
The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
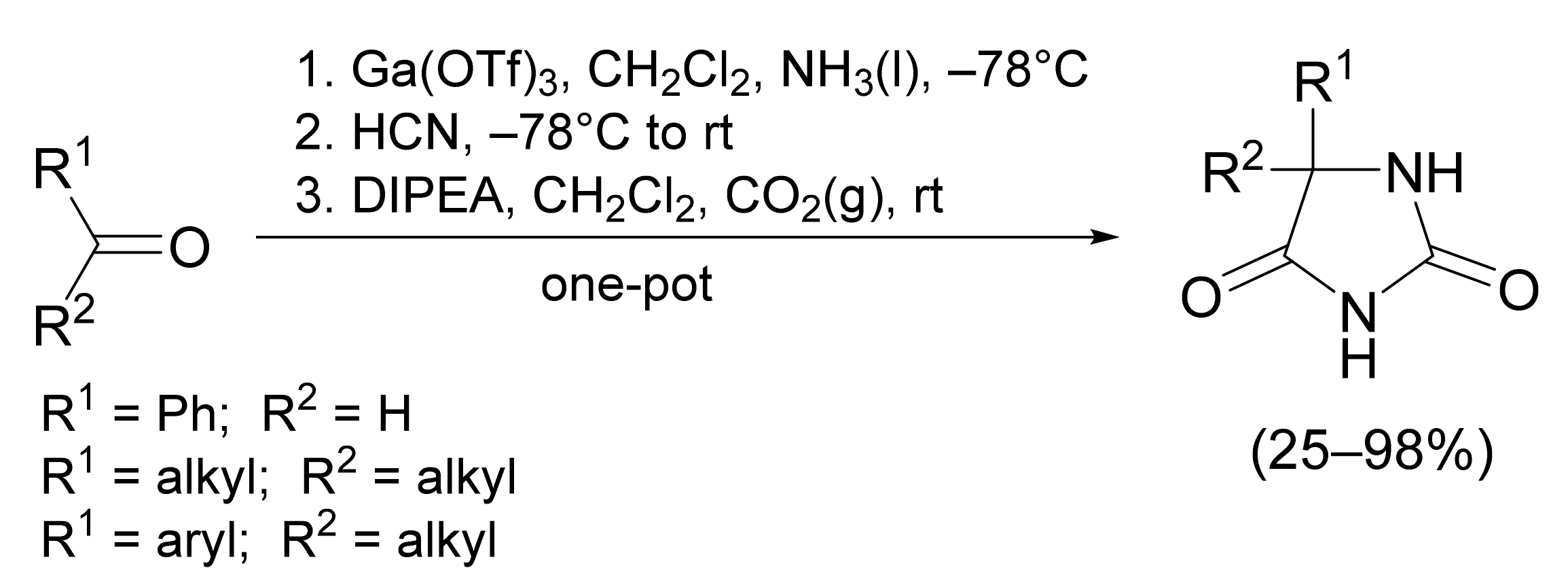{kind=link}
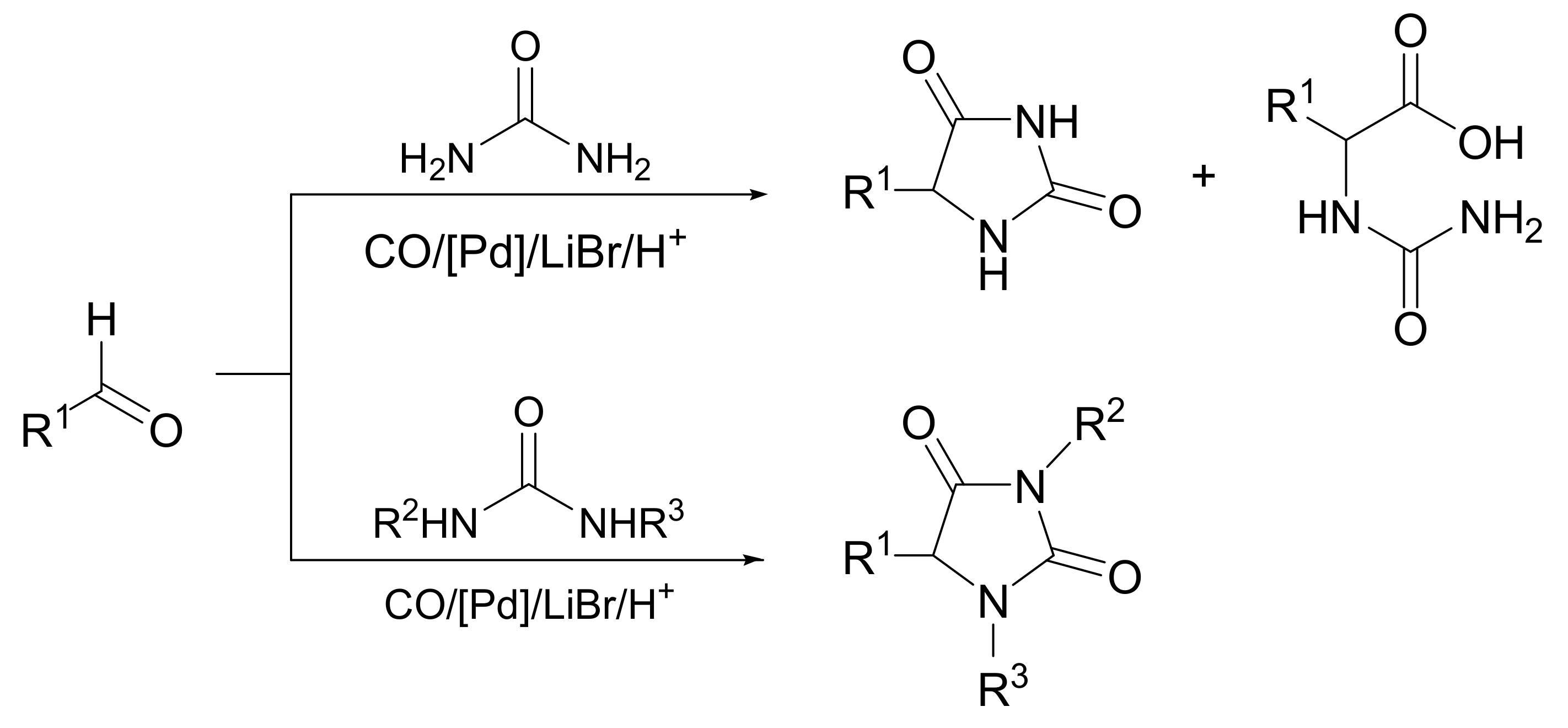{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
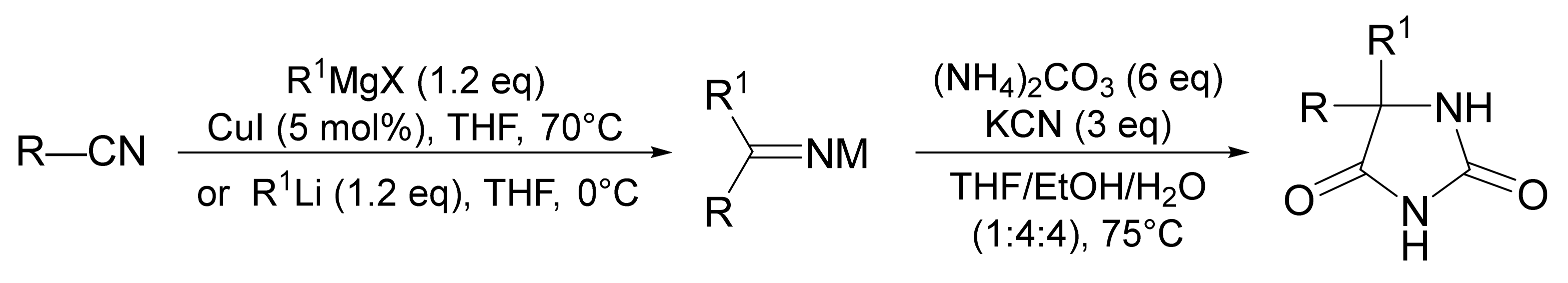{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanism and Stereochemistry
3. Scope and Limitations
4. Application to Synthesis
4.1. Overview
4.2. Applications in the Synthesis of Natural Products and Biologically Active Compounds
4.2.1. Sorbinil
4.2.2. Phenytoin
4.2.3. Aplysinopsins
4.2.4. Hydantocidin
5. Comparison with Other Methods Affording Hydantoins
6. Experimental Conditions
6.1. General Comments
6.2. Note
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergs, H. Verfahren zur Darstellung von Hydantoinen. German Patent DE566094, 14 December 1932. [Google Scholar]
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1905, 38, 1671–1675. [Google Scholar] [CrossRef]
- Bucherer, H.T.; Steiner, W. Syntheses of hydantoins. I. On reactions of α-hydroxy and α-amino nitriles. J. Prakt. Chem. 1934, 140, 291–316. [Google Scholar]
- Bucherer, H.T.; Fischbeck, H.T. Hexahydrodiphenylamine and its derivatives. J. Prakt. Chem. 1934, 140, 69–89. [Google Scholar]
- Bucherer, H.T.; Lieb, V.A. Syntheses of hydantoins. II. Formation of substituted hydantoins from aldehydes and ketones. J. Prakt. Chem. 1934, 141, 5–43. [Google Scholar] [CrossRef]
- Henze, H.R.; Long, L.M. Researches on phenylhydantoins. J. Am. Chem. Soc. 1941, 63, 1936–1938. [Google Scholar] [CrossRef]
- Henze, H.R.; Long, L.M. 5-(4-Biphenylyl)-5-R-hydantoins and bis-5-[(4-phenyl)-5-R-hydantoin]s. J. Am. Chem. Soc. 1941, 63, 1941–1943. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Li, T.; Li, H.; Liu, J. An efficient and convenient procedure for the synthesis of 5,5-disubstituted hydantoins under ultrasound. Ultrason. Sonochem. 1996, 3, S141–S143. [Google Scholar] [CrossRef]
- Hoyer, H.L. Über das camphan-2-spiro-hydantoin. Chem. Ber. 1950, 83, 491–500. [Google Scholar] [CrossRef]
- Henze, H.R.; Speer, R.J. Identification of carbonyl compounds through conversion into hydantoins. J. Am. Chem. Soc. 1942, 64, 522–523. [Google Scholar] [CrossRef]
- Oh, C.-H.; Kim, H.J.; Hong, S.-Y.; Lee, Y.-H.; Cho, J.K.; Cho, J.-H. New 1β-methylcarbapenems having a hydantoin moiety. Neue 1β-methylcarbapeneme mit hydantoin-substitution. Arch. Pharm. 1995, 328, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Marchand-Brynaert, J.; Arnadei, E.; Ghosez, L. Functionalized hydantoins as potential antibiotics. Bull. Soc. Chim. Belg. 1994, 103, 213–218. [Google Scholar] [CrossRef]
- Oliveira, S.M.; Silva, J.B.P.; Hernandes, M.Z.; Lima, M.C.A.; Galdino, S.L.; Pitta, I.R. Structure, reactivity, and biological properties of hidantoines. Quim. Nova 2008, 31, 614–622. [Google Scholar] [CrossRef]
- Ali, O.M.; Amer, H.H.; Mosaad, A.A.; Abdel-Rahman, A.A.-H. Synthesis and antimicrobial activity of new phenytoin derivatives and their acyclic nucleoside analogs. Chem. Heterocycl. Compd. 2012, 48, 1043–1049. [Google Scholar] [CrossRef]
- Ali, O.M.; El-Sayed, W.A.; Eid, S.A.; Abdelwahed, N.A.M.; Abdel-Rahman, A.A.-H. Antimicrobial activity of new synthesized [(oxadiazolyl)methyl]phenytoin derivatives. Acta Polon. Pharm. 2012, 69, 657–667. [Google Scholar]
- Kim, D.; Wang, L.; Caldwell, C.G.; Chen, P.; Finke, P.E.; Oates, B.; MacCoss, M.; Mills, S.G.; Malkowitz, L.; Gould, S.L.; et al. Discovery of human CCR5 antagonists containing hydantoins for the treatment of HIV-1 infection. Bioorg. Med. Chem. Lett. 2001, 11, 3099–3102. [Google Scholar] [CrossRef]
- Verlinden, Y.; Cuconati, A.; Wimmer, E.; Rombaut, B. The antiviral compound 5-(3,4-dichlorophenyl) methylhydantoin inhibits the post-synthetic cleavages and the assembly of poliovirus in a cell-free system. Antivir. Res. 2000, 48, 61–69. [Google Scholar] [CrossRef]
- El-Barbary, A.A.; Khodair, A.I.; Pedersen, E.B.; Nielsen, C. S-Glucosylated hydantoins as new antiviral agents. J. Med. Chem. 1994, 37, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J. The role of antiandrogen monotherapy in the treatment of prostate cancer. BJU Int. 2003, 91, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kassouf, W.; Tanguay, S.; Aprikian, A.G. Nilutamide as second line hormone therapy for prostate cancer after androgen ablation fails. J. Urol. 2003, 169, 1742–1744. [Google Scholar] [CrossRef]
- Struck, R.F.; Kirk, M.C.; Rice, L.S.; Suling, W.J. Isolation, synthesis and antitumor evaluation of spirohydantoin aziridine, a mutagenic metabolite of spirohydantoin mustard. J. Med. Chem. 1986, 29, 1319–1321. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Regan, M.M.; Lifsey, D.; Kantoff, P.W.; Taplin, M.-E.; Sartor, O.; Oh, W.K. Efficacy of nilutamide as secondary hormonal therapy in androgen-independent prostate cancer. BJU Int. 2005, 96, 783–786. [Google Scholar] [CrossRef]
- Ciechanowicz-Rutkowska, M.; Stadnicka, K.; Kiec-Kononowicz, K.; Byrtus, H.; Filipek, B.; Zygmunt, M.; Maciag, D. Structure-activity relationship of some new anti-arrhythmic phenytoin derivatives. Arch. Pharm. 2000, 333, 357–364. [Google Scholar] [CrossRef]
- Kieć-Kononowicz, K.; Stadnicka, K.; Mitka, A.; Pekala, E.; Filipek, B.; Sapa, J.; Zygmunt, M. Synthesis, structure and antiarrhythmic properties evaluation of new basic derivatives of 5,5-diphenylhydantoin. Eur. J. Med. Chem. 2003, 38, 555–566. [Google Scholar] [CrossRef]
- Knabe, J.; Baldauf, J.; Ahlhem, A. Racemates and enantiomers of basic substituted 5-phenylhydantoins. Syntheses and antiarrhythmic activity. (Razemate und enantiomere basisch substituierter 5-phenylhydantoine, synthese und antiarrhythmische wirkung). Pharmazie 1997, 52, 912–919. [Google Scholar]
- Matsukura, M.; Daiku, Y.; Ueda, K.; Tanaka, S.; Igarashi, T.; Minami, N. Synthesis and antiarrhythmic activity of 2,2-dialkyl-1′-(N-substituted aminoalkyl)-spiro-[chroman-4,4′-imidazolidine]-2′,5′-diones. Chem. Pharm. Bull. 1992, 40, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Thenmozhiyal, J.C.; Wong, P.T.-H.; Chui, W.-K. Anticonvulsant activity of phenylmethylenehydantoins: A structure–activity relationship study. J. Med. Chem. 2004, 47, 1527–1535. [Google Scholar] [CrossRef]
- LeTiran, J.; Stables, J.P.; Kohn, H. Functionalized amino acid anticonvulsants: Synthesis and pharmacological evaluation of conformationally restricted analogues. Bioorg. Med. Chem. 2001, 9, 2693–2708. [Google Scholar] [CrossRef]
- Anger, T.; Madge, D.J.; Mulla, M.; Riddall, D. Medicinal chemistry of neuronal voltage-gated sodium channel blockers. J. Med. Chem. 2001, 44, 115–137. [Google Scholar] [CrossRef]
- Scholl, S.; Koch, A.; Henning, D.; Kempter, G.; Kleinpeter, E. The influence of structure and lipophilicity of hydantoin derivatives on anticonvulsant activity. Struct. Chem. 1999, 10, 355–366. [Google Scholar] [CrossRef]
- Brouillette, W.J.; Jestkov, V.P.; Brown, M.L.; Akhtar, M.S.; DeLorey, T.M.; Brown, G.B. Bicyclic hydantoins with a bridgehead nitrogen. Comparison of anticonvulsant activities with binding to the neuronal voltage-dependent sodium channel. J. Med. Chem. 1994, 37, 3289–3293. [Google Scholar] [CrossRef]
