3.1.2. Experimental Procedure and Characterization Data for Compounds 2–9
General Procedure and Spectral Data for The Addition of 3,5-Dinitrobenzoyl Chloride to Functionalized Cyanoaziridines
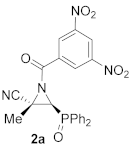
3,5-Dinitrobenzoyl chloride (1.4 g, 6 mmol, 1.2 eq) and Et3N (2.8 mL, 20 mmol, 4 eq) were added to a 0 °C solution of cyanoaziridine (5 mmol, 1 eq) in CH2Cl2 (25 mL). The reaction mixture was stirred at 0 °C until TLC showed the disappearance of the starting cyanoaziridine. The crude product was washed three times with a saturated NaCl solution (15 mL) and water (15 mL) and extracted with CH2Cl2 (15 mL). The organic layers were dried over anhydrous MgSO4, filtered and concentrated to dryness in vacuum conditions, and the resulting residue was purified by crystallization from Et2O/pentane or washed with pentane.
(
E)-(2
S*,3
S*)-1-(3,5-Dinitrobenzoyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carbonitrile (
2a), (1.97 g, 83%) was obtained as a grey solid from cyanoaziridine
1a (1.41 g, 5 mmol) after 24 h at 0 °C as described in the general procedure. The crude product was purified by crystallization from Et
2O/pentane (50:50) to give the title compound
2a; mp 120–122 °C; IR (neat)
vmax 3067, 2948, 2237, 1699, 1546, 1346, 1252, 1210, 1152 cm
−1;
1H NMR (300 MHz, CDCl
3)
δ 9.23 (t,
4JHH = 2.0 Hz, 1H, Ar
H), 9.08 (d,
4JHH = 2.0 Hz, 2H, Ar
H), 7.85–7.48 (m, 10H, Ar
H), 3.86 (d,
2JPH = 20.5 Hz, 1H, C
H-P), 2.17 (s, 3H, C
H3) ppm;
13C {
1H} NMR (75 MHz, CDCl
3)
δ 171.8 (d,
3JPC = 3.0 Hz, C=O), 149.0 (C
quat), 135.1(C
quat), 133.5, 133.5, 133.4, 133.3, 131.3, 131.1, 131.1, 131.0, 129.7, 129.5, 129.4, 129.2, 129.0 (C
Ar),120.4 (C
quat), 116.6 (CN), 43.4 (d,
1JPC = 90.7 Hz, CH-P), 37.0 (d,
2JPC = 2.5 Hz, C
quat), 16.9 (CH
3) ppm;
31P NMR (120 MHz, CDCl
3)
δ 21.9 ppm; ESI-HRMS (CI)
m/
z calculated for C
23H
18N
4O
6P ([M + H]
+) 477.0964 found 477.0971 (See
Supplementary Materials).
![Molecules 26 04265 i012]()
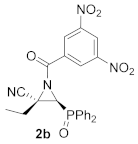
(E)-(2S*,3S*)-1-(3,5-Dinitrobenzoyl)-3-(diphenylphosphoryl)-2-ethylaziridine-2-carbonitrile (2b), (1.95 g, 80%) was obtained as a grey solid from cyanoaziridine 1b (1.48 g, 5 mmol) after 24 h at 0 °C as described in the general procedure. The crude product was purified by crystallization from Et2O/pentane (50:50) to give the title compound 2b; mp 201–203 °C; IR (neat) vmax 3101, 2884, 2237, 1710, 1630, 1544, 1460, 1441, 1344, 1294, 1202 1147, 1122 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.24 (t, 4JHH = 2.1 Hz, 1H, ArH), 9.11 (d, 4JHH = 2.1 Hz, 2H, ArH), 7.90–7.47 (m, 10H, ArH), 3.90 (d, 2JPH = 20.4 Hz, 1H, CH-P), 2.61–2.46 (m, 2H, CH2), 1.08 (t, 3JHH = 7.4 Hz, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 172.4 (d, 3JPC = 3.3 Hz, C=O), 148.9 (Cquat), 135.1 (Cquat), 133.5, 133.4, 133.3, 133.2, 131.2, 131.2, 131.1, 131.0, 129.6, 129.5, 129.3, 129.2, 129.1, 123.4 (CAr), 115.6 (CN), 43.6 (d, 1JPC = 90.6 Hz, CH-P), 43.2 (d, 2JPC = 2.6 Hz, Cquat), 24.2 (CH2), 10.5 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 21.4 ppm; ESI-HRMS (CI) m/z calculated for C24H20N4O6P ([M + H]+) 491.1120 found 491.1135.
![Molecules 26 04265 i013]()
Diethyl (E)-[(2S*,3S*)-3-cyano-1-(3,5-dinitrobenzoyl)-3-methylaziridin-2-yl]phosphonate (2c), (1.92 g, 93%) was obtained as a brown oil from cyanoaziridine 1c (1.09 g, 5 mmol) after 24 h at 0 °C as described in the general procedure. The crude product was washed with pentane to give the title compound 2c; Rf: 0.5 (AcOEt); IR (neat) vmax 3112, 2984, 2246, 1710, 1627, 1552, 1344, 1294, 1255, 1041, 1022 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.25 (t, 4JHH = 2.1 Hz, 1H, ArH), 9.05 (d, 4JHH = 2.1 Hz, 2H, ArH), 4.30–4.17 (m, 4H, OCH2CH3), 3.37 (d, 2JPH = 11.9 Hz, 1H, CH-P), 2.09 (s, 3H, CH3), 1.42–1.34 (m, 6H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 171.4 (d, 3JPC = 5.0 Hz, C=O), 149.1 (Cquat), 134.8 (Cquat), 128.9, 123.4 (CAr), 116.3 (3JPC = 2.1 Hz, CN), 64.2 (d, 2JPC = 6.1 Hz, OCH2), 63.7 (d, 2JPC = 6.5 Hz, OCH2) 40.4 (d, 1JPC = 202.3 Hz, CH-P), 35.8 (d, 2JPC = 2.7 Hz, Cquat), 17.6 (CH3), 16.5 (d, 3JPC = 5.9 Hz, OCH2CH3), 16.4 (d, 3JPC = 5.9 Hz, OCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 12.3 ppm; ESI-HRMS (CI) m/z calculated for C15H18N4O8P ([M + H]+) 413.0862 found 413.0857.
![Molecules 26 04265 i014]()
General Procedure and Spectral Data for Compound 3a
To a stirred solution of N-functionalized cyanoaziridine 2a (5 mmol, 1 eq) in THF (15 mL), NaI (0.02 g, 1 mmol, 0.2 eq) was added dropwise. The mixture was heated at 60 °C for 24 h until TLC showed the disappearance of the starting cyanoaziridine. The reaction mixture was concentrated to dryness in vacuum conditions to remove THF. The crude product was washed three times with water (15 mL) and extracted with CH2Cl2 (15 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to dryness in vacuum conditions. The crude product was purified by flash-column chromatography.
