
Structure–Activity Relationships of the Antimalarial Agent Artemisinin 10. Synthesis and Antimalarial Activity of Enantiomers of rac-5β-Hydroxy-d-Secoartemisinin and Analogs: Implications Regarding the Mechanism of Action


 , ,
, ,
Abstract
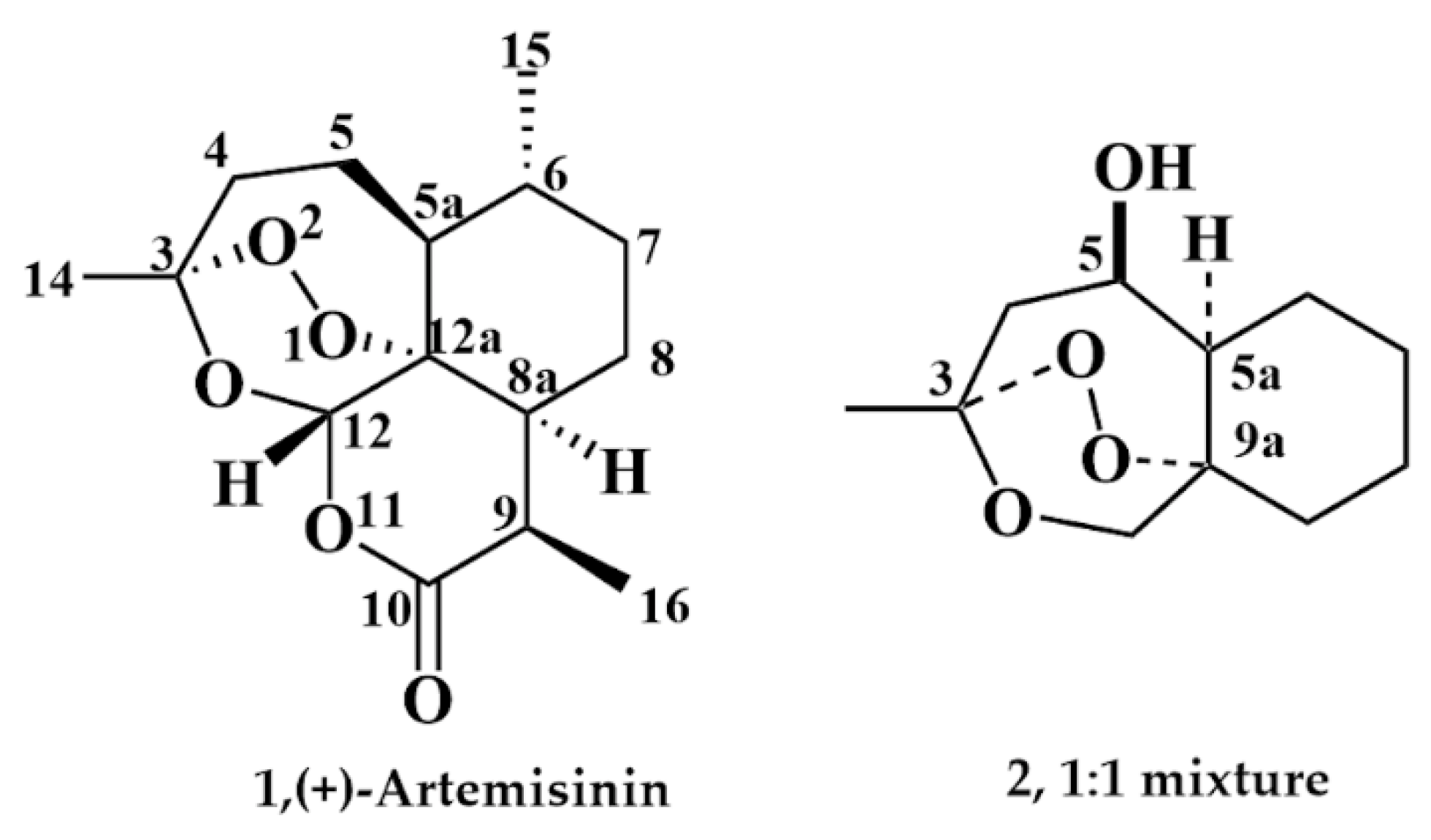
:1. Introduction
2. Results
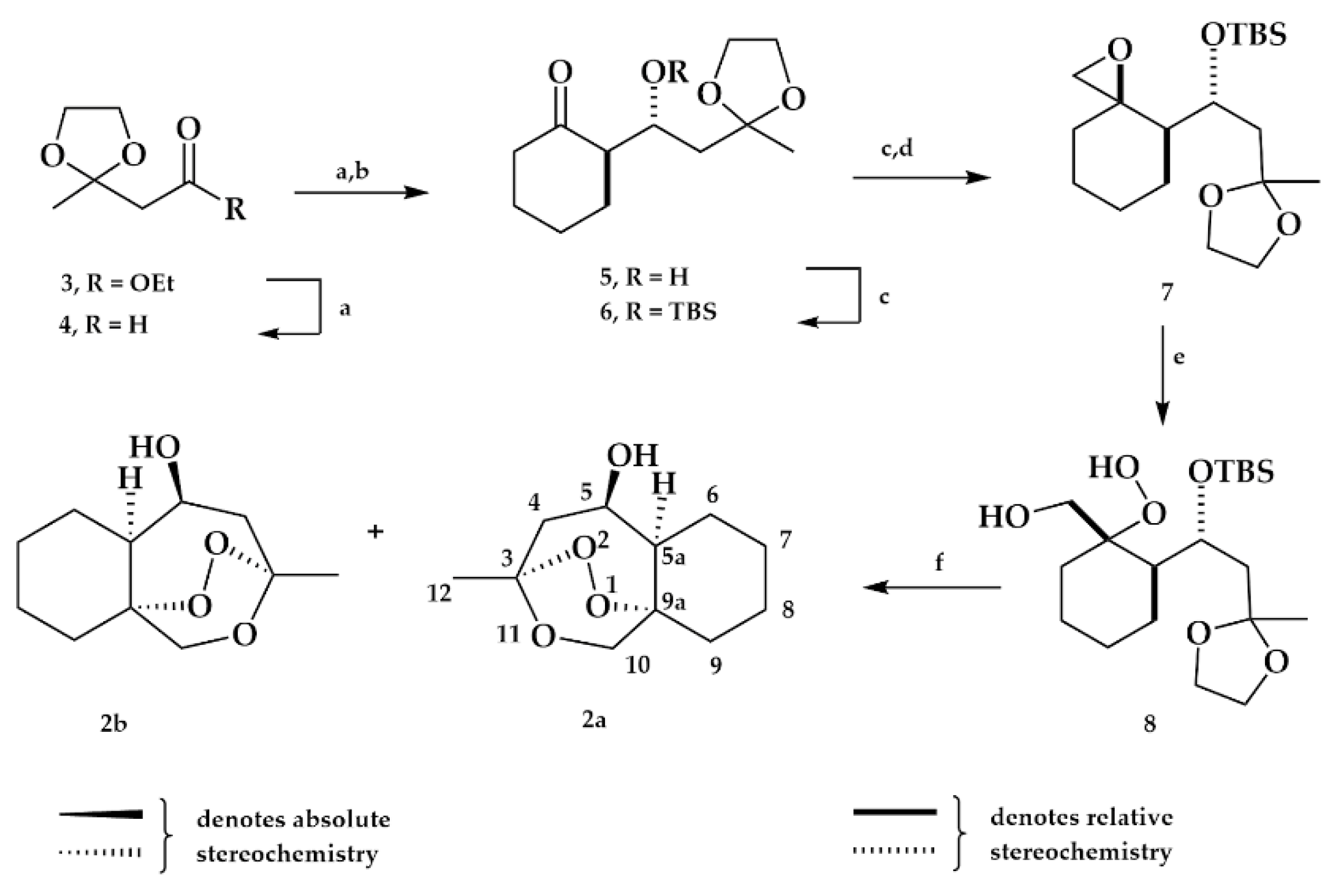
2.1. Chemistry
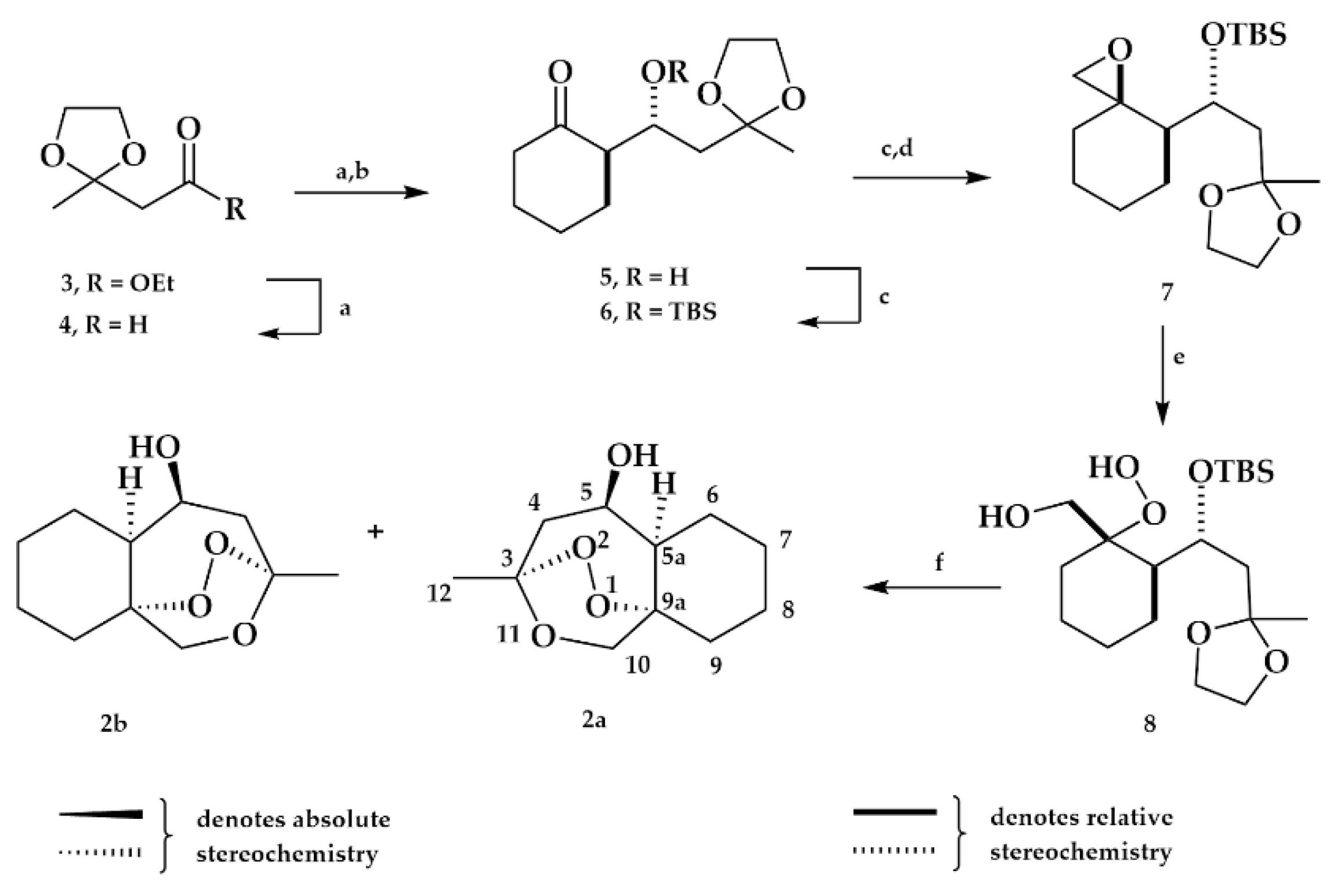
2.1.1. Ester Derivatives
2.1.2. Ketone Derivative
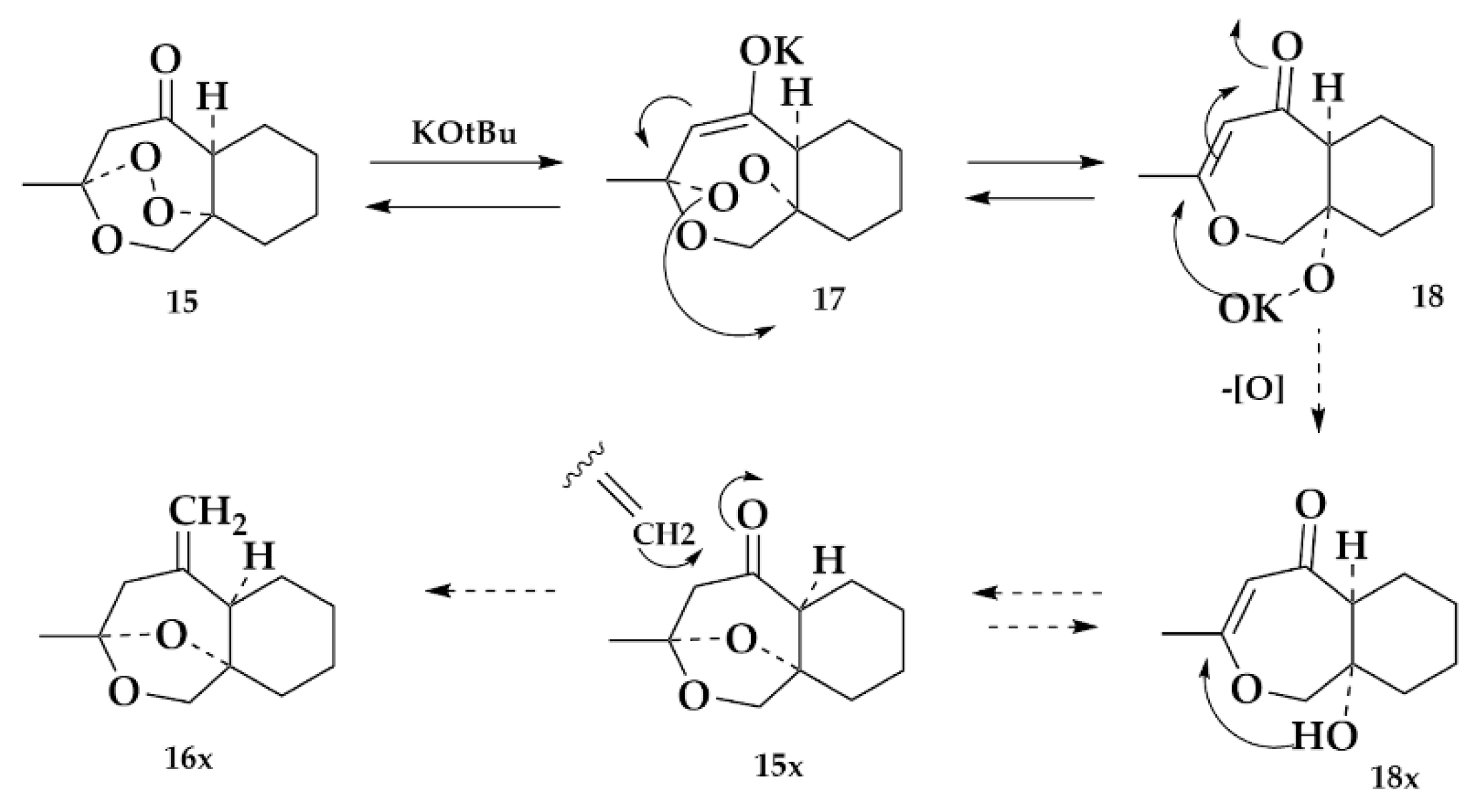
2.1.3. Exomethylene Derivative
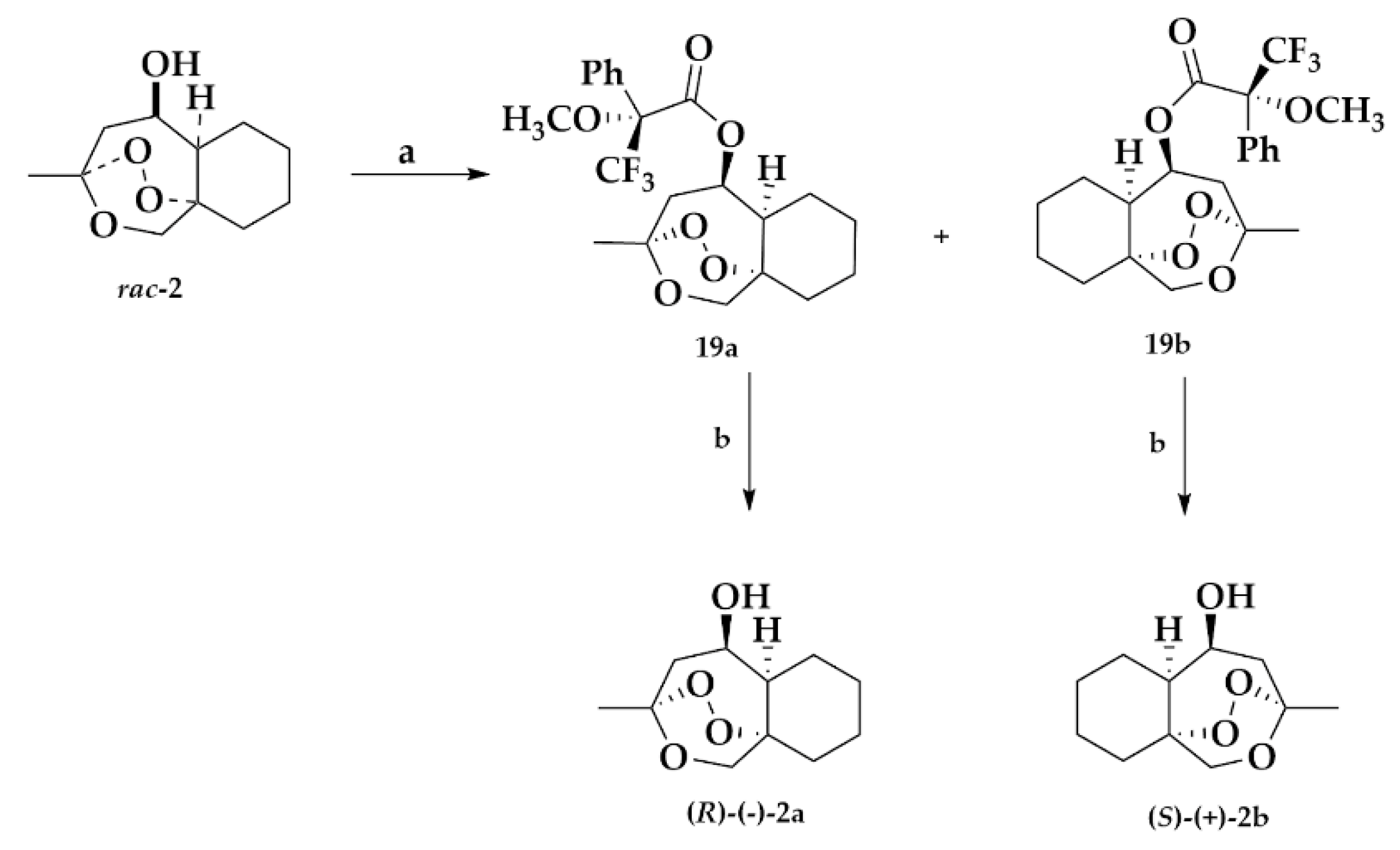
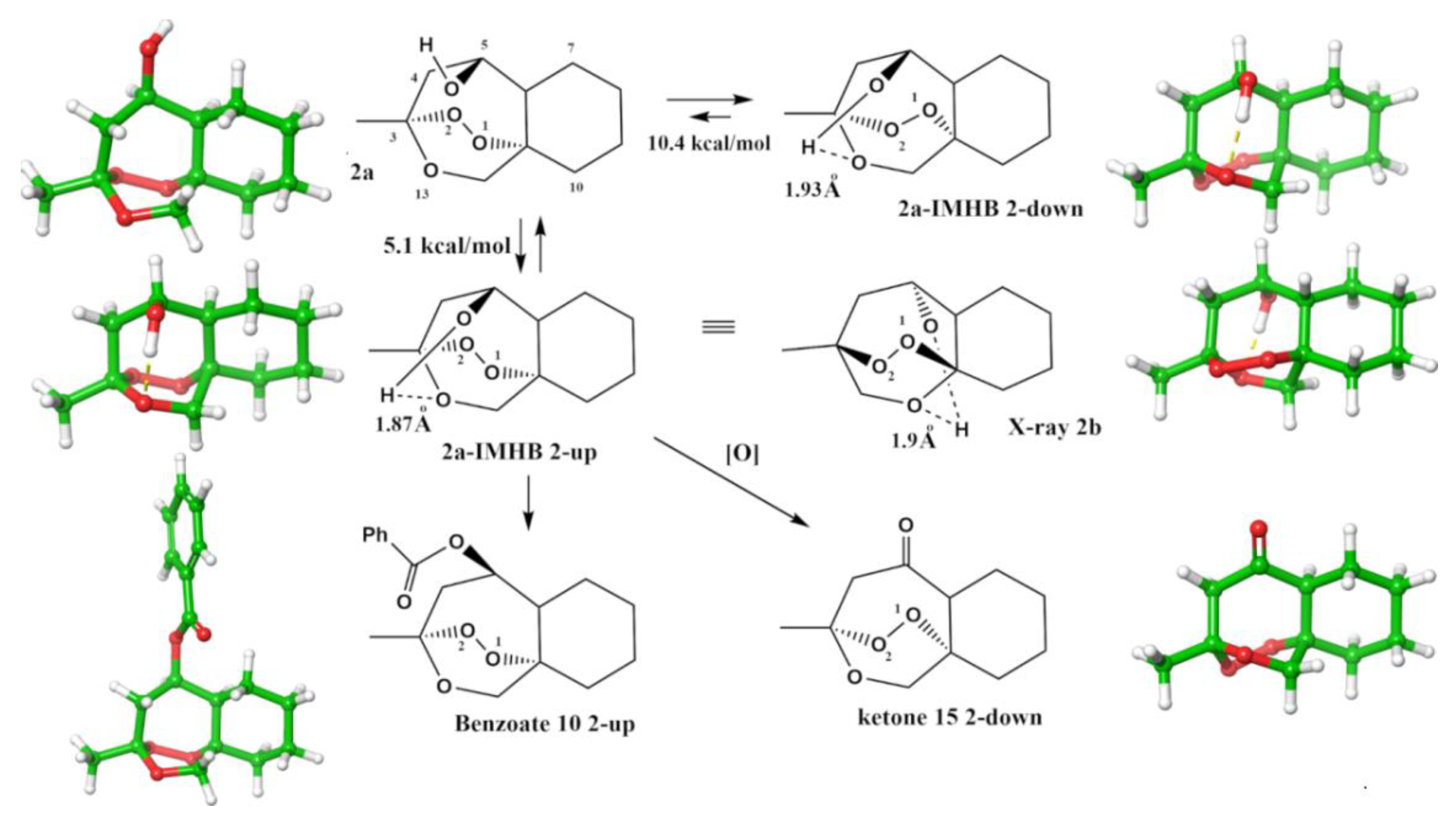
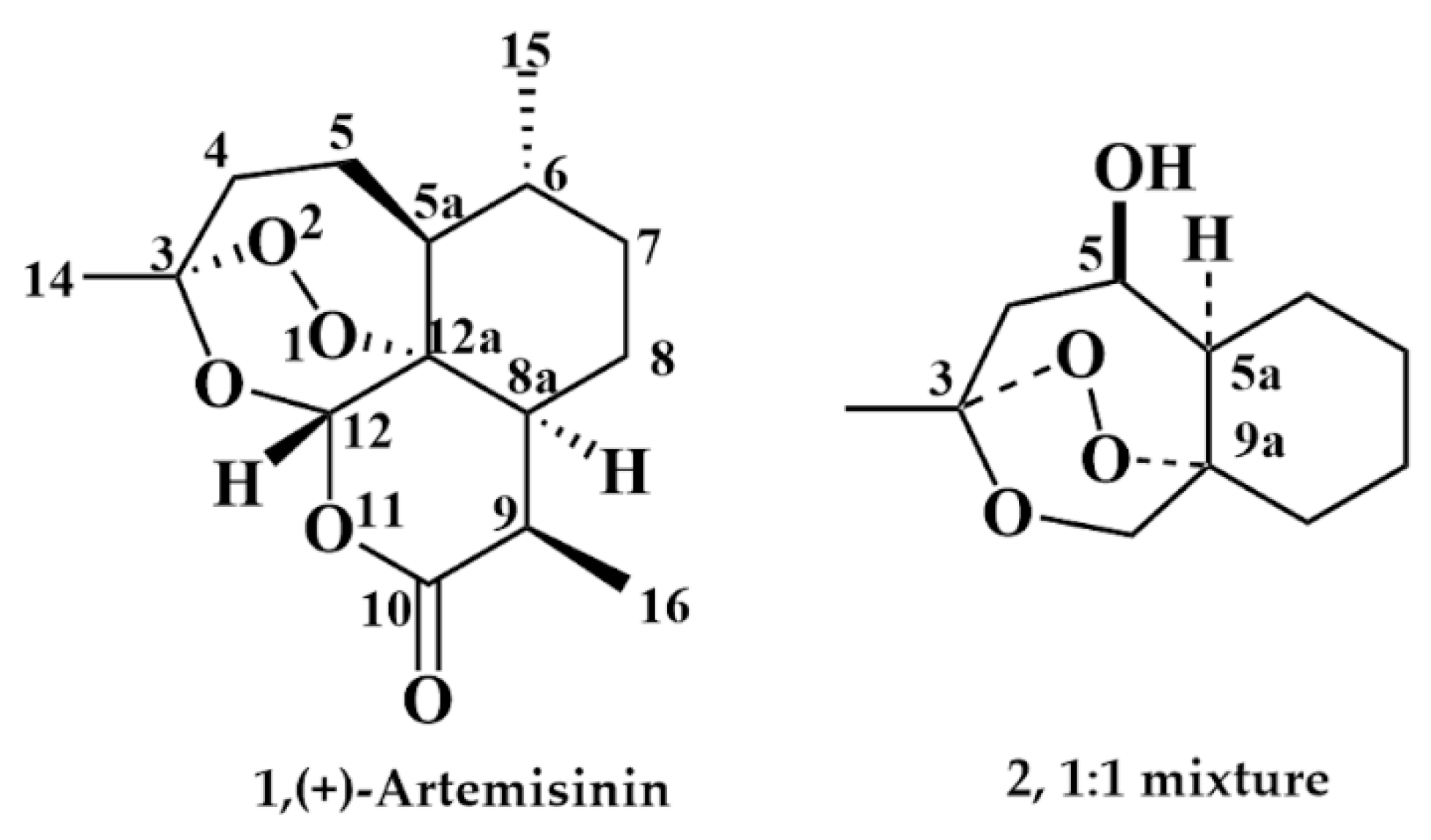
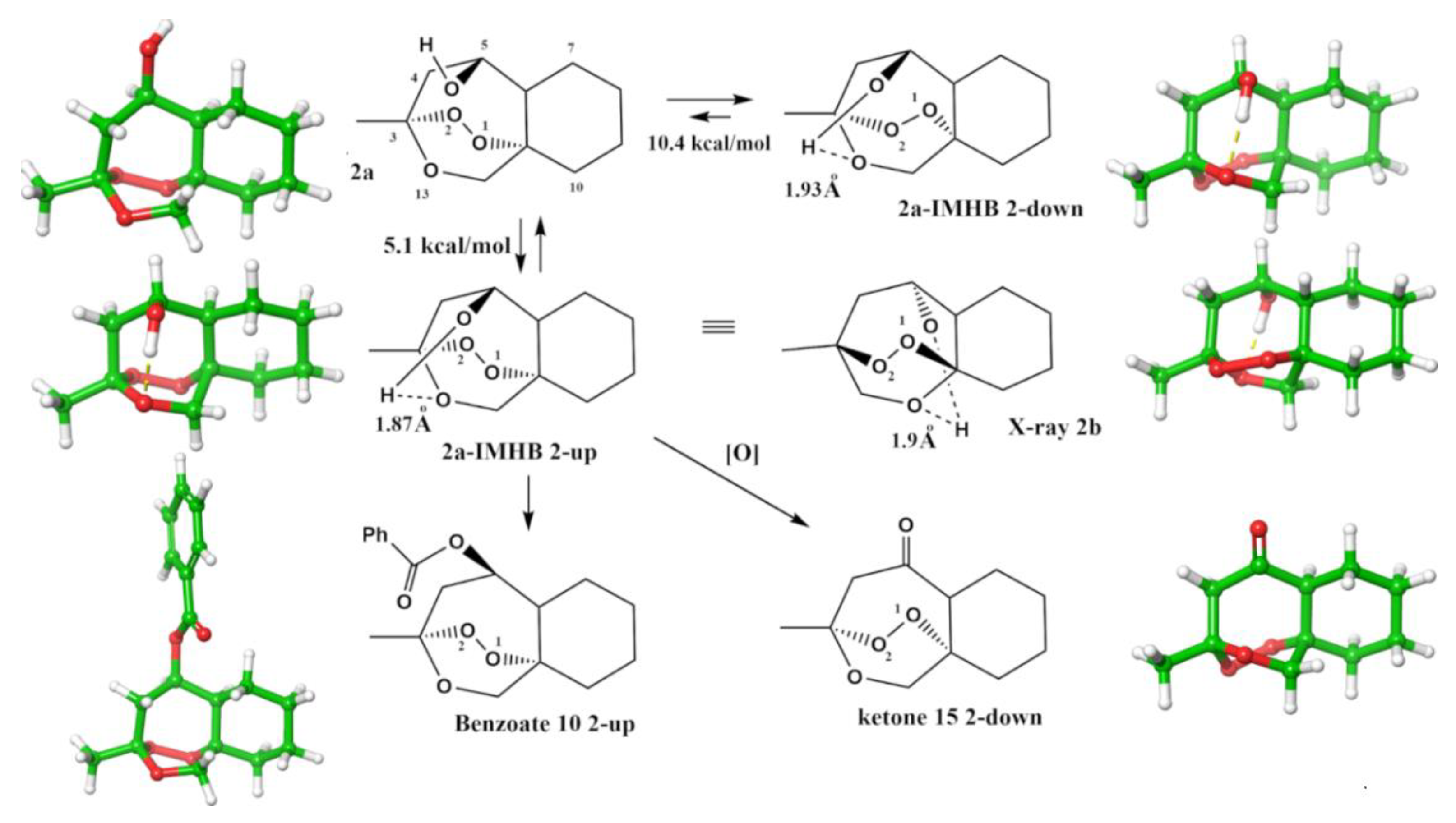
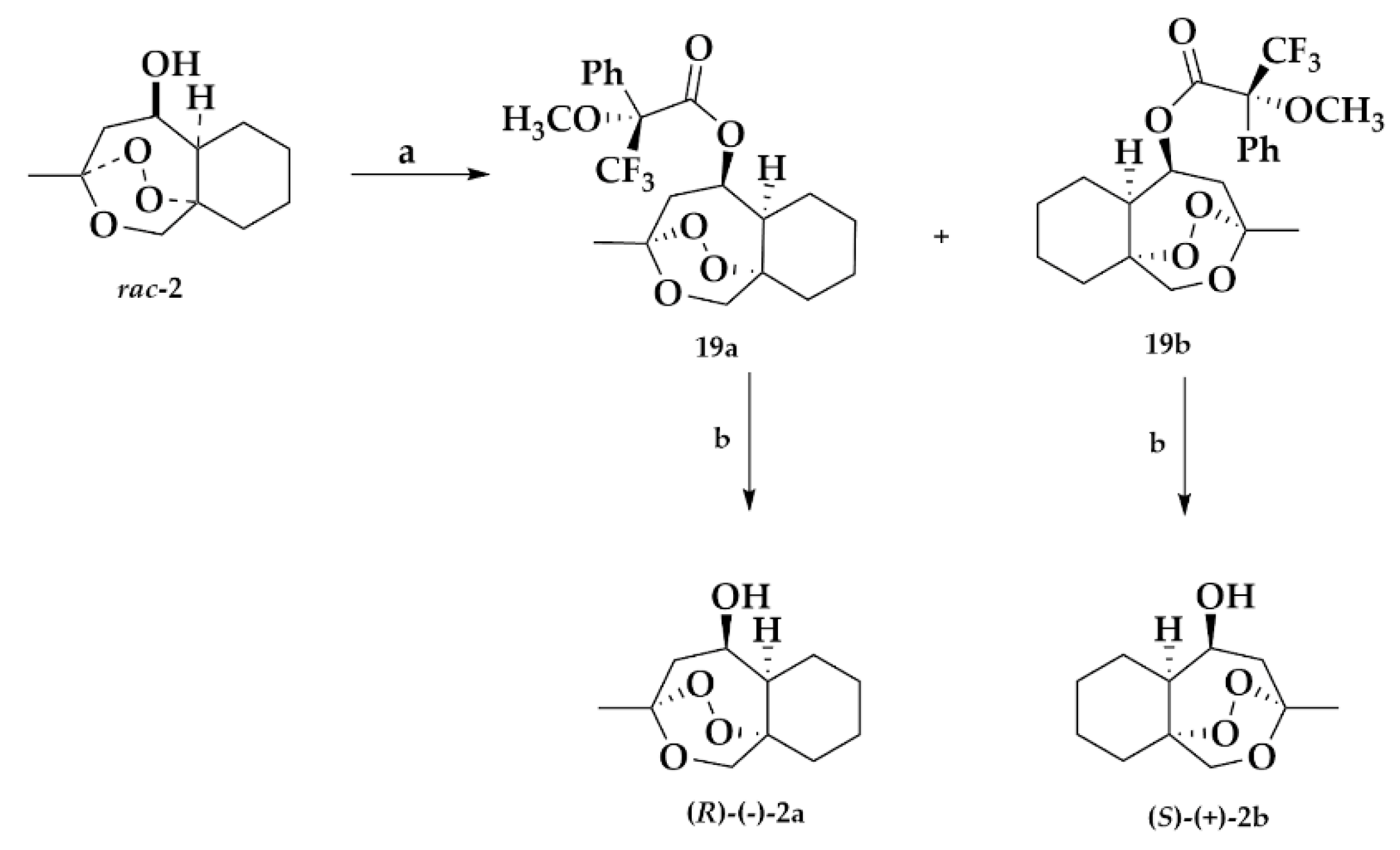
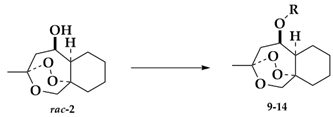
2.1.4. Separation of the Two Enantiomers 2a and 2b
Mosher Ester Analysis
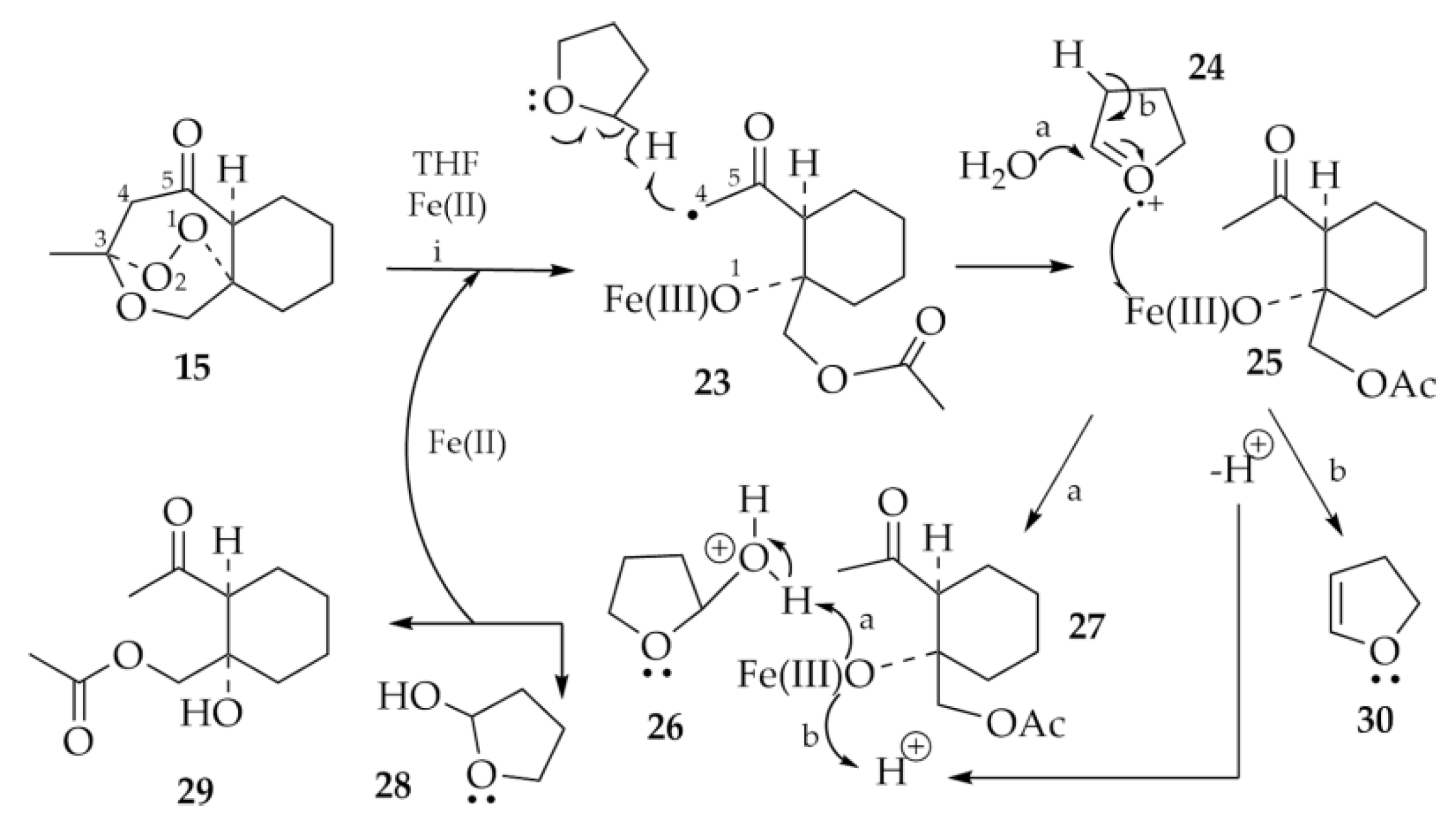
2.1.5. Rearrangement Chemistry
2.2. Antimalarial Activity
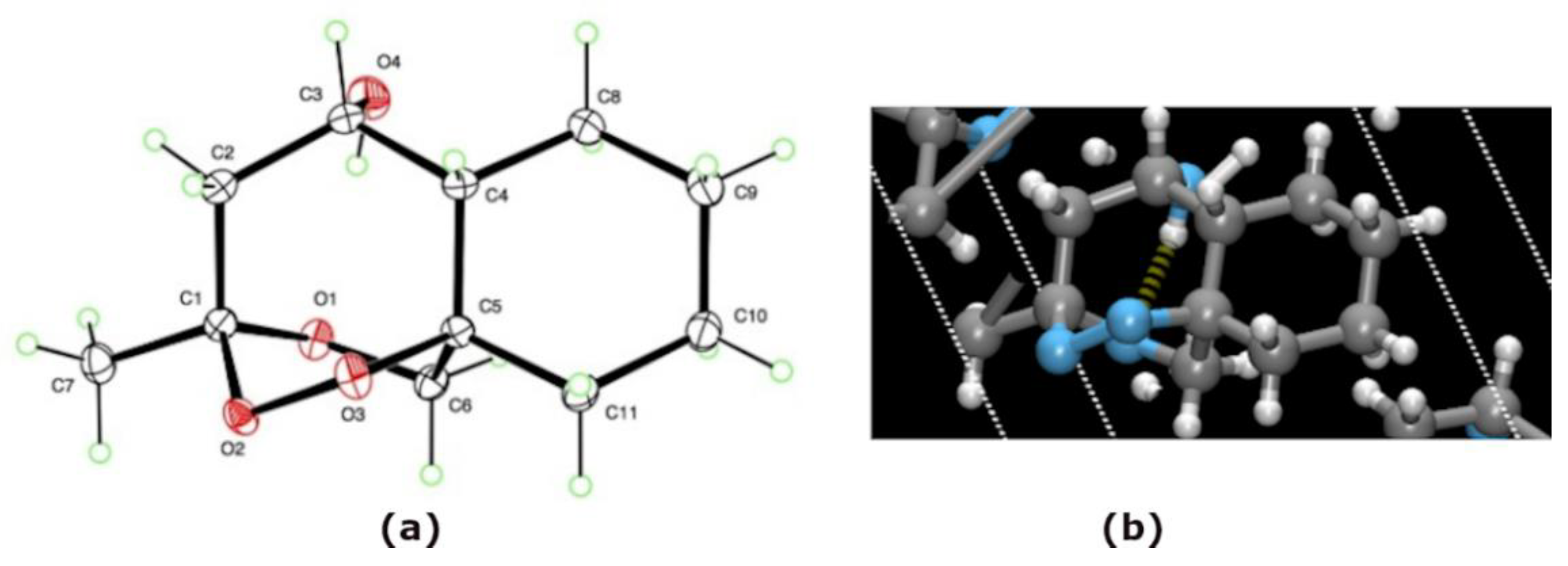
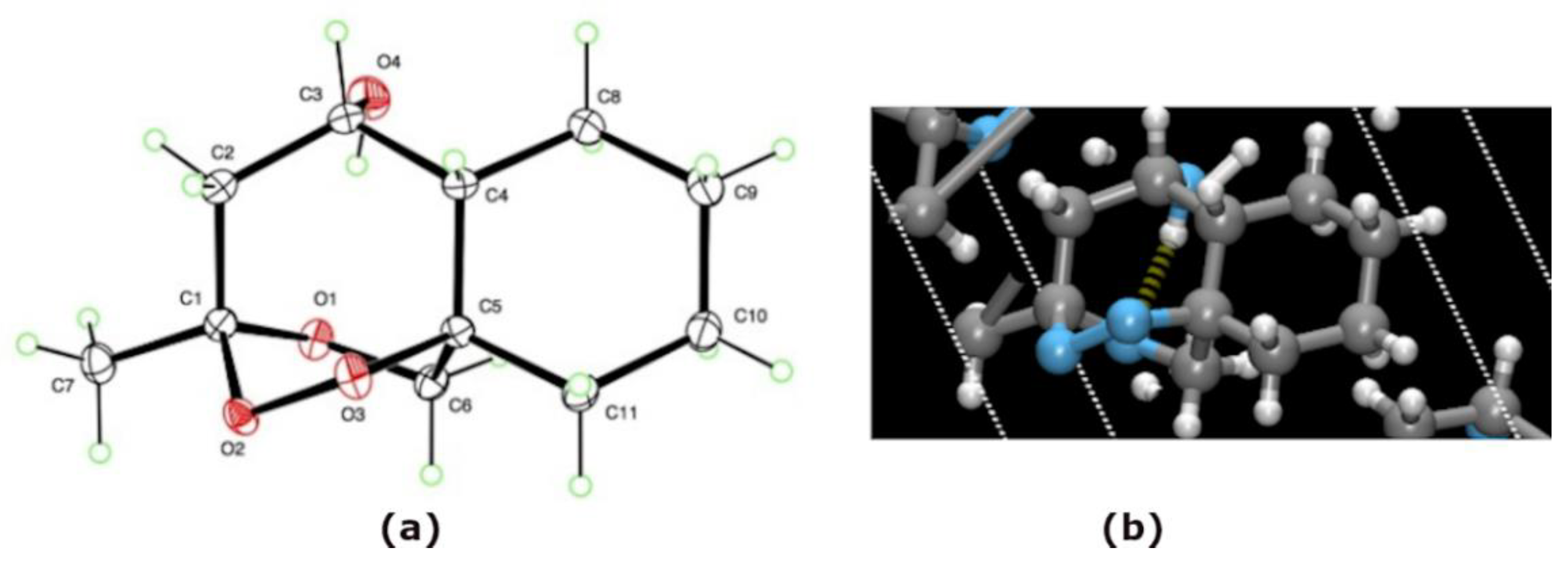
2.3. X-ray Crystallography
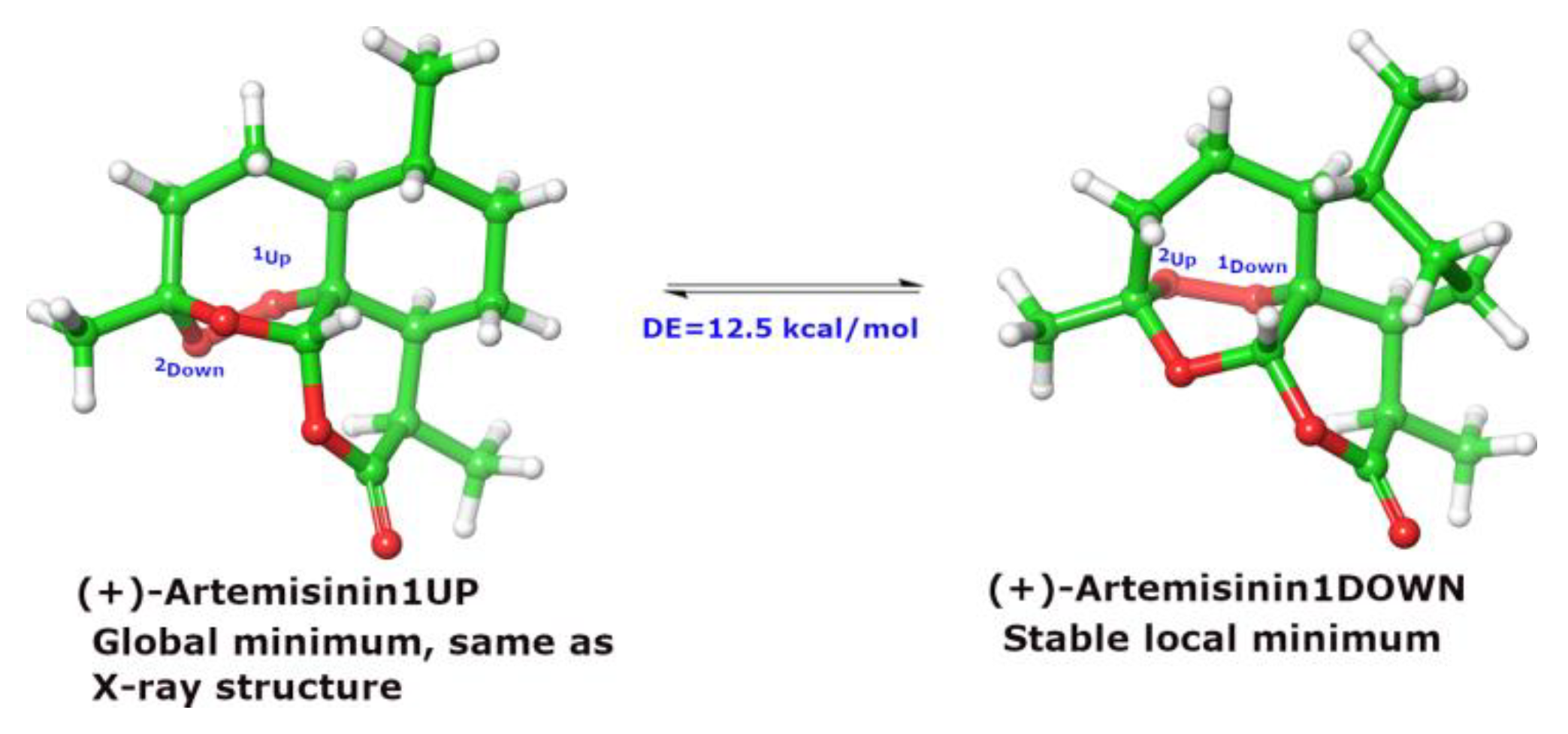
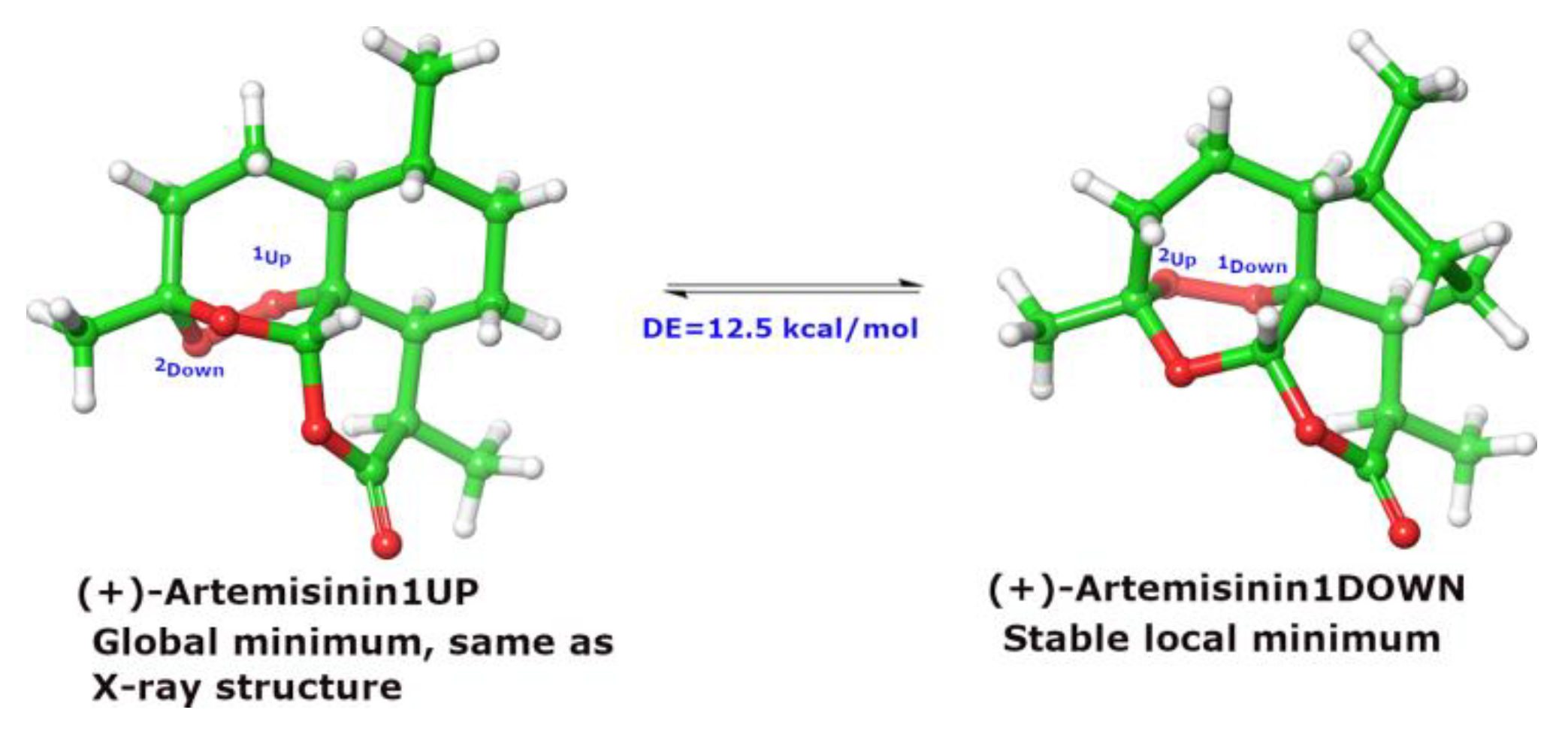
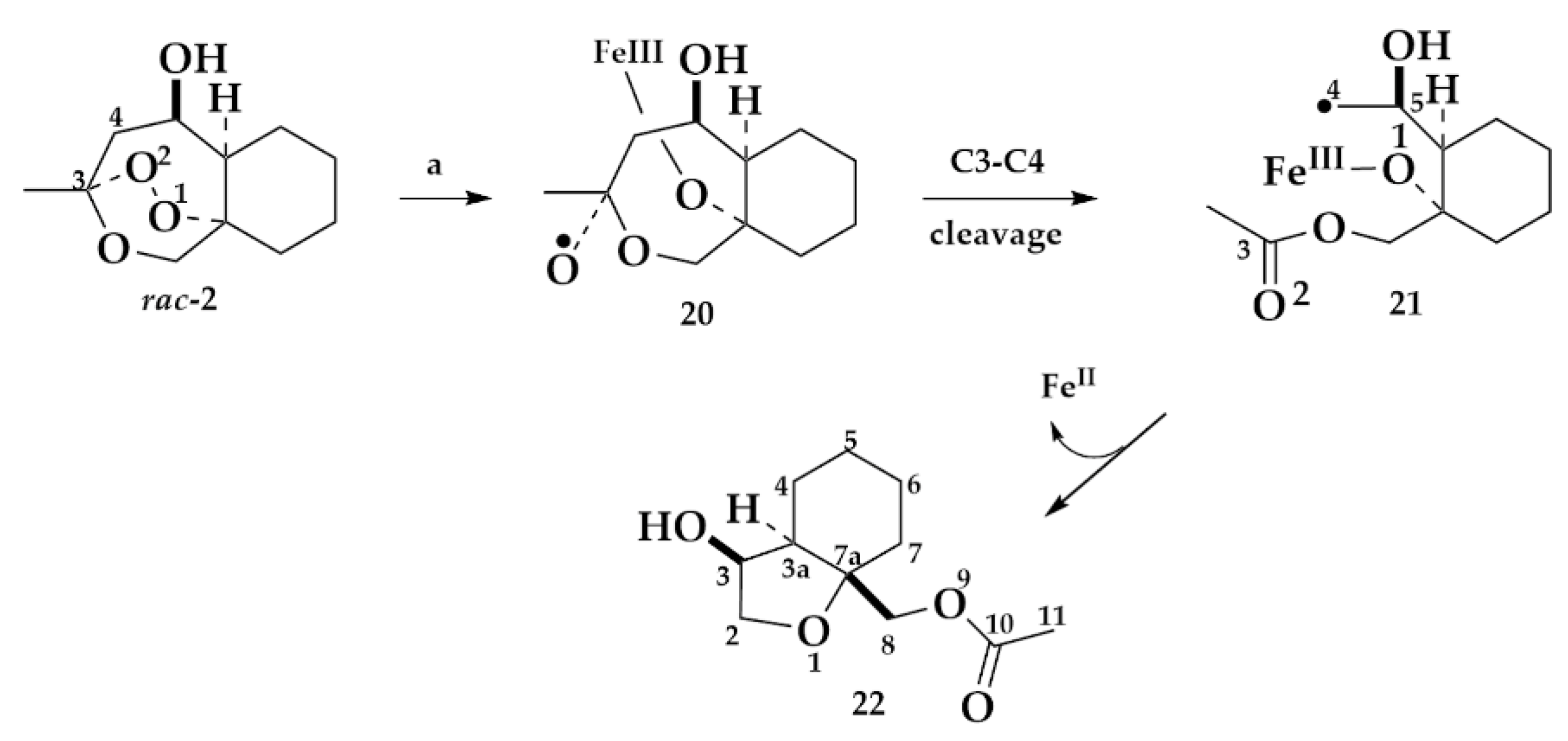
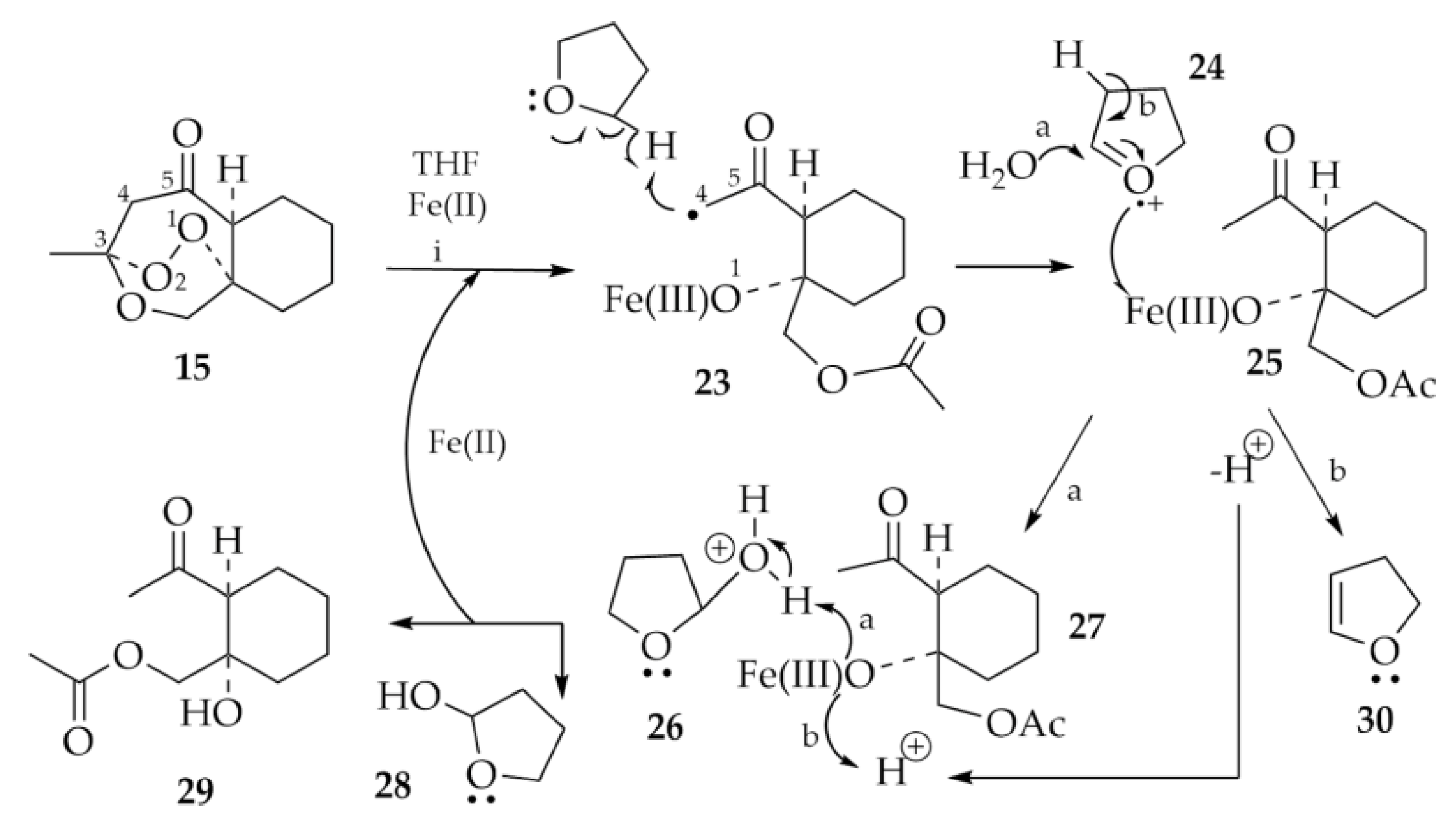
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Information
4.1.2. Preparation and Characterization of Synthesized Compounds
Ethyl 2-(2-methyl-1,3-dioxolan-2-yl) acetate (3)
2-(2-Methyl-1,3-dioxolan-2-yl)acetaldehyde (4)
2-(1-Hydroxy-2-(2-methyl-1,3-dioxolan-2-yl)ethyl)cyclohexan-1-one (5)
2-(1-((Tert-butyldimethylsilyl)oxy)-2-(2-methyl-1,3-dioxolan-2-yl)ethyl)cyclohexan-1-one (6)
Tert-butyldimethyl(2-(2-methyl-1,3-dioxolan-2-yl)-1-(1-oxaspiro[2.5]octan-4-yl)ethoxy)silane (7)
(2-(1-((tert-butyldimethylsilyl)oxy)-2-(2-methyl-1,3-dioxolan-2-yl)ethyl)-1hydroperoxycyclohexyl)methanol (8)
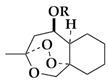
3-Methyloctahydro-1h-3,9a-epidioxybenzo[c]oxepin-5-ol, (rac-2)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl pentanoate (9)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl benzoate (10)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl 4-fluorobenzoate (11)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl 4-methoxybenzoate (12)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl cinnamate (13)
3-Methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl 4-chlorobenzoate (14)
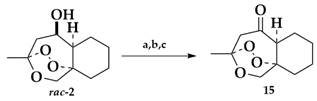
3-Methylhexahydro-1H-3,9a-epidioxybenzo[c]oxepin-5(5aH)-one (15)
(3R,5R,5aR,9aS)-3-methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl(R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (19a) and (3S,5S,5aS,9aR)-3-methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-yl(R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (19b)
(3R,5R,5aR,9aS)-3-methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-ol (2a)
(3S,5S,5aS,9aR)-3-methyloctahydro-1H-3,9a-epidioxybenzo[c]oxepin-5-ol (2b)
(3-Hydroxyhexahydrobenzofuran-7a(2H)-yl)methyl acetate (22)
(2-Acetyl-1-hydroxycyclohexyl)methyl acetate (29)