- Kwon, C.H.; Iqbal, M.T.; Wurpel, J.N.D. Synthesis and anticonvulsant activity of 2-iminohydantoins. J. Med. Chem. 1991, 34, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Botros, S.; Khalil, N.A.; Naguib, B.H.; El-Dash, Y. Synthesis and anticonvulsant activity of new phenytoin derivatives. Eur. J. Med. Chem. 2013, 60, 57–63. [Google Scholar] [CrossRef]
- Deodhar, M.; Sable, P.; Bhosale, A.; Juvale, K.; Dumbare, R.; Sakpal, P. Synthesis and evaluation of phenytoin derivatives as anticonvulsant agents. Turk. J. Chem. 2009, 33, 367–373. [Google Scholar]
- Edmunds, J.J.; Klutchko, S.; Hamby, J.M.; Bunker, A.M.; Connolly, C.J.C.; Winters, R.T.; Quin III, J.; Sircar, I.; Hodges, J.C.; Panek, R.L.; et al. Derivatives of 5-[[1-4(4-carboxybenzyl)imidazolyl]methylidene]hydantoins as orally active angiotensin II receptor antagonists. J. Med. Chem. 1995, 38, 3759–3771. [Google Scholar] [CrossRef]
- Somsák, L.; Kovács, L.; Tóth, M.; Ösz, E.; Szilágyi, L.; Györgydeák, Z.; Dinya, Z.; Docsa, T.; Tóth, B.; Gergely, P. Synthesis of and a comparative study on the inhibition of muscle and liver glycogen phosphorylases by epimeric pairs of D-gluco- and D-xylopyranosylidene-spiro-(thio)hydantoins and N-(D-glucopyranosyl) amides. J. Med. Chem. 2001, 44, 2843–2848. [Google Scholar] [CrossRef]
- Oka, M.; Matsumoto, Y.; Sugiyama, S.; Tsuruta, N.; Matsushima, M. A potent aldose reductase inhibitor, (2S,4S)-6-fluoro-2′,5′-dioxospiro[chroman-4,4′-imidazolidine]-2-carboxamide (Fidarestat): Its absolute configuration and interactions with the aldose reductase by X-ray crystallography. J. Med. Chem. 2000, 43, 2479–2483. [Google Scholar] [CrossRef]
- Murakami, N.; Ohta, M.; Kato, K.; Nakayama, K.; Mizota, M.; Miwa, I.; Okuda, J. Effects of 1-(3-bromobenzofuran-2-ylsulfonyl)hydantoin on human aldose reductase examined by a new application of HPLC system for measuring tissue polyol. Arzneimittelforschung/Drug Res. 1997, 47, 1222–1225. [Google Scholar]
- Sarges, R.; Oates, P.J. Aldose reductase inhibitors: Recent developments. Prog. Drug Res. 1993, 40, 99–161. [Google Scholar]
- Haruyama, H.; Takayama, T.; Kinoshita, T.; Kondo, M.; Nakajima, M.; Haneishi, T. Structural elucidation and solution conformation of the novel herbicide hydantocidin. J. Chem. Soc. Perkin Trans. 1 1991, 1637–1640. [Google Scholar] [CrossRef]
- Siehl, D.L.; Subramanian, M.V.; Walters, E.W.; Lee, S.F.; Anderson, R.J.; Toschi, A.G. Adenylosuccinate synthetase: Site of action of hydantocidin, a microbial phytotoxin. Plant. Physiol. 1996, 110, 753–758. [Google Scholar] [CrossRef]
- Heim, D.R.; Gerwick, B.C.; Murdoch, M.G.; Green, S.B. Hydantocidin: A possible proherbicide inhibiting purine biosynthesis at the site of adenylosuccinate synthetase. Pest. Biochem. Physiol. 1995, 53, 138–145. [Google Scholar] [CrossRef]
- Mizuno, T.; Kino, T.; Takatoshi, I.; Miyata, T. Synthesis of aromatic urea herbicides by the selenium-assisted carbonylation using carbon monoxide with sulfur. Synth. Commun. 2000, 30, 1675–1688. [Google Scholar] [CrossRef]
- Fischer, H.-P.; Buser, H.-P.; Chemla, P.; Huxley, P.; Lutz, W.; Mirza, S.; Tombo, G.M.R.; van Lommen, G.; Sipido, V. Synthesis and chirality of novel heterocyclic compounds designed for crop protection. Bull. Soc. Chim. Belg. 1994, 103, 565–581. [Google Scholar] [CrossRef]
- Sano, H.; Sugai, S. Synthesis of (±)-carbocyclic analogue of spirohydantoin nucleoside. Tetrahedron 1995, 51, 4635–4646. [Google Scholar] [CrossRef]
- Bazil, C.W. Sleep, sleep apnea, and epilepsy. Curr. Treat. Options Neurol. 2004, 6, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Roca, T.; Domènech, J.; Suriol, M. Synthesis of water-soluble phenytoin prodrugs. Bioorg. Med. Chem. Lett. 1999, 9, 1859–1862. [Google Scholar] [CrossRef]
- Bac, P.; Maurois, P.; Dupont, C.; Pages, N.; Stables, J.P.; Gressens, P.; Evrard, P. Magnesium deficiency-dependent audiogenic seizures (MDDASs) in adult mice: A nutritional model for discriminatory screening of anticonvulsant drugs and original assessment of neuroprotection properties. J. Neurosci. 1998, 18, 4363–4373. [Google Scholar] [CrossRef]
- Krall, R.L.; Penry, J.K.; White, B.G.; Kupferberg, H.J.; Swinyard, E.A. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 1978, 19, 409–428. [Google Scholar] [CrossRef]
- Reagan, L.P.; McKittrick, C.R.; McEwen, B.S. Corticosterone and phenytoin reduce neuronal nitric oxide synthase messenger RNA expression in rat hippocampus. Neuroscience 1999, 91, 211–219. [Google Scholar] [CrossRef]
- Taylor, C.P. Voltage-gated Na+ channels as targets for anticonvulsant, analgesic and neuroprotective drugs. Curr. Pharm. Des. 1996, 2, 375–388. [Google Scholar]
- Eadie, M.J. Phenytoin. In The Treatment of Epilepsy, 2nd ed.; Shorvon, S., Perucca, E., Fish, D., Dodson, E., Eds.; Blackwell Publishing: Oxford, UK, 2004; pp. 475–488. [Google Scholar]
- Brendstrup, L.; Hjelt, K.; Petersen, K.E.; Petersen, S.; Andersen, E.A.; Daugbjerg, P.S.; Stagegaard, B.R.; Nielsen, O.H.; Vejlsgaard, R.; Schou, G.; et al. Nitrofurantoin versus trimethoprim prophylaxis in recurrent urinary tract infection in children. A randomized, double-blind study. Acta Paediatr. Scand. 1990, 79, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.F. Nitrofuratoin. Drug Intell. Clin. Pharm. 1985, 19, 540–547. [Google Scholar] [CrossRef]
- Richards, W.A.; Riss, E.; Kass, E.H.; Finland, M. Nitrofurantoin: Clinical and laboratory studies in urinary tract infections. AMA Arch. Intern. Med. 1955, 96, 437–450. [Google Scholar] [CrossRef]
- Sarges, R.; Howard, H.R.; Kelbaugh, P.R. Synthesis of optically active spirohydantoins by asymmetric induction. Hydantoin formation from amino nitriles and chlorosulfonyl isocyanate. J. Org. Chem. 1982, 47, 4081–4085. [Google Scholar] [CrossRef]
- Cohen, R.A.; Hennekens, C.H.; Christen, W.G.; Krolewski, A.; Nathan, D.M.; Peterson, M.J.; LaMotte, F.; Manson, J.E. Determinants of retinopathy progression in type 1 diabetes mellitus. Am. J. Med. 1999, 107, 45–51. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Plurad, S.B.; Coleman, B.D.; Williamson, J.R.; Tilton, R.G. Effects of sorbinil, dietary myo-inositol supplementation, and insulin on resolution of neuroaxonal dystrophy in mesenteric nerves of streptozocin-induced diabetic rats. Diabetes 1991, 40, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Krause, T.; Gerbershagen, M.U.; Fiege, M.; Weisshorn, R.; Wappler, F. Dantrolene—A review of its pharmacology, therapeutic use and new developments. Anaesthesia 2004, 59, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Dorian, P.; Borggrefe, M.; Al-Khalidi, H.R.; Hohnloser, S.H.; Brum, J.M.; Tatla, D.S.; Brachmann, J.; Myerburg, R.J.; Cannom, D.S.; van der Laan, M.; et al. Placebo-controlled, randomized clinical trial of azimilide for prevention of ventricular tachyarrhythmias in patients with an implantable cardioverter defibrillator. Circulation 2004, 110, 3646–3654. [Google Scholar] [CrossRef]
