
Development of an Accurate Mass Retention Time Database for Untargeted Metabolomic Analysis and Its Application to Plasma and Urine Pediatric Samples

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Accurate Mass Retention Time Library
2.2. Analysis on Real Samples
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Library of Standards
4.3. Urine and Plasma Samples
4.4. Mass Spectrometric Conditions and Spectral Library
4.5. Selection of Chromatographic Conditions and Retention Time Assignment
4.6. Untargeted Metabolomic Analysis of Real Samples
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A Global Biochemical Approach to Drug Response and Disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.D.; Aghaeepour, N.; Ahn, A.H.; Angst, M.S.; Borsook, D.; Brenton, A.; Burczynski, M.E.; Crean, C.; Edwards, R.; Gaudilliere, B.; et al. Discovery and validation of biomarkers to aid the development of safe and effective pain therapeutics: Challenges and opportunities. Nat. Rev. Neurol. 2020, 16, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward personalized medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.; Moser, A.J.; Sarangarajan, R.; Narain, N.R.; Kiebish, M.A. Current Status of Metabolomic Biomarker Discovery: Impact of Study Design and Demographic Characteristics. Metabolites 2020, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- Gorrochategui, E.; Jaumot, J.; Lacorte, S.; Tauler, R. Data analysis strategies for targeted and untargeted LC-MS metabolomic studies: Overview and workflow. TrAC Trends Anal. Chem. 2016, 82, 425–442. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Chen, J.; Chen, J.; Wu, W.; Wang, X.; Wang, Y. The beneficial effects of betaine on dysfunctional adipose tissue and N6-methyladenosine mRNA methylation requires the AMP-activated protein kinase α1 subunit. J. Nutr. Biochem. 2015, 26, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Cowan, T.M.; Matern, D. Acylcarnitine profile analysis. Genet. Med. 2008, 10, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, T.; Vidova, V.; Bienertova-Vasku, J.; Janku, P.; Almasi, M.; Klanova, J.; Spacil, Z. Urinary intermediates of tryptophan as indicators of the gut microbial metabolism. Anal. Chim. Acta 2017, 987, 72–80. [Google Scholar] [CrossRef]
- Playdon, M.C.; Moore, S.C.; Derkach, A.; Reedy, J.; Subar, A.F.; Sampson, J.N.; Albanes, D.; Gu, F.; Kontto, J.; Lassale, C.; et al. Identifying biomarkers of dietary patterns by using metabolomics. Am. J. Clin. Nutr. 2017, 105, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Fresno, R.; Llorach, R.; Urpi-Sarda, M.; Lupianez-Barbero, A.; Estruch, R.; Corella, D.; Fitó, M.; Arós, F.; Ruiz-Canela, M.; Salas-Salvadó, J.; et al. Metabolomic Pattern Analysis after Mediterranean Diet Intervention in a Nondiabetic Population: A 1- and 3-Year Follow-up in the PREDIMED Study. J. Proteome Res. 2015, 14, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Grissa, D.; Pétéra, M.; Brandolini, M.; Napoli, A.; Comte, B.; Pujos-Guillot, E. Feature Selection Methods for Early Predictive Biomarker Discovery Using Untargeted Metabolomic Data. Front. Mol. Biosci. 2016, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folberth, J.; Begemann, K.; Jöhren, O.; Schwaninger, M.; Othman, A. MS2 and LC libraries for untargeted metabolomics: Enhancing method development and identification confidence. J. Chromatogr. B 2020, 1145, 122105. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a Liquid Chromatography–High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Bonini, P.; Kind, T.; Tsugawa, H.; Barupal, D.K.; Fiehn, O. Retip: Retention Time Prediction for Compound Annotation in Untargeted Metabolomics. Anal. Chem. 2020, 92, 7515–7522. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, P.; Teo, G.; Ow, J.R.; Lau, A.; Kaldis, P.; Tate, S.; Choi, H. MetaboKit: A comprehensive data extraction tool for untargeted metabolomics. Mol. Omi. 2020, 16, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; González-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography–high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Bougioukas, K.I.; Kapogiannis, D. Effects of creatine supplementation on cognitive function of healthy individuals: A systematic review of randomized controlled trials. Exp. Gerontol. 2018, 108, 166–173. [Google Scholar] [CrossRef]
- Koeberl, D.D.; Young, S.P.; Gregersen, N.; Vockley, J.; Smith, W.E.; Benjamin, D.K.; An, Y.; Weavil, S.D.; Chaing, S.H.; Bali, D.; et al. Rare Disorders of Metabolism with Elevated Butyryl- and Isobutyryl-Carnitine Detected by Tandem Mass Spectrometry Newborn Screening. Pediatr. Res. 2003, 54, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Panfoli, I.; Ravera, S.; Podestà, M.; Cossu, C.; Santucci, L.; Bartolucci, M.; Bruschi, M.; Calzia, D.; Sabatini, F.; Bruschettini, M.; et al. Exosomes from human mesenchymal stem cells conduct aerobic metabolism in term and preterm newborn infants. FASEB J. 2016, 30, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.; Santucci, L.; Ravera, S.; Candiano, G.; Bartolucci, M.; Calzia, D.; Lavarello, C.; Inglese, E.; Ramenghi, L.A.; Petretto, A.; et al. Human urinary exosome proteome unveils its aerobic respiratory ability. J. Proteom. 