
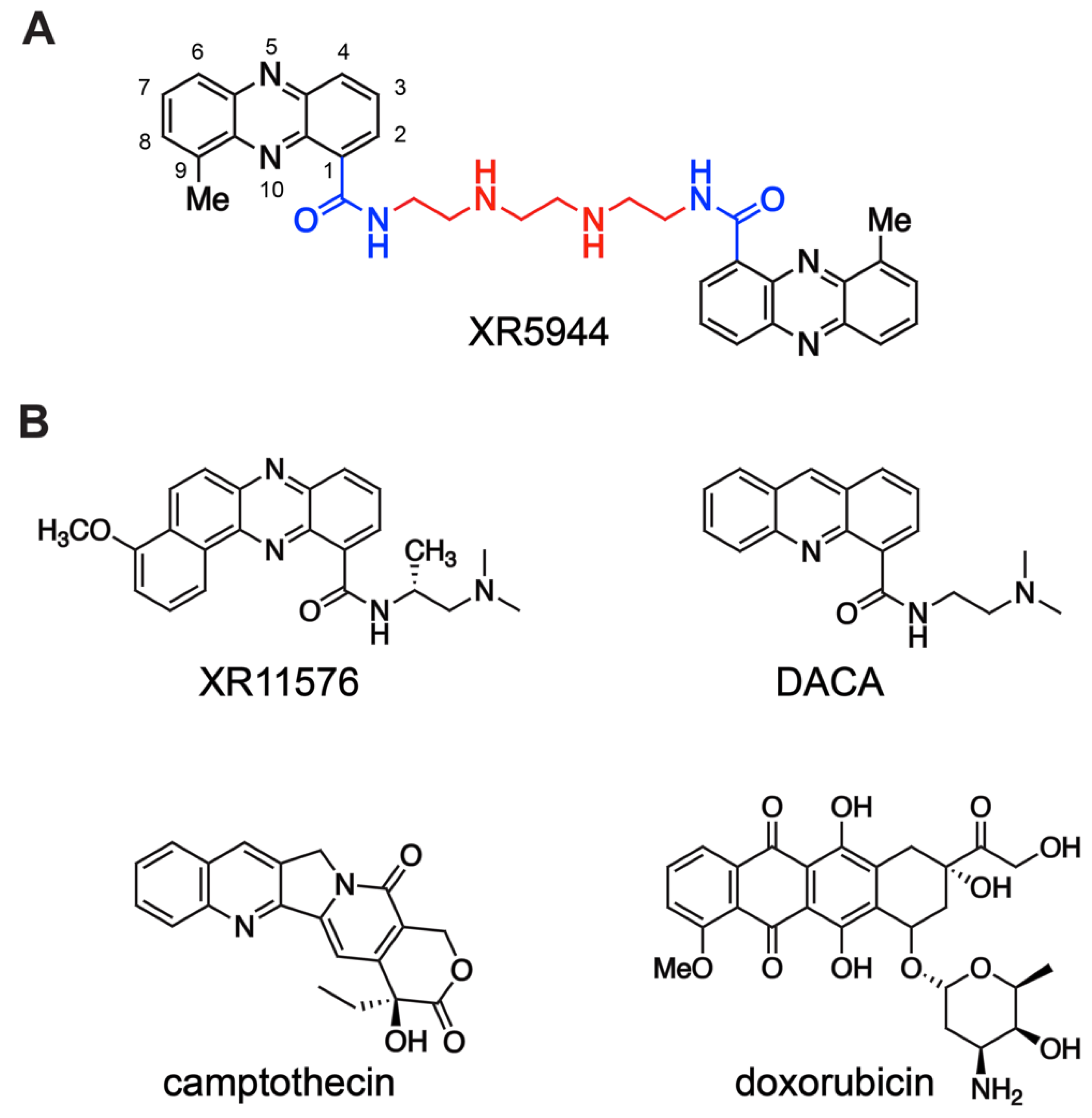
Novel DNA Bis-Intercalator XR5944 as a Potent Anticancer Drug—Design and Mechanism of Action


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
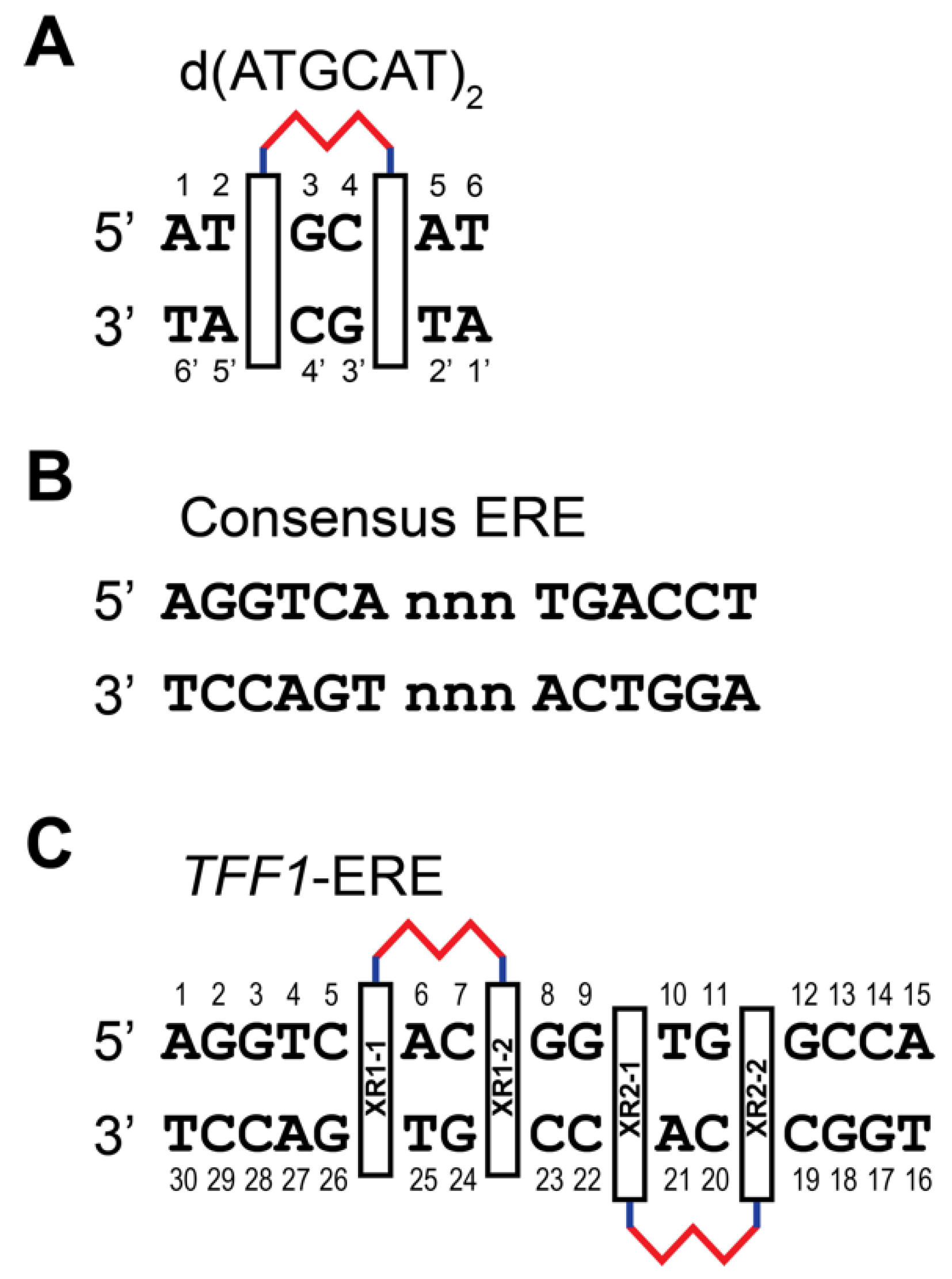
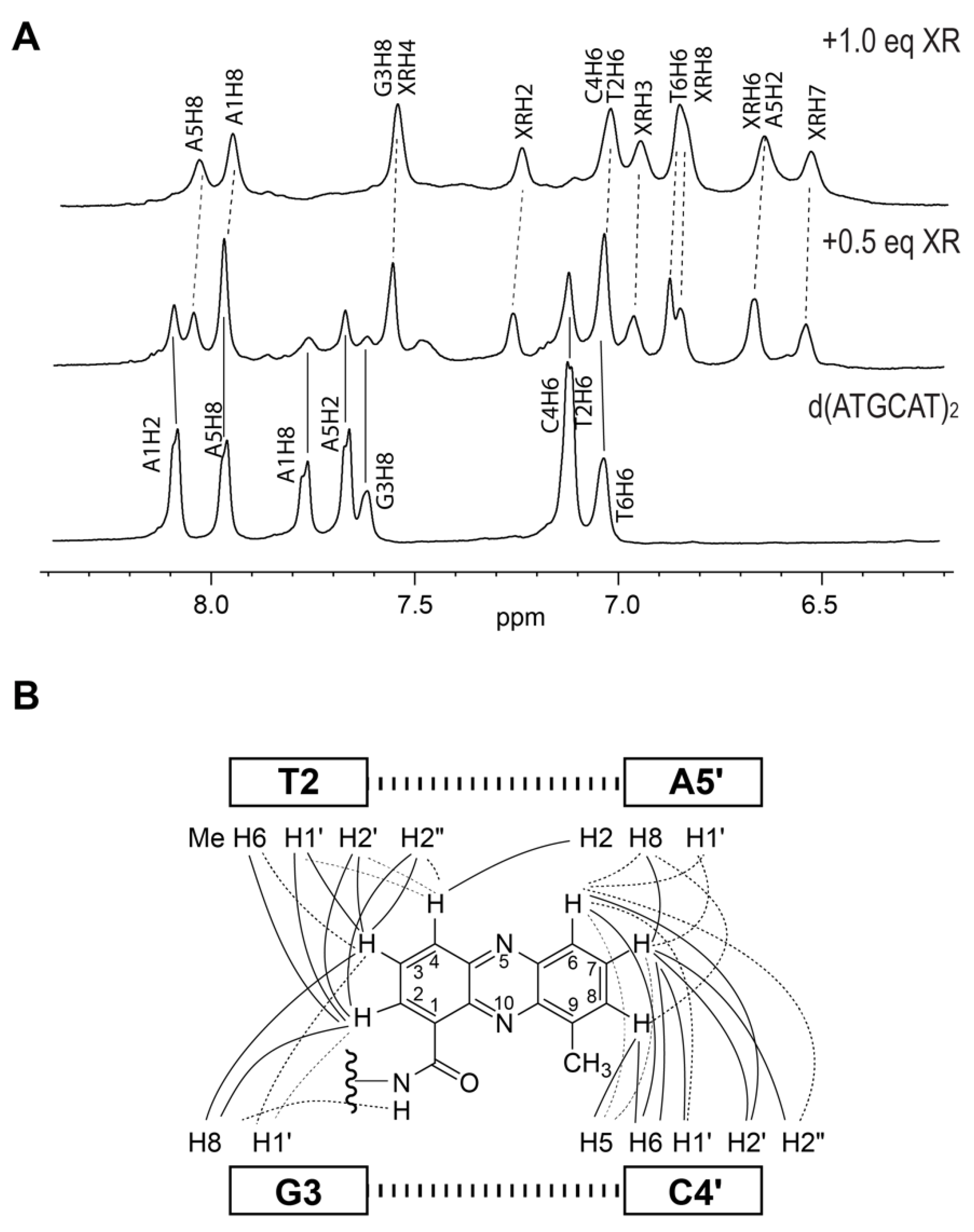
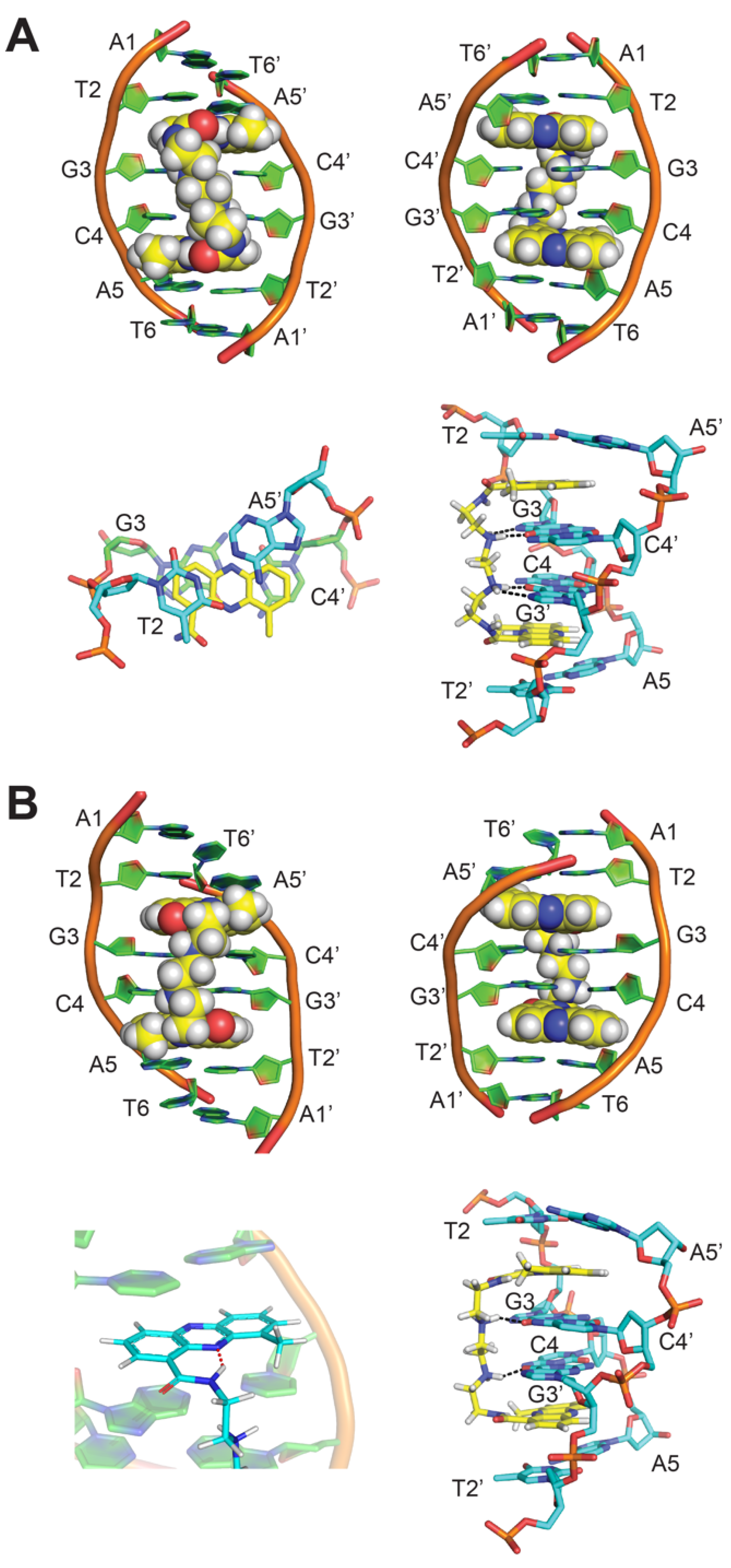
2. NMR Solution Structure of XR5944 in Complex with the d(ATGCAT)2 Duplex DNA
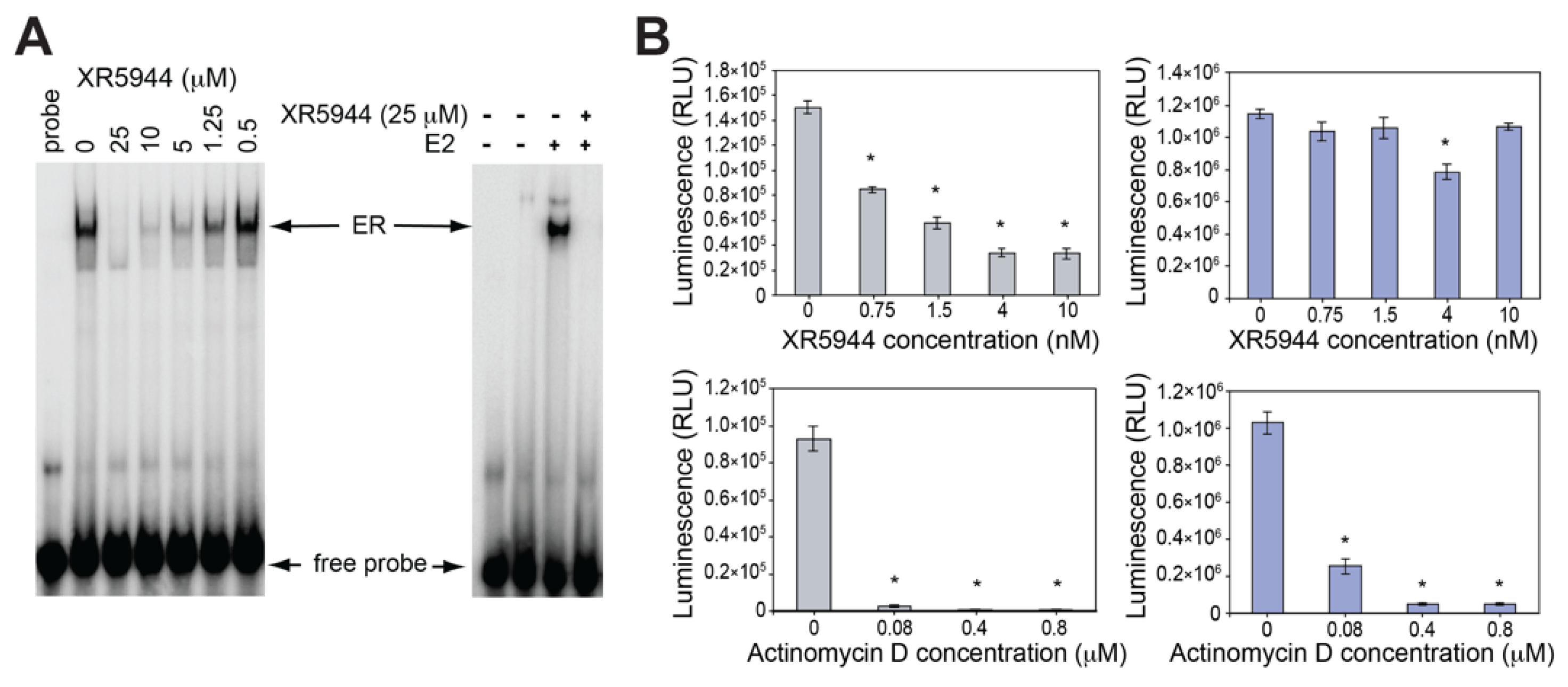
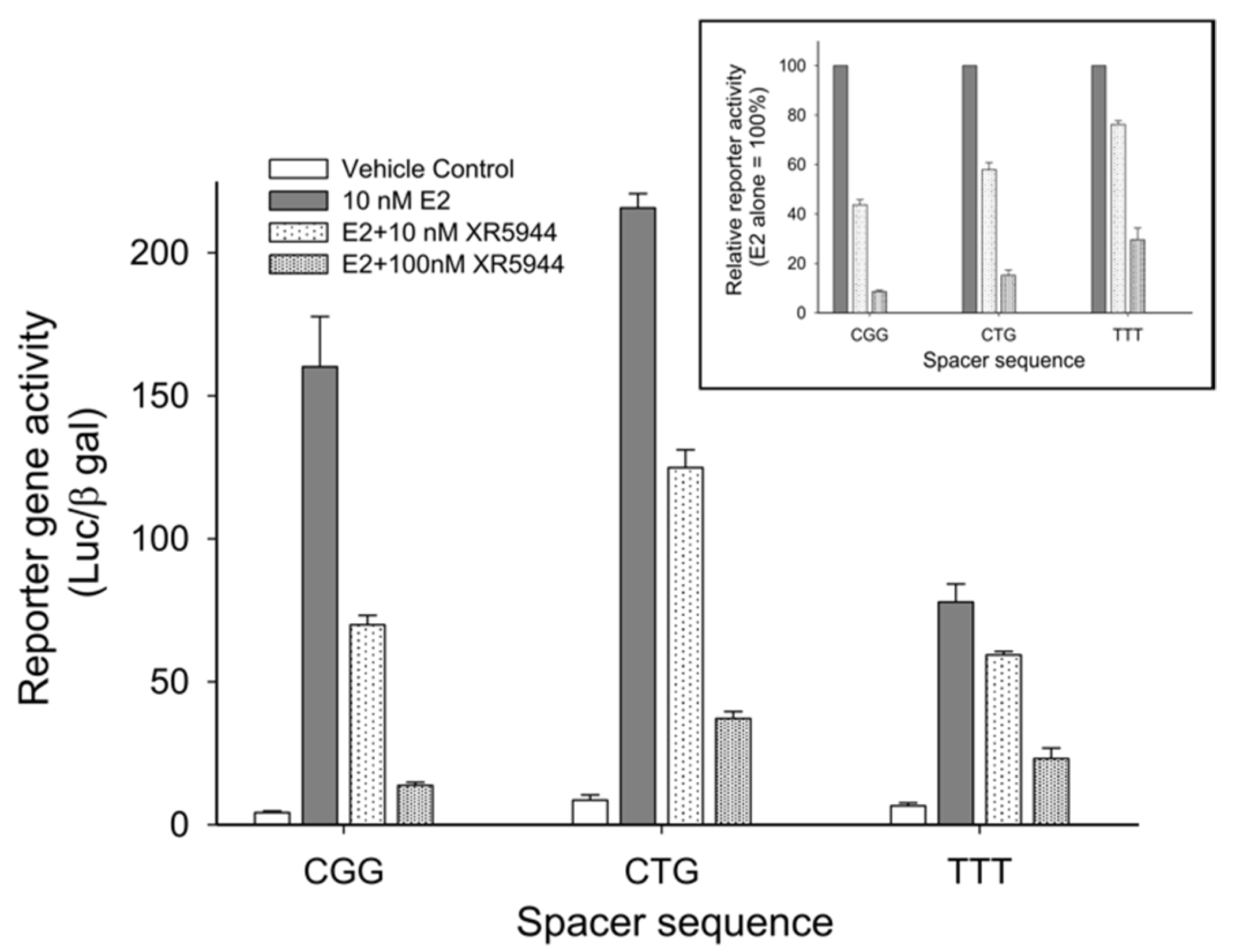
3. XR5944 Binds the ERE Sequence and Inhibits ERα-ERE Interaction
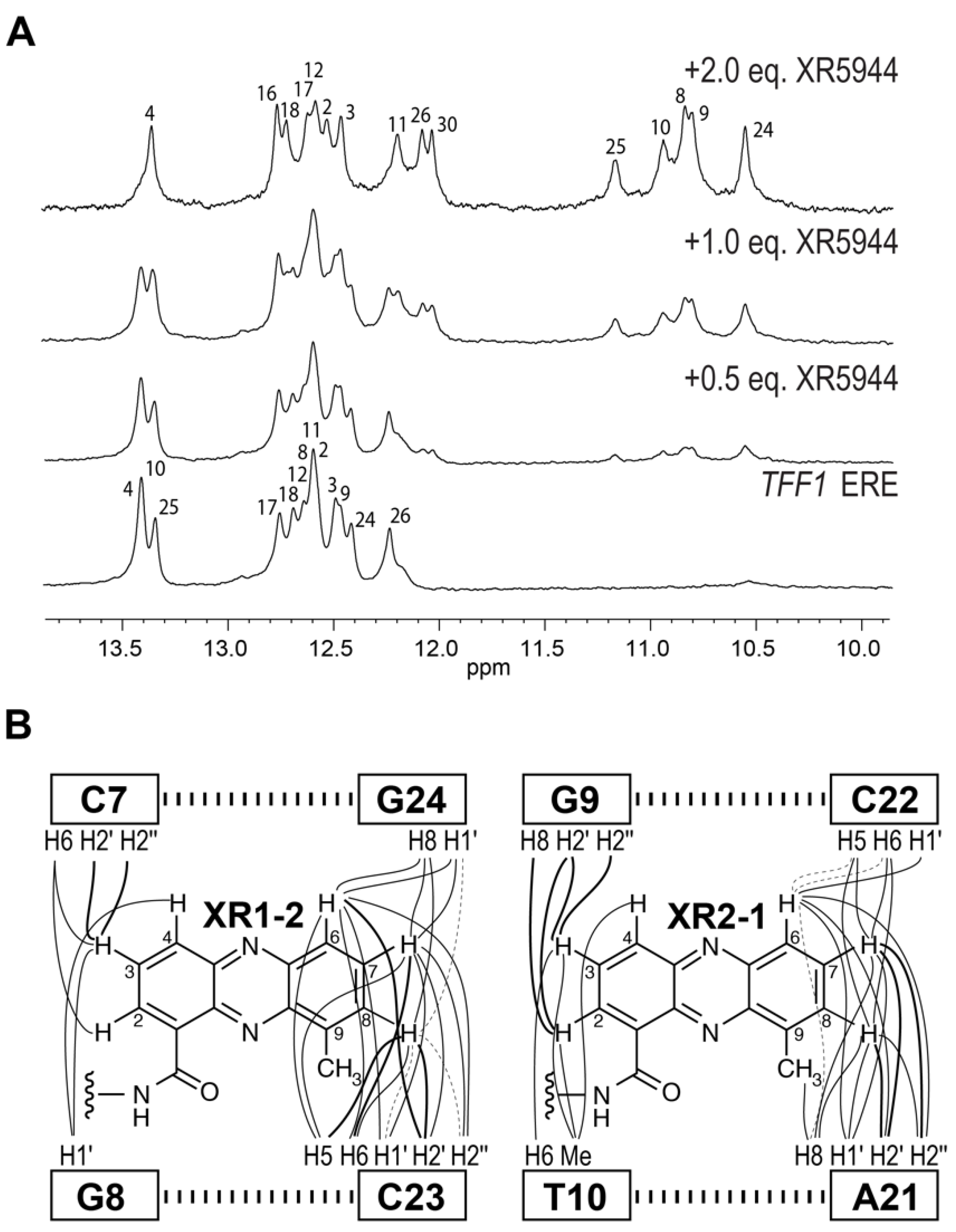
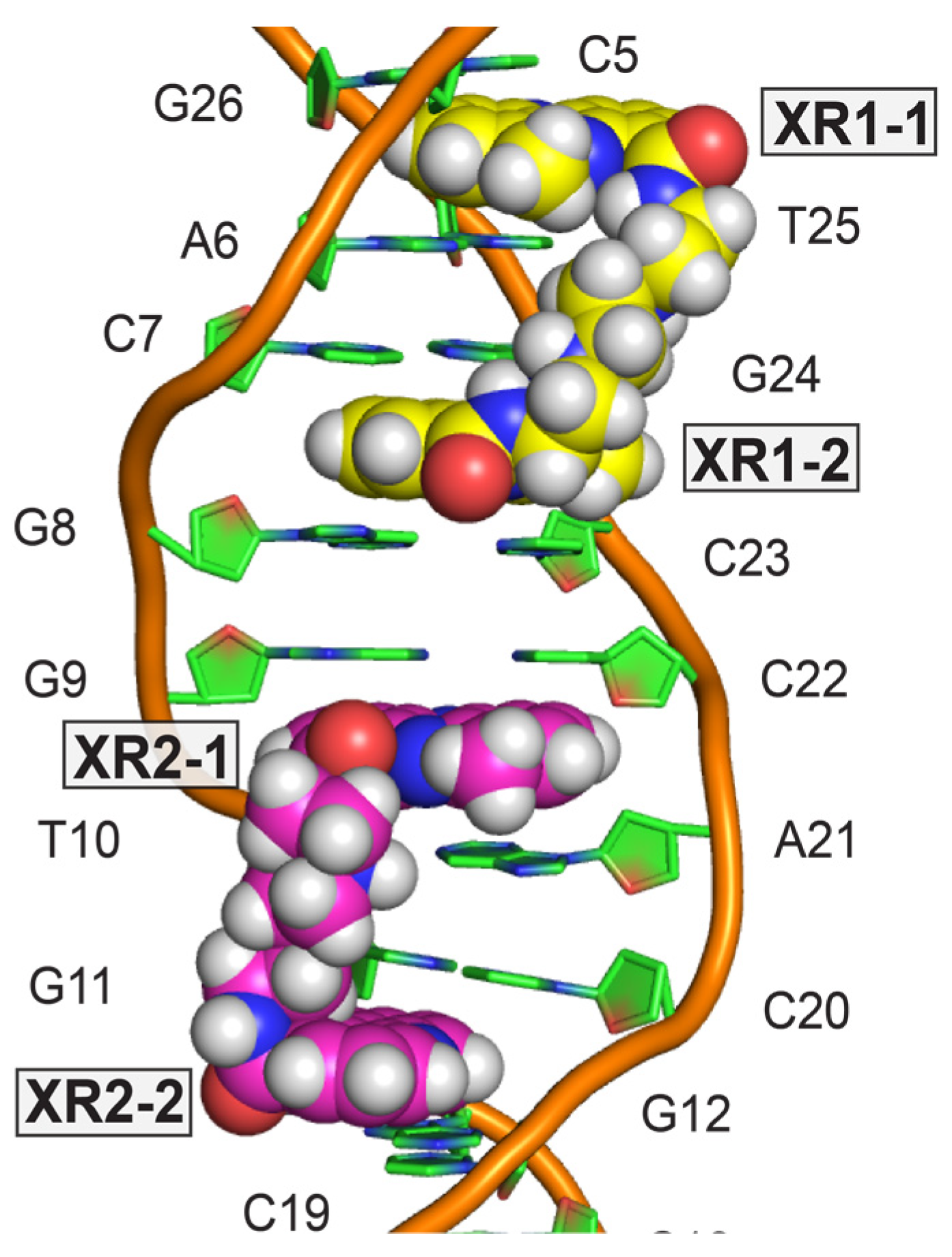
4. Solution Structure of 2:1 Complex of XR5944 with the Naturally Occurring ERE Sequence of TFF1 Gene
5. Disrupting ERE-ERα Complex Formation: A Potential Alternative to Antiestrogen Therapeutics
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cuya, S.M.; Bjornsti, M.-A.; van Waardenburg, R.C.A.M. DNA Topoisomerase-Targeting Chemotherapeutics: What’s New? Cancer Chemother. Pharmacol. 2017, 80, 1–14. [Google Scholar] [CrossRef]
- Portugal, J. Challenging Transcription by DNA-Binding Antitumor Drugs. Biochem. Pharmacol. 2018, 155, 336–345. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA Bis-Intercalation by MLN944, a Potent Clinical Bisphenazine Anticancer Drug. J. Biol. Chem. 2004, 279, 46096–46103. [Google Scholar] [CrossRef]
