
Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment



Abstract
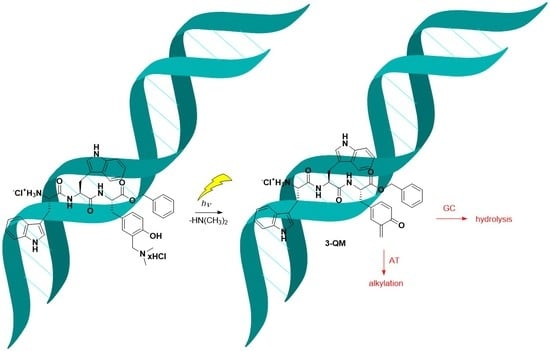
:
1. Introduction
2. Results and Discussion
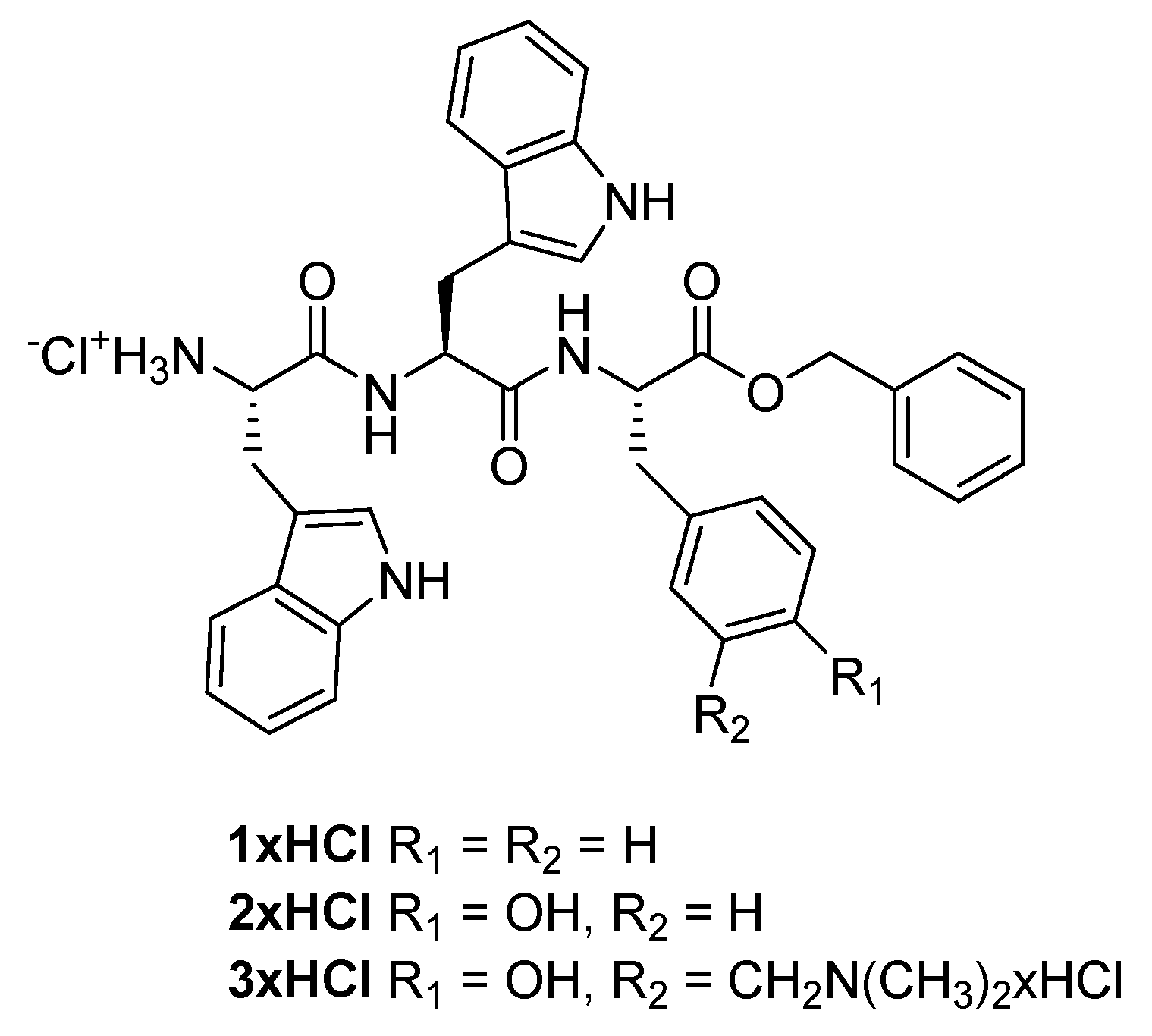
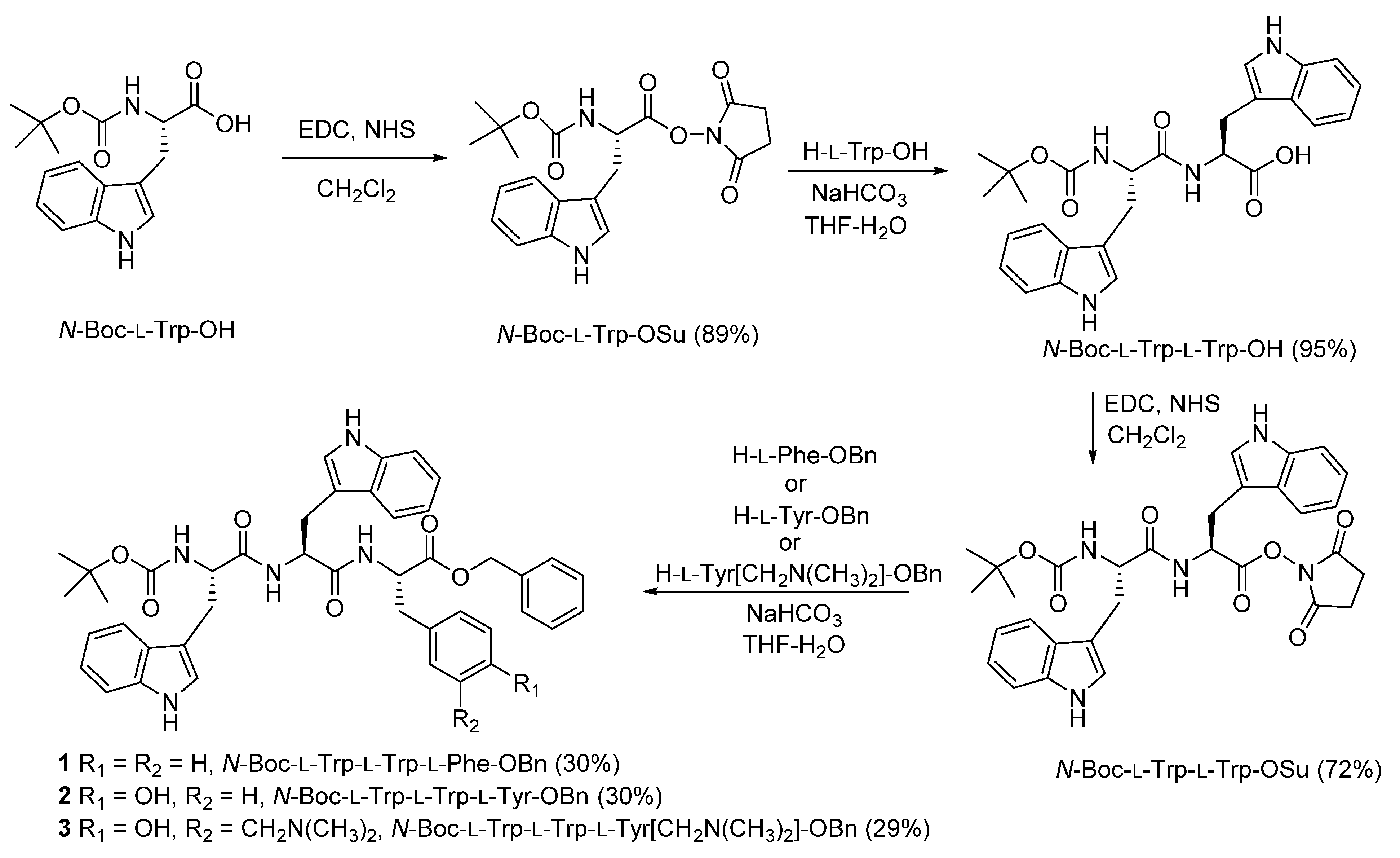
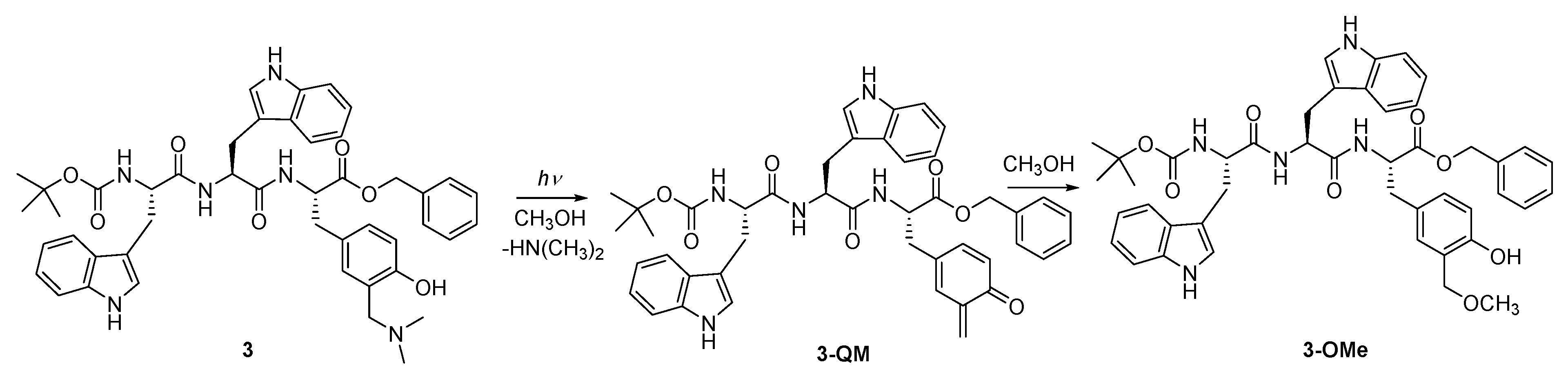
2.1. Synthesis
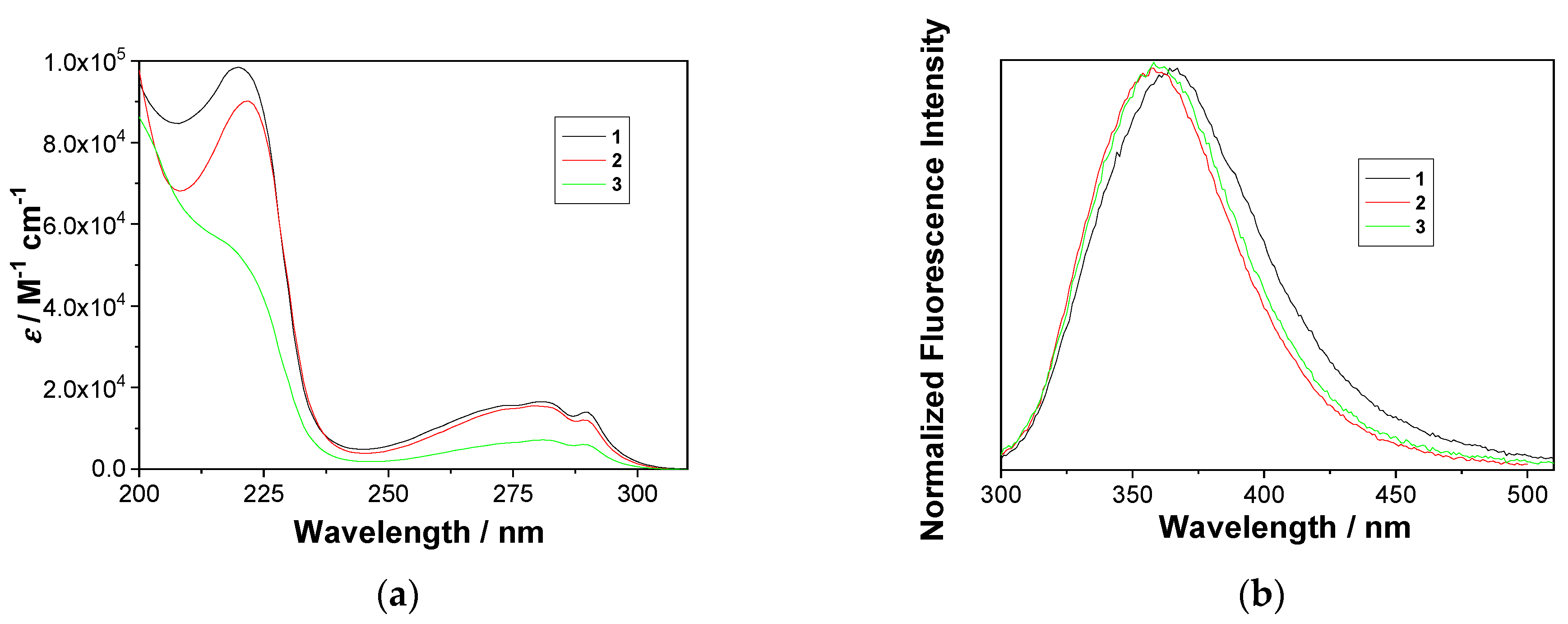
2.2. Photophysical Properties
2.3. Photochemistry
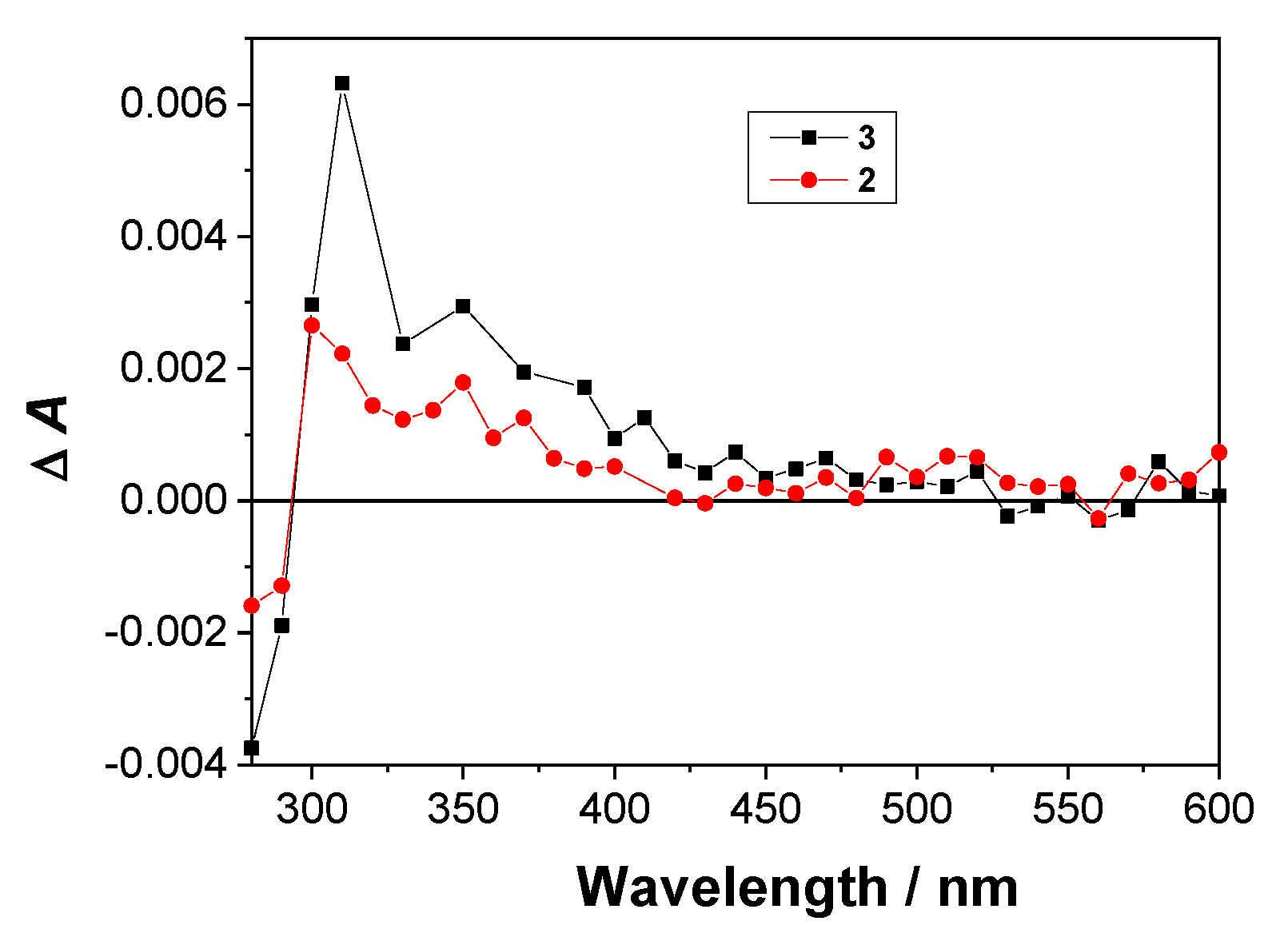
2.4. Laser Flash Photolysis (LFP)
2.5. Non-Covalent Binding to Polynucleotides
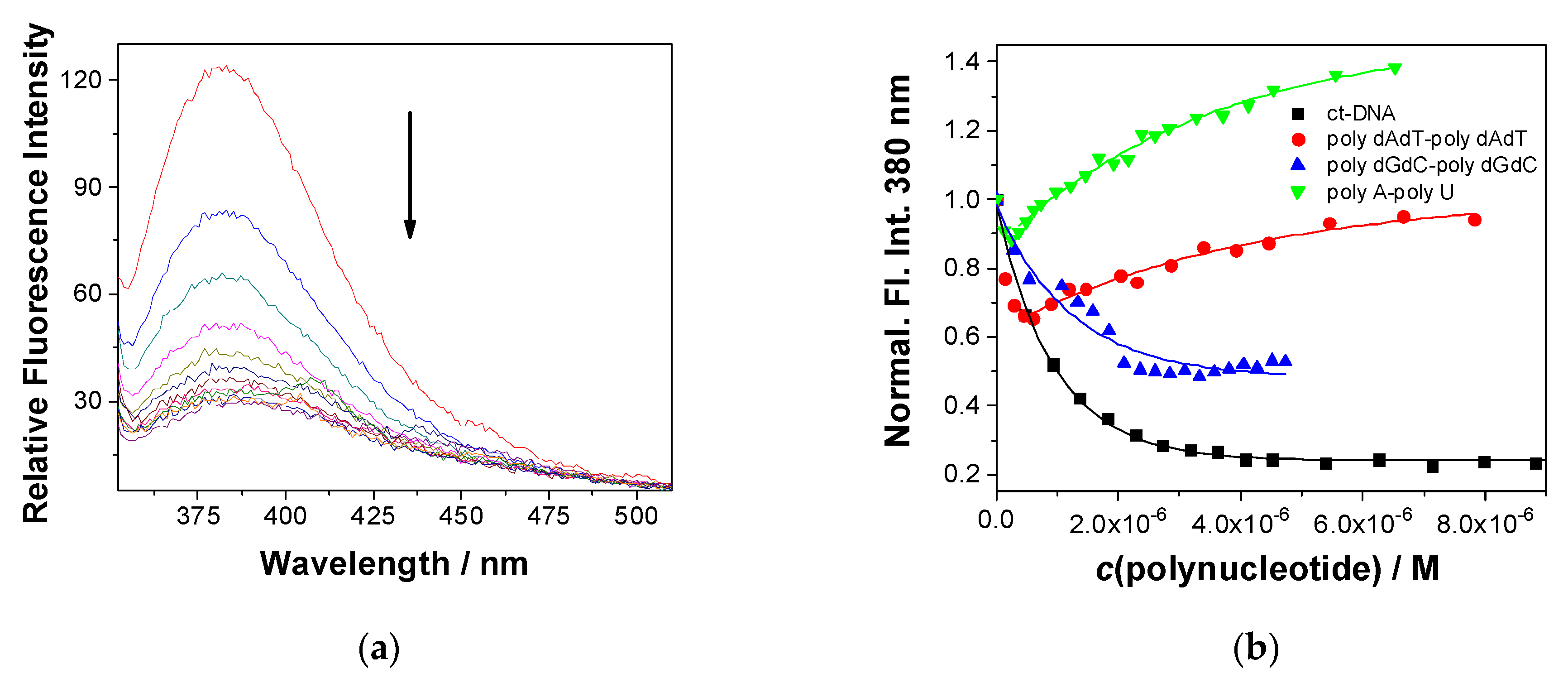
2.5.1. Fluorescence Titrations
2.5.2. Thermal Denaturation
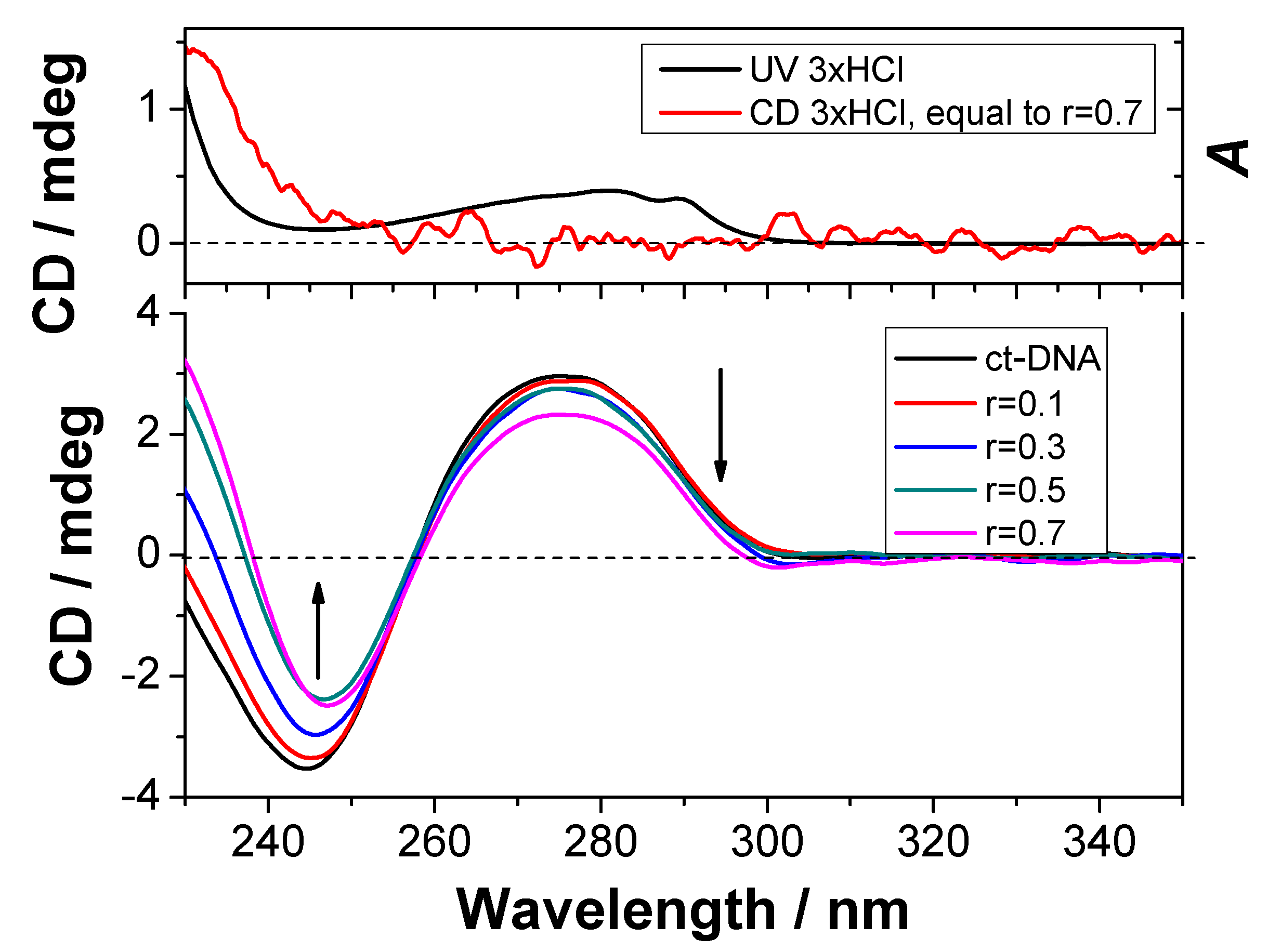
2.5.3. CD Experiments
2.6. Covalent Binding to Polynucleotides