4.2. Single Crystal X-ray Diffraction Experiment
Compound rac-2
4.3. Antimalarial Activity Assay
4.3.1. Reagents and Materials
4.3.2. In Vitro Antimalarial Assay
4.3.3. In Vitro Cytotoxicity Assay
Neutral Red Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avery, M.A.; Weldon, D.; Muraleedharan, K. Drugs for Parasitic Infections: Advances in the Discovery of New Antimalarials; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 765–8142. [Google Scholar]
- Robert, A.; Dechy-Cabaret, O.; Cazelles, J.; Meunier, B. From Mechanistic Studies on Artemisinin Derivatives to New Modular Antimalarial Drugs. Acc. Chem. Res. 2002, 35, 167–174. [Google Scholar] [CrossRef]
- Wu, Y. How Might Qinghaosu (Artemisinin) and Related Compounds Kill the Intraerythrocytic Malaria Parasite? A Chemist’s View. Acc. Chem. Res. 2002, 35, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Cazelles, J.; Robert, A.; Meunier, B. Alkylating Capacity and Reaction Products of Antimalarial Trioxanes after Activation by a Heme Model. J. Org. Chem. 2002, 67, 609–619. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Wu, Y.-L. A possible antimalarial action mode of qinghaosu (artemisinin) series compounds. Alkylation of reduced glutathione by C-centered primary radicals produced from antimalarial compound qinghaosu and 12-(2,4-dimethoxyphenyl)-12-deoxoqinghaosu. Chem. Commun. 2000, 22, 2193–2194. [Google Scholar] [CrossRef]
- Pandey, A.V.; Tekwani, B.L.; Singh, R.L.; Chauhan, V.S. Artemisinin, an Endoperoxide Antimalarial, Disrupts the Hemoglobin Catabolism and Heme Detoxification Systems in Malarial Parasite. J. Biol. Chem. 1999, 274, 19383–19388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-M.; Wu, Y.; Wu, Y.-L.; Yao, Z.-J.; Zhou, C.-M.; Li, Y.; Shan, F. Unified mechanistic framework for the Fe(II)-induced cleavage of qinghaosu and derivatives/analogues. The first spintrapping evidence for the previously postulated secondary C-4 radical. J. Am. Chem. Soc. 1998, 120, 3316–3325. [Google Scholar] [CrossRef]
- Paitayatat, S.; Tarmchompoo, B.; Thebtatranonth, Y.; Yuthavong, Y. Correlation of antimalarial activity of artemisinin derivatives with binding affinity with ferroprotoporphyrin IX. J. Med. Chem. 1997, 40, 633–638. [Google Scholar] [CrossRef]
- Haynes, R.K.; Vonwiller, S.C. ChemInform Abstract: From Qinghao, Marvelous Herb of Antiquity, to the Antimalarial Trioxane Qinghaosu-and Some Remarkable New Chemistry. Acc. Chem. Res. 2010, 28, 73–79. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Yang, Y.-Z.; Lima, V.; Kuypers, F.; Kamchonwongpaisan, S.; Yuthavong, Y. Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu). Antimicrob. Agents Chemother. 1993, 37, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Haynes, R.K.; Hung-On, P.H.; Voerste, A. Ring opening of artemisinin (qinghaosu) and dihydroartemisinin and interception of the open hydroperoxides with formation of N-oxides-a chemical model for antimalarial action. Tetrahedron Lett. 1999, 40, 4715–4718. [Google Scholar] [CrossRef]