(E)-(4S*,5S*)-2-(3,5-Dinitrophenyl)-5-(diphenylphosphoryl)-4-methyl-4,5-dihydrooxazole-4-carbonitrile (3a), (1.56 g, 65%) was obtained as a yellow solid from cyanoaziridine 2a (2.38 g, 5 mmol) after 24 h of heating in THF as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 25:75) to give the title compound 3a; mp 129–131 °C; IR (neat) vmax 3103, 2934, 2243, 1655, 1546, 1438, 1352, 1197, 1119 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.19 (t, 4JHH = 2.1 Hz, 1H, ArH), 8.99 (d, 4JHH = 2.1 Hz, 2H, ArH), 7.98–7.52 (m, 10H, ArH), 5.60 (d, 2JPH = 6.7 Hz, 1H, CH-P), 1.80 (s, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 161.7 (d, 3JPC = 5.6 Hz, Cquat), 148.8, 133.8, 133.8, 133.4, 133.3, 131.4, 131.3, 131.2, 131.1, 129.9, 129.7, 129.4, 129.3, 128.8, 122.2 (CAr),119.8 (d, 3JPC = 8.9 Hz, CN), 108.1 (Cquat), 83.2 (d, 1JPC = 76.7 Hz, CH-P), 68.3 (d, 2JPC = 2.0 Hz, Cquat), 23.0 (d, 3JPC = 5.2 Hz, CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 22.5 ppm; ESI-HRMS (CI) m/z calculated for C23H18N4O6P ([M + H]+) 477.0964 found 477.0965.
![Molecules 26 04265 i015]()
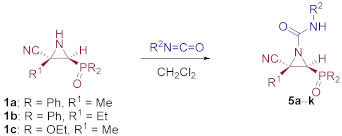
General Procedures and Spectral Data for The Addition of Isocyanates to Functionalized Cyanoaziridines 1
Method A. To a 0 °C solution of cyanoaziridine (5 mmol, 1 eq) in CH2Cl2 (25 mL) the corresponding isocyanate (6 mmol, 1.2 eq) was added dropwise. The reaction mixture was allowed to reach room temperature and stirred for 6–24 h. The crude products were concentrated to dryness in vacuum conditions and were purified by crystallization. Method B. To a 0 °C solution of cyanoaziridine (5 mmol, 1 eq) in CH2Cl2 (25 mL) phenyl isocyanate (15 mmol, 3 eq) and Sc (OTf)3 (0.49 g, 1 mmol, 0.2 eq) were added dropwise. The reaction mixture was stirred at 0 °C for 5 h until TLC showed the disappearance of the starting cyanoaziridine. The reaction mixture was washed with water (3 × 15 mL) and extracted with CH2Cl2 (15 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to dryness in vacuum conditions. The crude product was purified by crystallization from Et2O. Method C. To a solution of cyanoaziridine (5 mmol, 1 eq) in CH2Cl2 (25 mL) the corresponding aliphatic isocyanate (10 mmol, 2 eq) and ZnCl2 (0.85 g, 6.25 mmol, 1.25 eq) were added dropwise. The reaction mixture was stirred at room temperature for 5–48 h until TLC showed the disappearance of the starting cyanoaziridine. The reaction mixture was washed with saturated NH4Cl (1 × 15 mL) and water (3 × 15 mL) and extracted with CH2Cl2 (15 mL). The organic layers were dried over anhydrous MgSO4, filtered, and concentrated to dryness in vacuum conditions. The crude product was purified by crystallization.
(E)-(2S*,3S*)-2-Cyano-3-(diphenylphosphoryl)-2-methyl-N-phenylaziridine-1-carboxamide (5a), (1.27 g, 63%) was obtained as a white solid from cyanoaziridine 1a (1.41 g, 5 mmol) and phenylisocyanate (0.65 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from Et2O to give the title compound 5a. (1.42 g, 71%) obtained as an orange pale solid from cyanoaziridine 1a (1.41 g, 5 mmol), phenylisocyanate (0.65 mL, 6 mmol, 1.2 eq) and Sc(OTf)3 (1 mmol, 0.49 g) as described in the general procedure (method B). The crude product was purified by crystallization from Et2O to give the title compound 5a; mp 179–181 °C; IR (neat) vmax 3220, 3053, 2926, 2256, 1710, 1596, 1544, 1435, 1252, 1202, 1127 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.11 (bs, 1H, NH), 7.88–7.07 (m, 15H, ArH), 3.76 (d, 2JPH = 23.1 Hz, 1H, CH-P), 1.92 (s, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 157.2 (C=O), 137.8 (Cquat), 133.1, 133.1, 133.1, 133.0, 131.6, 131.4, 131.2, 131.2, 131.0, 130.1, 130.0, 129.5, 129.4, 129.3, 129.2, 129.1, 124.6, 120.1 (CAr), 117.1 (CN), 41.3 (d, 1JPC = 102.1 Hz, CH-P), 37.4 (Cquat), 17.9 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 23.4 ppm; ESI-HRMS (CI) m/z calculated for C23H21N3O2P ([M + H]+) 402.1371 found 402.1374.
![Molecules 26 04265 i016]()
Diethyl (E)-[(2S*,3S*)-3-cyano-3-methyl-1-(phenylcarbamoyl)aziridin-2-yl]phosphonate (5b), (1.65 g, 98%) was obtained as a white solid from cyanoaziridine 1c (1.09 g, 5 mmol) and phenylisocyanate (0.65 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from CH2Cl2/pentane to give the title compound 5b; mp 137–139 °C; IR (neat) vmax 3253, 3065, 2984, 2240, 1716, 1607, 1544, 1499, 1444, 1249, 1044, 1024 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.30 (bs, 1H, NH), 7.48–7.09 (m, 5H, ArH), 4.25–4.15 (m, 4H, OCH2CH3), 3.29 (d, 2JPH = 13.6 Hz, 1H, CH-P), 1.92 (s, 3H, CH3), 1.40–1.32 (m, 6H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 156.6 (C=O), 137.3 (Cquat), 129.2, 124.8, 120.0 (CAr), 116.9 (CN), 63.9 (d, 2JPC = 5.4 Hz, OCH2), 63.3 (d, 2JPC = 6.0 Hz, OCH2) 38.7 (d, 1JPC = 207.4 Hz, CH-P), 36.0 (Cquat), 18.2 (CH3), 16.6 (d, 3JPC = 4.9 Hz, OCH2CH3), 16.5 (d, 3JPC = 4.7 Hz, OCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 14.6 ppm; ESI-HRMS (CI) m/z calculated for C15H21N3O4P ([M + H]+) 338.1270 found 338.1264.
![Molecules 26 04265 i017]()
(E)-(2S*,3S*)-2-Cyano-3-(diphenylphosphoryl)-2-methyl-N-(p-tolyl) aziridine-1-carboxamide (5c), (1.70 g, 82%) was obtained as a yellow solid from cyanoaziridine 1a (1.41 g, 5 mmol) and p-tolyl isocyanate (0.76 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from Et2O to give the title compound 5c; mp 193–195 °C; IR (neat) vmax 3237, 3056, 2917, 2254, 1710, 1599, 1546, 1539, 1408, 1249, 1197 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.17 (bs, 1H, NH), 7.86–7.06 (m, 14H, ArH), 3.76 (d, 2JPH = 23.0 Hz, 1H, CH-P), 2.28 (s, 3H, CH3), 1.92 (s, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 157.1 (d, 3JPC = 4.2 Hz, C=O), 135.2 (Cquat), 134.2 (Cquat), 133.0, 133.0, 131.3, 131.2, 131.1, 131.0, 129.5, 129.4, 129.3, 129.1, 120.2, 120.0, (CAr), 117.1 (CN), 41.2 (d, 1JPC = 100.5 Hz, CH-P), 37.3 (d, 2JPC = 3.1 Hz, Cquat), 21.0 (CH3), 17.9 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 23.6 ppm; ESI-HRMS (CI) m/z calculated for C24H23N3O2P ([M + H]+) 416.1528 found 416.1532.