3. Materials and Methods
3.1. General
3.2. Preparation of N-Boc-L-Trp-L-Trp-L-Tyr[CH2N(CH3)2]-OBn (3)
3.3. Preparation of HCl×H-L-Trp-L-Trp-L-Tyr[CH2N(CH3)2]-OBn (3×HCl)
3.4. Irradiation of N-Boc-L-Trp-L-Trp-L-Tyr[CH2N(CH3)2]-OBn (3)
3.5. Quantum Yield of Methanolysis
3.6. Irradiation of N-Boc-L-Trp-L-Trp-L-Tyr[CH2N(CH3)2]-OBn (3) in the Presence of Nucleotides
3.7. Absorption and Fluorescence Measurements
3.8. Preparation of the Annealed Double Stranded Oligonucleotides
3.9. Thermal Denaturation Experiments
3.10. Fluorescence Titrations
3.11. Circular Dichroism Spectroscopy
3.12. Photochemical Alkylation ct-DNA and of Oligonucleotides
3.13. Laser Flash Photolysis (LFP)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mitchell, P.J.; Tjian, R. Transcriptional Regulationin Mamnalian Cells by Sequence-Specific DNA Binding Proteins. Science 1989, 245, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Sauer, R.T. Transcription Factors: Structural Families and Principles of DNA Recognition. Annu. Rev. Biochem. 1992, 61, 1053–1095. [Google Scholar] [CrossRef] [PubMed]
- Eugenio Vazquez, M.; Caamano, A.M.; Mascarenas, J.L. Fromtrans Cription Factors to Designed Sequence-Specific DNABinding Peptides. Chem. Soc. Rev. 2003, 32, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Matić, J.; Tumir, L.