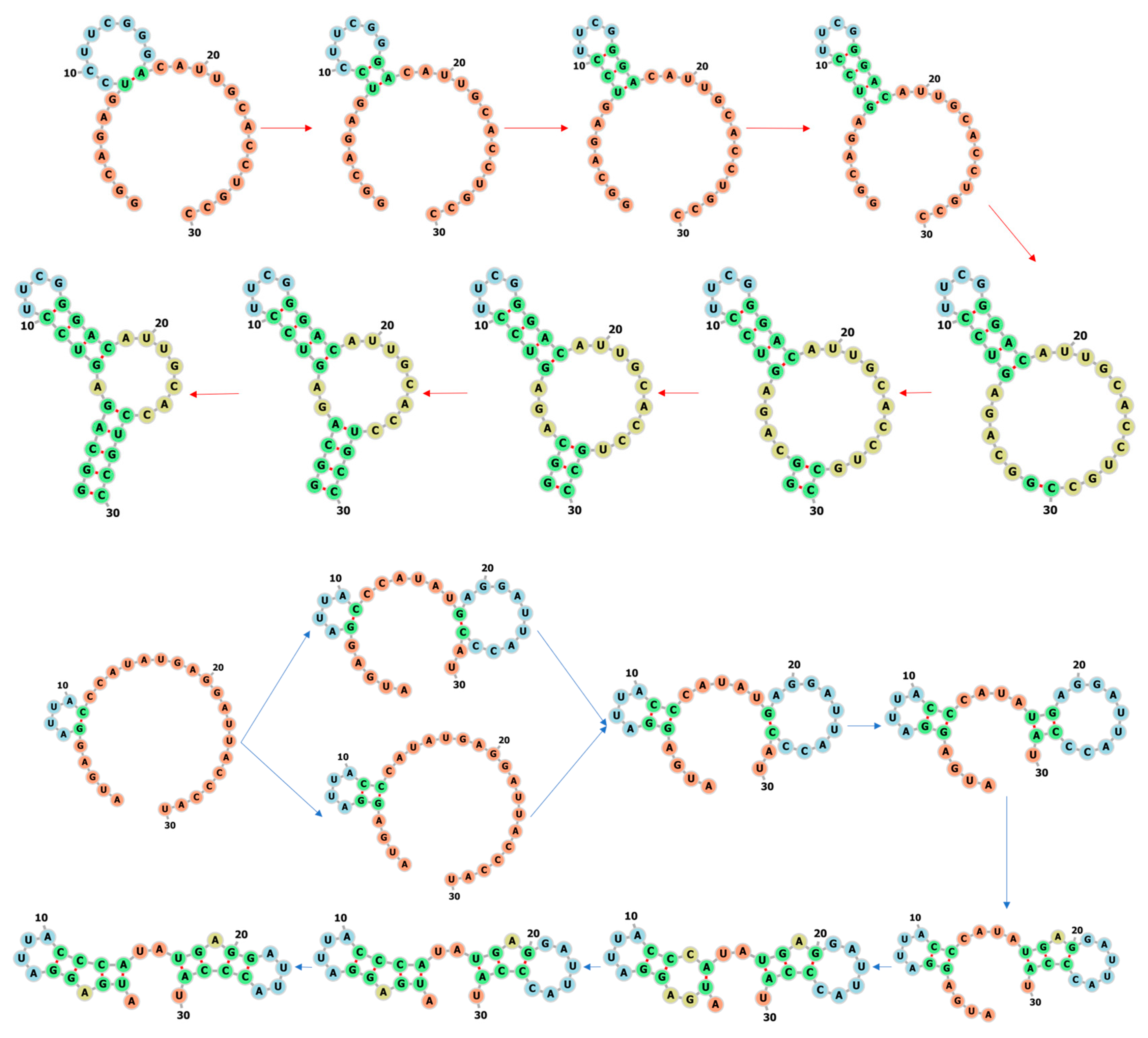
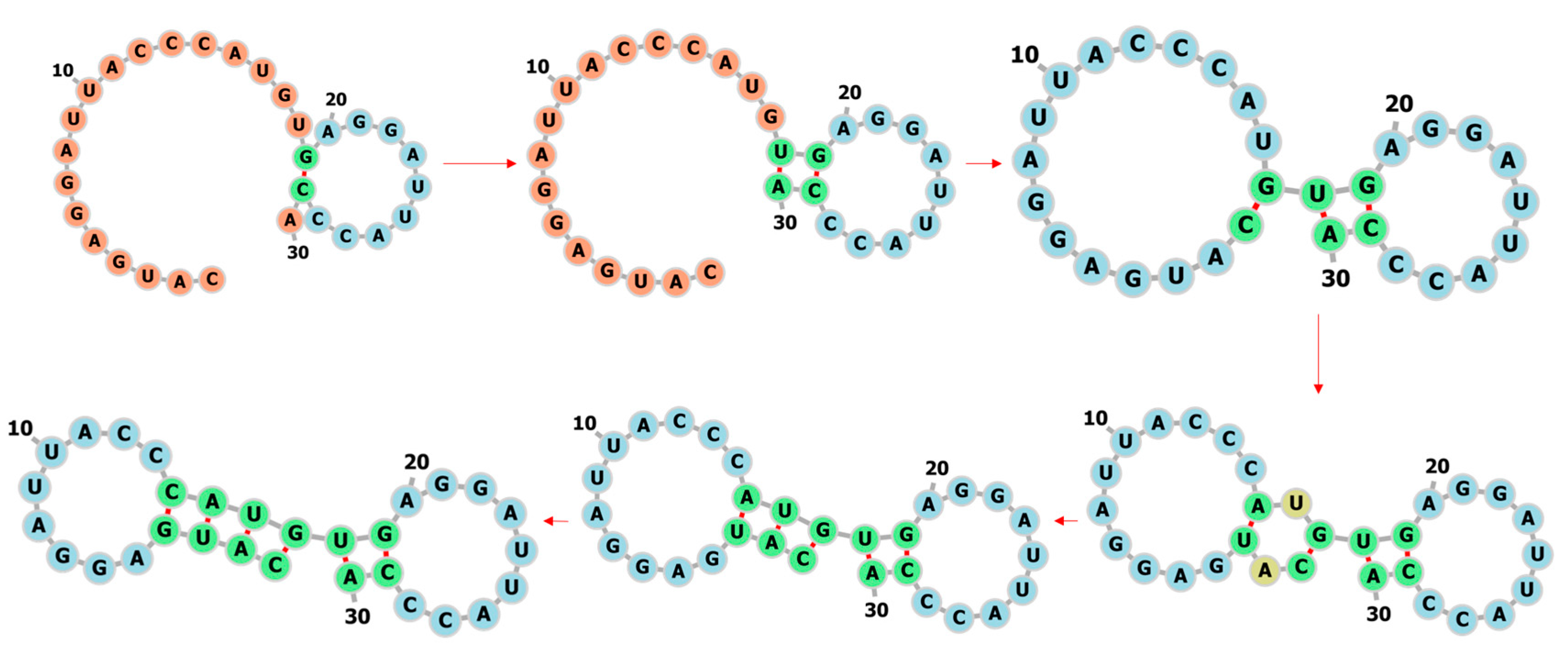
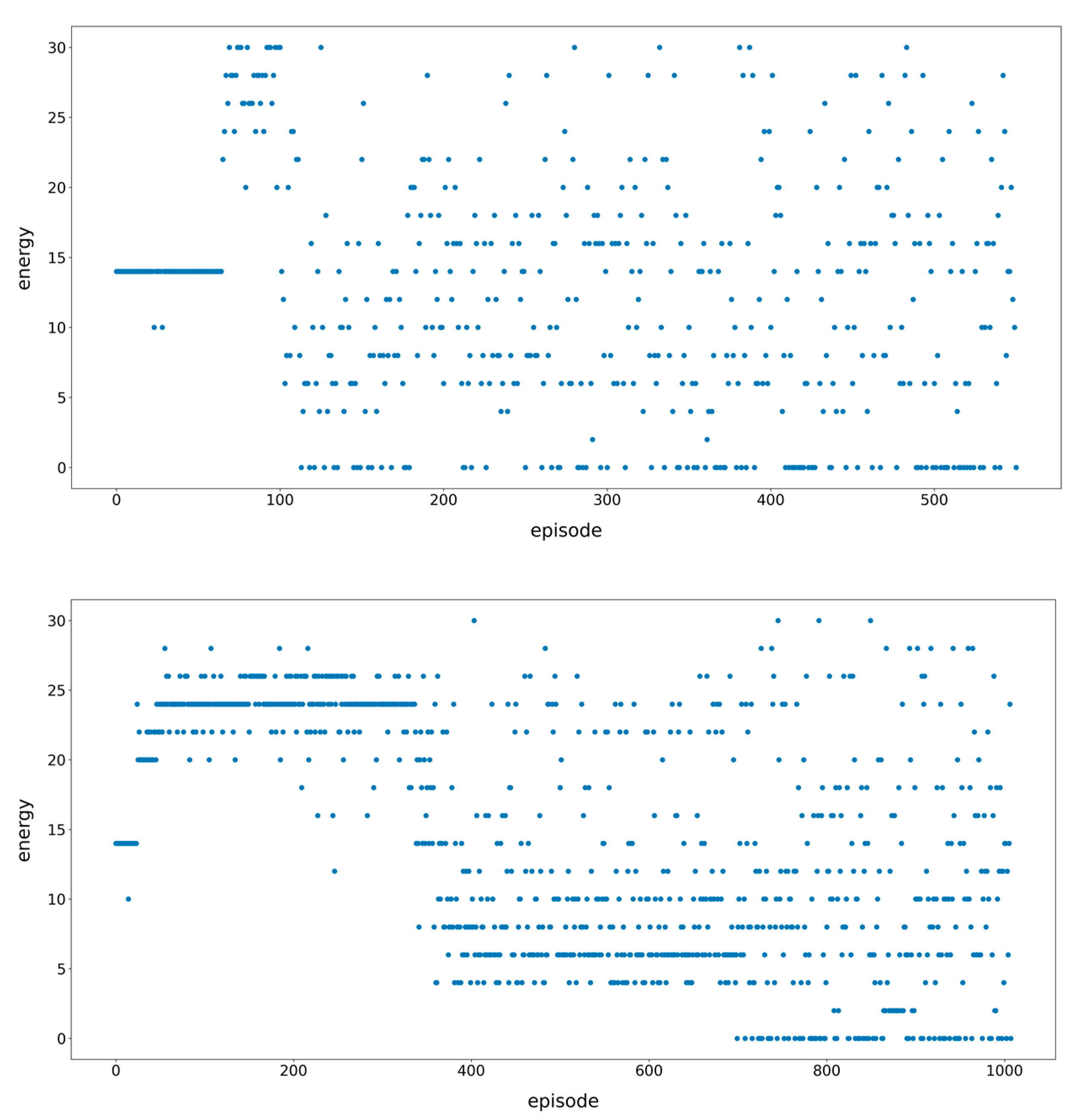
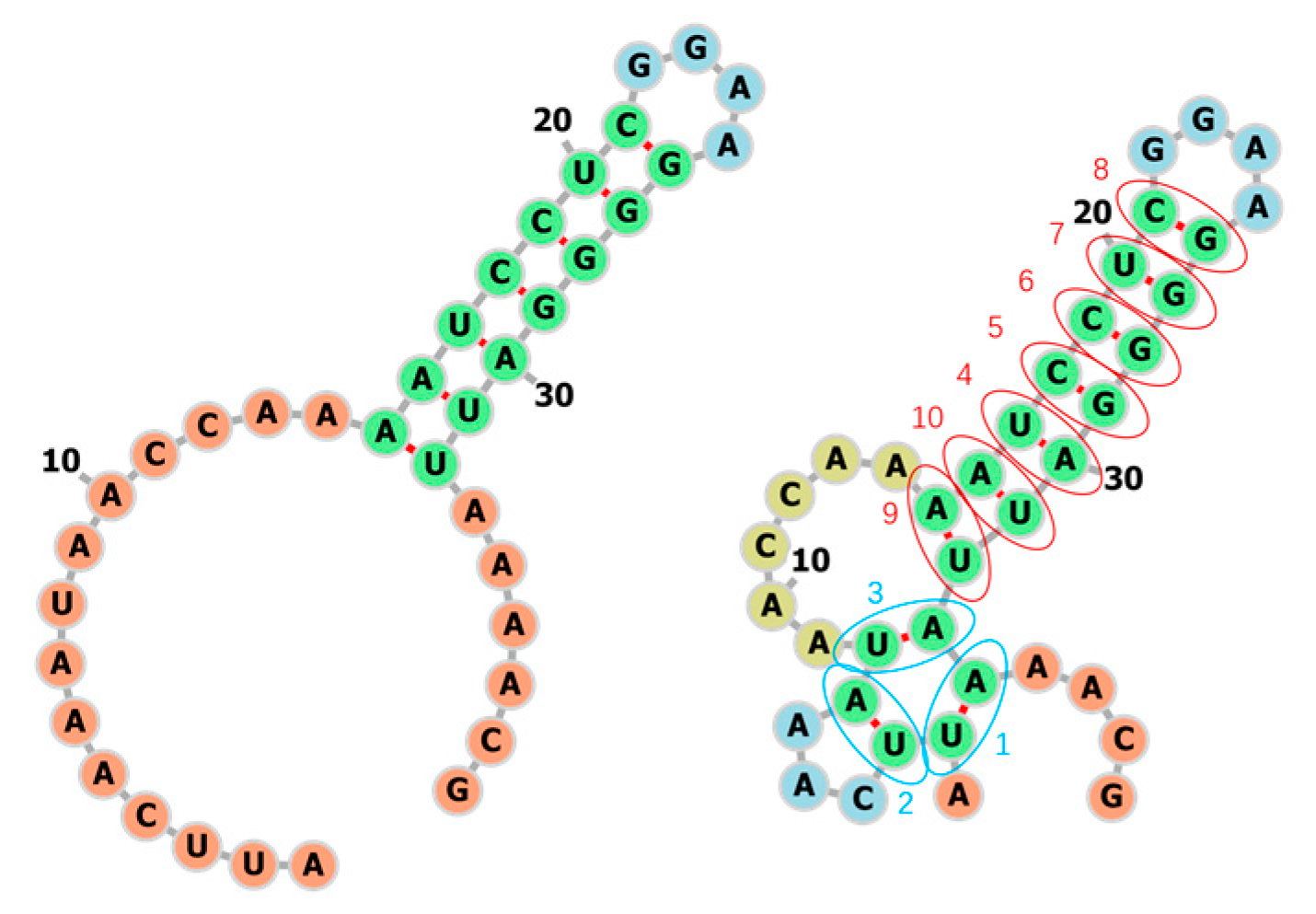
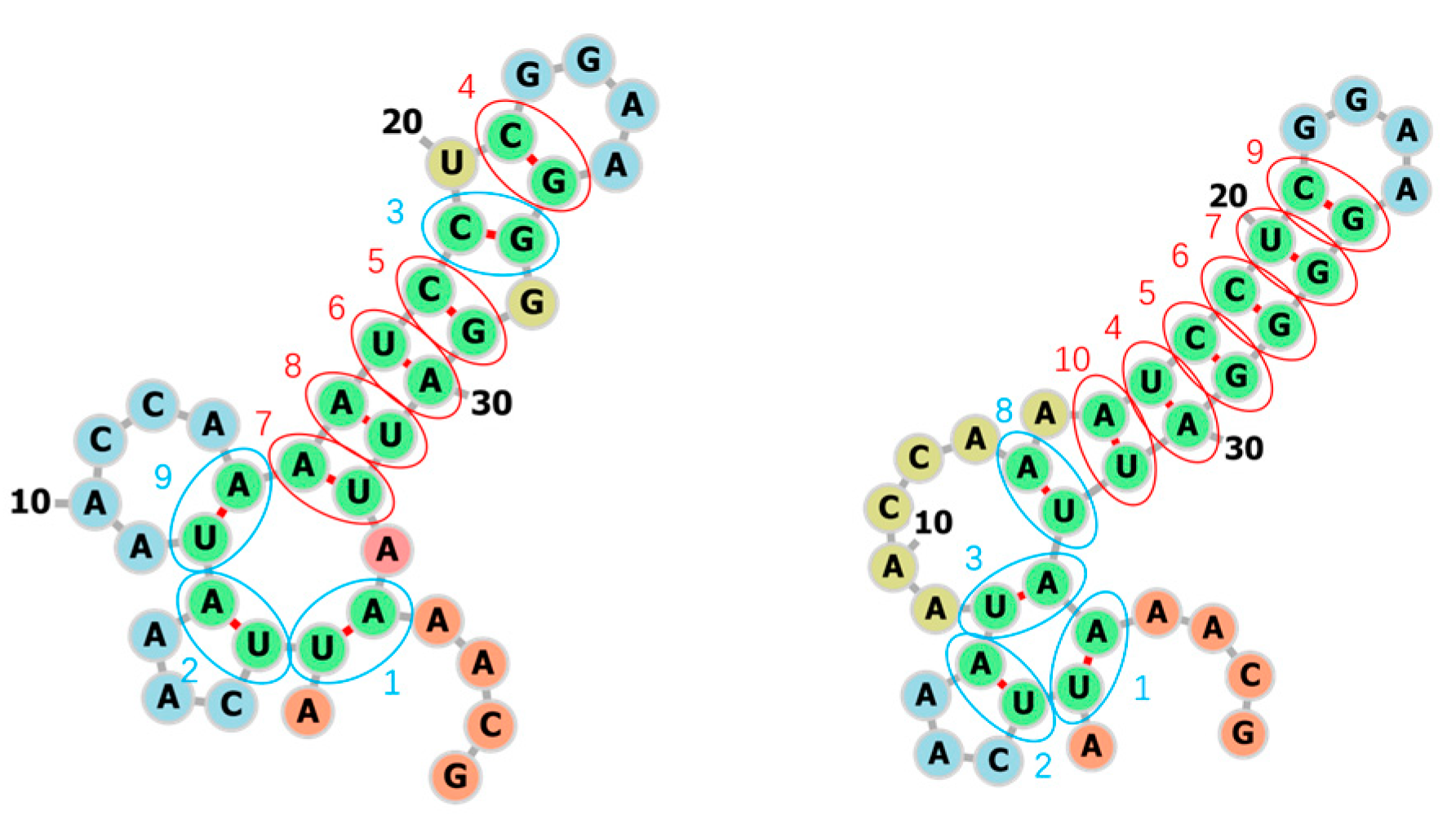
3.1. Learning the Fastest Folding Paths of Several Short RNAs
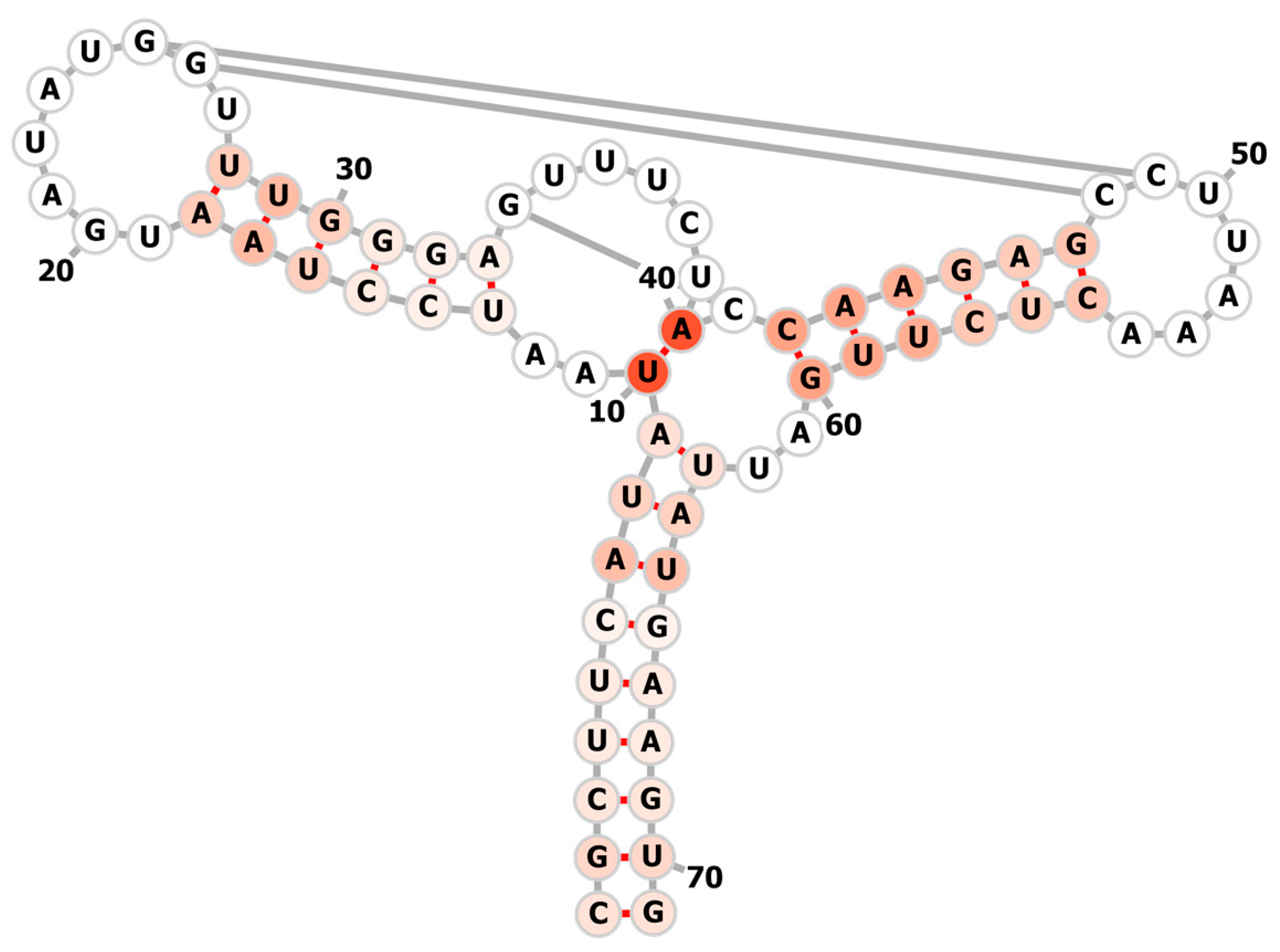
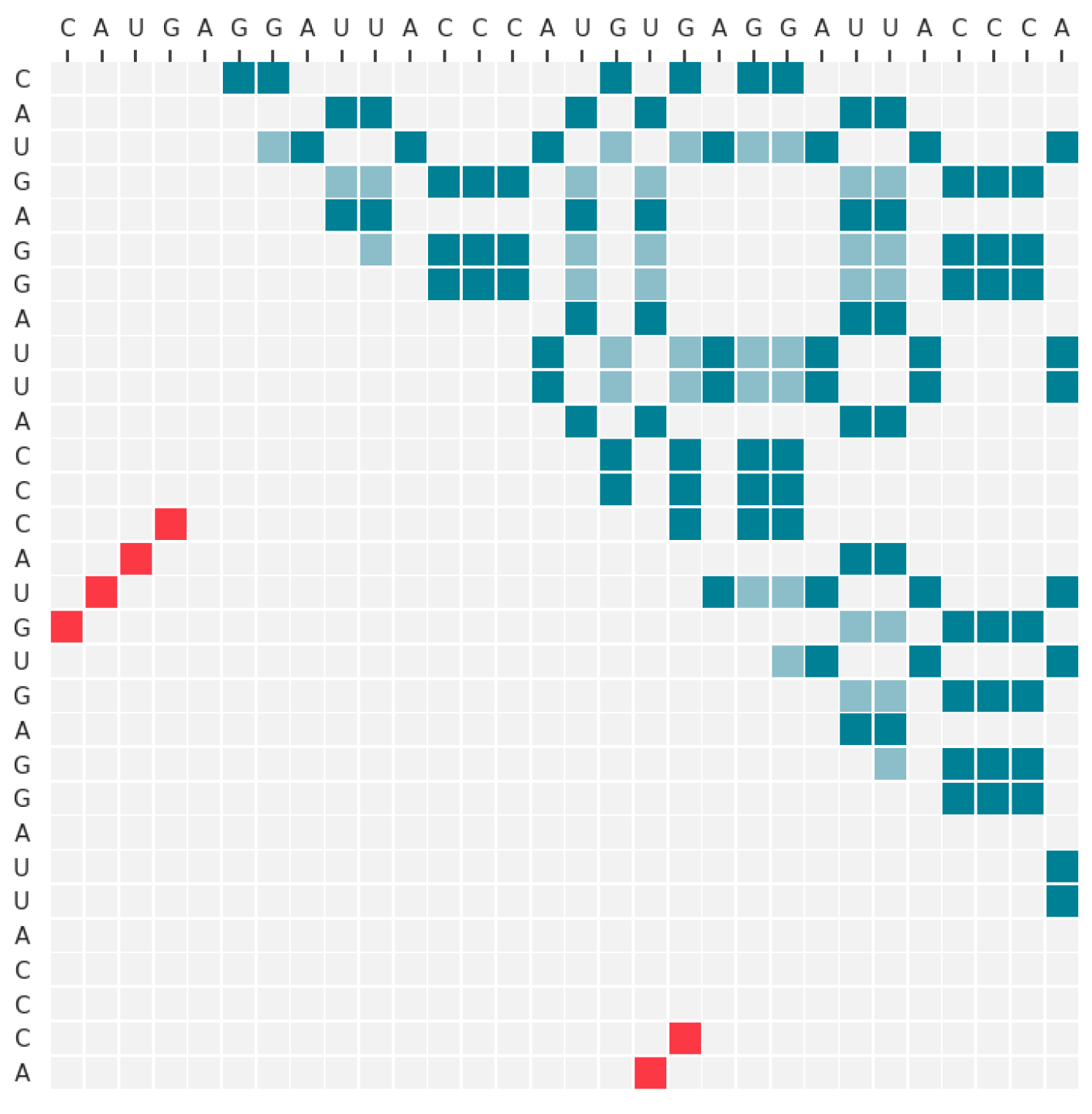
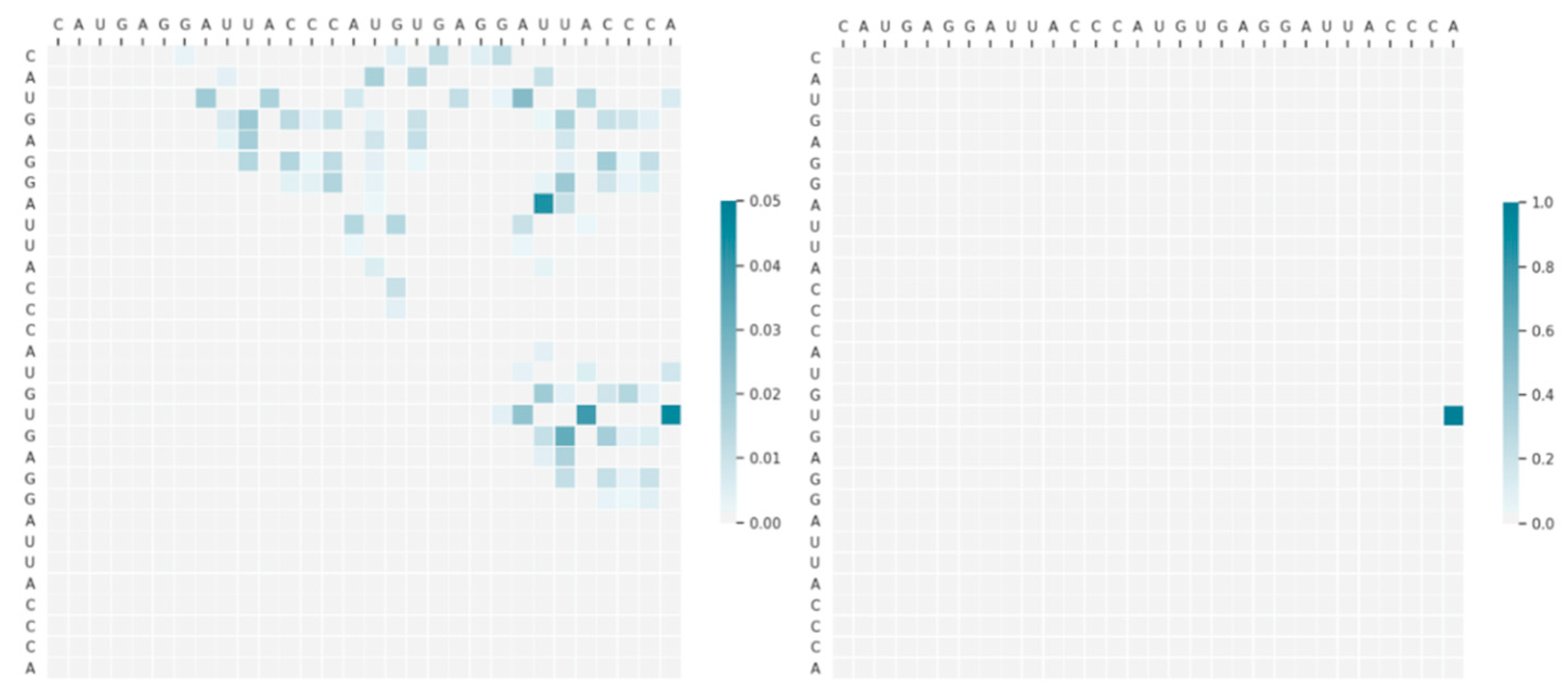
3.2. Learning the Fastest Folding Path of a Riboswitch RNA
4. Discussion
4.1. The Impact of Monte Carlo Tree Search
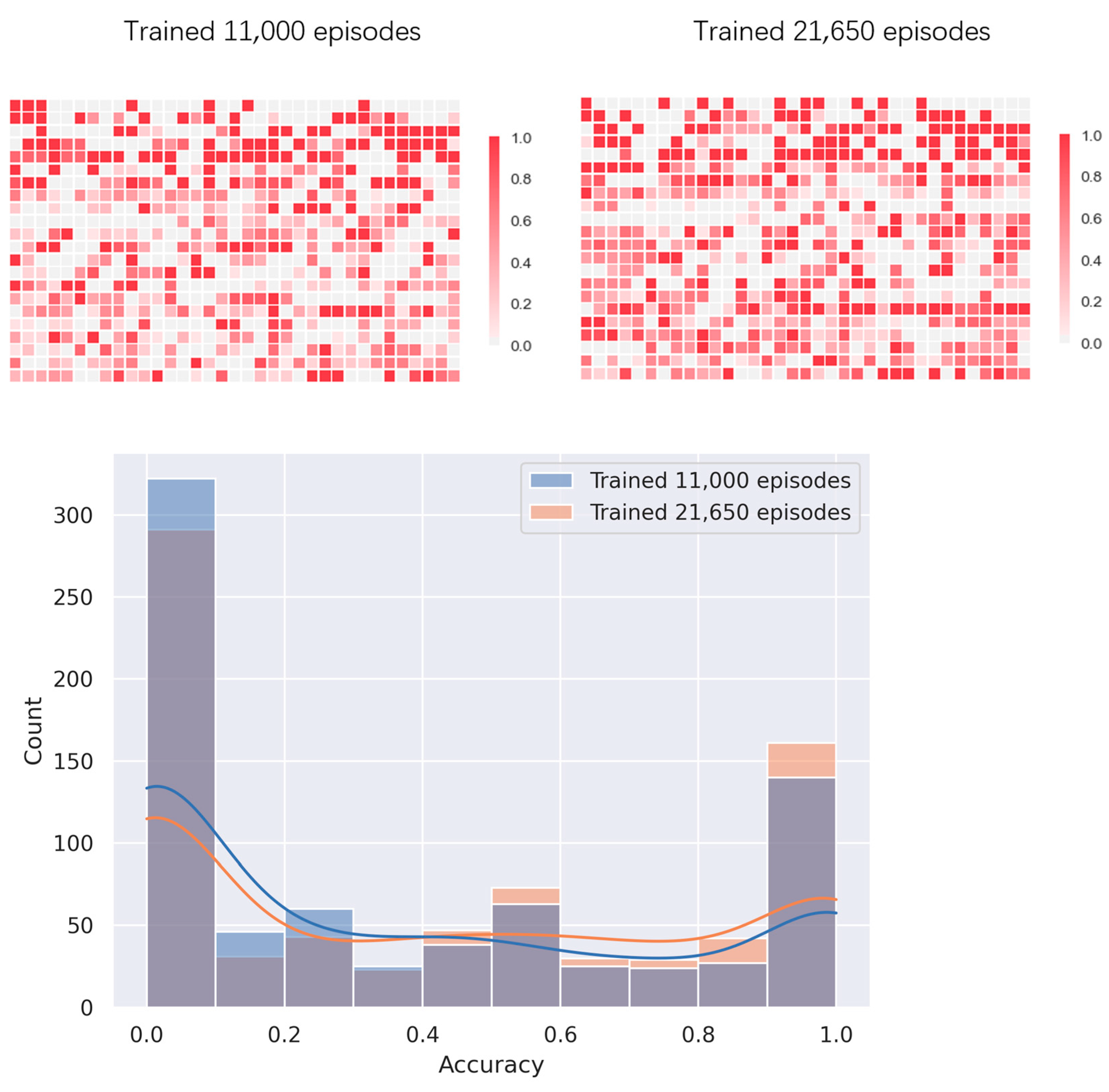
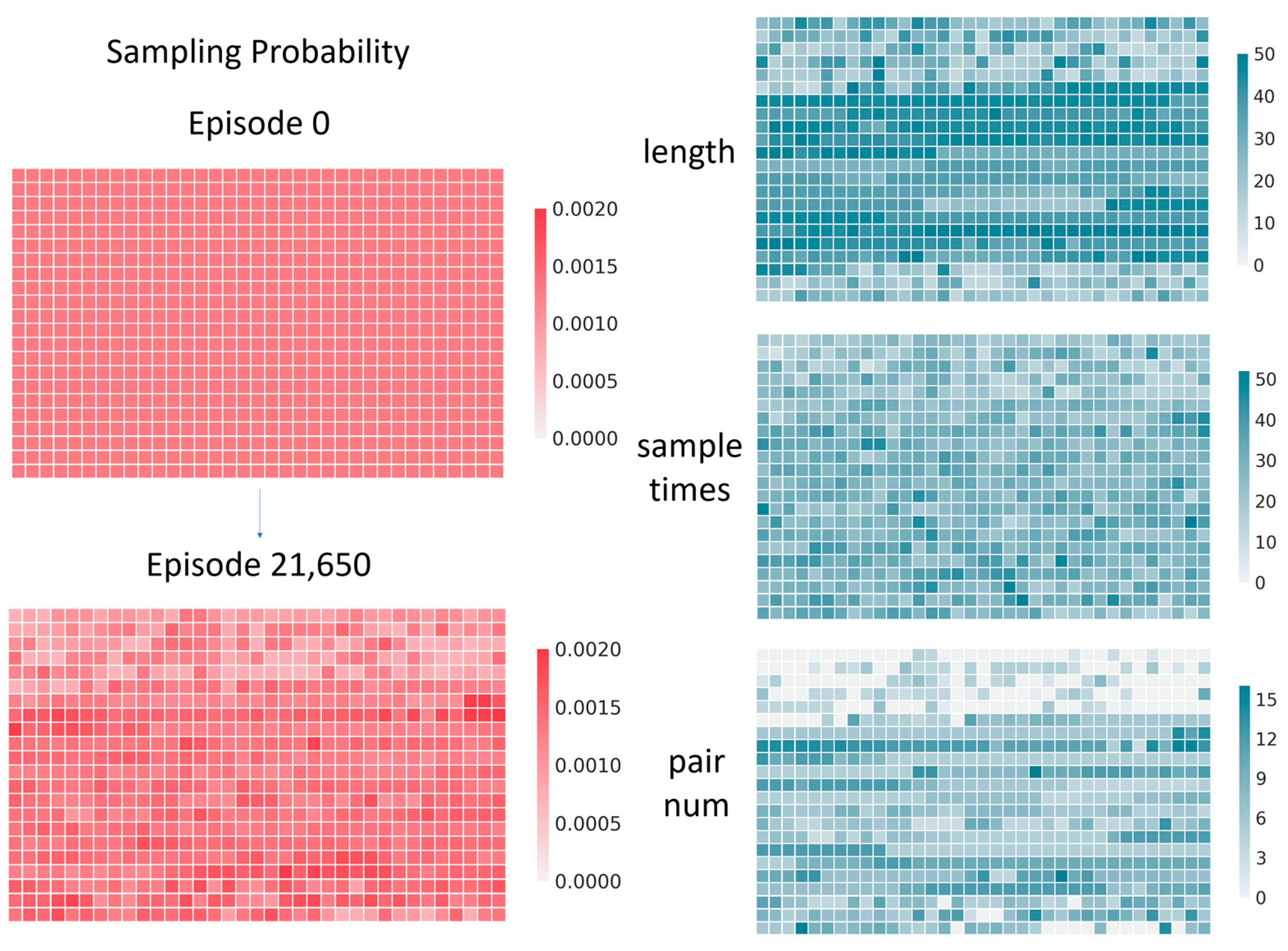
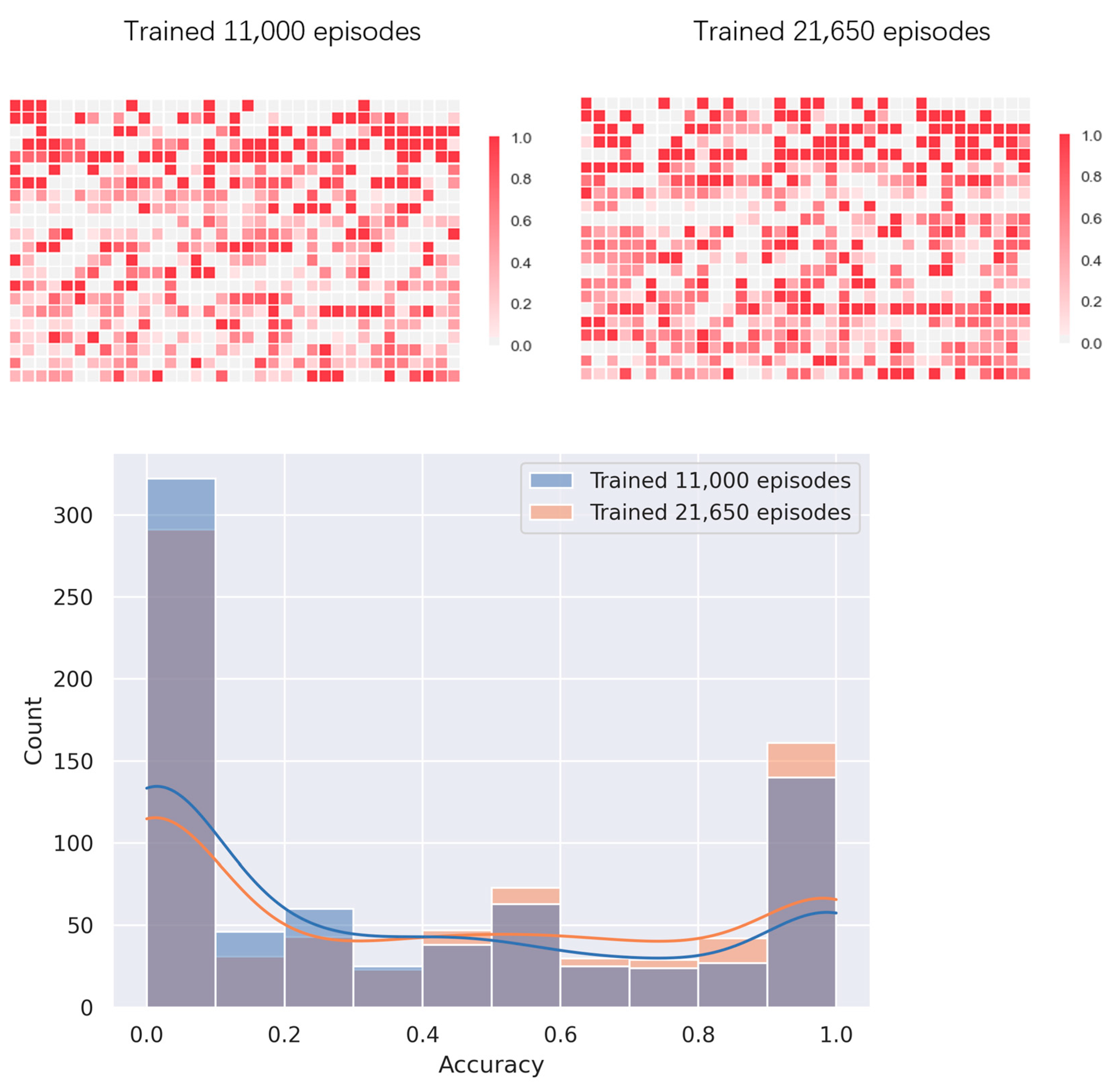
4.2. Multi-Threaded Accelerated Training and Its Impact
4.3. Prediction of the Fastest Folding Path and Secondary Structure of RNA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Myhrvold, C.; Silver, P.A. Using synthetic RNAs as scaffolds and regulators. Nat. Struct. Mol. Biol. 2015, 22, 8–10. [Google Scholar] [CrossRef]