- Haynes, R.K.; Cheu, K.W.; N’Da, D.; Coghi, P.; Monti, D. Considerations on the mechanism of action of artemisinin antimalarials: Part 1—The ‘carbon radical’ and ‘heme’ hypotheses. Infect. Disord. Drug Targets. 2013, 13, 217–277. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The Molecular Mechanism of Action of Artemisinin—The Debate Continues. Molecules 2010, 15, 1705–1721. [Google Scholar] [CrossRef]
- Haynes, R.K.; Chan, W.-C.; Wong, H.-N.; Li, K.-Y.; Wu, W.-K.; Fan, K.-M.; Sung, H.Y.; Williams, I.D.; Prosperi, D.; Melato, S.; et al. Facile Oxidation of Leucomethylene Blue and Dihydroflavins by Artemisinins: Relationship with Flavoenzyme Function and Antimalarial Mechanism of Action. ChemMedChem 2010, 5, 1282–1299. [Google Scholar] [CrossRef]
- Robert, A.; Meunier, B. Charactarization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968–5969. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Rawe, S.L.; Borstnik, K.; Miller, A.; Ward, S.A.; Bray, P.G.; Davies, J.; Oh, C.H.; Posner, G.H. Enantiomeric 1,2,4-Trioxanes Display Equivalent in vitro Antimalarial Activity Versus Plasmodium falciparum Malaria Parasites: Implications for the Molecular Mechanism of Action of the Artemisinins. ChemBioChem 2005, 6, 2048–2054. [Google Scholar] [CrossRef]
- Jefford, C.W. Synthetic Peroxides as Potent Antimalarials. News and Views. Curr. Top. Med. Chem. 2012, 12, 373–399. [Google Scholar] [CrossRef]
- Wu, Y.; Yue, Z.-Y.; Wu, Y.-L. Interaction of Qinghaosu (Artemisinin) with Cysteine Sulfhydryl Mediated by Traces of Non-Heme Iron. Angew. Chem. Int. Ed. 1999, 38, 2580–2582. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Wu, Y.-L.; Wu, Y.; Liang, J.; Li, Y. Further Evidence for the Participation of Primary Carbon-Centered Free Radicals in the Antimalarial Action of the Qinghaosu (Artemisinin) Series of Compounds. ChemInform Abstr. 2010, 32, 135. [Google Scholar]
- Wu, Y.; Yue, Z.-Y.; Liu, H.-H. New Insights into the Degradation of Qinghaosu (Artemisinin) Mediated by Non-Heme-Iron Chelates, and Their Relevance to the Antimalarial Mechanism. Helv. Chim. Acta 2001, 84, 928–932. [Google Scholar] [CrossRef]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthvarakul, S.; Wong, L.M.H.; Hemingway, J.; Baigini, G.A.; O’Neill, P.M.; Ward, S.A. Artemisinin activity-based probes identify multiple molecular targets witihin the asexual stage of the malarial parasites of Plasmodium falciparum 3D7. Proc. Nat. Acad. Sci. USA 2016, 113, 2080–2085. [Google Scholar] [CrossRef] [Green Version]
- Ecksteinludwig, U.; Webb, R.J.; Van Goethem, I.D.A.; East, J.M.; Lee, A.G.; Kimura, M.; O’Neill, P.M.; Bray, P.G.; Ward, S.; Krishna, S. Artemisinins target the SERCA of Plasmodium falciparum. Nat. Cell Biol. 2003, 424, 957–961. [Google Scholar] [CrossRef]
- Bhisutthibhan, J.; Pan, X.Q.; Hossler, P.A.; Walker, D.J.; Yowell, C.A.; Carlton, J.; Dame, J.B.; Meshnick, S.R. The Plasmodium falciparum translationally controlled tumor protein homologue and its reaction with the antimalarial drug artemisinin. J. Biol. Chem. 1998, 273, 16192–16198. [Google Scholar] [CrossRef] [Green Version]