-M.; Radić Stojković, M.; Piantanida, I. Advances in Peptide-based DNA/RNA-Intercalators. Curr. Protein Pept. Sci. 2016, 17, 127–134. [Google Scholar] [CrossRef]
- Boga, S.; Bouzada, D.; Garcia Pena, D.; Vazquez Lopez, M.; Eugenio Vazquez, M. Sequence-Specific DNA Recognition with Designed Peptides. Eur. J. Org. Chem. 2018, 2018, 249–261. [Google Scholar] [CrossRef]
- Klug, A.; Schwabe, J.W.R. Zinc Fingers. FASEB J. 1995, 9, 597–604. [Google Scholar] [CrossRef] [PubMed]
- O’neil, K.T.; Hoess, R.H.; Degrado, W.F. Design of DNA-Binding Peptides Based on the Leucine Zipper Motif. Science 1990, 249, 774–778. [Google Scholar] [CrossRef]
- Youngquist, R.S.; Dervan, P.B. Sequence-specific Recognition of B-DNA by Oligo(N-methylpyrrolecarboxamide)s. Proc. Natl. Acad. Sci. USA 1985, 82, 2565–2569. [Google Scholar] [CrossRef] [Green Version]
- Chenoweth, D.M.; Dervan, P.B. Structural Basis for Cyclic Py-Im Polyamide Allosteric Inhibition of Nuclear Receptor Binding. J. Am. Chem. Soc. 2010, 132, 14521–14529. [Google Scholar] [CrossRef]
- Caamano, A.M.; Vazquez, M.E.; Martinez-Costas, J.; Castedo, L.; Mascarenas, J.L. A Light-Modulated Sequence-Specific DNABinding Peptide. Angew. Chem. 2000, 112, 3234–3237. [Google Scholar] [CrossRef]
- Toseland, C.P. Fluorescent Labeling and Modification of Proteins. J. Chem. Biol. 2013, 6, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9780470027318.a1611 (accessed on 23 June 2021).
