
New Strategies for the Simple and Sensitive Voltammetric Direct Quantification of Se(IV) in Environmental Waters Employing Bismuth Film Modified Glassy Carbon Electrode and Amberlite Resin

Abstract
1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. Reagents and Solutions
2.3. Voltammetric Procedure
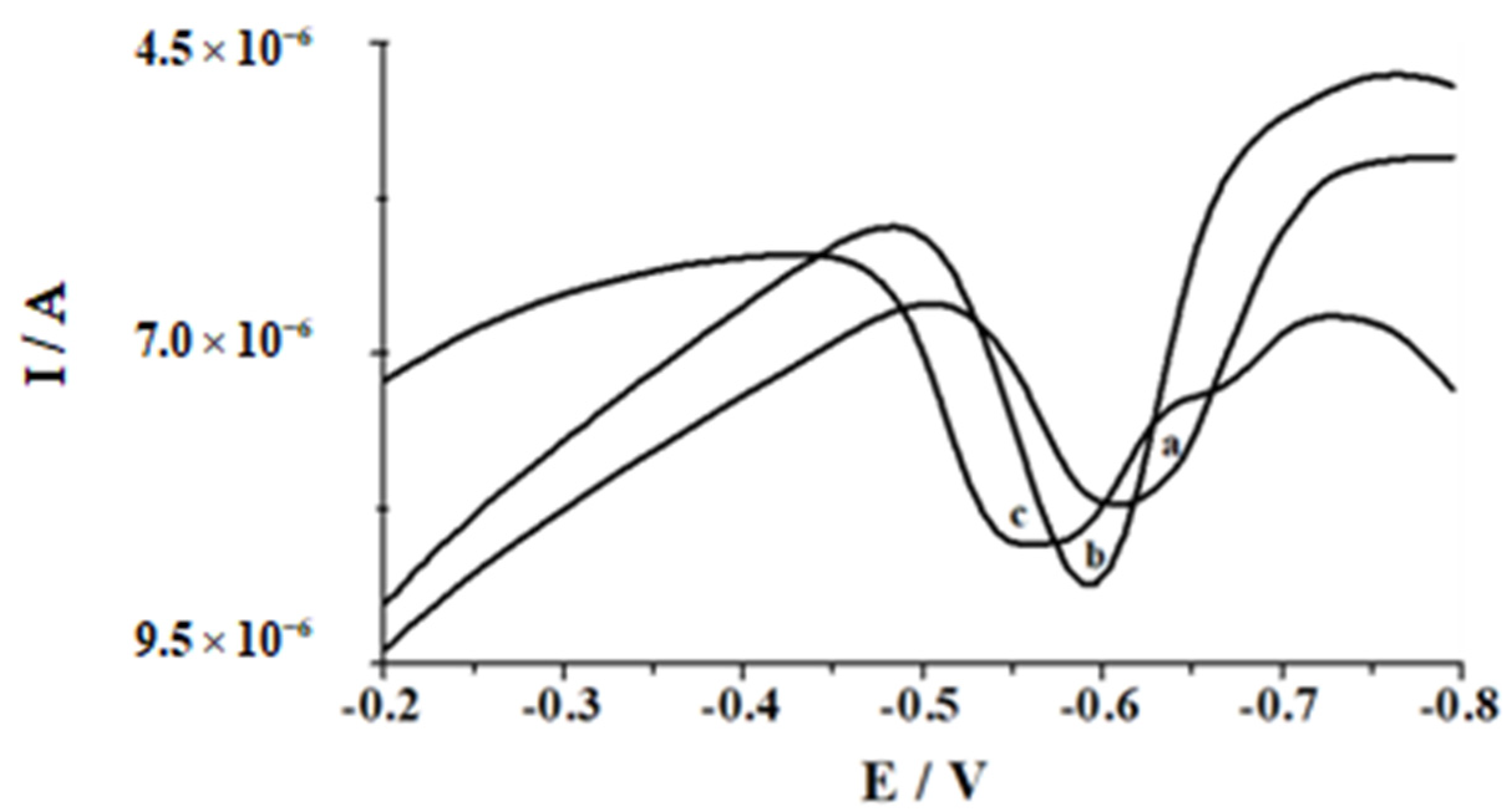
3. Results and Discussion
3.1. Influence of pH and Supporting Electrolyte Concentration
3.2. Examination of Stability of Se(IV) Signal Value in the Presence of XAD-7 Resin
3.3. Examination of the Se(IV) Signal Intensity Depending on the Temperature
3.4. Choice of Conditions Affecting the Morphology of the Bismuth Film Formed on the Glassy Carbon
3.4.1. Influence of Bismuth Concentration
3.4.2. Conditions of the Accumulation Step
3.5. Calibration Data
3.6. Selectivity
3.7. Influence of Organic Compounds
3.8. Analytical Applications
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Letavayová, L.; Vlasáková, D.; Spallholz, J.E.; Brozmanová, J.; Chovanec, M. Toxicity and mutagenicity of selenium compounds in saccharomyces cerevisiae. Mutat. Res. 2008, 638, 1–10. [Google Scholar] [CrossRef]
- Ferri, T.; Petruzzelli, G.; Pezzarossa, B.; Santaroni, P.; Brunori, C.; Morabito, R. Study of the influence of carboxymethylcellulose on the absorption of selenium (and selected metals) in a target plant. Microchem. J. 2003, 74, 257–265. [Google Scholar] [CrossRef]
- Lemly, A.D.; Finger, S.E.; Nelson, M.K. Sources and impacts of irrigation drain water contaminants in arid wetlands. Environ. Toxicol. Chem. 1992, 12, 2265–2279. [Google Scholar] [CrossRef]
- Campanella, L.; Ferri, T. Voltammetric behaviour of selenium(IV) at hanging mercury drop electrodes in acetate buffer. J. Electroanal. Chem. 1984, 165, 241–249. [Google Scholar] [CrossRef]
- Ferri, T.; Sangiorgio, P. Determination of selenium speciation in river waters by adsorption on iron(III)-Chelex-100 resin and differential pulse cathodic stripping voltammetry. Anal. Chim. Acta 1996, 321, 185–193. [Google Scholar] [CrossRef]
- Jarzabek, G.; Kublik, Z. Cyclic and stripping voltammetry of Se(+4) and Se(−2) at the HMDE in acidic media. J. Electroanal. Chem. 1982, 137, 247–259. [Google Scholar] [CrossRef]
- Mattsson, G.; Nyholm, L.; Olin, A. Cathodic stripping voltammetry of HgSe. J. Electroanal. Chem. 1994, 377, 149–162. [Google Scholar] [CrossRef]
- Bryce, D.W.; Izquierdo, A.; Castro, M.D. Sequential speciation of selenium by flow injection cathodic stripping voltammetry. Fresenius Z. Anal. Chem. 1995, 351, 433–437. [Google Scholar] [CrossRef]
- Baltensperger, U.; Hertz, J. Parameter evaluation for the determination of selenium by cathodic stripping voltammetry at the hanging mercury drop electrode. Anal. Chim. Acta 1985, 172, 49–56. [Google Scholar] [CrossRef]
- Zelic, M.; Branica, M. Study of electrochemical reduction of selenium (+4) using square wave voltammetry. Electroanalysis 1990, 2, 455–462. [Google Scholar] [CrossRef]