- Lacroix, L.; Laurent, M.; Buys, M. Iprodione. In Analytical Methods for Pesticides and Plant Growth Regulators: Vol. II.; Zweig, G., Sherma, J., Eds.; Academic Press: London, UK, 1980; pp. 247–261. [Google Scholar]
- Shiozaki, M. Synthesis of hydantocidin and C-2-thioxo-hydantocidin. Carbohydr. Res. 2001, 335, 147–150. [Google Scholar] [CrossRef]
- Shiozaki, M. Syntheses of hydantocidin and C-2-thioxohydantocidin. Carbohydr. Res. 2002, 337, 2077–2088. [Google Scholar] [CrossRef]
- Renard, A.; Lhomme, J.; Kotera, M. Synthesis and properties of spiro nucleosides containing the barbituric acid moiety. J. Org. Chem. 2002, 67, 1302–1307. [Google Scholar] [CrossRef]
- Walter, M.W. Structure-based design of agrochemicals. Nat. Prod. Rep. 2002, 19, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Itoi, K.; Takamatsu, Y.; Kinoshita, T.; Okazaki, T.; Kawakubo, K.; Shindo, M.; Honma, T.; Tohjigamori, M.; Haneishi, T. Hydantocidin: A new compound with herbicidal activity from Streptomyces hygroscopicus. J. Antibiot. 1991, 44, 293–300. [Google Scholar] [CrossRef]
- Bichard, C.J.F.; Mitchel, E.P.; Wormald, M.R.; Watson, K.A.; Johnson, L.N.; Zographos, S.E.; Koutra, D.D.; Oikonomakos, N.G.; Fleet, G.W.J. Potent inhibition of glycogen phosphorylase by a spirohydantoin of glucopyranose: First pyranose analogues of hydantocidin. Tetrahedron Lett. 1995, 36, 2145–2148. [Google Scholar] [CrossRef]
- Ösz, E.; Somsák, L.; Szilágyi, L.; Kovács, L.; Docsa, T.; Tóth, B.; Gergely, P. Efficient inhibition of muscle and liver glycogen phosphorylases by a new glucopyranosylidene-spiro-thiohydantoin. Bioorg. Med. Chem. Lett. 1999, 9, 1385–1390. [Google Scholar] [CrossRef]
- Ware, E. The chemistry of the hydantoins. Chem. Rev. 1950, 46, 403–470. [Google Scholar] [CrossRef]
- Bateman, J.H. Hydantoin and derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology, 3rd ed.; Grayson, M., Eckroth, D., Eds.; Wiley-Interscience: New York, NY, USA, 1980; Volume 12, pp. 692–711. [Google Scholar]
- López, C.A.; Trigo, G.G. The chemistry of hydantoins. Adv. Heterocycl. Chem. 1985, 38, 177–228. [Google Scholar]
- Meusel, M.; Gütschow, M. Recent developments in hydantoin chemistry: A review. Org. Prep. Proced. Int. 2004, 36, 391–443. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Recent advances in the synthesis of hydantoins: The state of the art of a valuable scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. Bucherer–Bergs and Strecker multicomponent reactions. In Multicomponent Reactions: Concepts and Applications for Design and Synthesis; Herrera, R.P., Marqués-López, E., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 331–357. [Google Scholar]
- Uhrich, K.; Olson, E.; Worman, J. Aqueous, room temperature synthesis of a 3(N) substituted hydantoin. Synth. Commun. 1986, 16, 1387–1392. [Google Scholar] [CrossRef]
- O’Brien, R.A.; Worman, J.J.; Olson, E.S. Carbon dioxide in organic synthesis: Preparation and mechanism of formation of N-(3)-substituted hydantoins. Synth. Commun. 1992, 22, 823–828. [Google Scholar] [CrossRef]
- Jacobson, R.A. N-substituted α-aminoisobutyronitriles from acetone cyanohydrin. J. Am. Chem. Soc. 1945, 67, 1996–1998. [Google Scholar] [CrossRef]
- Carrington, H.C. Thiohydantoins. Part, I. Preparation of 5: 5-disubstituted 2: 4-dithiohydantoins from the corresponding ketones. J. Chem. Soc. 1947, 681–683. [Google Scholar] [CrossRef]
- Munday, L. Amino-acids of the cyclohexane series. Part I. J. Chem. Soc. 1961, 4372–4379. [Google Scholar] [CrossRef]
- Cremlyn, R.J.W.; Chisholm, M. The configuration of some decalin spiro-hydantoins and amino-acids. J. Chem. Soc. C 1967, 2269–2273. [Google Scholar] [CrossRef]
- Maki, Y.; Masugi, T. Studies of alicyclic α-amino acids: II: Synthesis and unequivocal assignment of stereochemistry of 1-amino-trans- and cis-4-hydroxycyclohexane-l-carboxylic acids. Chem. Pharm. Bull. 1973, 21, 685–691. [Google Scholar] [CrossRef]
- Edward, J.T.; Jitrangsri, C. Stereochemistry of the Bucherer–Bergs and Strecker reactions of 4-tert-butylcyclohexanone. Can. J. Chem. 1975, 53, 3339–33350. [Google Scholar] [CrossRef]
- Mičová, J.; Steiner, B.; Koóš, M.; Langer, V.; Gyepesová, D. Synthesis of 4-carbamoyl-2-oxazolidinones C-4-linked with a saccharide moiety via Bucherer–Bergs reaction of hexofuranos-5-uloses. Synlett 2002, 2002, 1715–1717. [Google Scholar]
- Kuszmann, J.; Márton-Merész, M.; Jerkovich, G. Application of the Bucherer reaction to carbohydrate derivatives. Carbohydr. Res. 1988, 175, 249–264. [Google Scholar] [CrossRef]
- Mičová, J.; Steiner, B.; Koóš, M.; Langer, V.; Gyepesová, D. Characterisation and X-ray crystallography of products from the Bucherer–Bergs reaction of methyl 2,3-O-isopropylidene-α-D-lyxo-pentodialdo-1,4-furanoside. Carbohydr. Res. 2003, 338, 1917–1924. [Google Scholar] [CrossRef]
- Lamberth, C.; Blarer, S. Short synthesis of a new showdomycin analogue. Synlett 1994, 7, 489–490. [Google Scholar] [CrossRef]
- Montagne, C.; Shipman, M. Modified Bucherer–Bergs reaction for the one-pot synthesis of 5,5′-disubstituted hydantoins from nitriles and organometallic reagents. Synlett 2006, 2203–2206. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Ashford, S.W.; Conway, B.; Havens, J.L.; Taylor, B.; Hritzko, B.; Xiang, Y.; Zakarias, P.S. A scalable synthesis of the INOS inhibitor PHA-399733. Org. Process. Res. Dev. 2009, 13, 331–335. [Google Scholar] [CrossRef]
- Waser, M.; Moher, E.D.; Borders, S.S.K.; Hansen, M.M.; Hoard, D.W.; Laurila, M.E.; LeTourneau, M.E.; Miller, R.D.; Phillips, M.L.; Sullivan, K.A.; et al. Process development for a key synthetic intermediate of LY2140023, a clinical candidate for the treatment of schizophrenia. Org. Process. Res. Dev. 2011, 15, 1266–1274. [Google Scholar] [CrossRef]
- Sarges, R. Hydantoin Therapeutic Agents. U.S. Patent 4,130,714, 19 December 1978. [Google Scholar]
- Sysko, R.J. Sorbinil by Optical Resolution with Aminopinane Derivatives. European Patent 0,109,231, 22 October 1986. [Google Scholar]
- Howard, H.R.; Evans, M.; Sarges, R. Synthesis of 2H-, 3H- and 14C-labelled CP-45,634 (sorbinil). J. Labelled Compd. Radiopharm. 1991, 29, 703–708. [Google Scholar] [CrossRef]
- Sarges, R. Hydantoin Derivatives as Therapeutic Agents. U.S. Patent 4,117,230, 26 September 1978. [Google Scholar]
- Ueda, K.; Tanaka, S.; Kunii, T.; Kagei, K.; Sato, T.; Ono, H.; Ohtsuka, I.; Kawase, M.; Ohgoh, T.; Wakabayashi, T. Hydantoin Derivatives for Treating Complications of Diabetes. U.S. Patent 4,780,472, 25 October 1988. [Google Scholar]
- Lipinski, C.A. Spiro-3-hetero-azolones for Treatment of Diabetic Complications. U.S. Patent 4,556,670, 3 December 1985. [Google Scholar]
- Kurono, M.; Kondo, Y.; Yamaguchi, T.; Miura, K.; Usui, T.; Terada, N.; Asano, K.; Mizuno, K.; Matsubara, A.; Kato, N.; et al. Hydantoin Derivatives for Treating Complications of Diabetes. U.S. Patent 5,447,946, 5 September 1995. [Google Scholar]
- Biltz, H. Über die Konstitution der Einwirkungsprodukte von substituierten Harnstoffen auf Benzil und über einige neue Methoden zur Darstellung der 5,5-Diphenyl-hydantoine. Chem. Ber. 1908, 41, 1379–1393. [Google Scholar] [CrossRef]