- Byers, S.A.; Schafer, B.; Sappal, D.S.; Brown, J.; Price, D.H. The Antiproliferative Agent MLN944 Preferentially Inhibits Transcription. Mol. Cancer Ther. 2005, 4, 1260–1267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Verborg, W.; Thomas, H.; Bissett, D.; Waterfall, J.; Steiner, J.; Cooper, M.; Rankin, E.M. First-into-Man Phase I and Pharmacokinetic Study of XR5944.14, a Novel Agent with a Unique Mechanism of Action. Br. J. Cancer 2007, 97, 844–850. [Google Scholar] [CrossRef][Green Version]
- Stewart, A.J.; Mistry, P.; Dangerfield, W.; Bootle, D.; Baker, M.; Kofler, B.; Okiji, S.; Baguley, B.C.; Denny, W.A.; Charlton, P.A. Antitumor Activity of XR5944, a Novel and Potent Topoisomerase Poison. Anti-Cancer Drugs 2001, 12, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.A.; Spicer, J.A.; Finlay, G.J.; Stewart, A.J.; Charlton, P.; Baguley, B.C.; Denny, W.A. Dicationic Bis(9-Methylphenazine-1-Carboxamides): Relationships between Biological Activity and Linker Chain Structure for a Series of Potent Topoisomerase Targeted Anticancer Drugs. J. Med. Chem. 2001, 44, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Knight, L.A.; Whitehouse, P.A.; Mercer, S.J.; Sharma, S.; Charlton, P.A.; Norris, D.; Cree, I.A. The Ex Vivo Characterization of XR5944 (MLN944) against a Panel of Human Clinical Tumor Samples. Mol. Cancer Ther. 2004, 3, 1631–1637. [Google Scholar] [PubMed]
- Harris, S.M.; Mistry, P.; Freathy, C.; Brown, J.L.; Charlton, P.A. Antitumour Activity of XR5944 in Vitro and in Vivo in Combination with 5-Fluorouracil and Irinotecan in Colon Cancer Cell Lines. Br. J. Cancer 2005, 92, 722–728. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harris, S.M.; Scott, J.A.; Brown, J.L.; Charlton, P.A.; Mistry, P. Preclinical Anti-Tumor Activity of XR5944 in Combination with Carboplatin or Doxorubicin in Non-Small-Cell Lung Carcinoma. Anti-Cancer Drugs 2005, 16, 945–951. [Google Scholar] [CrossRef]
- Millennium and Xenova Initiate Phase I Clinical Trial Of MLN944. Available online: https://www.takedaoncology.com/media/news-media/news-releases/Millennium-and-Xenova-Initiate-Phase-I-Clinical-Trial-Of-MLN-7341/Print (accessed on 17 June 2021).
- Atwell, G.J.; Cain, B.F.; Baguley, B.C.; Finlay, G.J.; Denny, W.A. Potential Antitumor Agents. Part 43. Synthesis and Biological Activity of Dibasic 9-Aminoacridine-4-Carboxamides, a New Class of Antitumor Agent. J. Med. Chem. 1984, 27, 1481–1485. [Google Scholar] [CrossRef]