- Bhisutthibhan, J.; Meshnick, S.R. Immunprecipitation of [3H]Dihydroartemisinin Translationally Controlled Tumor Protein (TCTP) Adducts from Plasmodium falciparum-Infected Erythrocytes by Using Anti-TCTP Antibodies. Antimicrob. Agents Chemother. 2001, 45, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, T.; Winter, D.; Büchele, B.; Dirdjaja, N.; Frank, M.; Lehmann, W.-D.; Mertens, R.; Krauth-Siegel, R.L.; Simmet, T.; Granzin, J.; et al. Molecular interaction of artemisinin with translationally controlled tumor protein (TCTP) of Plasmodium falciparum. Biochem. Pharmacol. 2013, 85, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Vroman, J.; Alvim-Gaston, M.; Avery, M.A. Current progress in the chemistry, medicinal chemistry and drug design of artemisinin based antimalarials. Curr. Pharm. Design 1999, 5, 101–138. [Google Scholar] [CrossRef]
- Janse, C.J.; Waters, A.; Kos, J.; Lugt, C. Comparison of in vivo and in vitro antimalarial activity of artemisinin, dihydroartemisinin and sodium artesunate in the Plasmodium berghei-rodent model. Int. J. Parasitol. 1994, 24, 589–594. [Google Scholar] [CrossRef]
- Balint, G.A. Artemisinin and its derivatives: An important new class of antimalarial agents. Pharmacol. Ther. 2001, 90, 261–265. [Google Scholar] [CrossRef]
- Baker, J.K.; McChesney, J.D.; Chi, H.T. Decomposition of Arteether in Simulated Stomach Acid Yielding Compounds Retaining Antimalarial Activity. Pharm. Res. 1993, 10, 662–666. [Google Scholar] [CrossRef]
- Brewer, T.G.; Grate, S.J.; Peggins, J.O.; Weina, P.J.; Petras, J.M.; Levine, B.S.; Heiffer, M.H.; Schuster, B.G. Fatal Neurotoxicity of Arteether and Artemether. Am. J. Trop. Med. Hyg. 1994, 51, 251–259. [Google Scholar] [CrossRef]
- Lin, A.J.; Klayman, D.L.; Milhous, W.K. Antimalarial activity of new water-soluble dihydroartemisinin derivatives. J. Med. Chem. 1987, 30, 2147–2150. [Google Scholar] [CrossRef]
- Nagelschmitz, J.; Voith, B.; Wensing, G.; Roemer, A.; Fugmann, B.; Haynes, R.K.; Kotecka, B.M.; Rieckmann, K.H.; Edstein, M.D. First Assessment in Humans of the Safety, Tolerability, Pharmacokinetics, and Ex Vivo Pharmacodynamic Antimalarial Activity of the New Artemisinin Derivative Artemisone. Antimicrob. Agents Chemother. 2008, 52, 3085–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, P.M.; Higson, A.P.; Taylor, S.; Irving, E. Preparation of Dihydroartemisinin Derivatives as Antimalarial and Antitumor agents. WO Patent Application 2003/048167, 16 April 2003. [Google Scholar]
- Magueur, G.; Crousse, B.; Charneau, S.; Grellier, P.; Bégué, J.-P.; Bonnet-Delpon, D. Fluoroartemisinin: Trifluoromethyl Analogues of Artemether and Artesunate. J. Med. Chem. 2004, 47, 2694–2699. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; Jeon, H.B.; Ploypradith, P.; Paik, I.-H.; Borstnik, K.; Xie, S.; Shapiro, T.A. Orally active, water-soluble antimalarial 3-aryltrioxanes: Short synthesis and preclinical efficacy testing in rodents. J. Med. Chem. 2002, 45, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.; Mehrotra, S.; Bonk, J.D.; Vroman, J.A.; Goins, A.D.K.; Miller†, R. Structure–ActivityRelationships of the Antimalarial Agent Artemisinin. 4. Effect of Substitution at C-3. J. Med. Chem. 1996, 39, 2900–2906. [Google Scholar] [CrossRef]