![Molecules 26 04265 i018]()
Diethyl (E)-[(2S*,3S*)-3-cyano-3-methyl-1-(p-tolylcarbamoyl)aziridin-2-yl]phosphonate (5d), (1.40 g, 80%) was obtained as a pale yellow solid from cyanoaziridine 1c (1.09 g, 5 mmol) and p-tolyl isocyanate (0.76 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from CH2Cl2/pentane to give the title compound 5d; mp 140–142 °C; IR (neat) vmax 3256, 3040, 2987, 2240, 1710, 1607, 1538, 1444, 1516, 1321, 1247, 1039 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.15 (bs, 1H, NH), 7.33 (d, 3JHH = 7.9 Hz, 2H, ArH), 7.09 (d, 3JHH = 7.9 Hz, 2H, ArH), 4.24–4.14 (m, 4H, OCH2CH3), 3.28 (d, 2JPH = 13.5 Hz, 1H, CH-P), 2.28 (s, 3H, CH3), 1.91 (s, 3H, CH3), 1.39–1.32 (m, 6H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 156.6 (d, 3JPC = 5.9 Hz, C=O), 134.9 (Cquat), 134.3 (Cquat), 129.5, 120.1 (CAr), 116.9 (CN), 63.9 (d, 2JPC = 6.2 Hz, OCH2), 63.2 (d, 2JPC = 6.5 Hz, OCH2), 38.4 (d, 1JPC = 206.8 Hz, CH-P), 36.0 (d, 2JPC = 2.2 Hz, Cquat), 20.9 (CH3), 18.1 (CH3), 16.5 (d, 3JPC = 6.0 Hz, OCH2CH3), 16.4 (d, 3JPC = 6.0 Hz, OCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 14.8 ppm; ESI-HRMS (CI) m/z calculated for C16H23N3O4P ([M + H]+) 352.1426 found 352.1419.
![Molecules 26 04265 i019]()
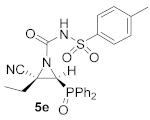
(E)-(2S*,3S*)-2-Cyano-3-(diphenylphosphoryl)-2-ethyl-N-tosylaziridine-1-carboxamide (5e), (2.12 g, 86%) was obtained as a white solid from cyanoaziridine 1b (1.48 g, 5 mmol) and p-toluenesulfonyl isocyanate (0.92 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from Et2O to give the title compound 5e; mp 201–203 °C; IR (neat) vmax 3257, 2931, 2245, 1743, 1605, 1444, 1360, 1242, 1124, 1094 cm−1; 1H NMR (400 MHz, MeOD) δ 7.92–7.38 (m, 14H, ArH), 3.39 (d, 2JPH = 23.4 Hz, 1H, CH-P), 1.97 (m, 2H, CH2), 2.44 (s, 3H, CH3), 1.02 (t, 3JHH = 7.4 Hz, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, MeOD) δ 153.5 (C=O), 146.0 (Cquat), 137.8 (Cquat), 134.2, 134.2, 133.9, 133.9, 132.2, 132.1, 132.1, 132.0, 130.5, 130.4, 130.3, 130.2, 130.0, 129.0 (CAr), 120.4 (CN), 39.5 (d, 1JPC = 101.6 Hz, CH-P), 36.6 (Cquat), 25.2 (CH2), 21.5 (CH3), 11.1 (CH3) ppm; 31P NMR (120 MHz, MeOD) δ 27.2 ppm; ESI-HRMS (CI) m/z calculated for C25H25N3O4PS ([M + H]+) 494.1303 found 494.1292.
![Molecules 26 04265 i020]()
Diethyl (E)-[(2S*,3S*)-3-cyano-3-methyl-1-(tosylcarbamoyl)aziridin-2-yl]phosphonate (5f), (1.64 g, 79%) was obtained as a waxy white solid from cyanoaziridine 1c (1.09 g, 5 mmol) and p-toluenesulfonyl isocyanate (0.92 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from CH2Cl2/pentane to give the title compound 5f; Rf: 0.4 (AcOEt). IR (neat) vmax 3248, 3092, 2992, 2237, 1738, 1649, 1596, 1446, 1335, 1247, 1160, 1047, 1027 cm−1; 1H NMR (300 MHz, CDCl3) δ 10.50 (bs, 1H, NH), 7.89–7.78 (m, 4H, ArH), 4.24–4.10 (m, 4H, OCH2CH3), 3.04 (d, 2JPH = 13.2 Hz, 1H, CH-P), 2.38 (s, 3H, CH3), 1.83 (s, 3H, CH3), 1.37–1.28 (m, 6H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 155.3 (d, 3JPC = 6.6 Hz, C=O), 145.3 (Cquat), 135.1 (Cquat), 129.8, 129.7, 128.6, 128.1, 126.5 (CAr), 116.2 (CN), 64.3 (d, 2JPC = 6.0 Hz, OCH2), 63.8 (d, 2JPC = 6.4 Hz, OCH2), 38.9 (d, 1JPC = 206.6 Hz, CH-P), 36.3 (d, 2JPC = 3.0 Hz, Cquat), 21.8 (CH3), 17.7 (CH3), 16.5 (d, 3JPC = 6.3 Hz, OCH2CH3), 16.4 (d, 3JPC = 6.2 Hz, OCH2CH3) ppm; 31P NMR (120 MHz) δ 13.3 ppm; ESI-HRMS (CI) m/z calculated for C16H23N3O6PS ([M + H]+) 416.1045 found 416.1038.
![Molecules 26 04265 i021]()
(E)-(2S*,3S*)-2-Cyano-3-(diphenylphosphoryl)-N-ethyl-2-methylaziridine-1-carboxamide (5g), (1.51 g, 86%) was obtained as a pale pink solid from cyanoaziridine 1a (1.41 g, 5 mmol) and ethyl isocyanate (0.79 mL, 10 mmol, 2 eq) as described in the general procedure (method C). The crude product was purified by crystallization from Et2O/pentane 50:50 to give the title compound 5g; mp 182–184 °C; IR (neat) vmax 3253, 3053, 2976, 2240, 1702, 1544, 1438, 1283, 1258, 1191, 1124 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.84–7.46 (m, 10H, ArH), 5.98 (t, 3JHH = 5.9 Hz, 1H, NH), 3.65 (d, 2JPH = 22.1 Hz, 1H, CH-P), 3.37–3.18 (m, 2H, NHCH2CH3), 1.85 (s, 3H, CH3), 1.13 (t, 3JHH = 7.3 Hz, NHCH2CH3) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 159.1 (d, 3JPC = 4.6 Hz, C=O), 133.0, 132.9, 132.8, 132.8, 131.6, 131.4, 131.3, 131.1, 131.1, 130.6, 130.3, 129.3, 129.2, 129.1, 129.0 (CAr), 117.1 (CN), 41.6 (d, 1JPC = 100.1 Hz, CH-P), 36.7 (d, 2JPC = 3.4 Hz, Cquat), 36.4 (NHCH2CH3), 18.1 (CH3), 15.1 (NHCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 23.1 ppm; ESI-HRMS (CI) m/z calculated for C19H21N3O2P ([M + H]+) 354.1371 found 354.1372.