- Henze, H.R. Method for Obtaining Hydantoins. U.S. Patent 2,409,754, 22 October 1946. [Google Scholar]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Aplysinopsin, a new tryptophan derivative from a sponge. Tetrahedron Lett. 1977, 18, 61–64. [Google Scholar] [CrossRef]
- Djura, P.; Faulkner, D.J. Metabolites of the marine sponge Dercitus sp. J. Org. Chem. 1980, 45, 735–737. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Zibrowius, H.; Pietra, F. Novel Aplysinopsin-type alkaloids from scleractinian corals of the family Dendrophylliidae of the Mediterranean and the Philippines. Configurational-assignment criteria, stereospecific synthesis, and photoisomerization. Helv. Chim. Acta 1988, 71, 773–782. [Google Scholar] [CrossRef]
- Hollenbeak, K.H.; Schmitz, F.J. Aplysinopsin: Antineoplastic tryptophan derivative from the marine sponge Verongia spengelii. Lloydia 1977, 40, 479–481. [Google Scholar]
- Baker, J.T.; Wells, R.J. Natural Products as Medicinal Reagents; Beal, J.L., Reinhard, E., Eds.; Hippokrates Verlag: Stuttgart, Germany, 1981; pp. 299–303. [Google Scholar]
- White, H.C.; Wysong, D.V. Production of Hydantoin and Glycine. U.S. Patent 2,663,713, 22 December 1953. [Google Scholar]
- Nefzi, A.; Dooley, C.; Ostresh, J.M.; Houghten, R.A. Combinatorial chemistry: From peptides and peptidomimetics to small organic and heterocyclic compounds. Bioorg. Med. Chem. Lett. 1998, 8, 2273–2278. [Google Scholar] [CrossRef]
- Mio, S.; Ichinose, R.; Goto, K.; Sugai, S.; Sato, S. Synthetic studies on (+)-hydantocidin (1): A total synthesis of (+)-hydantocidin, a new herbicidal metabolite from microorganism. Tetrahedron 1991, 47, 2111–2120. [Google Scholar] [CrossRef]
- Mio, S.; Shiraishi, M.; Sugai, S.; Haruyama, H.; Sato, S. Synthetic studies on (+)-hydantocidin (2): Aldol addition approaches toward the stereoisomers of (+)-hydantocidin. Tetrahedron 1991, 47, 2121–2132. [Google Scholar] [CrossRef]
- Mio, S.; Kumagawa, Y.; Sugai, S. Synthetic studies on (+)-hydantocidin (3): A new synthetic method for construction of the spiro-hydantoin ring at the anomeric position of D-ribofuranose. Tetrahedron 1991, 47, 2133–2144. [Google Scholar] [CrossRef]
- Mio, S.; Ueda, M.; Hamura, M.; Kitagawa, J.; Sugai, S. Synthetic studies on (+)-hydantocidin (4): Synthesis of stereoisomers of (+)-hydantocidin. Tetrahedron 1991, 47, 2145–2154. [Google Scholar] [CrossRef]
- Mio, S.; Sano, H.; Shindou, M.; Honma, T.; Sugai, S. Synthesis and herbicidal activity of deoxy derivatives of (+)-hydantocidin. Agric. Biol. Chem. 1991, 55, 1105–1109. [Google Scholar]
- Chemla, P. Stereoselective synthesis of (+)-hydantocidin. Tetrahedron Lett. 1993, 34, 7391–7394. [Google Scholar] [CrossRef]
- Harrington, M.P.; Jung, M.E. Stereoselective bromination of β-ribofuranosyl amide. Enantioselective synthesis of (+)-hydantocidin. Tetrahedron Lett. 1994, 35, 5145–5148. [Google Scholar] [CrossRef]
- Mirza, S. New 3-hydroxy-butylidene hydantoin cpds.-and new D-ribose-1-spiro-5′-hydantoin derivs. for prodn. of herbicidal ribose-sprio-hydantoin derivs. DE Patent 4129728 A1, 6 September 1991. [Google Scholar]
- Read, W.T. Researches on hydantoins. Synthesis of the soporific, 4, 4-phenylethyl-hydantoin (nirvanol). J. Am. Chem. Soc. 1922, 44, 1746–1755. [Google Scholar] [CrossRef]
- Reitz, B.E.; Baxter, E.W.; Bennett, D.J.; Codd, E.E.; Jordan, A.D.; Malloy, E.A.; Maryanoff, B.E.; McDonnell, M.E.; Ortegon, M.E.; Renzi, M.J.; et al. N-Aryl-N’-benzylpiperazines as potential antipsychotic agents. J. Med. Chem. 1995, 38, 4211–4222. [Google Scholar] [CrossRef]
- Urech, F. XXI. Ueber Lacturaminsäure und Lactylharnstoff. Justus Liebigs Ann. Chem. 1873, 165, 99–103. [Google Scholar] [CrossRef]
- Smith, R.J.; Bratovanov, S.; Bienz, S. Synthesis of silicon-containing α-amino acids and hydantoins. Tetrahedron 1997, 53, 13695–13702. [Google Scholar] [CrossRef]
- Postel, D.; Nguyen Van Nhien, A.; Villa, P.; Ronco, G. Novel spirohydantoins of D-allose and D-ribose derived from glyco-α-aminonitriles. Tetrahedron Lett. 2001, 42, 1499–1502. [Google Scholar] [CrossRef]
- Kato, N.; Suzuki, M.; Kanai, M.; Shibasaki, M. General and practical catalytic enantioselective Strecker reaction of ketoimines: Significant improvement through catalyst tuning by protic additives. Tetrahedron Lett. 2004, 45, 3147–3151. [Google Scholar] [CrossRef]
- Murray, R.G.; Whitehead, D.M.; Le Strat, F.; Conway, S.J. Facile one-pot synthesis of 5-substituted hydantoins. Org. Biomol. Chem. 2008, 6, 988–991. [Google Scholar] [CrossRef]
- Beller, M.; Eckert, M.; Moradi, W.A.; Neumann, H. Palladium-catalyzed synthesis of substituted hydantoins—A new carbonylation reaction for the synthesis of amino acid derivatives. Angew. Chem. Int. Ed. Engl. 1999, 38, 1454–1457. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Poupaert, J.H.; Wouters, J.; Norberg, B.; Poppitz, W.; Scribad, G.K.E.; Lambert, D.M. A rapid and efficient microwave-assisted synthesis of hydantoins and thiohydantoins. Tetrahedron 2003, 59, 1301–1307. [Google Scholar] [CrossRef]
- Poupaert, J.H.; De Keyser, J.L.; Vandervorst, D.; Dumont, P. Phase-transfer catalysis by poly(ethyleneglycol) 600 in the Biltz synthesis of phenytoin. Bull. Soc. Chim. Belg. 1984, 93, 493–496. [Google Scholar] [CrossRef]
- Napolitano, E.; Farina, V. Crystallization-induced asymmetric transformations and self-regeneration of stereocenters (SROSC): Enantiospecific synthesis of α-benzylalanine and hydantoin BIRT-377. Tetrahedron Lett. 2001, 42, 3231–3234. [Google Scholar] [CrossRef]
- Evindar, G.; Batey, R.A. Peptide heterocycle conjugates: A diverted Edman degradation protocol for the synthesis of N-terminal 2-iminohydantoins. Org. Lett. 2003, 5, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xing, X.; Cuny, G.D. Synthesis of hydantoins from enantiomerically pure α-amino amides without epimerization. J. Org. Chem. 2006, 71, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, I.; Takai, H.; Hirata, T.; Teranishi, M.; Ohno, T.; Kubo, K. Studies on cadiotonic agents. IV. Synthesis of novel 1-(6,7-dimethoxy-4-quinazolinyl)piperidine derivatives carrying substituted hydantoin and 2-thiohydantoin rings. Chem. Pharm. Bull. 1990, 38, 3014–3019. [Google Scholar] [CrossRef]
- Yamaguchi, J.-I.; Harada, M.; Kondo, T.; Noda, T.; Suyama, T. A facile method for preparation of optically active hydantoin. Chem. Lett. 2003, 32, 372–373. [Google Scholar] [CrossRef]
- Selič, L.; Jakše, R.; Lampič, K.; Golič, L.; Golič-Grdadolnik, S.; Stanovnik, B. A simple stereoselective synthesis of aplysinopsin analogs. Helv. Chim. Acta 2000, 83, 2802–2811. [Google Scholar] [CrossRef]
- Nefzi, A.; Giulianotti, M.A.; Truong, L.; Rattan, S.; Ostresh, J.M.; Houghten, R.A. Solid-phase synthesis of linear ureas tethered to hydantoins and thiohydantoins. J. Comb. Chem. 2002, 4, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Bhalay, G.; Cowell, D.; Hone, N.D.; Scobie, M.; Baxter, A.D. Multiple solid-phase synthesis of hydantoins and thiohydantoins. Mol. Divers. 1998, 3, 195–198. [Google Scholar] [CrossRef]