- van den Berg, C.M.G.; Khan, S.H. Determination of selenium in sea water by adsorptive cathodic stripping voltammetry. Anal. Chim. Acta 1990, 231, 221–229. [Google Scholar] [CrossRef]
- Mattsson, G.; Nyholm, L.; Olin, A.; Ornemark, U. Determination of selenium in freshwaters by cathodic stripping voltammetry after UV irradiation. Talanta 1995, 42, 817–825. [Google Scholar] [CrossRef]
- Papoff, P.; Bocci, E.; Lanza, F. Speciation of selenium in natural waters and snow by DPCSV at the hanging mercury drop electrode. Microchem. J. 1998, 59, 50–76. [Google Scholar] [CrossRef]
- Rurikova, D.; Tothova, E. Determination of inorganic selenium species in natural waters by preconcentration and cathodic stripping voltammetry. Chem. Pap. 1999, 53, 26–33. [Google Scholar]
- Piech, R.; Kubiak, W.W. Determination of trace selenium on hanging copper amalgam drop electrode. Electrochim. Acta 2007, 53, 584–589. [Google Scholar] [CrossRef]
- Piech, R. Determination of selenium traces on cyclic renewable mercury film silver electrode in presence of copper ions using cathodic stripping voltammetry. Electroanalysis 2008, 20, 2475–2481. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J. Ultratrace measurements of selenium by cathodic stripping voltammetry in the presence of rhodium. Anal. Chim. Acta 1993, 274, 219–224. [Google Scholar] [CrossRef]
- Lange, B.; van den Berg, C.M.G. Determination of selenium by catalytic cathodic stripping voltammetry. Anal. Chim. Acta 2000, 418, 33–42. [Google Scholar] [CrossRef]
- Ishiyama, T.; Tanaka, T. Cathodic stripping voltammetry of selenium(IV) at a silver disk electrode. Anal. Chem. 1996, 68, 3789–3792. [Google Scholar] [CrossRef]
- Bas, B.; Jedlinska, K.; Wegiel, K. New electrochemical sensor with the renewable silver annular band working electrode: Fabrication and application for determination of selenium(IV) by cathodic stripping voltammetry. Electrochem. Commun. 2014, 49, 79–82. [Google Scholar] [CrossRef]
- Suznjevic, D.; Blagojevic, S.; Vidic, J.; Erceg, M.; Vucelic, D. Determination of selenium(IV) by cathodic stripping voltammetry using a copper microelectrode. Microchem. J. 1997, 57, 255–260. [Google Scholar]
- Grabarczyk, M.; Korolczuk, M. Development of a simple and fast voltammetric procedure for determination of trace quantity of Se(IV) in natural lake and river water samples. J. Hazard. Mater. 2010, 175, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Merino, I.E.; Stegmann, E.; Aliaga, M.E.; Gomez, M.; Arancibia, V.; Rojas-Romo, C. Determination of Se(IV) concentration via cathodic stripping voltammetry in the presence of Cu(II) ions and ammonium diethyl dithiophosphate. Anal. Chim. Acta 2019, 1048, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.W.; Johnson, D.C. Voltammetric deposition and stripping of selenium(IV) at a rotating gold-disk electrode in 0.1 M perchloric acid. Anal. Chem. 1975, 47, 294–299. [Google Scholar] [CrossRef]