2016, 136, 25–34. [Google Scholar] [CrossRef]
- Tang, Z.-Z.; Chen, G.; Hong, Q.; Huang, S.; Smith, H.M.; Shah, R.D.; Scholz, M.; Ferguson, J.F. Multi-Omic Analysis of the Microbiome and Metabolome in Healthy Subjects Reveals Microbiome-Dependent Relationships Between Diet and Metabolites. Front. Genet. 2019, 10, 454. [Google Scholar] [CrossRef] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [Green Version]

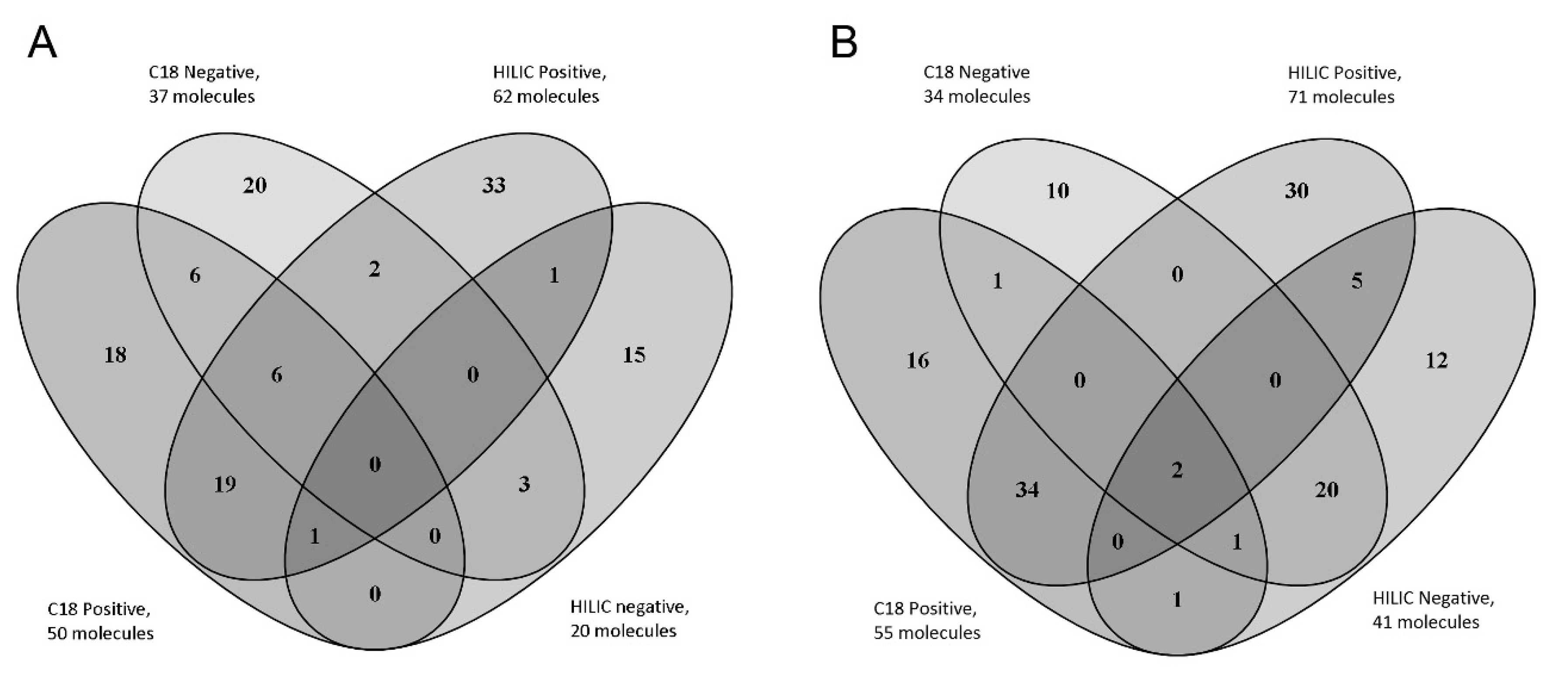

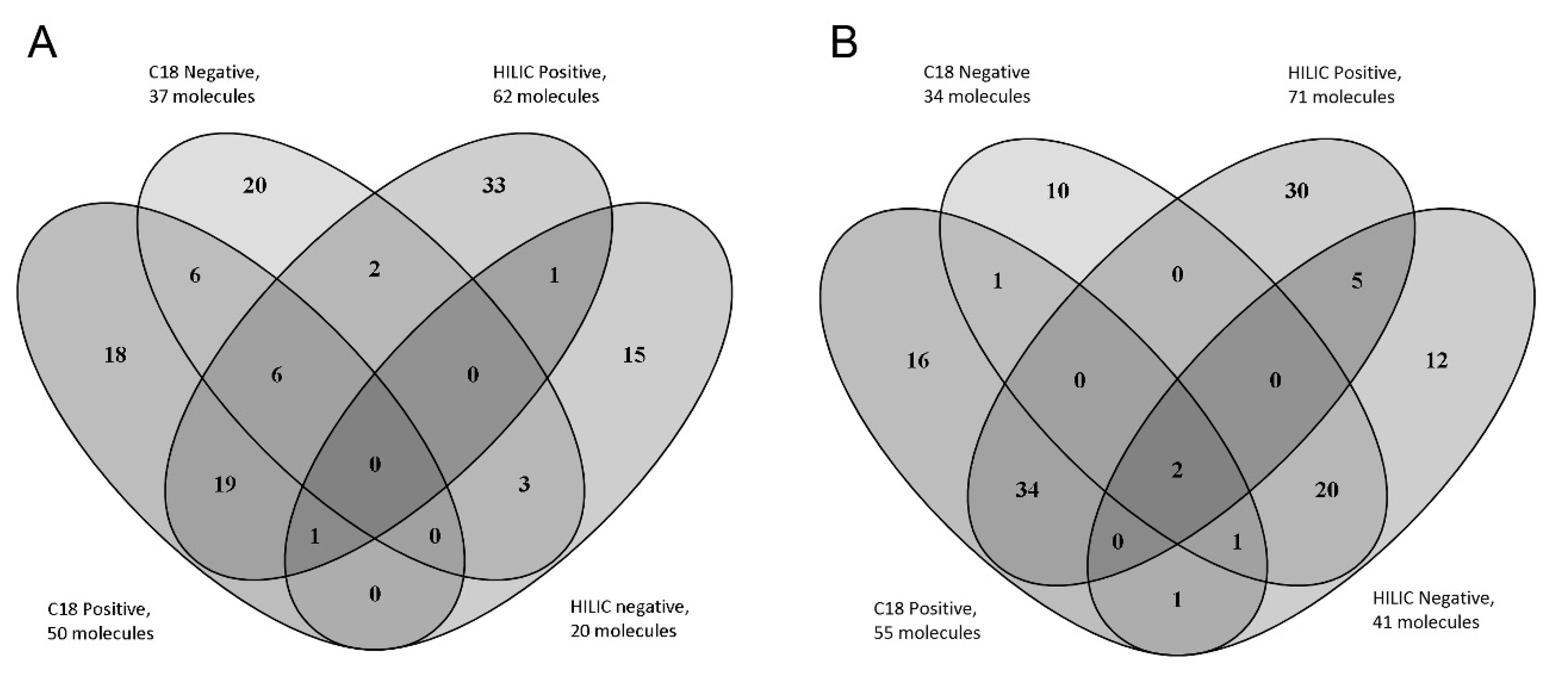
{kind=link}
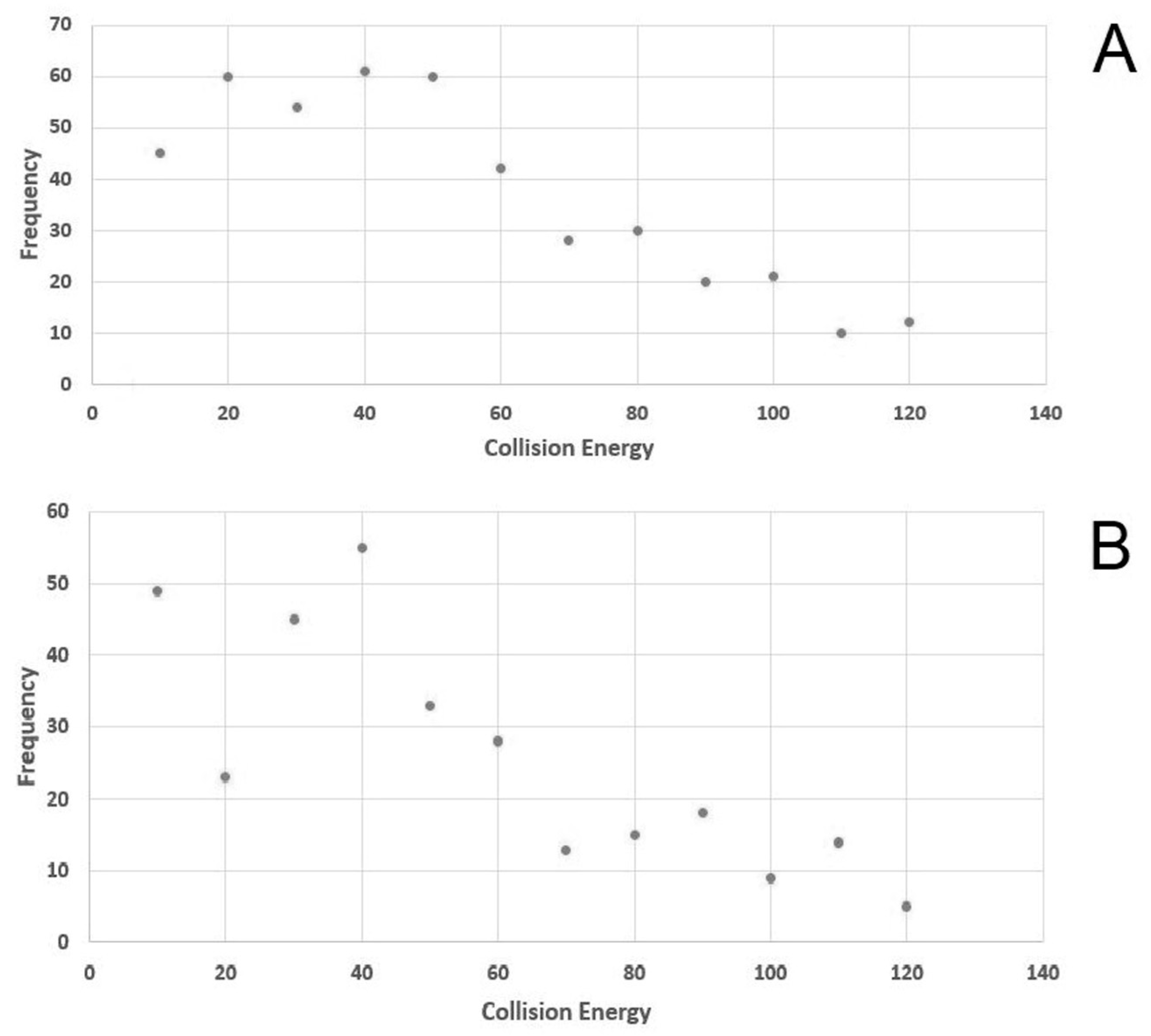
{kind=link}
| Column | Positive | Negative | |||||
|---|---|---|---|---|---|---|---|
| Reversed Phase | Phase B—Solvent | Symmetry | Width | Symmetry + Width | Symmetry | Width | Symmetry + Width |
| ACQUITY BEH C18 | ACN | 112 | 119 | 50 | 136 | 100 | 67 |
| MeOH | 72 | 129 | 29 | 91 | 101 | 40 | |
| Hypersil GOLD CN | ACN | 62 | 108 | 14 | 63 | 75 | 24 |
| MeOH | 56 | 99 | 12 | 59 | 59 | 12 | |
| Hypersil GOLD aQ | ACN | 82 | 137 | 42 | 110 | 82 | 49 |
| MeOH | 60 | 125 | 19 | 76 | 60 | 24 | |
| Accucore Polar Premium | ACN | 84 | 120 | 30 | 105 | 56 | 28 |
| MeOH | 64 | 108 | 19 | 92 | 76 | 26 | |
| HILIC | Phase A—pH | Symmetry | Width | Symmetry + Width | Symmetry | Width | Symmetry + Width |
| ACQUITY BEH Amide | pH 2 | 75 | 120 | 25 | 63 | 72 | 25 |
| pH 8 | 87 | 133 | 26 | 38 | 77 | 21 | |
| Accucore Amide HILIC | pH 2 | 73 | 114 | 22 | 20 | 34 | 6 |