![Molecules 26 04265 i022]()
Diethyl (E)-[(2S*,3S*)-3-cyano-1-(ethylcarbamoyl)-3-methylaziridin-2-yl]phosphonate (5h), (0.85 g, 59%) was obtained as a waxy solid from cyanoaziridine 1c (1.09 g, 5 mmol) and ethyl isocyanate (0.79 mL, 10 mmol, 2 eq) as described in the general procedure (method C). The crude product was purified by crystallization from Et2O/pentane 50:50 to give the title compound 5h; Rf: 0.3 (AcOEt); IR (neat) vmax 3281, 3062, 2987, 2243, 1699, 1541, 1455, 1385, 1371, 1252, 1160, 1044, 1039 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.03 (bs, 1H, NH), 4.19–4.09 (m, 4H, OCH2CH3), 3.36–3.18 (m, 2H, NHCH2CH3), 3.12 (d, 2JPH = 13.8 Hz, 1H, CH-P), 1.79 (s, 3H, CH3), 1.34–1.28 (m, 6H, OCH2CH3) 1.13 (t, 3JHH = 7.2 Hz, NHCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 158.8 (d, 3JPC = 6.4 Hz, C=O), 116.9 (CN), 63.7 (d, 2JPC = 5.7 Hz, OCH2), 63.0 (d, 2JPC = 6.2 Hz, OCH2), 38.6 (d, 1JPC = 207.0 Hz, CH-P), 36.2 (NHCH2CH3), 35.3 (d, 2JPC = 3.2 Hz, Cquat), 18.2 (CH3), 16.4 (d, 3JPC = 5.2 Hz, OCH2CH3), 16.4 (d, 3JPC = 5.3 Hz, OCH2CH3), 15.0 (NHCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 15.1 ppm; ESI-HRMS (CI) m/z calculated for C11H21N3O4P ([M + H]+) 290.1270 found 290.1275.
![Molecules 26 04265 i023]()
(E)-(2S*,3S*)-N-(tert-Butyl)-2-cyano-3-(diphenylphosphoryl)-2-methylaziridine-1-carboxamide (5i), (1.39 g, 73%) was obtained as a white solid from cyanoaziridine 1a (1.41 g, 5 mmol) and tert-butyl isocyanate (1.14 mL, 10 mmol, 2 eq) as described in the general procedure (method C). The crude product was purified by crystallization from Et2O/pentane 50:50 to give the title compound 5i; mp 166–168 °C; IR (neat) vmax 3259, 3056, 2976, 2237, 1707, 1541, 1452, 1441, 1369, 1285, 1208, 1127 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.82–7.48 (m, 10H, ArH), 5.53 (s, 1H, NH), 3.62 (d, 2JPH = 22.1 Hz, 1H, CH-P), 1.85 (s, 3H, CH3), 1.33 (s, 9H, C(CH3)3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 157.4 (d, 3JPC = 4.6 Hz, C=O), 132.9, 132.9, 132.8, 132.8, 131.9, 131.6, 131.4, 131.3, 131.1, 131.0, 130.5, 130.2, 129.3, 129.2, 129.1, 129.0 (CAr), 117.0 (CN), 52.1 (C(CH)3)3), 41.2 (d, 1JPC = 99.9 Hz, CH-P), 36.5 (d, 2JPC = 3.5 Hz, Cquat), 28.7 (C(CH)3), 18.1 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 23.1 ppm; ESI-HRMS (CI) m/z calculated for C21H25N3O2P ([M + H]+) 382.1684 found 382.1687.
![Molecules 26 04265 i024]()
Diethyl (E)-[(2S*,3S*)-1-(tert-butylcarbamoyl)-3-cyano-3-methylaziridin-2-yl]phosphonate (5j), (1.19 g, 75%) was obtained as a white solid from cyanoaziridine 1c (1.09 g, 5 mmol) and tert-butyl isocyanate (1.14 mL, 10 mmol, 2 eq) as described in the general procedure (method C). The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 20:80) to give the title compound 5j; mp 96–98 °C; IR (neat) vmax 3284, 3051, 2979, 2246, 1705, 1538, 1477, 1457, 1394, 1369, 1260, 1160, 1038 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.45 (bs, 1H, NH), 4.21–4.11 (m, 4H, OCH2CH3), 3.13 (d, 2JPH = 13.6 Hz, 1H, CH-P), 1.81 (s, 3H, CH3), 1.36–1.30 (m, 15H, C(CH3)3 + OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 157.1 (d, 3JPC = 6.7 Hz, C=O), 116.9 (d, 3JPC = 2.4 Hz, CN), 63.7 (d, 2JPC = 6.1 Hz, OCH2), 63.0 (d, 2JPC = 6.5 Hz, OCH2), 52.1 (C(CH)3)3), 38.3 (d, 1JPC = 207.1 Hz, CH-P), 35.3 (d, 2JPC = 3.4 Hz, Cquat), 28.7 (C(CH)3), 18.4 (CH3), 16.5 (d, 3JPC = 5.9 Hz, OCH2CH3), 16.4 (d, 3JPC = 5.6 Hz, OCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 15.5 ppm; ESI-HRMS (CI) m/z calculated for C13H24N3NaO4P ([M + Na]+) 340.1402 found 340.1400.
![Molecules 26 04265 i025]()
(E)-(2S*,3S*)-N-(tert-Butyl)-2-cyano-3-(diphenylphosphoryl)-2-ethylaziridine-1-carboxamide (5k), (1.27 g, 64%) was obtained as a white solid from cyanoaziridine 1b (1.48 g, 5 mmol) and tert-butyl isocyanate (1.14 mL, 10 mmol, 2 eq) as described in the general procedure (method C). The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5k; mp 197–199 °C; IR (neat) vmax 3262, 2976, 2240, 1718, 1499, 1457, 1438, 1369, 1274, 1199, 1122 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.83–7.45 (m, 10H, ArH), 5.43 (s, 1H, NH), 3.67 (d, 2JPH = 22.2 Hz, 1H, CH-P), 2.34–2.24 (m, 1H, CH2CH3), 2.01–1.99 (m, 1H, CH2CH3), 1.33 (s, 9H, C(CH3)3, 1.09 (t, 3JHH = 1.9 Hz, 3H, CH2CH3) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 157.5 (C=O), 132.9, 132.8, 131.3, 131.2, 131.1, 131.0, 129.3, 129.2, 129.1, 129.0 (CAr), 116.1 (CN), 52.0 (C(CH3)3), 42.3 (d, 2JPC = 1.7 Hz, Cquat), 41.9 (d, 1JPC = 117.2 Hz, CH-P), 28.7 (C(CH)3), 24.6 (CH2CH3), 10.8 (CH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 22.8 ppm; ESI-HRMS (CI) m/z calculated for C22H27N3O2P ([M + H]+) 396.1841 found 396.1847.