- Bryce, D.W.; Izquierdo, A.; Castro, M.D. Flow-injection anodic stripping voltammetry at a gold electrode for selenium(IV) determination. Anal. Chim. Acta 1995, 308, 96–101. [Google Scholar] [CrossRef]
- Cai, Q.; Khoo, S.B. Poly(3,3′-diaminobenzidine) film on a gold electrode for selective preconcentration and stripping analysis of selenium(IV). Anal. Chem. 1994, 66, 4543–4550. [Google Scholar] [CrossRef]
- Pereira, C.F.; Gonzaga, F.B.; Guaritá-Santos, A.M.; Souza De, J.R. Determination of Se(IV) by anodic stripping voltammetry using gold electrodes made from recordable CDs. Talanta 2006, 69, 877–881. [Google Scholar] [CrossRef]
- Beni, V.; Collins, G.; Arrigan, D.W.M. Investigation into the voltammetric behaviour and detection of selenium(IV) at metal electrodes in diverse electrolyte media. Anal. Chim. Acta 2011, 699, 127–133. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento, F.H.; Masini, J.C. An electrochemical sequential injection method to investigate the adsorption of selenite on Fe(III) polyhydroxy cations intercalated vermiculite. Water Sci. Technol. 2017, 1, 134–143. [Google Scholar] [CrossRef]
- Ochab, M.; Gęca, I.; Korolczuk, M. Determination of trace Se(IV) by anodic stripping voltammetry following double deposition and stripping steps. Talanta 2017, 165, 364–368. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, K.; Boyd, D.; Chi, H.; Smyth, R. Anodic stripping voltammetry of selenium(IV) at a gold fiber working electrode. Electroanalysis 1992, 4, 689–693. [Google Scholar] [CrossRef]
- Tan, S.H.; Kounaves, S.P. Determination of selenium(IV) at a microfabricated gold ultramicroelectrode array using square wave anodic stripping voltammetry. Electroanalysis 1998, 10, 364–368. [Google Scholar] [CrossRef]
- Segura, R.; Pizarro, J.; Díaz, K.; Placencio, A.; Godoy, F.; Pino, E.; Recio, F. Development of electrochemical sensors for the determination of selenium using gold nanoparticles modified electrodes. Sens. Actuators B 2015, 220, 263–269. [Google Scholar] [CrossRef]
- Idris, A.O.; Mabuba, N.; Arotiba, O.A. Electroanalysis of selenium in water on an electrodeposited gold-nanoparticle modified glassy carbon electrode. J. Electroanal. Chem. 2015, 758, 7–11. [Google Scholar] [CrossRef]
- Wei, H.; Pan, D.; Cui, Y.; Liu, H.; Gao, G.; Xia, J. Anodic Stripping Determination of Selenium in Seawater Using an Electrode Modified with Gold Nanodendrites/Perforated Reduced Graphene Oxide. Int. J. Electrochem. Sci. 2020, 15, 1669–1680. [Google Scholar] [CrossRef]
- Ramadan, A.A.; Mandil, H.; Shikh-Debes, A. Differential Pulse Anodic Stripping Voltammetric Analysis of Selenium (IV) at a Gold Electrode Modified with O-Phenylenediamine-Nafion. Res. J. Pharm. Tech. 2018, 11, 2030–2035. [Google Scholar] [CrossRef]