- Woolfrey, J.R.; Avery, M.A.; Doweyko, A.M. Comparison of 3D quantitative structure-activity relationship methods: Analysis of the in vitro antimalarial activity of 154 artemisinin analogues by hypothetical active-site lattice and comparative molecular field analysis. J. Comput. Mol. Des. 1998, 12, 165–181. [Google Scholar] [CrossRef]
- Avery, M.A.; Mehrotra, S.; Johnson, T.L.; Bonk, J.D.; Vroman, A.J.A.; Miller†, R. Structure–ActivityRelationships of the Antimalarial Agent Artemisinin. 5. Analogs of 10-Deoxoartemisinin Substituted at C-3 and C-9. J. Med. Chem. 1996, 39, 4149–4155. [Google Scholar] [CrossRef]
- Grellepois, F.; Chorki, F.; Ourévitch, M.; Charneau, S.; Grellier, P.; McIntosh, K.A.; Charman, W.N.; Pradines, B.; Crousse, B.; Bonnet-Delpon, D.; et al. Orally Active Antimalarials: Hydrolytically Stable Derivatives of 10-Trifluoromethyl Anhydrodihydroartemisinin†. J. Med. Chem. 2004, 47, 1423–1433. [Google Scholar] [CrossRef]
- Cumming, J.N.; Wang, D.; Park, S.B.; Shapiro, T.A.; Posner, G.H. Design, Synthesis, Derivatization, and Structure-Activity Relationships of Simplified, Tricyclic, 1,2,4-Trioxane Alcohol Analogues of the Antimalarial Artemisinin. J. Med. Chem. 1998, 41, 952–964. [Google Scholar] [CrossRef]
- Bonk, J.D. Structure-Activity Relationships of the Antimalarial Agent Artemisinin: Synthesis and Evaluation of 11-Aza and 5-Cyano Analogs and Partial Synthesis of the Antifungal Natural Product Pseudolaric Acid. Ph.D. Thesis, The University of Mississippi, University, MS, USA, 1997. [Google Scholar]
- Avery, M.A.; Bonk, J.D.; Chong, W.K.M.; Mehrotra, S.; Miller, R.; Milhous, W.; Goins, D.K.; Venkatesan, S.; Wyandt, C. Structure-Activity Relationships of the Antimalarial Agent Artemisinin. 2. Effect of Heteroatom Substitution at O-11: Synthesis and Bioassay of N-Alkyl-11-aza-9-desmethylartemisinins. J. Med. Chem. 1995, 38, 5038–5044. [Google Scholar] [CrossRef]
- Avery, M.A.; Chong, W.; Jennings-White, C. Stereoselective total synthesis of (+)-artemisinin, the antimalarial constituent of Artemisia annua L. J. Am. Chem. Soc. 1992, 114, 974–979. [Google Scholar] [CrossRef]
- Avery, B.A.; Pabbisetty, D.; Li, L.; Sharma, A.; Gundluru, M.K.; Chittiboyina, A.G.; Williamson, J.S.; Avery, M. A pharmacokinetic comparison of homodimer ARB-92 and heterodimer ARB-89: Novel, potent antimalarial candidates derived from 7β-hydroxyartemisinin. J. Pharm. Investig. 2017, 48, 585–593. [Google Scholar] [CrossRef]
- Avery, M.A.; Gao, F.; Chong, W.K.; Hendrickson, T.F.; Inman, W.D.; Crews, P. Synthesis, conformational analysis, and antimalarial activity of tricyclic analogs of artemisinin. Tetrahedron 1994, 50, 957–972. [Google Scholar] [CrossRef]