- Maity, D. Selected Peptide-based Fluorescent Probes for Biological Applications. J. Org. Chem. 2020, 16, 2971–2982. [Google Scholar]
- Maity, D.; Li, M.; Ehlers, M.; Schmuck, C. A Metal-Free Fluorescence Turn-on Molecular Probe for Detection of Nucleoside Triphosphates. Chem. Commun. 2017, 53, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zou, Y.; Li, C.; Sicking, W.; Piantanida, I.; Yi, T.; Schmuck, C. A Molecular Peptide Beacon for the Ratiometric Sensing of Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Maity, D.; Jiang, J.; Ehlers, M.; Wu, J.; Schmuck, C. A FRET-Enabled Molecular Peptide Beacon with a Significant Red Shift for the Ratiometric Detection of Nucleic Acids. Chem. Commun. 2016, 52, 6134–6137. [Google Scholar] [CrossRef]
- Maity, D.; Matković, M.; Li, S.; Ehlers, M.; Wu, J.; Piantanida, I.; Schmuck, C. Peptide-Based Probes with an Artificial Anion-Binding Motif for Direct Fluorescence “Switch-On” Detection of Nucleic Acid in Cells. Chem. Eur. J. 2017, 23, 17356–17362. [Google Scholar] [CrossRef]
- Matić, J.; Šupljika, F.; Tandarić, T.; Dukši, M.; Piotrowski, P.; Vianello, R.; Brozović, A.; Piantanida, I.; Schmuck, C.; Radić Stojković, M. DNA/RNA Recognition Controlled by the Glycine Linker and the Guanidine Moiety of Phenanthridine Peptides. Int. J. Biol. Macromol. 2019, 134, 422–434. [Google Scholar] [CrossRef]
- Saftić, D.; Radić Stojković, M.; Žinić, B.; Glavaš-Obrovac, L.; Jukić, M.; Piantanida, I.; Tumir, L.-M. Impact of Linker between Triazolyluracil and Phenanthridine on Recognitionof DNA and RNA. Recognition of Uracil-Containing RNA. New J. Chem. 2017, 41, 13240–13252. [Google Scholar] [CrossRef]
- Ban, Ž.; Žinić, B.; Matković, M.; Tomašić Paić, A.; Crnolatac, I.; Piantanida, I. Pyrrolocytosine-Pyrene Conjugates as Fluorescent and CD Probes for the Fine Sensing of ds-Polynucleotide Secondary Structure and Specific Recognition of poly G. New J. Chem. 2019, 43, 8204–8214. [Google Scholar] [CrossRef]
- Chen, H.; Gao, P.; Zhang, M.; Liao, W.; Zhang, J. Synthesis and Biological Evaluation of a Novel Classof β-Carboline Derivatives. New J. Chem. 2014, 38, 4155–4166. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhao, M.; Liu, L.; Zheng, M.; Wang, Y.; Wu, J.; Peng, S. A Class of Trp-Trp-AA-OBzl: Synthesis, in vitro anti-Proliferation/in vivo anti-Tumor Evaluation, Intercalation-Mechanism Investigation and 3D QSAR Analysis. Eur. J. Med. Chem. 2011, 46, 3410–3419. [Google Scholar] [CrossRef] [PubMed]
- Rokita, S.E. (Ed.) Quinone Methides; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Singh, M.S.; Nagaraju, A.; Anand, N.; Chowdhury, S. ortho-Quinone Methide (o-QM): A Highly Reactive, Ephemeral and Versatile Intermediate in Organic Synthesis. RSC Adv. 2014, 4, 55924–55959. [Google Scholar] [CrossRef]
- Bai, W.-J.; David, J.G.; Feng, Z.-G.; Weaver, M.G.; Wu, K.-L.; Pettus, T.R.R. The Domestication of ortho-Quinone Methides. Acc. Chem. Res. 2014, 47, 3655–3664. [Google Scholar] [CrossRef] [Green Version]
- Frecero, M. Quinone Methides as Alkylating and Cross-Linking Agents. Mini Rev. Org. Chem. 2004, 1, 403–415. [Google Scholar] [CrossRef]
- Wang, P.; Song, Y.; Zhang, L.; He, H.; Zhou, X. Quinone Methide Derivatives: Important Intermediates to DNA Alkylating and DNA Cross-Linking Actions. Curr. Med. Chem. 2005, 12, 2893–2913. [Google Scholar] [CrossRef] [PubMed]
- Basarić, N.; Mlinarić-Majerski, K.; Kralj, M. Quinone Methides: Photochemical Generation and its Application in Biomedicine. Curr. Org. Chem. 2014, 18, 3–18. [Google Scholar] [CrossRef]
- Percivalle, C.; Doria, F.; Freccero, M. Quinone Methides as DNA Alkylating Agents: An Overview on Efficient Activation Protocols for Enhanced Target Selectivity. Curr. Org. Chem. 2014, 18, 19–43. [Google Scholar] [CrossRef]
- McCracken, P.G.; Bolton, J.L.; Thatcher, G.R.J. Covalent Modification of Proteins and Peptides by the Quinone Methide from 2-tert-Butyl-4,6-dimethylphenol: Selectivity and Reactivity with Respect to Competitive Hydration. J. Org. Chem. 1997, 62, 1820–1825. [Google Scholar] [CrossRef]
- Arumugam, S.; Guo, J.; Mbua, N.E.; Friscourt, F.; Lin, N.; Nekongo, E.; Boons, G.-J.; Popik, V.V. Selective and Reversible Photochemical Derivatization of Cysteine Residues in Peptides and Proteins. Chem. Sci. 2014, 5, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Ruiz, R.; Molins-Molina, O.; Lence, E.; Gonzalez-Bello, C.; Miranda, M.A.; Consuelo Jimenez, M. Photogeneration of Quinone Methides as Latent Electrophiles for Lysine Targeting. J. Org. Chem. 2018, 83, 13019–13029. [Google Scholar] [CrossRef]
- Pande, P.; Shearer, J.; Yang, J.; Greenberg, W.A.; Rokita, S.E. Alkylation of Nucleic Acids by a Model Quinone Methide. J. Am. Chem. Soc. 1999, 121, 6773–6779. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Shallop, A.J.; Jones, R.A.; Rokita, S.E. Thermodynamic versus Kinetic Products of DNA Alkylation as Modeled by Reaction of Deoxyadenosine. J. Am. Chem. Soc. 2001, 123, 11126–11132. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Pande, P.; Rokita, S.E. A Transient Productof DNA Alkylation Can Be Stabilized by Binding Localization. J. Am. Chem. Soc. 2003, 125, 14005–14013. [Google Scholar] [CrossRef]