- Kolliopoulos, A.V.; Metters, J.P.; Banks, C.E. Electroanalytical sensing of selenium(IV) utilising screen printed graphite macro electrodes. Anal. Methods 2013, 5, 851–856. [Google Scholar] [CrossRef]
- Idris, A.O.; Mabuba, N.; Nkosi, D.; Maxakato, N.; Arotiba, O.A. Electrochemical detection of selenium using glassy carbon electrode modified with reduced graphene oxide. Int. J. Environ. Anal. Chem. 2017, 97, 534–547. [Google Scholar] [CrossRef]
- Sharifian, P.; Aliakbar, A. Determination of Se(IV) as a 5-nitropiazselenol complex by adsorptive stripping voltammetry at an in situ plated bismuth film electrode. Anal. Methods 2015, 7, 4321–4327. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, X.; Shi, H.; Yuan, Z. Determination of trace selenium by differential pulse adsorptive stripping voltammetry at a bismuth film electrode. Electrochim. Acta 2010, 55, 4717–4721. [Google Scholar] [CrossRef]
- Inam, R.; Somer, G. Adsorptive stripping voltammetry of selenium(IV) in the presence of thioglycolic acid. Anal. Sci. 1997, 13, 653–656. [Google Scholar] [CrossRef]
- Ashournia, M.; Aliakbar, A. Determination of selenium in natural waters by adsorptive differential pulse cathodic stripping voltammetry. J. Hazard. Mater. 2009, 168, 542–547. [Google Scholar] [CrossRef]
- Sharifian, P.; Aliakbar, A. Determination of Se(IV) by adsorptive cathodic stripping voltammetry at a Bi/Hg film electrode. Anal. Methods 2015, 7, 2121–2128. [Google Scholar] [CrossRef]
- Stozhko, N.Y.; Morosanova, E.I.; Kolyadina, L.I.; Fomina, S.V. Ceramic composite electrode for the determination of selenium(IV) by stripping voltammetry. J. Anal. Chem. 2006, 61, 158–165. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Hocevar, S.B.; Farias, P.A.M.; Ogorevc, B. Bismuth-coated carbon electrodes for anodic stripping voltammetry. Anal. Chem. 2000, 72, 3218. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, J.; Kirgöz, Ü.A.; Hocevar, S.B.; Ogorevc, B. Insights into the anodic stripping voltammetric behavior of bismuth film electrodes. Anal. Chim. Acta 2001, 434, 29–34. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Adamczyk, M. Bismuth film electrode and chloranilic acid as a new alternative for simple, fast and sensitive Ge(IV) quantification by adsorptive stripping voltammetry. RSC Adv. 2018, 8, 15215–15221. [Google Scholar] [CrossRef]
- Kawde, A.N. Electroanalytical determination of heavy metals in drinking waters in the eastern province of Saudi Arabia. Desalin. Water Treat. 2016, 57, 15697–15705. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Adamczyk, M. Simple, responsive and cost effective simultaneous quantification of Ga(III) and In(III) in environmental water samples. Int. Agrophys. 2019, 33, 161–166. [Google Scholar] [CrossRef]
- Baldrianova, L.; Svancara, I.; Vlcek, M.; Economou, A.; Sotiropoulos, S. Effect of Bi(III) concentration on the stripping voltammetric response of in situ bismuth-coated carbon paste and gold electrodes. Electrochim. Acta 2006, 52, 481–490. [Google Scholar] [CrossRef]