- Avery, M.; Fan, P.; Karle, J.M.; Bonk, J.D.; Miller, A.R.; Goins, D.K. Structure–ActivityRelationships of the Antimalarial Agent Artemisinin. 3. Total Synthesis of (+)-13-Carbaartemisinin and Related Tetra- and Tricyclic Structures. J. Med. Chem. 1996, 39, 1885–1897. [Google Scholar] [CrossRef]
- Avery, M.; Gao, F.; Chong, W.; Mehrotra, S.; Milhous, W.K. Structure-activity relationships of the antimalarial agent artemisinin. 1. Synthesis and comparative molecular field analysis of C-9 analogs of artemisinin and 10-deoxoartemisinin. J. Med. Chem. 1993, 36, 4264–4275. [Google Scholar] [CrossRef]
- Avery, M.A.; Muraleedharan, K.M.; Desai, P.V.; Bandyopadhyaya, A.K.; Furtado, A.M.M.; Tekwani, B.L. Structure–ActivityRelationships of the Antimalarial Agent Artemisinin. 8. Design, Synthesis, and CoMFA Studies toward the Development of Artemisinin-Based Drugs against Leishmaniasis and Malaria†. J. Med. Chem. 2003, 46, 4244–4258. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.A.; Alvim-Gaston, M.; Rodrigues, C.R.; Barreiro, E.J.; Cohen, F.E.; Sabnis, Y.A.; Woolfrey, J.R. Structure–ActivityRelationships of the Antimalarial Agent Artemisinin. 6. The Development of Predictive In Vitro Potency Models Using CoMFA and HQSAR Methodologies. J. Med. Chem. 2002, 45, 292–303. [Google Scholar] [CrossRef]
- Li, Y.; Hao, H.-D.; Wittlin, S.; Wu, Y. Simple Analogues of Qinghaosu (Artemisinin). Chem. Asian J. 2012, 7, 1881–1886. [Google Scholar] [CrossRef]
- Tan, J.S.; Ciufolini, M.A. Total Synthesis of Topopyrones B and D. Org. Lett. 2006, 8, 4771–4774. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Hao, H.-D.; Li, Y.; Han, W.-B.; Wu, Y. A Hydrogen Peroxide Based Access to Qinghaosu (Artemisinin). Org. Lett. 2011, 13, 4212–4215. [Google Scholar] [CrossRef]
- Ohtawa, M.; Yamazaki, H.; Ohte, S.; Matsuda, D.; Ohshiro, T.; Rudel, L.L.; Omura, S.; Tomoda, H.; Nagamitsu, T. Synthesis and structure–activity relationship of pyripyropene A derivatives as potent and selective acyl-CoA:cholesterol acyltransferase 2 (ACAT2) inhibitors: Part 1. Bioorganic Med. Chem. Lett. 2013, 23, 1285–1287. [Google Scholar] [CrossRef]
- Nakatsuji, H.; Morimoto, M.; Misaki, T.; Tanabe, Y. Mild, powerful, and robust methods for esterification, amide formation, and thioesterification between acid chlorides and alcohols, amines, thiols, respectively. Tetrahedron 2007, 63, 12071–12080. [Google Scholar] [CrossRef]
- Bolm, C.; Magnus, A.S.; Hildebrand, J.P. Catalytic synthesis of aldehydes and ketones under mild conditions using TEMPO/Oxone. Org. Lett. 2000, 2, 1173–1175. [Google Scholar] [CrossRef]
- Corey, E.; Suggs, J. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975, 16, 2647–2650. [Google Scholar] [CrossRef]
- Sumii, Y.; Kotoku, N.; Fukuda, A.; Kawachi, T.; Sumii, Y.; Arai, M.; Kobayashi, M. Enantioselective synthesis of dictyoceratin-A (smenospondiol) and -C, hypoxia-selective growth inhibitors from marine sponge. Bioorganic Med. Chem. 2015, 23, 966–975. [Google Scholar] [CrossRef]