- Richter, S.N.; Maggi, S.; Colloredo Mels, S.; Palumbo, M.; Freccero, M. Binol Quinone Methides as Bisalkylating and DNA Cross-Linking Agents. J. Am. Chem. Soc. 2004, 126, 13973–13979. [Google Scholar] [CrossRef] [PubMed]
- Verga, D.; Nadai, M.; Doria, F.; Percivalle, C.; Di Antonio, M.; Palumbo, M.; Richter, S.N.; Freccero, M. Photogeneration and Reactivity of Naphthoquinone Methides as Purine Selective DNA Alkylating Agents. J. Am. Chem. Soc. 2010, 132, 14625–14637. [Google Scholar] [CrossRef] [PubMed]
- Di Antonio, M.; Doria, F.; Richter, S.N.; Bertipaglia, C.; Mella, M.; Sissi, C.; Palumbo, M.; Freccero, M. Quinone Methides Tethered to Naphthalene Diimides as Selective G-Quadruplex Alkylating Agents. J. Am. Chem. Soc. 2009, 131, 13132–13141. [Google Scholar] [CrossRef]
- Nadai, M.; Doria, F.; Di Antonio, M.; Sattin, G.; Germani, L.; Percivalle, C.; Palumbo, M.; Richter, S.N.; Freccero, M. Naphthalene Diimide Scaffolds with Dual Reversible and Covalent Interaction Properties towards G-Quadruplex. Biochimie 2011, 93, 1328–1340. [Google Scholar] [CrossRef]
- Doria, F.; Nadai, M.; Folini, M.; Di Antonio, M.; Germani, L.; Percivalle, C.; Sissi, C.; Zaffaroni, N.; Alcaro, S.; Artese, A.; et al. Hybrid Ligand-Alkylating Agents Targeting Telomeric G-Quadruplex Structures. Org. Biomol. Chem. 2012, 10, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Doria, F.; Nadai, M.; Folini, M.; Scalabrin, M.; Germani, L.; Sattin, G.; Mella, M.; Palumbo, M.; Zaffaroni, N.; Fabris, D.; et al. Targeting Loop Adeninesin G-Quadruplexby a Selective Oxirane. Chem. Eur. J. 2013, 19, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wahi, M.S.; Rokita, S.E. Immortalizing a Transient Electrophile for DNA Cross-Linking. Angew. Chem. Int. Ed. 2008, 47, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rokita, S.E. Dynamic Cross-Linking is Retained in Duplex DNA after Multiple Exchange of Strands. Angew. Chem. Int. Ed. 2010, 49, 5957–5960. [Google Scholar] [CrossRef]
- Rossiter, C.S.; Modica, E.; Kumar, D.; Rokita, S.E. Few Constraints Limit the Design of Quinone Methide-Oligonucleotide Self-Adducts for Directing DNA Alkylation. Chem. Commun. 2011, 47, 1476–1478. [Google Scholar] [CrossRef] [Green Version]
- Li, V.S.; Kohn, H. Studies on the Bonding Specificity for Mitomycin C-DNA Monoalkylation Processes. J. Am. Chem. Soc. 1991, 113, 275–283. [Google Scholar] [CrossRef]
- Han, I.; Russell, D.J.; Kohn, H. Studies on the Mechanism of Mitomycin C(1) Electrophilic Transformations: Structure-Reactivity Relationships. J. Org. Chem. 1992, 57, 1799–1807. [Google Scholar] [CrossRef]
- Tomasz, M.; Das, A.; Tang, K.S.; Ford, M.G.J.; Minnock, A.; Musser, S.M.; Waring, M.J. The Purine 2-Amino Group as the Critical Recognition Element for Sequence-Specific Alkylation and Cross-Linking of DNA by Mitomycin C. J. Am. Chem. Soc. 1998, 120, 11581–11593. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Kinetics and Mechanisms of Hydration of o-Quinone Methides in Aqueous Solution. J. Am. Chem. Soc. 2000, 122, 9854–9855. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Flash Photolytic Generation of ortho-Quinone Methide in Aqueous Solution and Study of Its Chemistry in that Medium. J. Am. Chem. Soc. 2001, 123, 8089–8094. [Google Scholar] [CrossRef] [PubMed]
- Toteva, M.M.; Richard, J.P. The Generation and Reactions of Quinone Methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar]
- Arumugam, S.; Popik, V.V. Photochemical Generation and the Reactivity of o-Naphthoquinone Methides in Aqueous Solutions. J. Am. Chem. Soc. 2009, 131, 11892–11899. [Google Scholar] [CrossRef]
- Husak, A.; Noichl, B.P.; Šumanovac Ramljak, T.; Sohora, M.; Škalamera, Đ.; Budiša, N.; Basarić, N. Photochemical Formation of Quinone Methides from Peptides Containing Modified Tyrosine. Org. Biomol. Chem. 2016, 14, 10894–10905. [Google Scholar] [CrossRef] [Green Version]
- Škalamera, Đ.; Bohne, C.; Landgraf, S.; Basarić, N. Photodeamination Reaction Mechanism in Aminomethyl p-Cresol Derivatives: Different Reactivity of Amines and Ammonium Salts. J. Org. Chem. 2015, 80, 10817–10828. [Google Scholar] [CrossRef]
- Ma, J.; Šekutor, M.; Škalamera, Đ.; Basarić, N.; Phillips, D.L. Formation of Quinone Methides by Ultrafast Photodeamination: A Spectroscopic and Computational Study. J. Org. Chem. 2019, 84, 8630–8637. [Google Scholar] [CrossRef] [PubMed]
- Siddique, B.; Duhamel, J. Effect of Polypeptide Sequence on Polypeptide Self-Assembly. Langmuir 2011, 27, 6639–6650. [Google Scholar] [CrossRef] [PubMed]
- Van Duuren, B.L. Solvent Effects in the Fluorescence of Indole and Substituted Indoles. J. Org. Chem. 1961, 26, 2954–2960. [Google Scholar] [CrossRef]
- Strickland, E.H.; Billups, C.; Kay, E. Effect of Hydrogen Bonding and Solvents upon the Tryptophan 1La Absorption Band. Studies using 2,3-dimethylindole. Biochemistry 1972, 11, 3657–3662. [Google Scholar] [CrossRef] [PubMed]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Valeur, B.; Weber, G. Resolution of the Fluorescence Excitation Spectrum of Indole into The 1La and 1Lb Excitation Bands. Photochem. Photobiol. 1977, 25, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Rabani, J. The Ferrioxalate and Iodide-Iodate Actinometers in the UV Region. J. Photochem. Photobiol. 2008, 193, 50–55. [Google Scholar] [CrossRef]