- Garst, A.D.; Edwards, A.L.; Batey, R.T. Riboswitches: Structures and mechanisms. Cold Spring Harbor Perspect. Biol. 2011, 3, a003533. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [Green Version]
- Thirumalai, D.; Hyeon, C. Theory of RNA Folding: From Hairpins to Ribozymes. In Non-Protein Coding RNAs; Walter, N.G., Woodson, S.A., Batey, R.T., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 27–47. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. In Bioinformatics: Structure, Function and Applications; Keith, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 3–31. [Google Scholar]
- Mathews, D.H. Revolutions in RNA secondary structure prediction. J. Mol. Biol. 2006, 359, 526–532. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Fu, Y.; Sharma, G.; Mathews, D.H. TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucleic Acids Res. 2017, 45, 11570–11581. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Lindgreen, S.; Gardner, P.P.; Krogh, A. MASTR: Multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics 2007, 23, 3304–3311. [Google Scholar] [CrossRef]
- Gong, Z.; Yang, S.; Yang, Q.; Zhu, Y.; Jiang, J.; Tang, C. Refining RNA solution structures with the integrative use of label-free paramagnetic relaxation enhancement NMR. Biophys. Rep. 2019, 5, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Wang, J.; Xiao, Y. Prediction of RNA secondary structure with pseudoknots using coupled deep neural networks. Biophys. Rep. 2020, 6, 146–154. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xiao, Y. 3dRNA: Building RNA 3D structure with improved template library. Comput. Struct. Biotechnol. J. 2020, 18, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Jie, P.; Deras, M.L.; Woodson, S.A. Fast folding of a ribozyme by stabilizing core interactions: Evidence for multiple folding pathways in RNA. J. Mol. Biol. 2000, 296, 133–144. [Google Scholar]