- Kumar, B.S.; Mishra, G.P.; Rao, B.V. Synthesis of (+)/(−) pentenomycins via Me2AlCl induced cascade reaction. Tetrahedron Lett. 2013, 54, 2845–2848. [Google Scholar] [CrossRef]
- Che, C.-T.; McPherson, D.D.; Cordell, G.A.; Fong, H.H.S. Pulcherralpin, a New Diterpene Ester from Caesalpinia pulcherrima. J. Nat. Prod. 1986, 49, 561–569. [Google Scholar] [CrossRef]
- Makler, M.T.; Piper, R.; Williams, J.A.; Gibbins, B.L.; Hinrichs, D.J.; Ries, J.M.; Bancroft, J.E. Parasite Lactate Dehydrogenase as an Assay for Plasmodium falciparum Drug Sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef]
- Makler, M.T.; Hinrichs, D.J. Measurement of the LDH Activity of Plasmodium falciparum as an Assessment of Parasitemia. Am. J. Trop. Med. Hyg. 1993, 48, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.J.B.; de Figueiredo, A.F.; Lobato, M.da.; de Miranda, R.M.; de Almeida, R.C.O.; Pinheiro, J.C. A study on antimalarial artemisinin derivatives using MEP maps and multivariate QSAR. J. Mol. Model. 2007, 14, 39–48. [Google Scholar] [CrossRef]
- dos Santos, C.B.R.; Lobato, C.C.; Vieira, J.B.; Brasil, D.S.B.; Brito, A.U.; Macêdo, W.J.C.; Carvalho, J.C.T.; Pinheiro, J.C. Evaluation of Quantum Chemical Methods and Basis Sets Applied in the Molecular Modeling of Artemisinin. Comput. Mol. Biosci. 2013, 3, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Macêdo, W.J.C.; Braga, F.S.; Santos, C.F.; Costa, J.d.S.; de Melo, G.S.; de Mello, M.N.; Sousa, D.S.; Carvalho, J.C.T.; Socorro, D.d.; Brasil, B.; et al. Antimalarial Artemisinins Derivatives Study: Molecular Modeling and Multivariate Analysis (PCA, HCA, KNN, SIMCA and SDA). J. Comput. Theor. Nanosci. 2015, 12, 3443–3458. [Google Scholar] [CrossRef]
- Gu, J.; Chen, K.; Jiang, H.; Ji, R. A DFT study of Artemisinin and 1,2,4-trioxane. J. Mol. Struct. 1999, 459, 103–111. [Google Scholar] [CrossRef]
- Gu, J.-D.; Chert, K.-X.; Jiang, H.-L.; Zhu, W.-L.; Chen, J.-Z.; Ji, R.-Y. A quantum chemistry study of Qinghaosu. Chem. Phys. Lett. 1997, 277, 234–238. [Google Scholar] [CrossRef]
- Gironés, X.; Gallegos, A.; Carbó-Dorca, R. Antimalarial activity of synthetic 1,2,4-trioxanes and cyclic peroxy-ketals, a quantum similarity study. J. Comput. Aided Mol. Des. 2001, 15, 1053–1063. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.C.; Carvalho, J.C.T. A SAR and QSAR Study of New Artemisinin Compounds with Antimalarial Activity. Molecules 2013, 19, 367–399. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, O.; Minor, W. International Tables for Crystallography Volume F: Macromolecular. In Crystallography, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 226–235. [Google Scholar]
- Mustafa, J.; Khan, S.I.; Ma, G.; Walker, L.A.; Khan, I.A. Synthesis and anticancer activities of fatty acid analogs of podophyllotoxin. Lipids 2004, 39, 167–172. [Google Scholar] [CrossRef]
- Repetto, G.; del Paso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
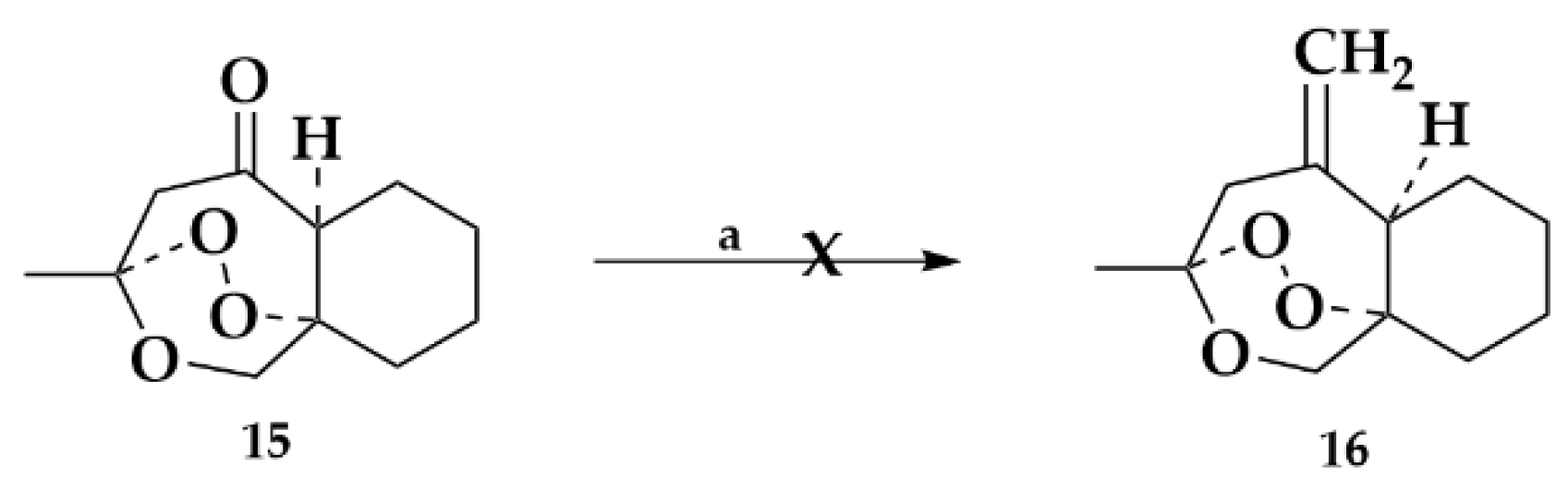



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
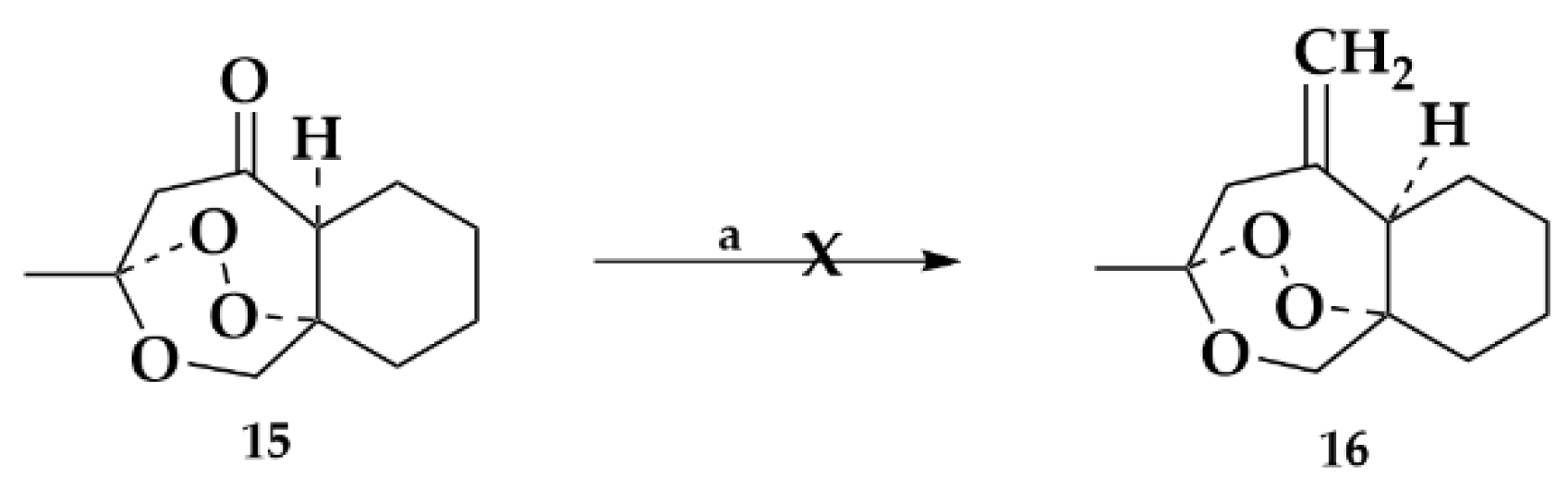{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | % Yield | Compound | R | % Yield |