- Lou, Y.; Fu, D.; Fabre, B.; Fourcade, F.; Amrane, A.; Pasturel, M.; Bourzami, R.; Merdrignac-Conanec, O.; Labasque, T.; Geneste, F. Bismuth coated graphite felt modified by silver particles for selective electroreduction of CO2 into formate in a flow cell. Electrochim. Acta 2021, 371, 137821. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Hocevar, S.B.; Ogorevc, B. Bismuth-coated screen-printed electrodes for stripping voltammetric measurements of trace lead. Electroanalysis 2001, 13, 13–16. [Google Scholar] [CrossRef]
- Long, J.; Nagaosa, Y. Determination of Selenium(IV) by Catalytic Stripping Voltammetry with an in situ Plated Bismuth-film Electrode. Anal. Sci. 2007, 23, 1343–1346. [Google Scholar] [CrossRef] [PubMed][Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Working Electrode | Voltammetric Method | Detection Limit [nmol L−1] | Linear Range [nmol L−1] | Accumulation Time [s] | Analytical Application | Ref. |
|---|---|---|---|---|---|---|
| CCE-DAN | AdSV | 0.25 | 1.27–250 | 90 | natural and mineral water | [52] |
| GEMO-PN | ASV | 0.60 | 50–1000 | 200 | pharmaceuticals | [44] |
| BiFE | AdSV | 0.63 | 25–633 | 90 | - | [47] |
| BiFE | ASV | 0.80 | 3–3000 | 65 | SPS-SW1 and TM-25.5 certified reference materials, river, tap, mineral, and rain water | This work |
| AuµE and AuE | ASV | 0.85 | 5–100 | 300 | SPS-SW1 certified reference material | [37] |
| AuNDs/P-rGO | ASV | 0.90 | 3–300 | - | seawater and standard artificial seawater | [43] |
| BiFE | AdSV | 1.3 | 25–380 | 300 | - | [48] |
| GCE- AuNPs | ASV | 1.5 | 126–633 | 120 | seawater | [41] |
| RAgABE | CSV | 1.9 | 12.7–127 | 300 | SPS-SW1 and SPS-SW2 certified reference materials | [28] |
| AgE | CSV | 2.5 | 63–506 | 1800 | - | [27] |
| Au UME | ASV | 5.3 | 0–1266.5 | - | - | [40] |
| AuµE | ASV | 6.3 | 0–190 | 20 | - | [38] |
| GCE-AuNPs | ASV | 8.1 | - | 60 | real water samples | [42] |
| AuE | ASV | 9.9 | - | 10 | supplements | [33] |
| GCE-rGO | ASV | 10 | - | 240 | water samples | [46] |
| AuE | ASV | 60 | 200–1000 | 300 | river water | [36] |
| SPGE | ASV | 62 | 127–3165 | 300 | tap water | [45] |
| AuE | ASV | - | 6.3–3683.5 | 180 | - | [34] |
| CuµE | CSV | - | 5000–50,000 | 15 | - | [29] |
| Interfering Ions | Recovery of DPV Response of Selenium (%) | ||
|---|---|---|---|
| Interferent/Selenium Concentration | |||
| 1:1 | 5:1 | 10:1 | |
| Cd(II) | 73 | 39 | 27 |
| Cd(II) * | 100 | 100 | 100 |
| Co(II) | 78 | 48 | 34 |
| Co(II) * | 100 | 100 | 100 |
| Cr(VI) | 102 | 101 | 102 |
| Cu(II) | 17 | 10 | lack of signal |
| Cu(II) * | 100 | 44 | 10 |
| Fe(III) | 98 | 68 | 53 |
| Ga(III) | 100 | 82 | 78 |
| Ge(IV) | 102 | 101 | 100 |
| Hg(II) | 100 | 100 | 95 |
| Mn(II) | 100 | 100 | 98 |
| Mo(VI) | 101 | 69 | 52 |
| Ni(II) | 100 | 98 | 96 |
| Pb(II) | 67 | 38 | 22 |