![Molecules 26 04265 i026]()
General Procedures and Spectral Data for The Addition of Ethoxycarbonyl Isothiocyanate to Functionalized Cyanoaziridines 1
Method A. To a −30 °C solution of cyanoaziridine 1 (5 mmol, 1 eq) in CH2Cl2 (25 mL) ethoxycarbonyl isothiocyanate (6 mmol, 1.2 eq) was added dropwise. The reaction mixture was stirred at −30 °C for 6–8 h until TLC showed the disappearance of the starting cyanoaziridine. The crude products were concentrated to dryness in vacuum conditions and were purified by crystallization. Method B. To a 0 °C solution of cyanoaziridine 1 (5 mmol, 1 eq) in CH2Cl2 (25 mL) ethoxycarbonyl isothiocyanate (6 mmol, 1.2 eq) was added dropwise. The reaction mixture was allowed to reach room temperature and stirred for 6–24 h. The crude product was concentrated to dryness in vacuum conditions and was purified by crystallization.
Ethyl (E)-[(2S*,3S*)-2-cyano-3-(diphenylphosphoryl)-2-methylaziridine-1-carbonothioyl]carbamate (6a), (1.47 g, 71%) was obtained as an orange solid from cyanoaziridine 1a (1.41 g, 5 mmol) and ethoxycarbonyl isothiocyanate (0.71 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from Et2O to give the title compound 6a. (1.66 g, 80%) which was obtained as an orange solid from cyanoaziridine 1a (1.41 g, 5 mmol) and ethoxycarbonyl isothiocyanate (0.71 mL, 6 mmol, 1.2 eq) as described in the general procedure (method B). The crude product was purified by crystallization from Et2O to give the title compound 6a; mp 156–158 °C; IR (neat) vmax 3406, 3147, 2984, 2254, 1771, 1593, 1491, 1438, 1383, 1233, 1152, 1122, 1041 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.43 (bs, 1H, NH), 7.94–7.42 (m, 10H, ArH), 4.17 (q, 3JHH = 7.1 Hz, 2H, OCH2), 3.92 (d, 2JPH = 20.0 Hz, 1H, CH-P), 1.97 (s, 3H, CH3). A value of 1.23 (t, 3JHH = 7.1 Hz, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 190.5 (d, 3JPC = 4.6 Hz, C=S), 149.0 (C=O), 133.0, 133.0, 132.8, 132.8, 131.8, 131.7, 131.5, 131.2, 131.1, 130.9, 130.1, 129.4, 129.3, 129.1, 128.8, 128.6 (CAr), 116.5 (CN), 63.0 (CH2), 48.8 (d, 1JPC = 94.5 Hz, CH-P), 42.4 (d, 2JPC = 3.2 Hz, Cquat), 18.6 (CH3), 14.2 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 22.7 ppm; ESI-HRMS (CI) m/z calculated for C20H21N3O3PS ([M + H]+) 414.1041, found 414.1041.
![Molecules 26 04265 i027]()
Ethyl (E)-[(2S*,3S*)-2-cyano-3-(diphenylphosphoryl)-2-ethylaziridine-1-carbonothioyl]carbamate (6b), (1.82 g, 85%) was obtained as a pale yellow solid from cyanoaziridine 1b (1.48 g, 5 mmol) and ethoxycarbonyl isothiocyanate (0.71 mL, 6 mmol, 1.2 eq) as described in the general procedure (method A). The crude product was purified by crystallization from Et2O to give the title compound 6b; mp 179–181 °C; IR (neat) vmax 3409, 3062, 2981, 2254, 2237, 1752, 1541, 1438, 1230, 1197, 1163, 1044 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.96 (bs, 1H, NH), 7.97–7.44 (m, 10H, ArH), 4.20 (q, 3JHH = 7.2 Hz, 2H, OCH2), 3.96 (d, 2JPH = 20.2 Hz, 1H, CH-P), 2.59–2.50 (m, 1H, CH2), 2.37–2.28 (m, 1H, CH2). A value of 1.27 (t, 3JHH = 7.2 Hz, 3H, CH3), 0.99 (t, 3JHH = 7.5 Hz, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 190.1 (d, 3JPC = 4.4 Hz, C=S), 149.0 (C=O), 133.1, 133.0, 132.8, 132.8, 131.8, 131.6, 131.5, 131.4, 131.3, 131.2, 130.1, 129.9, 129.3, 129.2, 128.9, 128.7 (CAr), 115.4 (CN), 63.2 (OCH2), 48.8 (d, 1JPC = 94.4 Hz, CH-P), 48.0 (d, 2JPC = 3.1 Hz, Cquat), 24.6 (CH2), 14.3 (CH3), 10.4 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 21.8 ppm; ESI-HRMS (CI) m/z calculated for C21H23N3O3PS ([M + H]+) 428.1198, found 428.1204.
![Molecules 26 04265 i028]()
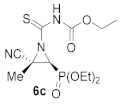
Ethyl (E)-[(2S*,3S*)-2-cyano-3-(diethoxyphosphoryl)-2-methylaziridine-1-carbonothioyl]carbamate (6c), (1.50 g, 86%) was obtained as an orange solid from cyanoaziridine 1c (1.09 g, 5 mmol) and ethoxycarbonyl isothiocyanate (0.71 mL, 6 mmol, 1.2 eq) as described in the general procedure (method B). The crude product was purified by crystallization from Et2O/pentane to give the title compound 6c; mp 116–118 °C; IR (neat) vmax 3395, 3162, 2987, 2251, 1774, 1491, 1385, 1241, 1158, 1041 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.03 (bs, 1H, NH), 4.30–4.14 (m, 6H, OCH2CH3 + CH2CH3), 3.41 (d, 2JPH = 12.3 Hz, 1H, CH-P), 1.98 (s, 3H, CH3), 1.38–1.27 (m, 9H, OCH2CH3 + CH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 190.3 (d, 3JPC = 6.7 Hz, C=S), 148.7 (C=O), 116.4 (d, 3JPC = 2.3 Hz, CN), 64.2 (d, 2JPC = 5.8 Hz, OCH2CH3), 63.3 (d, 2JPC = 6.9 Hz, OCH2CH3), 63.2 (CH2), 45.8 (d, 1JPC = 201.7 Hz, CH-P), 41.8 (d, 2JPC = 3.2 Hz, Cquat), 18.6 (CH3), 16.5 (d, 3JPC = 4.0 Hz, OCH2CH3), 16.4 (d, 3JPC = 6.0 Hz, OCH2CH3), 14.2 (CH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 13.3 ppm; ESI-HRMS (CI) m/z calculated for C12H21N3O5PS ([M + H]+) 350.0940 found 350.0932.