|---|---|---|---|---|---|
| 9 | CH3(CH2)3CO | a 11 | 12 | p-MeOC6H4CO | d 68 |
| 10 | C6H5CO | b 42,c 70, d 74 | 13 | C6H5(CH)2CO | d 36 |
| 11 | p-FC6H4CO | d 70 | 14 | p-ClC6H4CO | d 49 |
| Compound | Method | % Yield |
|---|---|---|
| 15 | a | 40 |
| b | 50 | |
| c | 55 |
| Structure | R | IC50 (ng/mL) | Relative Activity a | |||
|---|---|---|---|---|---|---|
| D-6 | W-2 | D-6 | W-2 | anal. (C, H) | ||
| 1 | (Artemisinin) | 7.05 | 5.08 | 100 | 100 | |
| rac-2 | H | 1716.22 | 1778.35 | 0.6 | 0.43 | C11H18O4 |
| 2a | H | 1255.56 | 884.89 | 0.43 | 0.44 | C11H18O4 |
| 2b | H | NA b | NA b | C11H18O4 | ||
| 9 | CH3(CH2)3CO | 149.19 | 208.86 | 5.00 | 2.60 | C16H26O5 |
| 10 | C6H5CO | 152.81 | 120.97 | 10.40 | 9.50 | C18H22O5 |
| 11 | p-FC6H4CO | 80.72 | 97.54 | 20.81 | 12.41 | C18H21FO5 |
| 12 | p-MeOC6H4CO | 114.96 | 167.22 | 15.13 | 7.50 | C19H24O6 |
| 13 | C6H5(CH)2CO | 130.87 | 148.09 | 13.14 | 8.37 | C20H24O5 |
| 14 | p-ClC6H4CO | 144.65 | 119.95 | 12.18 | 10.58 | C18H21ClO5 |
| 15 | Ketone | 31.83 | 23.34 | 33.30 | 32.72 | C11H16O4 |
| 16 | Exomethylene | 344.84 | 346.31 | 3.05 | 2.18 | C12H18O3 |
| 19a | C6H5C(OCH3)(CF3)CO | 150.64 | 357.24 | 7.13 | 2.17 | C21H25F3O6 |
| 19b | C6H5C(OCH3)(CF3)CO | 2169.31 | 2255.40 | 0.50 | 0.34 | C21H25F3O6 |
| 22 | Scheme 5 | NA b | NA b | C12H20O3 | ||
| 29 | Scheme 6 | NA b | NA b | C11H18O4 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahan, M.; Leon, F.; Fronczek, F.R.; Elokely, K.M.; Rimoldi, J.; Khan, S.I.; Avery, M.A. Structure–Activity Relationships of the Antimalarial Agent Artemisinin 10. Synthesis and Antimalarial Activity of Enantiomers of rac-5β-Hydroxy-d-Secoartemisinin and Analogs: Implications Regarding the Mechanism of Action. Molecules 2021, 26, 4163. https://doi.org/10.3390/molecules26144163
Jahan M, Leon F, Fronczek FR, Elokely KM, Rimoldi J, Khan SI, Avery MA. Structure–Activity Relationships of the Antimalarial Agent Artemisinin 10. Synthesis and Antimalarial Activity of Enantiomers of rac-5β-Hydroxy-d-Secoartemisinin and Analogs: Implications Regarding the Mechanism of Action. Molecules. 2021; 26(14):4163. https://doi.org/10.3390/molecules26144163
Chicago/Turabian StyleJahan, Mohamed, Francisco Leon, Frank R. Fronczek, Khaled M. Elokely, John Rimoldi, Shabana I. Khan, and Mitchell A. Avery. 2021. "Structure–Activity Relationships of the Antimalarial Agent Artemisinin 10. Synthesis and Antimalarial Activity of Enantiomers of rac-5β-Hydroxy-d-Secoartemisinin and Analogs: Implications Regarding the Mechanism of Action" Molecules 26, no. 14: 4163. https://doi.org/10.3390/molecules26144163
APA StyleJahan, M., Leon, F., Fronczek, F. R., Elokely, K. M., Rimoldi, J., Khan, S. I., & Avery, M. A. (2021). Structure–Activity Relationships of the Antimalarial Agent Artemisinin 10. Synthesis and Antimalarial Activity of Enantiomers of rac-5β-Hydroxy-d-Secoartemisinin and Analogs: Implications Regarding the Mechanism of Action. Molecules, 26(14), 4163. https://doi.org/10.3390/molecules26144163