- Thirumalai, D.; Hyeon, C. RNA and Protein Folding: Common Themes and Variations. Biochemistry 2005, 44, 4957–4970. [Google Scholar] [CrossRef]
- Hyeon, C.; Thirumalai, D. Mechanical unfolding of RNA hairpins. Proc. Natl. Acad. Sci. USA 2005, 102, 6789–6794. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Orden, A.V. A three-state mechanism for DNA hairpin folding characterized by multiparameter fluorescence fluctuation spectroscopy. J. Am. Chem. Soc. 2006, 128, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, C.; Thirumalai, D. Multiple probes are required to explore and control the rugged energy landscape of RNA hairpins. J. Am. Chem. Soc. 2008, 130, 1538–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; Van Den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- Silver, D.; Schrittwieser, J.; Simonyan, K.; Antonoglou, I.; Huang, A.; Guez, A.; Hubert, T.; Baker, L.; Lai, M.; Bolton, A. Mastering the game of go without human knowledge. Nature 2017, 550, 354–359. [Google Scholar] [CrossRef]
- Vinyals, O.; Babuschkin, I.; Czarnecki, W.M.; Mathieu, M.; Dudzik, A.; Chung, J.; Choi, D.H.; Powell, R.; Ewalds, T.; Georgiev, P. Grandmaster level in StarCraft II using multi-agent reinforcement learning. Nature 2019, 575, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Berner, C.; Brockman, G.; Chan, B.; Cheung, V.; Dębiak, P.; Dennison, C.; Farhi, D.; Fischer, Q.; Hashme, S.; Hesse, C. Dota 2 with large scale deep reinforcement learning. arXiv 2019, arXiv:1912.06680. [Google Scholar]
- Brown, N.; Sandholm, T. Superhuman AI for multiplayer poker. Science 2019, 365, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Runge, F.; Stoll, D.; Falkner, S.; Hutter, F. Learning to design RNA. arXiv 2018, arXiv:1812.11951. [Google Scholar]