![Molecules 26 04265 i029]()
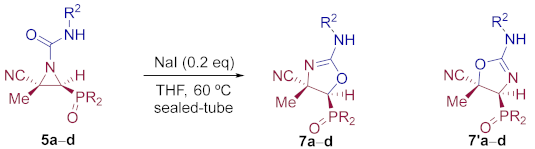
General Procedure and Spectral Data for The Reaction of NaI with N-Carbamoyl Cyanoaziridines 7
To a stirred solution of N-functionalized cyanoaziridine 5 (5 mmol, 1 eq) in THF (15 mL), NaI (0.02 g, 1 mmol, 0.2 eq) was added dropwise. The mixture was heated at 60 °C for 24 h until TLC showed the disappearance of the starting cyanoaziridine. NaI was filtered through a sintered glass vacuum filtration funnel with celite and washed with THF. The filtrate was concentrated to dryness in vacuum conditions and the resulting residue was purified by flash-column chromatography.
(4S*,5S*)-5-(Diphenylphosphoryl)-4-methyl-2-(phenylamino)-4,5-dihydrooxazole-4-carbonitrile (7a) and (4S*,5S*)-4-(diphenylphosphoryl)-5-methyl-2-(phenylamino)-4,5-dihydrooxazole-5-carbonitrile (7′a), (0.90 g, 45%) were obtained as yellow solids from N-functionalized cyanoaziridine 5a (2.00 g, 5 mmol) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 50:50) to give the minor regioisomer; mp 117–119 °C; IR (neat) vmax 3420, 3057, 2981, 2237, 1674, 1438, 1402, 1199, 1122 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.95–7.20 (m, 15H, ArH), 4.23 (bs, 1H, NH), 3.00 (d, 2JPH = 16.2 Hz, 1H, CH-P), 1.76 (s, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 164.6 (d, 3JPC = 5.9 Hz, C=N), 132.9, 132.1, 131.9, 131.2, 131.1, 130.4, 130.0, 129.4, 129.2, 129.1, 128.9, 126.8 (CAr), 127.6 (CN), 51.8 (d, 1JPC = 95.5 Hz, CH-P), 29.8 (Cquat), 12.2 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 21.0 ppm; ESI-HRMS (CI) m/z calculated for C23H21N3O2P ([M + H]+) 402.1371 found 402.1368.
![Molecules 26 04265 i030]()
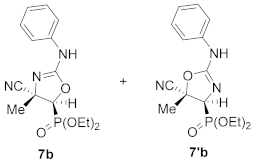
Diethyl (E)-[(4S*,5S*)-4-cyano-4-methyl-2-(phenylamino)-4,5-dihydroozazol-5-yl]phosphonate (7b) and diethyl (E)-[(4S*,5S*)-5-cyano-5-methyl-2-(phenylamino)-4,5-dihydroozazol-4-yl]phosphonate (7′b), (1.16 g, 69%) were obtained as waxy white solids from N-functionalized cyanoaziridine 5b (1.68 g, 5 mmol) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt) to give the title compound 7 as a mixture of two regioisomers 7b + 7′b; Rf: 0.1 (AcOEt); IR (neat) vmax 3370, 3061, 2990, 2240, 1666, 1499, 1402, 1255, 1158, 1052, 1019 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.10 (bs, 1H, NH), 7.52–7.12 (m, 11H, ArH + NH), 4.32–4.20 (m, 8H, OCH2), 2.65 (d, 2JPH = 11.0 Hz, 1H, CH-P)major, 2.62 (d, 2JPH = 11.1 Hz, 1H, CH-P)minor, 1.89 (s, 3H, CH3)major, 1.84 (s, 3H, CH3)minor, 1.40–1.33 (m, 12H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 166.5 (d, 3JPC = 3.7 Hz, C=N)minor, 161.8 (d, 3JPC = 4.5 Hz, C=N)major, 132.4 (Cquat), 130.9 (Cquat), 130.3, 129.7, 129.3, 128.7, 127.7, 127.0, 126.7 (CAr), 121.6 (CN)minor, 121.5 (CN)major, 64.3 (d, 2JPC = 6.0 Hz, OCH2)minor, 64.0 (d, 2JPC = 6.0 Hz, OCH2)major, 63.3 (d, 2JPC = 6.2 Hz, OCH2)major, 48.4 (d, 1JPC = 201.8 Hz, CH-P)major, 47.8 (d, 1JPC = 201.3 Hz, CH-P)minor, 47.5 (d, 2JPC = 3.4 Hz, Cquat)major, 46.0 (d, 2JPC = 3.00 Hz, Cquat)minor, 16.6, 16.5, 16.5, 16.4 (OCH2CH3), 12.5 (CH3)major, 12.1 (CH3)minor ppm; 31P NMR (120 MHz, CDCl3) δ 13.8major, 13.3minor ppm; ESI-HRMS (CI) m/z calculated for C15H21N3O4P ([M + H]+) 338.1270 found 338.1254.
![Molecules 26 04265 i031]()
(E)-(4S*,5S*)-5-(Diphenylphosphoryl)-4-methyl-2-(p-tolylamino)-4,5-dihydrooxazole-4-carbonitrile (7c) and (E)-(4S*,5S*)-4-(diphenylphosphoryl)-5-methyl-2-(p-tolylamino)-4,5-dihydrooxazole-5-carbonitrile (7′c), (1.56 g, 75%) were obtained as white solids from N-functionalized cyanoaziridine 5c (2.07 g, 5 mmol) as described in the general procedure. The crude product was purified by flash-column chromatography (AcOEt) to give the title compound 7 as a mixture of two regioisomers 7c +7′c; mp 128–130 °C; IR (neat) vmax 3425, 3059, 2959, 2235, 1617, 1516, 1438, 1405, 1197, 1119 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.01 (bs, 1H, NH), 7.96–7.01 (m, 29H, ArH + NH), 2.98 (d, 2JPH = 15.5 Hz, 1H, CH-P)minor, 2.98 (d, 2JPH = 16.7 Hz, 1H, CH-P)major, 2.37 (s, 3H, CH3)major, 2.33 (s, 3H, CH3)minor, 1.80 (s, 3H, CH3)major, 1.68 (s, 3H, CH3)minor ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 166.8 (C=N)minor, 162.1 (C=N)major, 140.0 (Cquat), 138.8 (Cquat), 132.9, 132.0, 131.9, 131.3, 131.2, 131.1, 131.0, 130.4, 130.0, 129.7, 129.3, 129.2, 129.0, 128.9, 128.2, 127.5, 126.8, 126.4 (CAr), 121.5 (d, 3JPC = 3.8 Hz, CN), 51.5 (d, 1JPC = 95.9 Hz, CH-P)major, 51.6 (d, 1JPC = 95.2 Hz, CH-P)minor, 48.8 (d, 2JPC = 3.1Hz Cquat)major, 47.1 (Cquat), 21.3 (CH3), 12.3 (CH3)major, 11.9 (CH3)minor ppm; 31P NMR (120 MHz, CDCl3) δ 21.0major, 20.8minor ppm; ESI-HRMS (CI) m/z calculated for C24H23N3O2P ([M + H]+) 416.1528 found 416.1544.