- Kujundžić, N.; Kovačević, K.; Jakovina, M.; Glunčić, B. Synthesis and antibacterial effect of derivatives of 5-(3,4,5-trimethoxy benzyl)-pyrimidine, -tetrahydropyrimidine, -hexahydropyrimidine and -hydantoine. Croat. Chim. Acta 1988, 61, 121–135. [Google Scholar]
- Fraser, W.; Suckling, C.J.; Wood, H.C.S. Latent inhibitors. Part 7. Inhibition of dihydro-orotate dehydrogenase by spirocyclopropanobarbiturates. J. Chem. Soc. Perkin Trans. 1990, 3137–3144. [Google Scholar] [CrossRef]
- Hulme, C.; Ma, L.; Romano, J.J.; Morton, G.; Tang, S.-Y.; Cherrier, M.-P.; Choi, S.; Salvino, J.; Labaudiniere, R. Novel applications of carbon dioxide/MeOH for the synthesis of hydantoins and cyclic ureas via the Ugi reaction. Tetrahedron Lett. 2000, 41, 1889–1893. [Google Scholar] [CrossRef]
- Ignacio, J.M.; Macho, S.; Marcaccini, S.; Pepino, R.; Torroba, T. A facile synthesis of 1,3,5-trisubstituted hydantoins via Ugi four-component condensation. Synlett 2005, 3051–3054. [Google Scholar] [CrossRef]
- Gütschow, M.; Hecker, T.K.; Eger, K. A new one-pot synthesis of 5,5-disubstituted hydantoins from diethyl acetamidomalonates and ureas. Synthesis 1999, 410–414. [Google Scholar] [CrossRef]
- Meusel, M.; Ambrozak, A.; Hecker, T.K.; Gütschow, M. The aminobarbituric acid-hydantoin rearrangement. J. Org. Chem. 2003, 68, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.-I.; Iwabuchi, H.; Sawanishi, H. Synthesis of homochiral 4-amino-4-carboxy-2-phosphonomethylpyrrolidines via a diastereoselective Bucherer–Bergs reaction of 4-oxopyrrolidine derivative: Novel conformationally restricted AP 5 analogues. Tetrahedron Asymmetry 1995, 6, 2271–2279. [Google Scholar] [CrossRef]
- Koóš, M.; Steiner, B.; Mičová, J.; Langer, V.; Ďurík, M.; Gyepesová, D. Synthesis and structure determination of some sugar amino acids related to alanine and 6-deoxymannojirimycin. Carbohydr. Res. 2001, 332, 351–361. [Google Scholar] [CrossRef]
- Steiner, B.; Mičová, J.; Koóš, M.; Langer, V.; Gyepesová, D. Some non-anomerically C–C-linked carbohydrate amino acids related to leucine—Synthesis and structure determination. Carbohydr. Res. 2003, 338, 1349–1357. [Google Scholar] [CrossRef]
- Mičová, J.; Steiner, B.; Koóš, M.; Langer, V.; Gyepesová, D. Synthesis and structure determination of some non-anomerically C–C-linked serine glycoconjugates structurally related to mannojirimycin. Carbohydr. Res. 2004, 339, 2187–2195. [Google Scholar] [CrossRef]
- Wermuth, U.D.; Jenkins, I.D.; Bott, R.C.; Byriel, K.A.; Smith, G. Some stereochemical aspects of the Strecker synthesis and the Bucherer–Bergs reaction. Aust. J. Chem. 2004, 57, 461–465. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Yao, M.L. Synthesis of a potential boron neutron capture therapy agent: 1-aminocyclobutane-1-carboxylic acid bearing a butylboronic acid side chain. Synthesis 2003, 2890–2893. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Das, B.C.; Das, S.; Li, G.S.; Srivastava, R.; Natarajan, N.; Khan, M.K. Synthesis of 1-amino-3-{2-[7-(6-deoxy-α/β-D-galactopyranos-6-yl)-1,7-dicarba-closo-dodecaboran(12)-1-yl]ethyl}cyclobutanecarboxylic acid hydrochloride. Collect. Czech. Chem. Commun. 2002, 67, 836–842. [Google Scholar] [CrossRef]
- Bessis, A.S.; Vadesne, G.; Bourrat, E.; Bertho, G.; Pin, J.P.; Acher, F.C. 3-Carboxy-4-phosphonocyclopentane amino acids: New metabotropic glutamate receptor ligands. Amino Acids 2003, 24, 303–310. [Google Scholar] [PubMed]
- Adediran, S.A.; Cabaret, D.; Drouillat, B.; Pratt, R.F.; Wakselman, M. The synthesis and evaluation of benzofuranones as β-lactamase substrates. Bioorg. Med. Chem. 2001, 9, 1175–1183. [Google Scholar] [CrossRef]
- Paik, S.; Kwak, H.S.; Park, T.H. A facile synthesis of (−)-cucurbitine. Bull. Korean Chem. Soc. 2000, 21, 131–132. [Google Scholar]
- Domínguez, C.; Ezquerra, J.; Baker, S.R.; Borrelly, S.; Prieto, L.; Espada, M.; Pedregal, C. Enantiospecific synthesis of (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid by a modified Corey–Link reaction. Tetrahedron Lett. 1998, 39, 9305–9308. [Google Scholar] [CrossRef]
- Domínguez, C.; Ezquerra, J.; Prieto, L.; Espada, M.; Pedregal, C. Asymmetric synthesis of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740). Tetrahedron Asymmetry 1997, 8, 511–514. [Google Scholar] [CrossRef]
- Tanaka, K.; Suzuki, H.; Sawanishi, H. Asymmetric syntheses of (2R,4S)-4-amino-4-carboxy-2-methylpyrrolidine and (2R,4S)-4-amino-2-carboxy-2-ethyl-pyrrolidine as novel 2-alkyl-substituted (−)-cucurbitine analogues. Heterocycles 1996, 43, 205–219. [Google Scholar] [CrossRef]
- Tanaka, K.; Sawanishi, H. Asymetric synthesis of all 4 isomers of 4-amino-4-carboxyproline: Novel conformationally restricted glutamic-acid analogs. Tetrahedron Asymmetry 1995, 6, 1641–1656. [Google Scholar] [CrossRef]
- Alonso, F.; Mico, I.; Najera, C.; Sansano, J.M.; Yus, M.; Ezquerra, J.; Yruretagoyena, B.; Gracia, I. Synthesis of 3-substituted and 4-substituted cyclic α-amino-acids structurally related to ACPD. Tetrahedron 1995, 51, 10259–10280. [Google Scholar] [CrossRef]
- Curry, K.; McLennan, H.; Rettig, S.J.; Trotter, J. The synthesis and X-ray structures of the geometric isomers of 1-amino-1,2-cyclopentanedicarboxylic acid. Can. J. Chem. 1993, 71, 76–83. [Google Scholar] [CrossRef]
- Chatterjie, N.; Alexander, G. Stereochemical results of the Bucherer–Bergs reaction in the 14-hydroxydihydromorphinone series. Res. Commun. Subst. Abuse 1991, 12, 132–143. [Google Scholar]
- Davis, A.L.; Tabb, D.L.; Swan, J.K.; McCord, T.J. Synthesis of the 3-methyl and 4-methyl derivatives of 3-amino-3,4-dihydro-1-hydroxycarbostyril and related compounds. J. Heterocycl. Chem. 1980, 17, 1405–1408. [Google Scholar] [CrossRef]
- Musson, D.G.; Karashima, D.; Rubiero, H.; Melmon, K.L.; Cheng, A.; Castagnoli, N. Synthetic and preliminary hemodynamic and whole animal toxicity studies on (R,S)-, (R)-, and (S)-2-methyl-3-(2,4,5-trihydroxyphenyl)alanine. J. Med. Chem. 1980, 23, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Voigt, B.; Lischke, I. Synthesis of anti-proteolytically active N-α-arylsulfonylated amidinophenylglycinamides. Pharmazie 1981, 36, 467–470. [Google Scholar]
- Farrington, G.K.; Kumar, A.; Wedler, F.C. Design and synthesis of phosphonate inhibitors of glutamine synthetase. J. Med. Chem. 1987, 30, 2062–2067. [Google Scholar] [CrossRef]
- Rosenthal, A.; Dodd, R.H. Branched-chain glycos-3-yl-α-amino acids. 9. Alternate synthesis of L-2- and D-2-(1,2:5,6-di-O-isopropylidene-α-D-allofuranos-3-yl)glycine via application of the Bucherer hydantoin procedure. J. Carbohydr. Nucleos. Nucleot. 1979, 6, 467–476. [Google Scholar]
- Abshire, C.J.; Berlinguet, L. Synthesis of α-alkyl-substituted amino acids and derivatives. Can. J. Chem. 1965, 43, 1232–1234. [Google Scholar] [CrossRef]
- Henze, H.R.; Thompson, T.R.; Speer, R.J. Mesityl oxide and diacetone alkohol. IX. The Bucherer synthesis of hydantoins. J. Org. Chem. 1943, 8, 17–28. [Google Scholar] [CrossRef]
- Trišović, N.; Valentić, N.; Ušćumlić, G. Solvent effects on the structure-propertyrelationship of anticonvulsant hydantoinderivatives: A solvatochromic analysis. Chem. Cent. J. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Thennarasu, S.; Perumal, P.T. 5-(1-Acetamido)benzyl-5-methylimidazolidin-2,4-dione. Molbank 2003, 2003, M326. [Google Scholar] [CrossRef]