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Potential Antitumor Agents. 50. In Vivo Solid-Tumor Activity of Derivatives of N-[2-(Dimethylamino)Ethyl]Acridine-4-Carboxamide. J. Med. Chem. 1987, 30, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Rewcastle, G.W.; Denny, W.A.; Baguley, B.C. Potential Antitumor Agents. 51. Synthesis and Antitumor Activity of Substituted Phenazine-1-Carboxamides. J. Med. Chem. 1987, 30, 843–851. [Google Scholar] [CrossRef]
- Palmer, B.D.; Rewcastle, G.W.; Atwell, G.J.; Baguley, B.C.; Denny, W.A. Potential Antitumor Agents. 54. Chromophore Requirements for in Vivo Antitumor Activity among the General Class of Linear Tricyclic Carboxamides. J. Med. Chem. 1988, 31, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.A.; Gamage, S.A.; Atwell, G.J.; Finlay, G.J.; Baguley, B.C.; Denny, W.A. Dimeric Analogues of Non-Cationic Tricyclic Aromatic Carboxamides Are a New Class of Cytotoxic Agents. Anticancer Drug Des. 1999, 14, 281–289. [Google Scholar]
- Spicer, J.A.; Gamage, S.A.; Rewcastle, G.W.; Finlay, G.J.; Bridewell, D.J.A.; Baguley, B.C.; Denny, W.A. Bis(Phenazine-1-Carboxamides): Structure−Activity Relationships for a New Class of Dual Topoisomerase I/II-Directed Anticancer Drugs. J. Med. Chem. 2000, 43, 1350–1358. [Google Scholar] [CrossRef]
- Wang, S.; Miller, W.; Milton, J.; Vicker, N.; Stewart, A.; Charlton, P.; Mistry, P.; Hardick, D.; Denny, W.A. Structure–Activity Relationships for Analogues of the Phenazine-Based Dual Topoisomerase I/II Inhibitor XR11576. Bioorganic Med. Chem. Lett. 2002, 12, 415–418. [Google Scholar] [CrossRef]
- Finlay, G.J.; Riou, J.-F.; Baguley, B.C. From Amsacrine to DACA (N-[2-(Dimethylamino)Ethyl]Acridine-4-Carboxamide): Selectivity for Topoisomerases I and II among Acridine Derivatives. Eur. J. Cancer 1996, 32, 708–714. [Google Scholar] [CrossRef]
- Sappal, D.S.; McClendon, A.K.; Fleming, J.A.; Thoroddsen, V.; Connolly, K.; Reimer, C.; Blackman, R.K.; Bulawa, C.E.; Osheroff, N.; Charlton, P.; et al. Biological Characterization of MLN944: A Potent DNA Binding Agent. Mol. Cancer Ther. 2004, 3, 47–58. [Google Scholar] [PubMed]
- Serobian, A.; Thomas, D.S.; Ball, G.E.; Denny, W.A.; Wakelin, L.P.G. The Solution Structure of Bis(Phenazine-1-Carboxamide)-DNA Complexes: MLN 944 Binding Corrected and Extended: MLN 944 Binding Corrected and Extended. Biopolymers 2014, 101, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.; Guss, J.M.; Collyer, C.A.; Denny, W.A.; Wakelin, L.P. A Novel Form of Intercalation Involving Four DNA Duplexes in an Acridine-4-Carboxamide Complex of d(CGTACG)(2). Nucleic Acids Res. 2000, 28, 4244–4253. [Google Scholar] [CrossRef][Green Version]
- Todd, A.K.; Adams, A.; Thorpe, J.H.; Denny, W.A.; Wakelin, L.P.G.; Cardin, C.J. Major Groove Binding and ‘DNA-Induced’ Fit in the Intercalation of a Derivative of the Mixed Topoisomerase I/II Poison N -(2-(Dimethylamino)Ethyl)Acridine-4- Carboxamide (DACA) into DNA: X-Ray Structure Complexed to d(CG(5-BrU)ACG) 2 at 1.3-Å Resolution. J. Med. Chem. 1999, 42, 536–540. [Google Scholar] [CrossRef]
- Bhaduri, S.; Ranjan, N.; Arya, D.P. An Overview of Recent Advances in Duplex DNA Recognition by Small Molecules. Beilstein J. Org. Chem. 2018, 14, 1051–1086. [Google Scholar] [CrossRef]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of Specificity in Protein-DNA Recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef]
- MacGregor, J.I.; Jordan, V.C. Basic Guide to the Mechanisms of Antiestrogen Action. Pharmacol. Rev. 1998, 50, 151–196. [Google Scholar]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. J. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Punchihewa, C.; De Alba, A.; Sidell, N.; Yang, D. XR5944: A Potent Inhibitor of Estrogen Receptors. Mol. Cancer. Ther. 2007, 6, 213–219. [Google Scholar] [CrossRef]
- Sidell, N.; Mathad, R.I.; Shu, F.; Zhang, Z.; Kallen, C.B.; Yang, D. Intercalation of XR5944 with the Estrogen Response Element Is Modulated by the Tri-Nucleotide Spacer Sequence between Half-Sites. J. Steroid Biochem. Mol. Biol. 2011, 124, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Shu, F.; Sidell, N.; Yang, D.; Kallen, C.B. The Tri-Nucleotide Spacer Sequence between Estrogen Response Element Half-Sites Is Conserved and Modulates ERα-Mediated Transcriptional Responses. J. Steroid Biochem. Mol. Biol. 2010, 120, 172–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, C.; Mathad, R.I.; Zhang, Z.; Sidell, N.; Yang, D. Solution Structure of a 2:1 Complex of Anticancer Drug XR5944 with TFF1 Estrogen Response Element: Insights into DNA Recognition by a Bis-Intercalator. Nucleic Acids Res. 2014, 42, 6012–6024. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.M. Prognostic and Predictive Factors for Breast Cancer. Breast Cancer 1995, 2, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for Estrogen Receptor Expression in Human Cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. The Oncol. 