![Molecules 26 04265 i032]()
Diethyl (E)-[(4S*,5S*)-4-cyano-4-methyl-2-(p-tolylamino)-4,5-dihydroozazol-5-yl]phosphonate (7d) and diethyl (E)-[(4S*,5S*)-5-cyano-5-methyl-2-(p-tolylamino)-4,5-dihydroozazol-5-yl]phosphonate (7′d), (0.95 g, 54%) were obtained as waxy white solids from N-functionalized cyanoaziridine 5d (1.75 g, 5 mmol) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt) to give the title compound 7d + 7′d as a mixture of two regioisomers; Rf: 0.1 (AcOEt); IR (neat) vmax 3356, 3037, 2987, 2237, 1671, 1516, 1444, 1402, 1321, 1260, 1160, 1127, 1049, 1024 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.05 (bs, 1H, NH)minor, 7.49 (bs, 1H, NH)major, 7.31–7.00 (m, 8H, ArH), 4.33–4.21 (m, 8H, OCH2), 2.62 (d, 2JPH = 11.1 Hz, 1H, CH-P)major, 2.60 (d, 2JPH = 11.3 Hz, 1H, CH-P)minor, 2.37 (s, 3H, CH3)major, 2.33 (s, 3H, CH3)minor, 1.89 (s, 3H, CH3)major, 1.85 (s, 3H, CH3)minor, 1.41–1.33 (m, 12H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 166.7 (d, 3JPC = 3.0 Hz, C=N)minor, 162.1 (d, 1JPC = 3.7 Hz, C=N)major, 140.0 (Cquat), 138.9 (Cquat), 131.0, 130.6, 130.1, 129.7, 128.2, 126.9, 126.5 (CAr), 127.6 (CN)major, 121.3 (d, 3JPC = 5.5 Hz, CN)minor, 64.3 (d, 2JPC = 6.0 Hz, OCH2)minor, 64.0 (d, 2JPC = 5.9 Hz, OCH2)minor, 63.3 (d, 2JPC = 6.2 Hz, OCH2)major, 48.4 (d, 1JPC = 201.8 Hz, CH-P)major, 47.9 (d, 1JPC = 201.8 Hz, CH-P)minor, 47.5 (d, 2JPC = 3.2 Hz, Cquat)major, 46.0 (Cquat)minor, 21.3 (CH3), 16.6, 16.6, 16.5, 16.5 (OCH2CH3), 12.5 (CH3)major, 12.2 (CH3)minor ppm; 31P NMR (120 MHz, CDCl3) δ 13.9major, 13.4minor ppm; ESI-HRMS (CI) m/z calculated for C16H23N3O4P ([M + H]+) 352.1426 found 352.1426.
![Molecules 26 04265 i033]()
General Procedure and Spectral Data for Compound 8a
To a −70 °C solution of 6a (5 mmol, 1 eq) in THF (25 mL) boron trifluoride diethyl etherate (25 mmol, 5 eq) was added dropwise. The reaction mixture was stirred at −70 °C for 24 h until TLC showed the disappearance of the starting N-functionalized cyanoaziridine. The crude product was washed three times with water (15 mL) and extracted with CH2Cl2 (15 mL). The organic layers were dried over anhydrous MgSO4, filtered, and concentrated to dryness in vacuum conditions, and the resulting residue was purified by flash-column chromatography.
Ethyl (Z)-[(4R*,5S*)-5-cyano-4-(diphenylphosphoryl)-5-methyl-4,5-dihydrothiazol-2-yl]carbamate (8a), (1.39 g, 67%) was obtained as a pale yellow solid from N-functionalized cyanoaziridine 6a (2.06 g, 5 mmol) and boron trifluoride diethyl etherate (3.1 mL, 25 mmol, 5 eq) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 8a; mp 208–210 °C; IR (neat) vmax 3145, 3065, 2937, 2254, 2232, 1724, 1624, 1507, 1438, 1244, 1174, 1113, 1094 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06–7.40 (m, 11H, ArH + NH), 4.58 (d, 2JPH = 12.8 Hz, 1H, CH-P), 4.22–4.16 (m, 2H, CH2), 2.11 (s, 3H, CH3), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 157.1 (d, 3JPC = 19.4 Hz, C=N), 152.8 (C=O), 134.0 (Cquat), 133.3, 133.2, 132.7, 132.6, 132.5, 132.4, 132.4, 131.3, 131.2, 129.2, 128.9, 128.7, 128.2, 128.1, 127.9 (CAr), 118.8 (d, 3JPC = 7.1 Hz, CN), 76.6 (d, 1JPC = 82.7 Hz, CH-P), 63.1 (CH2), 52.0 (d, 2JPC = 1.3 Hz, Cquat), 26.4 (CH3), 14.4 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 26.1 ppm; ESI-HRMS (CI) m/z calculated for C20H21N3O3PS ([M + H]+) 414.1041 found 414.1046.
![Molecules 26 04265 i034]()
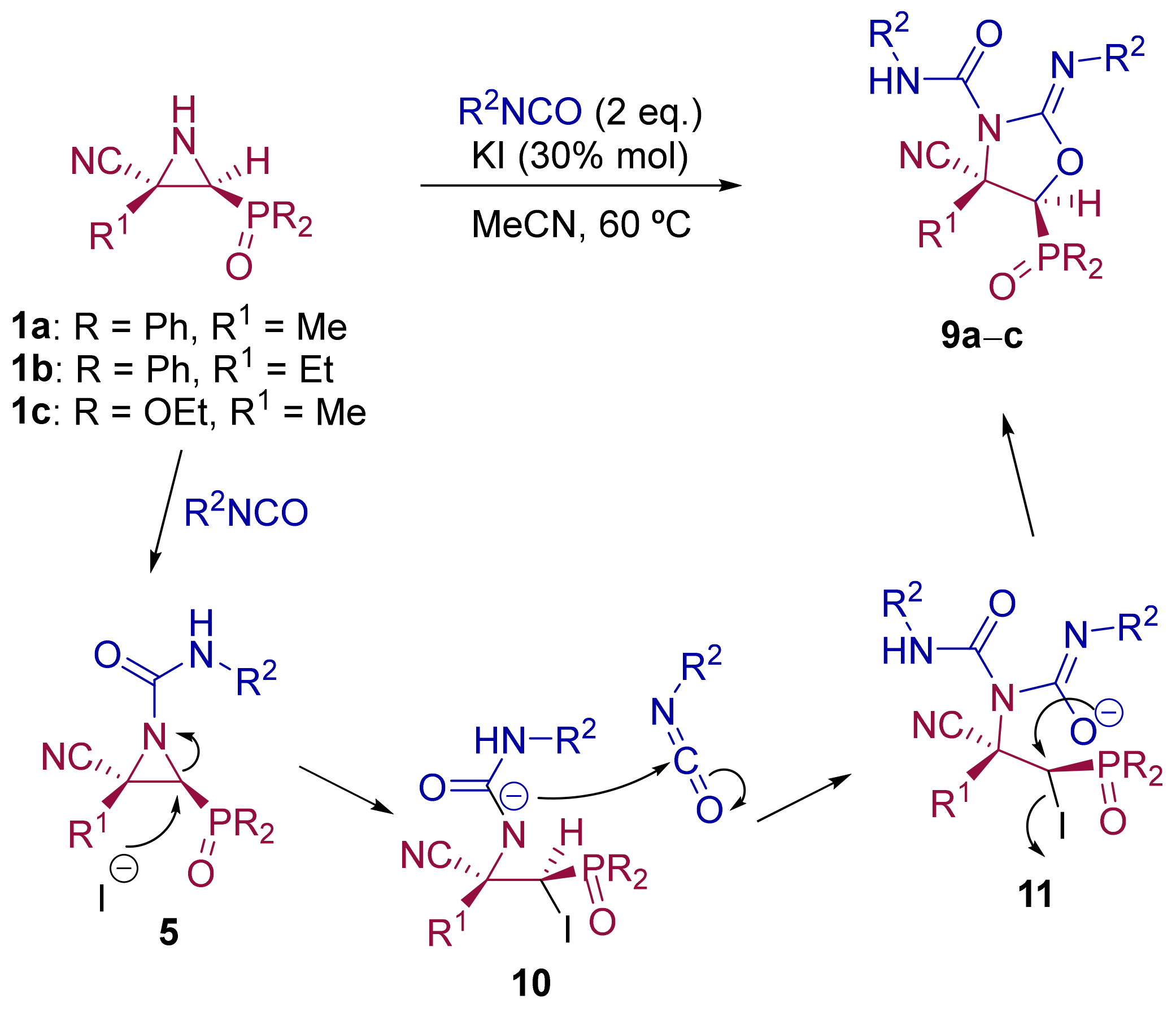
General Procedure and Spectral Data for The Reaction of Cyanoaziridines 1 and Isocyanates in The Ppresence of KI
A mixture of the corresponding isocyanate (2 mmol, 2 eq), KI (0.25 g, 0.3 mmol) and cyanoaziridine (1 mmol, 1 eq) in CH3CN (15 mL) was stirred at 60 °C until TLC showed the disappearance of the starting cyanoaziridine. After the completion of the reaction, the solvent was evaporated under reduced pressure and the crude product was washed three times with water (15 mL) and extracted with CH2Cl2 (15 mL). The organic layers were dried over anhydrous MgSO4, filtered and concentrated to dryness in vacuum conditions, and the resulting residue was purified by crystallization or by flash-column chromatography.