- Wheeler, W.J.; O’Bannon, D.D.; Kennedy, J.H.; Monn, J.A.; Tharp-Taylor, R.W.; Valli, M.J.; Kuo, F.J. The synthesis of isotopically labeled (+)-2-amino-bicyclo[3.1.0]hexane-2,6-carboxylic acid and its 2-oxa- and 2-thia-analogs. J. Labelled Compd. Radiopharm. 2005, 48, 605–620. [Google Scholar] [CrossRef]
- Unkovskii, B.V.; D’yakov, M.Y.; Cherkaev, G.V.; Sokolova, T.D. Synthesis and spatial structure of methyl substituted 2-phenylpiperidine-4-spiro-5′-imidazolidine-2′,4′-diones. Chem. Heterocycl. Compd. 1994, 30, 696–700. [Google Scholar] [CrossRef]
- Pankaskie, M.; Abdel-Monem, M.M. Inhibitors of polyamine biosynthesis. 8. Irreversible inhibition of mammalian S-adenosyl-L-methionine decarboxylase by substrate analogues. J. Med. Chem. 1980, 23, 121–127. [Google Scholar] [CrossRef]
- Trigo, G.G.; Avendaño, C.; Santos, E.; Edward, J.T.; Wong, S.C. Stereochemistry of the Bucherer–Bergs and Strecker reactions of tropinone, cis-bicyclo[3.3.0]octan-3-one and cis-3,4-dimethylcyclopentanone. Can. J. Chem. 1979, 57, 1456–1461. [Google Scholar] [CrossRef]
- Mahmoodi, N.O.; Khodaee, Z. One-pot diastereoselective synthesis of new racemic and achiral spirohydantoins. Mendeleev Commun. 2004, 14, 304–306. [Google Scholar] [CrossRef]
- Grunewald, G.L.; Kuttab, S.H.; Pleiss, M.A.; Mangold, J.B. Conformationally defined aromatic amino acids. Synthesis and stereochemistry of 2-endo- and 2-exo-amino-1,2,3,4-tetrahydro-1,4-ethanonaphthalene-2-carboxylic acids (2-endo- and 2-exo-aminobenzobicyclo[2.2.2]octene-2-carboxylic acids). J. Med. Chem. 1980, 23, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Maehr, H.; Yarmchuk, L.; Leach, M. Antimetabolites produced by microorganisms. XV. Synthesis of 2-methyl-L-arginine, 2-methyl-L-ornithine and their enantiomers. J. Antibiot. 1976, 29, 221–226. [Google Scholar] [CrossRef]
- Winn, M.; Rasmussen, R.; Minard, F.; Kyncl, J.; Plotnikoff, N. Homologs of dopa, α-methyldopa, and dopamine as potential cardiovascular drugs. J. Med. Chem. 1975, 18, 434–437. [Google Scholar] [CrossRef] [PubMed]
- El Masry, A.H.; EI Masry, S.E.; Hare, L.E.; Counsell, R.E. Aromatic amino acid hydroxylase inhibitors. 4. 3-Substituted α-methyltyrosines. J. Med. Chem. 1975, 18, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Ellington, J.J.; Honigberg, I.L. The synthesis of 2-methylproline and 2-methylornithin. J. Org. Chem. 1974, 39, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Monem, M.M.; Newton, N.E.; Weeks, C.E. Inhibitors of polyamine biosynthesis. 1. α-Methyl-(±)-ornithine, an inhibitor of ornithine decarboxylase. J. Med. Chem. 1974, 17, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Ames, M.M.; Castagnoli, N. The synthesis of 13C-enriched α-methyldopa. J. Label. Compd. 1974, 10, 195–205. [Google Scholar] [CrossRef]
- Tomohara, K.; Ito, T.; Hasegawa, N.; Kato, A.; Adachi, I. Direct chemical derivatization of natural plant extract: Straightforward synthesis of natural plant-like hydantoin. Tetrahedron Lett. 2016, 57, 924–927. [Google Scholar] [CrossRef]
- Xu, G.; Wang, J.; Zhou, Z.; Mao, L. A high-yield and cost-effective synthesis of spirotetramat. Russ. J. Org. Chem. 2020, 56, 1775–1778. [Google Scholar] [CrossRef]
- Charnay-Pouget, F.; Le Liepvre, M.; Eijsberg, H.; Guillot, R.; Ollivier, J.; Secci, F.; Frongia, A.; Aitken, D.J. A short synthesis of both enantiomers of 2-aminobicyclo[3.2.0]heptane-2,7-dicarboxylic acid. Tetrahedron Lett. 2021, 68, 152912. [Google Scholar] [CrossRef]
- Żesławska, E.; Kincses, A.; Spengler, G.; Nitek, W.; Wyrzuc, K.; Kieć-Kononowicz, K.; Handzlik, J. The 5-aromatic hydantoin-3-acetate derivatives as inhibitors of the tumour multidrug resistance efflux pump P-glycoprotein (ABCB1): Synthesis, crystallographic and biological studies. Bioorg. Med. Chem. 2016, 24, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Kashif, M.K.; Naseer, M.M.; Rana, U.A.; Hameed, S. Synthesis and in vivo hypoglycemic activity of new imidazolidine-2,4-dione derivatives. Res. Chem. Intermed. 2015, 41, 7313–7326. [Google Scholar] [CrossRef]
- Šmit, B.M.; Pavlović, R.Z. Three-step synthetic pathway to fused bicyclic hydantoins involving a selenocyclization step. Tetrahedron 2015, 71, 1101–1108. [Google Scholar] [CrossRef]
- Delgado, G.E.; Rodríguez, J.A.; Mora, A.J.; Bruno-Colmenárez, J.; Uzcáteguic, J.; Chacón, C. Supramolecular structure of 5-methyl-5-phenyl hydantoin and hydrogen-bonding patterns in 5,5′-substituted hydantoins. Mol. Cryst. Liq. Cryst. 2016, 629, 96–104. [Google Scholar] [CrossRef]
- Bisello, A.; Cardena, R.; Rossi, S.; Crisma, M.; Formaggio, F.; Santi, S. Hydrogen-bond-assisted, concentration-dependent molecular dimerization of ferrocenyl hydantoins. Organometallics 2017, 36, 2190–2197. [Google Scholar] [CrossRef]
- Hmuda, S.F.; Banjac, N.R.; Trišović, N.P.; Božić, B.Ð.; Valentić, N.V.; Uščumlić, G.S. Solvent effects on the absorption spectra of potentially pharmacologically active 5-alkyl-5-arylhydantoins: A structure-activity relationship study. J. Serb. Chem. Soc. 2013, 78, 627–637. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Zakurdaev, E.P.; Prusov, E.V.; Balenkova, E.S. A novel convenient approach to the synthesis of 2-substituted analogsof ornithine and homolysine. Russ. Chem. Bull. Int. Ed. 2004, 53, 2866–2870. [Google Scholar] [CrossRef]
- Tellier, F.; Acher, F.; Brabet, I.; Pin, J.-P.; Azerad, R. Aminobicyclo[2.2.1.]heptane dicarboxylic acids (ABHD), rigid analogs of ACPD and glutamic acid: Synthesis andpharmacological activity on metabotropic receptors mGluR1 and mGluR2. Bioorg. Med. Chem. 1998, 6, 195–208. [Google Scholar] [CrossRef]
- Oba, M.; Shimabukuro, A.; Ono, M.; Doi, M.; Tanaka, M. Synthesis of both enantiomers of cyclic methionine analogue: (R)- and (S)-3-aminotetrahydrothiophene-3-carboxylic acids. Tetrahedron Asymmetry 2013, 24, 464–467. [Google Scholar] [CrossRef]
- Hennion, G.F.; Reardon, J.E. Sterically crowded amines. VIII. The synthesis and reactions of some polysubstituted 2-imidazolidinones. J. Org. Chem. 1967, 32, 2819–2822. [Google Scholar] [CrossRef]
- Minoru, H.; Toshiyasu, I. The synthesis and the configuration of 3-aminotetrahydrothiophene-3-carboxylic acids. Bull. Chem. Soc. Jpn. 1973, 46, 2515–2519. [Google Scholar]
- Trigalo, F.; Acher, F.; Azerad, R. Synthesis and resolution of DHCGA, a new conformationally rigid 3,4-dehydroglutamic acid analogue. Tetrahedron 1990, 46, 5203–5212. [Google Scholar] [CrossRef]
- Natchev, I.A. Organophosphorus analogues and derivatives of the natural L-amino carboxylic acids and peptides. III. Synthesis and enzyme-substrate interactions of D-, DL-, and L-5-dihydroxyphosphinyl-3,4-didehydronorvaline and their cyclic analogues and derivatives. Bull. Chem. Soc. Jpn. 1988, 61, 3711–3715. [Google Scholar] [CrossRef]
- Šmit, B.; Pavlović, R.Z. Synthesis of novel 5-(alk-3-enyl)-hydantoins. Molecules (ECSOC-16) 2012. Available online: https://sciforum.net/paper/view/1066 (accessed on 12 May 2021).