| pH 8 | 76 | 118 | 32 | 21 | 42 | 15 | |
| Shodex Asahipak NH2P-50 2D | pH 2 | 51 | 100 | 10 | 27 | 84 | 5 |
| pH 8 | 71 | 116 | 32 | 45 | 116 | 8 | |
| SeQuant ZIC-pHILIC | pH 2 | 65 | 130 | 18 | 59 | 60 | 24 |
| pH 8 | 69 | 120 | 22 | 38 | 44 | 18 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavarello, C.; Barco, S.; Bartolucci, M.; Panfoli, I.; Magi, E.; Tripodi, G.; Petretto, A.; Cangemi, G. Development of an Accurate Mass Retention Time Database for Untargeted Metabolomic Analysis and Its Application to Plasma and Urine Pediatric Samples. Molecules 2021, 26, 4256. https://doi.org/10.3390/molecules26144256
Lavarello C, Barco S, Bartolucci M, Panfoli I, Magi E, Tripodi G, Petretto A, Cangemi G. Development of an Accurate Mass Retention Time Database for Untargeted Metabolomic Analysis and Its Application to Plasma and Urine Pediatric Samples. Molecules. 2021; 26(14):4256. https://doi.org/10.3390/molecules26144256
Chicago/Turabian StyleLavarello, Chiara, Sebastiano Barco, Martina Bartolucci, Isabella Panfoli, Emanuele Magi, Gino Tripodi, Andrea Petretto, and Giuliana Cangemi. 2021. "Development of an Accurate Mass Retention Time Database for Untargeted Metabolomic Analysis and Its Application to Plasma and Urine Pediatric Samples" Molecules 26, no. 14: 4256. https://doi.org/10.3390/molecules26144256
APA StyleLavarello, C., Barco, S., Bartolucci, M., Panfoli, I., Magi, E., Tripodi, G., Petretto, A., & Cangemi, G. (2021). Development of an Accurate Mass Retention Time Database for Untargeted Metabolomic Analysis and Its Application to Plasma and Urine Pediatric Samples. Molecules, 26(14), 4256. https://doi.org/10.3390/molecules26144256