- Lee, J.; Robinson, G.W.; Webb, S.P.; Philips, L.A.; Clark, J.H. Hydration Dynamics of Protons from Photon Initiated Acids. J. Am. Chem. Soc. 1986, 108, 6538–6542. [Google Scholar] [CrossRef]
- Robinson, G.W. Proton Charge Transfer Involving the Water Solvent. J. Phys. Chem. 1991, 95, 10386–10391. [Google Scholar] [CrossRef]
- Tolbert, L.M.; Haubrich, J.E. Photoexcited Proton Transfer from Enhanced Photoacids. J. Am. Chem. Soc. 1994, 116, 10593–10600. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Agmon, N.; Tolbert, L.M. Photochemistry of “Super” Photoacids. 2. Excited-State Proton Transfer in Methanol/Water Mixtures. J. Phys. Chem. A 2000, 104, 4658–4669. [Google Scholar] [CrossRef]
- Bent, D.V.; Hayon, E. Excited State Chemistry of Aromatic Amino Acids and Related Peptides. III. Tryptophan. J. Am. Chem. Soc. 1975, 97, 2612–2619. [Google Scholar] [CrossRef] [PubMed]
- Dudley Bryant, F.; Sanfusl, R.; Grossweiner, L.I. Laser Flash Photolysis of Aqueous Tryptophan. J. Phys. Chem. 1975, 79, 2711–2716. [Google Scholar] [CrossRef]
- Basarić, N.; Franco-Cea, A.; Alešković, M.; Mlinarić-Majerski, K.; Wan, P. Photochemical Deuterium Exchange in Phenyl-Substitute Dipyrroles and Indoles in CD3CN-D2O. Phtochem. Photobiol. Sci. 2010, 9, 779–790. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D.; von Hippel, P.H. Theoretical Aspects of DNA-Protein Interactions: Co-operative and Non-co-operative Binding of Large Ligands to a one-Dimensional Homogeneous Lattice. J. Mol. Biol. 1976, 103, 679–681. [Google Scholar] [CrossRef]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Wilson, W.D.; Ratmeyer, L.; Zhao, M.; Strekowski, L.; Boykin, D. The Search for Structure-Specific Nucleic Acid-Interactive Drugs: Effects of Compound Structure on RNA Versus DNA Interaction Strength. Biochemistry 1993, 32, 4098–4104. [Google Scholar] [CrossRef] [PubMed]
- Jana, P.; Šupljika, F.; Schmuck, C.; Piantanida, I. Naphthalene Diimide bis-Guanidinio-Carbonyl-Pyrrole as a pH-Switchable Threading DNA Intercalator. Beilstein J. Org. Chem. 2020, 16, 2201–2211. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Piantanida, I.; Palm, B.S.; Žinić, M.; Schneider, H.J. A New 4,9-Diazapyrenium Intercalator for Single- and Double-Stranded Nucleic Acids: Distinct Differences from Related Diazapyrenium Compounds and Ethidium Bromide. J. Chem. Soc. Perkin Trans. 2 2001, 9, 1808–1816. [Google Scholar] [CrossRef]
- Mergny, J.L.; Lacroix, L. Analysis of Thermal Melting Curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: Oxford, NY, USA, 1997. [Google Scholar]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization Spectroscopy Methods in the Determination of Interactions of Small Molecules with Nucleic Acids-Tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Norden, B. Linear and Circular Dichroism of Drug-Nucleic Acid Complexes. Methods Enzymol. 2001, 340, 68–98. [Google Scholar] [PubMed]
- Kumar Mishra, N.; Ballabh Joshi, K.; Verma, S. Inhibition of Human and Bovine Insulin Fibril Formation by Designed Peptide Conjugates. Mol. Pharm. 2013, 10, 3903–3912. [Google Scholar] [CrossRef] [PubMed]
- Škalamera, Đ.; Mlinarić-Majerski, K.; Martin-Kleiner, I.; Kralj, M.; Wan, P.; Basarić, N. Near-visible Light Generation of a Quinone Methide from 3-Hydroxymethyl-2-anthrol. J. Org. Chem. 2014, 79, 4390–4397. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Φf (CH3CN) 1 | τf (CH3CN) 2/ns | Compound | Φf (CH3CN-H2O) 3 | τf (CH3CN-H2O)/ns |
|---|---|---|---|---|---|
| 1 | 0.11 ± 0.03 | 0.12 ± 0.03 (4%) 3.49 ± 0.06 (96%) | 1×HCl | 0.06 ± 0.01 | - |
| 2 | 0.11 ± 0.02 | 0.14 ± 0.03 (5%) 2.69 ± 0.06 (95%) | 2×HCl | 0.06 ± 0.01 | 0.36 ± 0.03 (7%) 2.63 ± 0.07 (93%) |
| 3 | 0.06 ± 0.01 | 0.24 ± 0.03 (7%) 2.37 ± 0.06 (93%) | 3×HCl | 0.03 ± 0.01 | ≈0.03 (11%) 1.62 ± 0.05 (60%) 2.9 ± 0.1 (29%) |
| ct-DNA | p(dAdT) 1 | p(dGdC) 2 | pApU | |
|---|---|---|---|---|
| 1×HCl | 5.7/0.62 3 | - | - | - |
| 2×HCl | 6.8/0.69 | - 4 | 6.5/0.3 | 5.0/3 5 |
| 3×HCl | 6.5/0.24 | 5.9/1.02 | 6.4/0.48 | 6.0/1.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erben, A.; Sviben, I.; Mihaljević, B.; Piantanida, I.; Basarić, N. Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment. Molecules 2021, 26, 4315. https://doi.org/10.3390/molecules26144315
Erben A, Sviben I, Mihaljević B, Piantanida I, Basarić N. Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment. Molecules. 2021; 26(14):4315. https://doi.org/10.3390/molecules26144315
Chicago/Turabian StyleErben, Antonija, Igor Sviben, Branka Mihaljević, Ivo Piantanida, and Nikola Basarić. 2021. "Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment" Molecules 26, no. 14: 4315. https://doi.org/10.3390/molecules26144315
APA StyleErben, A., Sviben, I., Mihaljević, B., Piantanida, I., & Basarić, N. (2021). Non-Covalent Binding of Tripeptides-Containing Tryptophan to Polynucleotides and Photochemical Deamination of Modified Tyrosine to Quinone Methide Leading to Covalent Attachment. Molecules, 26(14), 4315. https://doi.org/10.3390/molecules26144315