- Greenfield, A.A.; Butera, J.A. Convenient synthesis and isolationof conformationally rigid glutamic acid analogues. Synth. Commun. 2004, 34, 3939–3947. [Google Scholar] [CrossRef]
- Cocker, J.N.; Kohlhase, W.L.; Martens, T.F.; Rogers, A.O.; Allan, G.G. A general route to hydantoins. J. Org. Chem. 1962, 27, 3201–3204. [Google Scholar] [CrossRef]
- Krysiak, J.; Midura, W.H.; Wieczorek, W.; Sieroń, L.; Mikołajczyk, M. Constrained cycloalkyl analogues of glutamic acid: Stereocontrolled synthesis of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740) and its 6-phosphonic acid analogue. Tetrahedron Asymmetry 2010, 21, 1486–1493. [Google Scholar] [CrossRef]
- Khodaee, Z.; Yahyazadeh, A.; Mahmoodi, N.O. One-pot synthesis and characterization of some new types of 5,5′-disubstituted bis(imidazolidine-2,4-diones). J. Heterocycl. Chem. 2013, 50, 288–292. [Google Scholar] [CrossRef]
- Matys, A.; Podlewska, S.; Witek, K.; Witek, J.; Bojarski, A.J.; Schabikowski, J.; Otrębska-Machaj, E.; Latacz, G.; Szymańska, E.; Kieć-Kononowicz, K.; et al. Imidazolidine-4-one derivatives in the search for novel chemosensitizers of Staphylococcus aureus MRSA: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2015, 101, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.D.; Hanrahan, J.R.; Hambley, T.W.; Johnston, G.A.R.; Mewett, K.N.; Mitrovic, A.D. Synthesis and activity of a potent N-methyl-D-aspartic acid agonist, trans-1-aminocyclobutane-1,3-dicarboxylic acid, and related phosphonic and carboxylic acids. J. Med. Chem. 1990, 33, 2905–2915. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Nakabayashi, N.; Matsumoto, K.; Shibuya, S. Lipase-catalyzed kinetic resolution of cis-1-diethylphosphonomethyl-2-hydroxymethylcyclohexane. Application to enantioselective synthesis of 1-diethylphosphonomethyl-2-(5′-hydantoinyl)cyclohexane. Tetrahedron Asymmetry 1995, 6, 3055–3062. [Google Scholar] [CrossRef]
- Rizzi, J.P.; Schnur, R.C.; Hutson, N.J.; Kraus, K.G.; Kelbaugh, P.R. Rotationally restricted mimics of rigid molecules: Nonspirocyclic hydantoin aldose reductase inhibitors. J. Med. Chem. 1989, 32, 1208–1213. [Google Scholar] [CrossRef]
- Šmit, B.; Rodić, M.; Pavlović, R.Z. Synthesis of angularly fused (homo)triquinane-type hydantoins as precursors of bicyclic prolines. Synthesis 2016, 48, 387–393. [Google Scholar] [CrossRef]
- Nique, F.; Hebbe, S.; Triballeau, N.; Peixoto, C.; Lefrançois, J.-M.; Jary, H.; Alvey, L.; Manioc, M.; Housseman, C.; Klaassen, H.; et al. Identification of a 4-(hydroxymethyl)diarylhydantoin as a selective androgen receptor modulator. J. Med. Chem. 2012, 55, 8236–8247. [Google Scholar] [CrossRef]
- Sarges, R.; Schnur, R.C.; Belletire, J.L.; Peterson, M.J. Spiro hydantoin aldose reductase inhibitors. J. Med. Chem. 1988, 31, 230–243. [Google Scholar] [CrossRef]
- Brown, M.L.; Zha, C.C.; van Dyke, C.C.; Brown, G.B.; Brouillette, W.J. Comparative molecular field analysis of hydantoin binding to the neuronal voltage-dependent sodium channel. J. Med. Chem. 1999, 42, 1537–1545. [Google Scholar] [CrossRef]
- Zhang, Q.-L.; Song, L.-J.; Wang, E.-S. Synthesis and antitussive effect of new hydantoin compounds. Chem. Res. Chin. Univ. 2013, 29, 76–81. [Google Scholar] [CrossRef]
- Kwon, S.-K.; Park, M.-S.; Nam, Y.-J. Synthesis of 5-alkylthio(or sulfonyl)methyl-5-m-methoxyphenylhydantoin-3-acetic acid derivatives. Arch. Pharm. Res. 1993, 16, 322–326. [Google Scholar] [CrossRef]
- Garcia, M.J.; Azerad, R. Production of ring-substituted D-phenylglycines by microbial or enzymatic hydrolysis/deracemisation of the corresponding DL-hydantoins. Tetrahedron Asymmetry 1997, 8, 85–92. [Google Scholar] [CrossRef]
- Sergent, D.; Wang, Q.; Sasaki, N.A.; Ouazzani, J. Synthesis of hydantoin analogues of (2S,3R,4S)-4-hydroxyisoleucine with insulinotropic properties. Bioorg. Med. Chem. Lett. 2008, 18, 4332–4335. [Google Scholar] [CrossRef]
- Abshire, C.J.; Planet, G. Preliminary biological studies of several aliphatic amino acid analogs. J. Med. Chem. 1972, 15, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Curry, K.; Peet, M.J.; Magnuson, D.S.K.; McLennan, H. Synthesis, resolutian, and absolute configuration of the isomers of the neuronal excitant l-amino-1,3-cyclopentanedicarboxylic acid. J. Med. Chem. 1988, 31, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Czopek, A.; Byrtus, H.; Kołaczkowski, M.; Pawłowski, M.; Dybała, M.; Nowak, G.; Tatarczyńska, E.; Wesołowska, A.; Chojnacka-Wójcik, E. Synthesis and pharmacological evaluation of new 5-(cyclo)alkyl-5-phenyl- and 5-spiroimidazolidine-2,4-dione derivatives. Novel 5-HT1A receptor agonist with potential antidepressant and anxiolytic activity. Eur. J. Med. Chem. 2010, 45, 1295–1303. [Google Scholar] [CrossRef]
- Allan, R.D.; Apostopoulos, C.; Hambley, T.W. The synthesis and structure of a cyclobutane analog of glutamic acid with an acetic acid side chain. Aust. J. Chem. 1995, 48, 919–928. [Google Scholar] [CrossRef]
- Madaiah, M.; Prashanth, M.K.; Revanasiddappa, H.D.; Veeresh, B. Synthesis and pharmacological evaluation of novel 1′-[2-(difluoromethoxy)benzyl]-2′H,5′H-spiro[8-azabicyclo[3.2.1]octane-3,4′-imidazolidine]-2′,5′-diones and their derivatives. Arch. Pharm. Chem. Life Sci. 2014, 347, 370–380. [Google Scholar] [CrossRef]
- Madaiah, M.; Prashanth, M.K.; Revanasiddappa, H.D.; Veeresh, B. Synthesis and structure–activity relationship studies on novel 8-amino-3-[2-(4-fluorophenoxy)ethyl]-1,3-diazaspiro[4.5]decane-2,4-dione derivatives as anticonvulsant agents. Med. Chem. Res. 2013, 22, 2633–2644. [Google Scholar] [CrossRef]
- Ananda Kumar, C.S.; Kavitha, C.V.; Vinaya, K.; Benaka Prasad, S.B.; Thimmegowda, N.R.; Chandrappa, S.; Raghavan, S.C.; Rangappa, K.S. Synthesis and in vitro cytotoxic evaluation of novel diazaspiro bicyclo hydantoin derivatives in human leukemia cells: A SAR study. Investig. New Drugs 2009, 27, 327–337. [Google Scholar] [CrossRef]
- Oh, C.-H.; Kang, Y.-K.; Park, S.-W.; Cho, J.-H. Synthesis of new hydantoin-3-acetic acid derivatives. Bull. Korean Chem. Soc. 1988, 9, 231–235. [Google Scholar]
- Sakagami, K.; Kumagai, T.; Taguchi, T.; Nakazato, A. Scalable synthesis of (+)-2-amino-3-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic acid as a potent and selective group II metabotropic glutamate receptor agonist. Chem. Pharm. Bull. 2007, 55, 37–43. [Google Scholar] [CrossRef]
- Nagasawa, H.; Elberling, J.; Shirota, F. 2-Aminoadamantane-2-carboxylic acid, a rigid, achiral, tricyclic α-amino acid with transport inhibitory properties. J. Med. Chem. 1973, 16, 823–826. [Google Scholar] [CrossRef]
- Giannakopoulou, E.; Pardali, V.; Skrettas, I.; Zoidis, G. Transesterification instead of N-alkylation: An intriguing reaction. ChemistrySelect 2019, 4, 3195–3198. [Google Scholar] [CrossRef]
- Hayes, R.L.; Washburn, L.C.; Wieland, B.W.; Sun, T.T.; Anon, J.B.; Butler, T.A.; Callahan, A.P. Synthesis and purification of 11C-carboxyl-labeled amino acids. Int. J. Appl. Radiat. Isot. 1978, 29, 186–187. [Google Scholar] [CrossRef]
- McCown, W.H.; Henze, H.R. Alkaline hydrolysis of fluorenone-spirohydantoin. J. Am. Chem. Soc. 1942, 64, 689–690. [Google Scholar] [CrossRef]
- Cremlyn, R.J.W.; Chisholm, M. Some terpene and steroid hydantoins. J. Chem. Soc. 1967, 1762–1764. [Google Scholar] [CrossRef]
- Sheppeck II, J.E.; Gilmore, J.L.; Tebben, A.; Xue, C.-B.; Liu, R.-Q.; Decicco, C.P.; Duan, J.J.-W. Hydantoins, triazolones, and imidazolones as selective non-hydroxamate inhibitors of tumor necrosis factor-α converting enzyme (TACE). Bioorg. Med. Chem. Lett. 2007, 17, 2769–2774. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Kinoshita, M.; Nakada, S.; Umezawa, S. Synthesis of cyclic α-amino acids. IV. Syntheses of adenine nucleosides of 3-amino-3-C-carboxy-3-deoxy-D-ribofuranose and 3-amino-3-C-carboxy-3-deoxy-D-ribopyranose. Bull. Chem. Soc. Jpn. 1970, 43, 246–252. [Google Scholar] [CrossRef]
- Stalker, R.A.; Munsch, T.E.; Tran, J.D.; Nie, X.P.; Warmuth, R.; Beatty, A.; Aakeroy, C.B. Asymmetric synthesis of two new conformationally constrained lysine derivatives. Tetrahedron 2002, 58, 4837–4849. [Google Scholar] [CrossRef]
- Martins, F.J.C.; Viljoen, A.M.; Kruger, H.G.; Fourie, L.; Roscher, J.; Joubert, A.J.; Wessels, P.L. Enantioselective synthesis of amino acids from pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione. Tetrahedron 2001, 57, 1601–1607. [Google Scholar] [CrossRef]
- Srivastava, R.R.; Singhaus, R.R.; Kabalka, G.W. 4-Dihydroxyborylphenyl analogues of 1-aminocyclobutane-carboxylic acids: Potential boron neutron capture therapy agents. J. Org. Chem. 1999, 64, 8495–8500. [Google Scholar] [CrossRef]
- Ezquerra, J.; Yruretagoyena, B.; Avendaño, C.; De la Cuesta, E.; González, R.; Prieto, L.; Pedregal, C.; Espada, M.; Prowse, W. Conformationally constrained ACPD analogues. Synthesis and resolution of 3-aminobicyclo[3,3,0]octane-1,3-dicarboxylic acids. Tetrahedron 1995, 51, 3271–3278. [Google Scholar] [CrossRef]
- Villacampa, M.; Martínez, M.; González-Trigo, G.; Söllhuber, M.M. Synthesis and stereochemistry of (3α)-6β,7β-dihydroxy- and 6β-hydroxy-8-alkyl-8-azabicyclo[3.2.1]octane-3-spiro-5′-imidazoline-2′,4′-diones. J. Heterocycl. Chem. 1992, 29, 1541–1544. [Google Scholar] [CrossRef]
- Villacampa, M.; Martínez, M.; González-Trigo, G.; Söllhuber, M.M. Synthesis and stereochemistry of tropane 6-spiro-hydantoins. Heterocycles 1992, 34, 1885–1895. [Google Scholar] [CrossRef]