2018, 23, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Bouker, K.B.; Wang, Y.; Xuan, J.; Clarke, R. Antiestrogen Resistance and the Application of Systems Biology. Drug Discov. Today Dis. Mech. 2012, 9, e11–e17. [Google Scholar] [CrossRef][Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buric, A.J.; Dickerhoff, J.; Yang, D. Novel DNA Bis-Intercalator XR5944 as a Potent Anticancer Drug—Design and Mechanism of Action. Molecules 2021, 26, 4132. https://doi.org/10.3390/molecules26144132
Buric AJ, Dickerhoff J, Yang D. Novel DNA Bis-Intercalator XR5944 as a Potent Anticancer Drug—Design and Mechanism of Action. Molecules. 2021; 26(14):4132. https://doi.org/10.3390/molecules26144132
Chicago/Turabian StyleBuric, Adam J., Jonathan Dickerhoff, and Danzhou Yang. 2021. "Novel DNA Bis-Intercalator XR5944 as a Potent Anticancer Drug—Design and Mechanism of Action" Molecules 26, no. 14: 4132. https://doi.org/10.3390/molecules26144132
APA StyleBuric, A. J., Dickerhoff, J., & Yang, D. (2021). Novel DNA Bis-Intercalator XR5944 as a Potent Anticancer Drug—Design and Mechanism of Action. Molecules, 26(14), 4132. https://doi.org/10.3390/molecules26144132