(E)-(4S*,5S*)-4-Cyano-5-(diphenylphosphoryl)-4-ethyl-N-phenyl-2-(phenylimino)oxazolidine-3-carboxamide (9a), (1.66 g, 62%) was obtained as a white solid from cyanoaziridine 5a (1.48 g, 5 mmol) and phenyl isocyanate (1.09 mL, 10 mmol, 2 eq) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 40:60) to give the title compound 9a; mp 208–210 °C; IR (neat) vmax 3267, 3062, 2973, 2246, 1779, 1560, 1502, 1435, 1383, 1316, 1260, 1225, 1119 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.01–7.07 (m, 21H, ArH + NH), 3.32 (d, 2JPH = 18.7 Hz, 1H, CH-P), 2.46–2.27 (m, 2H, CH2), 1.05 (t, 3JHH = 7.4 Hz, 3H, CH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 164.6 (d, 3JPC = 6.0 Hz, C=N), 157.9 (C=O), 138.0 (Cquat), 133.1, 133.0, 132.2, 132.1, 131.9, 131.8, 131.7, 129.0, 126.9, 124.2, 119.2 (CAr), 121.6 (CN), 55.1 (d, 1JPC = 96.5 Hz, CH-P), 54.5 (Cquat), 18.9 (CH2), 10.3 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 24.1 ppm; ESI-HRMS (CI) m/z calculated for C31H28N4O3P ([M + H]+) 535.1899 found 535.1899.
![Molecules 26 04265 i035]()
(E)-(4S*,5S*)-4-Cyano-5-(diphenylphosphoryl)-4-methyl-N-(p-tolyl)-2-(p-tolylimino)oxazolidine-3-carboxamide (9b), (1.51 g, 55%) was obtained as a white solid from cyanoaziridine 5c (1.41 g, 5 mmol) and p-tolyl isocyanate (1.26 mL, 10 mmol, 2 eq) as described in the general procedure. The crude product was purified by flash-column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 9b; mp 220–222 °C; IR (neat) vmax 3259, 3040, 2926, 2246, 1777, 1613, 1596, 1513, 1438, 1391, 1241, 1172, 1191, 1155 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.03–7.01(m, 19H, ArH + NH), 3.21 (d, 2JPH = 18.2 Hz, 1H, CH-P), 2.33 (s, 3H, CH3), 2.29 (s, 3H, CH3), 1.91 (s, 3H, CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 164.6 (d, 3JPC = 6.0 Hz, C=N), 158.1 (C=O), 139.4 (Cquat), 135.4 (Cquat), 133.8 (Cquat), 133.1, 132.9, 132.9, 132.0, 131.9, 131.8, 130.0, 129.7, 129.5, 129.3, 129.2, 128.9, 126.7, 120.9, 120.3, 119.2, (CAr + CN), 53.9 (d, 1JPC = 92.0 Hz, CH-P), 49.1 (d, 2JPC = 3.8 Hz, Cquat), 21.3 (CH3), 21.0 (CH3), 12.6 (CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 23.7 ppm; ESI-HRMS (CI) m/z calculated for C32H30N4O3P ([M + H]+) 549.2056 found 549.2056.
![Molecules 26 04265 i036]()
Diethyl (E)-[(4S*,5S*)-4-cyano-4-methyl-3-(phenylcarbamoyl)-2-(phenylimino)oxazolidin-5-yl]phosphonate (9c), (1.80 g, 79%) was obtained as a pale yellow solid from cyanoaziridine 1c (1.09 g, 5 mmol) and phenyl isocyanate (1.09 mL, 10 mmol, 2 eq) as described in the general procedure. The crude product was purified by crystallization from Et2O to give the title compound 9c; mp 169–171 °C; IR (neat) vmax 3231, 3140, 2985, 2249, 1716, 1610, 1580, 1499, 1488, 1313, 1249, 1194, 1052, 1024 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.47–7.02 (m, 10H, ArH), 6.58 (d, 1H, NH), 4.19–4.02 (m, 4H, OCH2), 3.98 (d, 2JPH = 12.4 Hz, 1H, CH-P), 1.98 (s, 3H, CH3), 1.24–1.16 (m, 3H, OCH2CH3) ppm; 13C {1H} NMR (75 MHz, CDCl3) δ 177.3 (d, 2JPC = 15.9 Hz, C=N), 158.5 (C=O) 151.2 (d, 3JPC = 12.4 Hz, Cquat), 145.2 (Cquat), 134.5 (Cquat), 129.5, 129.3, 129.1, 128.6, 128.5, 127.9, 127.7, 124.4, 123.7, 123.5 (CAr), 120.1 (CN), 80.3 (d, 2JPC = 6.1 Hz, Cquat), 64.4 (d, 2JPC = 6.8 Hz, OCH2), 63.7 (d, 2JPC = 6.7 Hz, OCH2), 52.0 (d, 1JPC = 156.6 Hz, CH-P), 19.2 (CH3), 16.3 (d, 2JPC = 5.8 Hz, OCH2CH3) ppm; 31P NMR (120 MHz, CDCl3) δ 15.1 ppm; ESI-HRMS (CI) m/z calculated for C22H26N4O5P ([M + H]+) 457.1641 found 457.1629.
![Molecules 26 04265 i037]()
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}