- Menéndez, J.C.; Díaz, M.P.; Bellver, C.; Söllhuber, M.M. Synthesis, anticonvulsant and antihypertensive activity of diastereomeric 9,10-dimethoxy-1,3,4,6,7,11b-hexahydrospiro[benzo[a]quinolizin-2,4′-imidazolidine]-2′,5′-diones. Eur. J. Med. Chem. 1992, 27, 61–66. [Google Scholar] [CrossRef]
- Sacripante, G.; Edward, J.T. Stereochemistry of the conventional and modified Bucherer–Bergs reactions of 2-substituted cyclohexanones. Can. J. Chem. 1982, 60, 1982–1987. [Google Scholar] [CrossRef]
- Christensen, H.N.; Handlogten, M.E.; Vadgama, J.V.; De la Cuesta, E.; Ballesteros, P.; Trigo, G.G.; Avendano, C. Synthesis and transport applications of 3-aminobicyclo[3.2.1]octane-3-carboxylic acids. J. Med. Chem. 1983, 26, 1374–1378. [Google Scholar] [CrossRef]
- Knizhnikov, V.O.; Voitenko, Z.V.; Golovko, V.B.; Gorichko, M.V. Diastereospecific ring cleavage of bornane-2,3-dione in the Bucherer–Bergs reaction. Tetrahedron Asymmetry 2012, 23, 1080–1083. [Google Scholar] [CrossRef]
- Monteiro, J.L.; Pieber, B.; Corrêa, A.G.; Kappe, C.O. Continuous synthesis of hydantoins: Intensifying the Bucherer–Bergs reaction. Synlett 2016, 27, 83–87. [Google Scholar]
- Trigo, G.G.; Avendaño, C.; Ballesteros, P.; Sastre, A. Synthesis of granatanine-3-spiro-5′-hydantoin-N-ω-hydroxyalkyl esters. J. Heterocycl. Chem. 1980, 17, 103–105. [Google Scholar] [CrossRef]
- Knizhnikov, V.O.; Voitenko, Z.V.; Gorichko, M.V. Hydantoins derived from ketopinic and 4-camphorcarboxylic acids. French-Ukrain. J. Chem. 2013, 1, 23–26. [Google Scholar]
- Cheong, J.E.; Pfeiffer, C.T.; Northrup, J.D.; Parker, M.F.L.; Schafmeister, C.E. An improved, scalable synthesis of bis-amino acids. Tetrahedron Lett. 2016, 57, 4882–4884. [Google Scholar] [CrossRef]
- Loughlin, W.A.; Schweiker, S.S.; Jenkins, I.D.; Henderson, L.C. Synthesis and evaluation of C8-substituted 4.5-spiro lactams as glycogen phosphorylase a inhibitors. Tetrahedron 2013, 69, 1576–1582. [Google Scholar] [CrossRef]
- Conway, S.J.; Miller, J.C.; Bond, A.D.; Clark, B.P.; Jane, D.E. Synthesis and biological evaluation of phospholane and dihydro-phosphole analogues of the glutamate receptor agonist AP4. J. Chem. Soc. Perkin Trans. 2002, 1625–1627. [Google Scholar] [CrossRef]
- Angeli, A.; Di Cesare Mannelli, L.; Ghelardini, C.; Peat, T.S.; Bartolucci, G.; Menicatti, M.; Carta, F.; Supuran, C.T. Benzensulfonamides bearing spyrohydantoin moieties act as potent inhibitors of human carbonic anhydrases II and VII and show neuropathic pain attenuating effects. Eur. J. Med. Chem. 2019, 177, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Biggers, M.S. General preparation of 7-substituted 4-chromanones: Synthesis of a potent aldose reductase inhibitor. J. Org. Chem. 1994, 59, 1216–1218. [Google Scholar] [CrossRef]
- Obrecht, D.; Spiegler, C.; Schönholzer, P.; Müller, K.; Heimgartner, H.; Stierli, F. A new general approach to enantiomerically pure cyclic and open-chain (R)- and (S)-α,α-disubstituted α-amino acids. Helv. Chim. Acta 1992, 75, 1666–1696. [Google Scholar] [CrossRef]
- Trigo, G.G.; Galvez, E.; Avendaño, C. 1H NMR structural analysis of azabicyclospirohydantoins. J. Heterocycl. Chem. 1978, 15, 907–912. [Google Scholar] [CrossRef]
- Fourie, L.; Govender, T.; Hariprakasha, H.K.; Kruger, H.G.; Raasch, T. Complete NMR elucidation of a novel trishomocubane hydantoin and its mono- and bis-t-Boc protected derivatives. Magn. Reson. Chem. 2004, 42, 617–623. [Google Scholar] [CrossRef]
- González, J.; Martínez-Otero, D.; Frontana-Uribe, B.A.; Cuevas-Yañez, E. Synthesis of chiral aza-bis(oxazolines) derived from (+)-camphor. Tetrahedron Asymmetry 2017, 28, 505–510. [Google Scholar] [CrossRef]
- Wysong, C.L.; Yokum, T.S.; Morales, G.A.; Gundry, R.L.; McLaughlin, M.L.; Hammer, R.P. 4-Aminopiperidine-4-carboxylic acid: A cyclic α,α-disubstituted amino acid for preparation of water-soluble highly helical peptides. J. Org. Chem. 1996, 61, 7650–7651. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.; Ten, A.; Baktybayeva, L.; Sagatbekova, I.; Praliyev, K.; Zolotareva, D.; Seilkhanov, T.; Zazybin, A. Synthesis and biological evaluation of 1,3,8-triazaspiro[4.5]decane-2,4-dione derivatives as myelostimulators. J. Chem. 2018. [Google Scholar] [CrossRef]
- Goodson, L.H.; Honigberg, I.L.; Lehman, J.; Burton, W. Potential growth antagonists. I. Hydantoins and disubstituted glycines. J. Org. Chem. 1960, 25, 1920–1924. [Google Scholar] [CrossRef]
- Chu, Y.; Lynch, V.; Iverson, B.L. Synthesis and DNA binding studies of bis-intercalators with a novel spiro-cyclic linker. Tetrahedron 2006, 62, 5536–5548. [Google Scholar] [CrossRef]
- Pellicciari, R.; Luneia, R.; Costantino, G.; Marinozzi, M.; Natalini, B.; Jakobsen, P.; Kanstrup, A.; Lombardi, G.; Moroni, F.; Thomsen, C. 1-Aminoindan-1,5-dicarboxylic acid: A novel antagonist at phospholipase C-linked metabotropic glutamate receptors. J. Med. Chem. 1995, 38, 3717–3719. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Pissarnitski, D.A.; Josien, H.B.; Bara, T.A.; Clader, J.W.; Li, H.; McBriar, M.D.; Rajagopalan, M.; Xu, R.; Terracina, G.; et al. Substituted 4-morpholine N-arylsulfonamides as γ-secretase inhibitors. Eur. J. Med. Chem. 2016, 124, 36–48. [Google Scholar] [CrossRef]
- Caturelli, J.; Martini, M.F.; Fabian, L.; Moltrasio, G.Y.; Moglioni, A.G. Synthesis and spectroscopic characterization of cyclobutyl hydantoins. J. Mol. Struct. 2018, 1171, 495–502. [Google Scholar] [CrossRef]
- Bolla, R.S.; Gandikota, N.M.; Viswanath, I.V.K. Synthesis of deuterium labeled 5,5-dimethyl-3-(α,α,α-trifluoro-4-nitro-m-tolyl) hydantoin. Curr. Radiopharm. 2019, 12, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Bae, S.M.; Shin, H.Y.; Jung, D.I.; Cho, J.H. Synthesis of spirohydantoins and schiff bases of indenoquinoxalinones and indenopyridopyrazinones. Asian J. Chem. 2020, 32, 1925–1930. [Google Scholar] [CrossRef]
- Sarges, R.; Goldstein, S.W.; Welch, W.M.; Swindel, A.C.; Siegel, T.W.; Bever, T.A. Spiro hydantoin aldose reductase inhibitors derived from 8-aza-4-chromanones. J. Med. Chem. 1990, 33, 1859–1865. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Li, T.; Wang, J. Ultrasound-promoted synthesis of 5-substituted and 5,5-disubstituted hydantoins. Indian J. Chem. 1998, 37B, 298–300. [Google Scholar]
- Li, J.-T.; Wang, S.-X.; Chen, G.-F.; Li, T.-S. Some applications of ultrasound irradiation in organic synthesis. Curr. Org. Synth. 2005, 2, 415–436. [Google Scholar] [CrossRef]
- Safari, J.; Gandomi-Ravandi, S.; Javadian, L. Microwave-promoted facile and rapid synthesis procedure for the efficient synthesis of 5,5-disubstituted hydantoins. Synth. Commun. 2013, 43, 3115–3120. [Google Scholar] [CrossRef]
- Prevet, H.; Flipo, M.; Roussel, P.; Deprez, B.; Willand, N. Microwave-assisted synthesis of functionalized spirohydantoins as 3-D privileged fragments for scouting the chemical space. Tetrahedron Lett. 2016, 57, 2888–2894. [Google Scholar] [CrossRef]
- Nencka, R.; Hřebabecký, H.; Dračínský, M. Model synthesis of six-membered carbocyclic spironucleosides. Collect. Czech. Chem. Commun. 2010, 75, 1259–1272. [Google Scholar] [CrossRef]
- Rivero, I.A.; Reynoso-Soto, E.A.; Ochoa-Terán, A. Microwave-assisted synthesis of cycloalkanespirohydantoins and piperidinespirohydantoins as precursors of restricted α-amino acids. Arkivoc 2011, 2, 260–271. [Google Scholar] [CrossRef]
- Chruma, J.J.; Liu, L.; Zhou, W.; Breslow, R. Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics. Bioorg. Med. Chem. 2005, 13, 5873–5883. [Google Scholar] [CrossRef]
- Safari, J.; Javadian, L. Montmorillonite K-10 as a catalyst in the synthesis of 5,5-disubstituted hydantoins under ultrasound irradiation. J. Chem. Sci. 2013, 125, 981–987. [Google Scholar] [CrossRef]
- Maddah, B. Highly efficient and rapid synthesis of diverse hydantoin derivatives using nano-ordered ZnO catalyst under mechanochemical ball milling. Iran. Chem. Commun. 2017, 5, 58–66. [Google Scholar]
- Safari, J.; Javadian, L. Fe3O4-chitosan nanoparticles as a robust magnetic catalyst for efficient synthesis of 5-substituted hydantoins using zinc cyanide. Iran. J. Catal. 2016, 6, 57–64. [Google Scholar]
- Santiago-Ruiz, S.; Torres-Pacheco, L.J.; Oropeza-Guzman, M.T.; Rivero, I.A. Pulsed Fe electro-oxidation for catalytic synthesis of hydantoin derivatives. Int. J. Electrochem. Sci. 2016, 11, 6324–6335. [Google Scholar] [CrossRef]
- López-López, L.I.; de Loera, D.; Rivera-Avalos, E.; Sáenz-Galindo, A. Green synthesis of hydantoins and derivatives. Mini Rev. Org. Chem. 2020, 17, 176–184. [Google Scholar] [CrossRef]






































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalník, M.; Gabko, P.; Bella, M.; Koóš, M. The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity. Molecules 2021, 26, 4024. https://doi.org/10.3390/molecules26134024
Kalník M, Gabko P, Bella M, Koóš M. The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity. Molecules. 2021; 26(13):4024. https://doi.org/10.3390/molecules26134024
Chicago/Turabian StyleKalník, Martin, Peter Gabko, Maroš Bella, and Miroslav Koóš. 2021. "The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity" Molecules 26, no. 13: 4024. https://doi.org/10.3390/molecules26134024
APA StyleKalník, M., Gabko, P., Bella, M., & Koóš, M. (2021). The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity. Molecules, 26(13), 4024. https://doi.org/10.3390/molecules26134024