| Pb(II) * | 100 | 100 | 100 |
| Pt(IV) | 103 | 104 | 104 |
| Sb(III) | 100 | 100 | 100 |
| Sn(II) | 100 | 100 | 99 |
| V(V) | 100 | 100 | 99 |
| Zn(II) | 75 | 54 | 38 |
| Zn(II) * | 100 | 100 | 100 |
| Humic Substances Added | Relative Signal of Se(IV) ± RSD [%] | ||
|---|---|---|---|
| Type of Substances | Concentration [mg L−1] | In the Absence of Resin | In the Presence of Resin |
| HA | 0.5 | 98.0 ± 3.3 | 99.9 ± 2.9 |
| 1 | 74.3 ± 5.0 | 97.7 ± 4.1 | |
| 2 | 50.9 ± 2.3 | 98.6 ± 4.5 | |
| 3 | 39.2 ± 3.8 | 108.5 ± 4.6 | |
| 5 | 35.0 ± 5.4 | 100.0 ± 4.0 | |
| 10 | 24.0 ± 4.3 | 98.7 ± 3.8 | |
| FA | 0.5 | 94.1 ± 3.9 | 100.9 ± 1.7 |
| 1 | 74.7 ± 5.1 | 108.3 ± 4.6 | |
| 2 | 66.6 ± 4.5 | 104.7 ± 3.9 | |
| 3 | 60.5 ± 3.6 | 106.0 ± 4.8 | |
| 5 | 52.0 ± 4.2 | 105.3 ± 3.6 | |
| 10 | 33.1 ± 4.5 | 99.6 ± 4.6 | |
| NOM | 0.5 | 88.9 ± 3.7 | 102.4 ± 2.5 |
| 1 | 79.7 ± 4.5 | 101.2 ± 4.7 | |
| 2 | 68.6 ± 5.3 | 104.7 ± 3.9 | |
| 3 | 58.8 ± 5.1 | 102.0 ± 5.3 | |
| 5 | 54.4 ± 3.8 | 97.8 ± 3.8 | |
| 10 | 49.5 ± 5.0 | 104.5 ± 5.6 | |
| Certified Reference Material | Measured Value ± SD (n = 3) [µg L−1] | Certified Value ± SD (n = 3) [µg L−1] |
|---|---|---|
| SPS SW-1 | 1.95 ± 0.11 | 2.00 ± 0.02 |
| TM-25.5 | 30.0 ± 4.1 | 29.2 ± 3.4 |
| Sample | Concentration of Added Se(IV) [nmol L−1] | Concentration of Found Se(IV) [nmol L−1] | Recovery [%] | RSD (n = 5) [%] |
|---|---|---|---|---|
| Bystrzyca riverwater | 50 | 54 | 108 | 6.1 |
| 100 | 109 | 109 | 5.2 | |
| Tapwater | 50 | 51 | 102 | 5.8 |
| 100 | 105 | 105 | 6.0 | |
| Mineralwater | 50 | 50 | 100 | 3.3 |
| 100 | 107 | 107 | 4.2 | |
| Rainwater | 50 | 51 | 102 | 3.5 |
| 100 | 102 | 102 | 4.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Adamczyk, M. New Strategies for the Simple and Sensitive Voltammetric Direct Quantification of Se(IV) in Environmental Waters Employing Bismuth Film Modified Glassy Carbon Electrode and Amberlite Resin. Molecules 2021, 26, 4130. https://doi.org/10.3390/molecules26144130
Grabarczyk M, Adamczyk M. New Strategies for the Simple and Sensitive Voltammetric Direct Quantification of Se(IV) in Environmental Waters Employing Bismuth Film Modified Glassy Carbon Electrode and Amberlite Resin. Molecules. 2021; 26(14):4130. https://doi.org/10.3390/molecules26144130
Chicago/Turabian StyleGrabarczyk, Małgorzata, and Marzena Adamczyk. 2021. "New Strategies for the Simple and Sensitive Voltammetric Direct Quantification of Se(IV) in Environmental Waters Employing Bismuth Film Modified Glassy Carbon Electrode and Amberlite Resin" Molecules 26, no. 14: 4130. https://doi.org/10.3390/molecules26144130
APA StyleGrabarczyk, M., & Adamczyk, M. (2021). New Strategies for the Simple and Sensitive Voltammetric Direct Quantification of Se(IV) in Environmental Waters Employing Bismuth Film Modified Glassy Carbon Electrode and Amberlite Resin. Molecules, 26(14), 4130. https://doi.org/10.3390/molecules26144130