- Sutton, R.S.; Barto, A.G. Reinforcement Learning: An Introduction; MIT Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Mnih, V.; Badia, A.P.; Mirza, M.; Graves, A.; Lillicrap, T.; Harley, T.; Silver, D.; Kavukcuoglu, K. Asynchronous methods for deep reinforcement learning. In Proceedings of the International Conference on Machine Learning, New York, NY, USA, 19–24 June 2016; pp. 1928–1937. [Google Scholar]
- Coulom, R. Efficient selectivity and backup operators in Monte-Carlo tree search. In Proceedings of the International Conference on Computers and Games, Turn, Italy, 29–31 May 2006; pp. 72–83. [Google Scholar]
- Chaslot, G.; Bakkes, S.; Szita, I.; Spronck, P. Monte-Carlo Tree Search: A New Framework for Game AI. In Proceedings of the AIIDE, Stanford, CA, USA, 22–24 October 2008. [Google Scholar]
- Kocsis, L.; Szepesvári, C. Bandit based monte-carlo planning. In Proceedings of the European Conference on Machine Learning, Berlin, Germany, 18–22 September 2006; pp. 282–293. [Google Scholar]
- Agarap, A.F. Deep learning using rectified linear units (relu). arXiv 2018, arXiv:1803.08375. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Brockman, G.; Cheung, V.; Pettersson, L.; Schneider, J.; Schulman, J.; Tang, J.; Zaremba, W. Openai gym. arXiv 2016, arXiv:1606.01540. [Google Scholar]
- Hagberg, A.; Swart, P.; Chult, D.S. Exploring Network Structure, Dynamics, and Function Using NetworkX; Los Alamos National Lab.(LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Tafer, H. In Silico Modelling of RNA-RNA Dimer and Its Application for Rational siRNA Design and ncRNA Target Search. Doctoral Dissertation, Universität Wien, Wien, Austria, 2011. [Google Scholar]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013, 41, 4307–4323. [Google Scholar] [CrossRef]
- Danaee, P.; Rouches, M.; Wiley, M.; Deng, D.; Huang, L.; Hendrix, D. bpRNA: Large-scale automated annotation and analysis of RNA secondary structure. Nucleic Acids Res. 2018, 46, 5381–5394. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, K.; Xiao, Y. Learning the Fastest RNA Folding Path Based on Reinforcement Learning and Monte Carlo Tree Search. Molecules 2021, 26, 4420. https://doi.org/10.3390/molecules26154420
Mao K, Xiao Y. Learning the Fastest RNA Folding Path Based on Reinforcement Learning and Monte Carlo Tree Search. Molecules. 2021; 26(15):4420. https://doi.org/10.3390/molecules26154420
Chicago/Turabian StyleMao, Kangkun, and Yi Xiao. 2021. "Learning the Fastest RNA Folding Path Based on Reinforcement Learning and Monte Carlo Tree Search" Molecules 26, no. 15: 4420. https://doi.org/10.3390/molecules26154420
APA StyleMao, K., & Xiao, Y. (2021). Learning the Fastest RNA Folding Path Based on Reinforcement Learning and Monte Carlo Tree Search. Molecules, 26(15), 4420. https://doi.org/10.3390/molecules26154420