
Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity

 ,
,  , ,
, , 
 , ,
, , 
 , and
, and 
Abstract
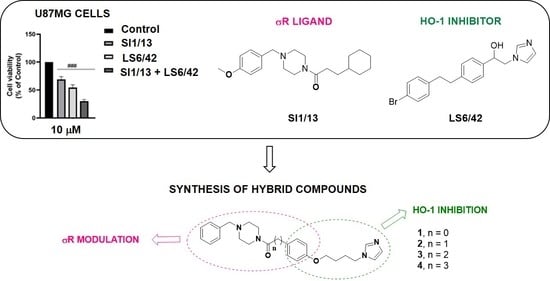
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
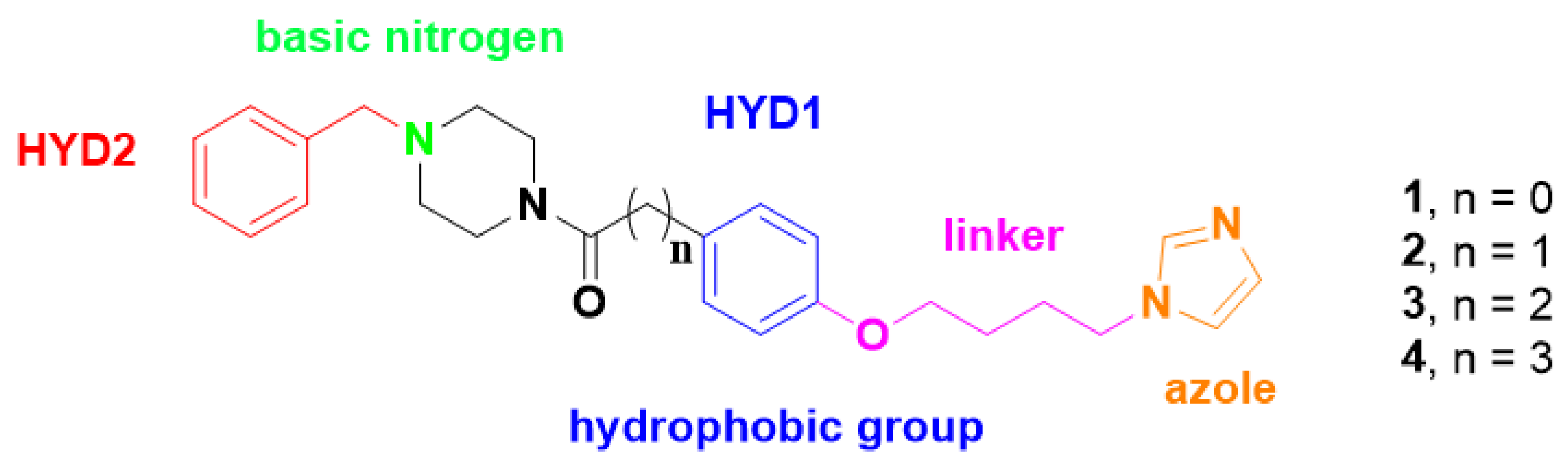
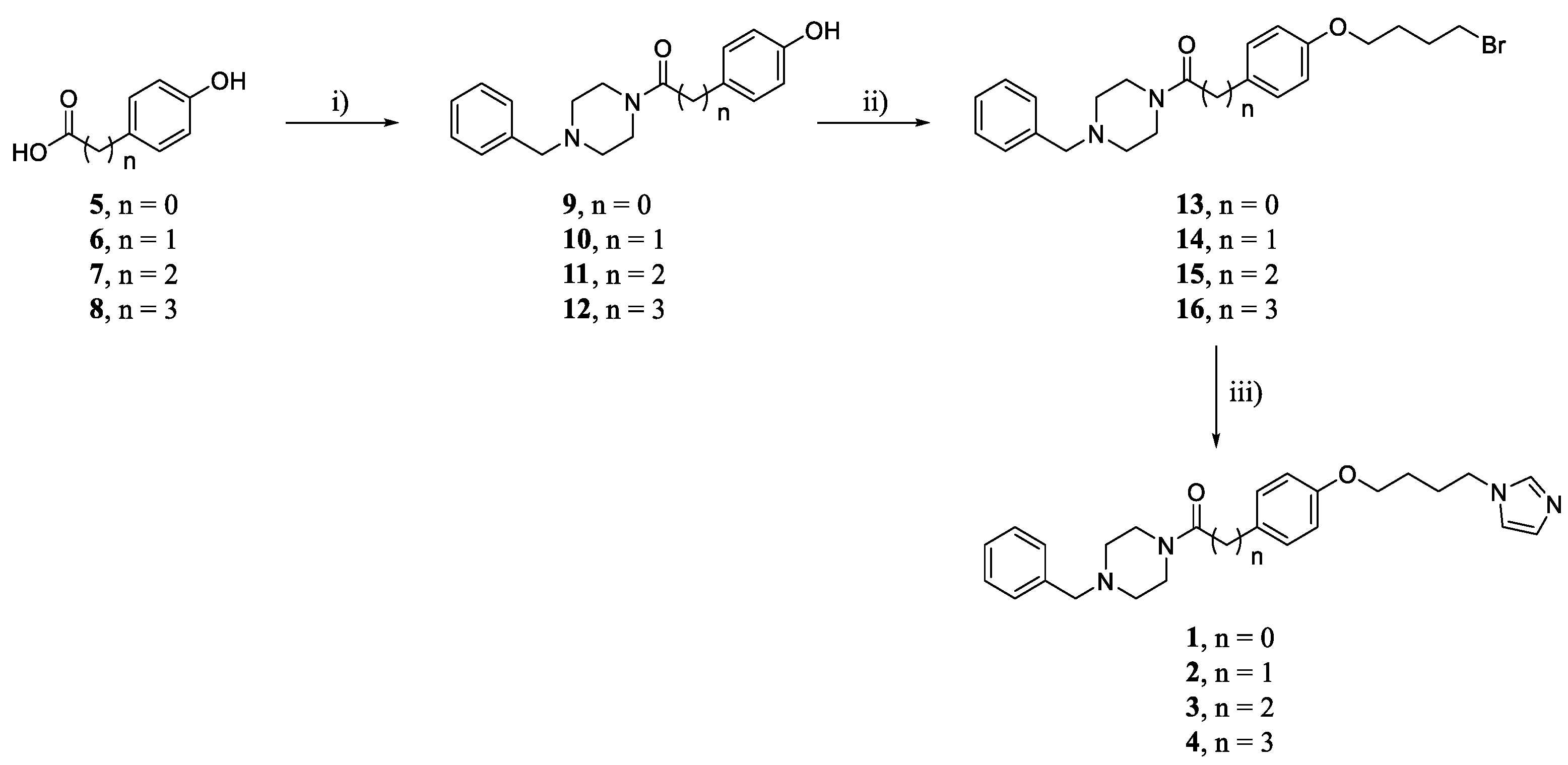
2.1. Chemistry
2.2. Biological Activity
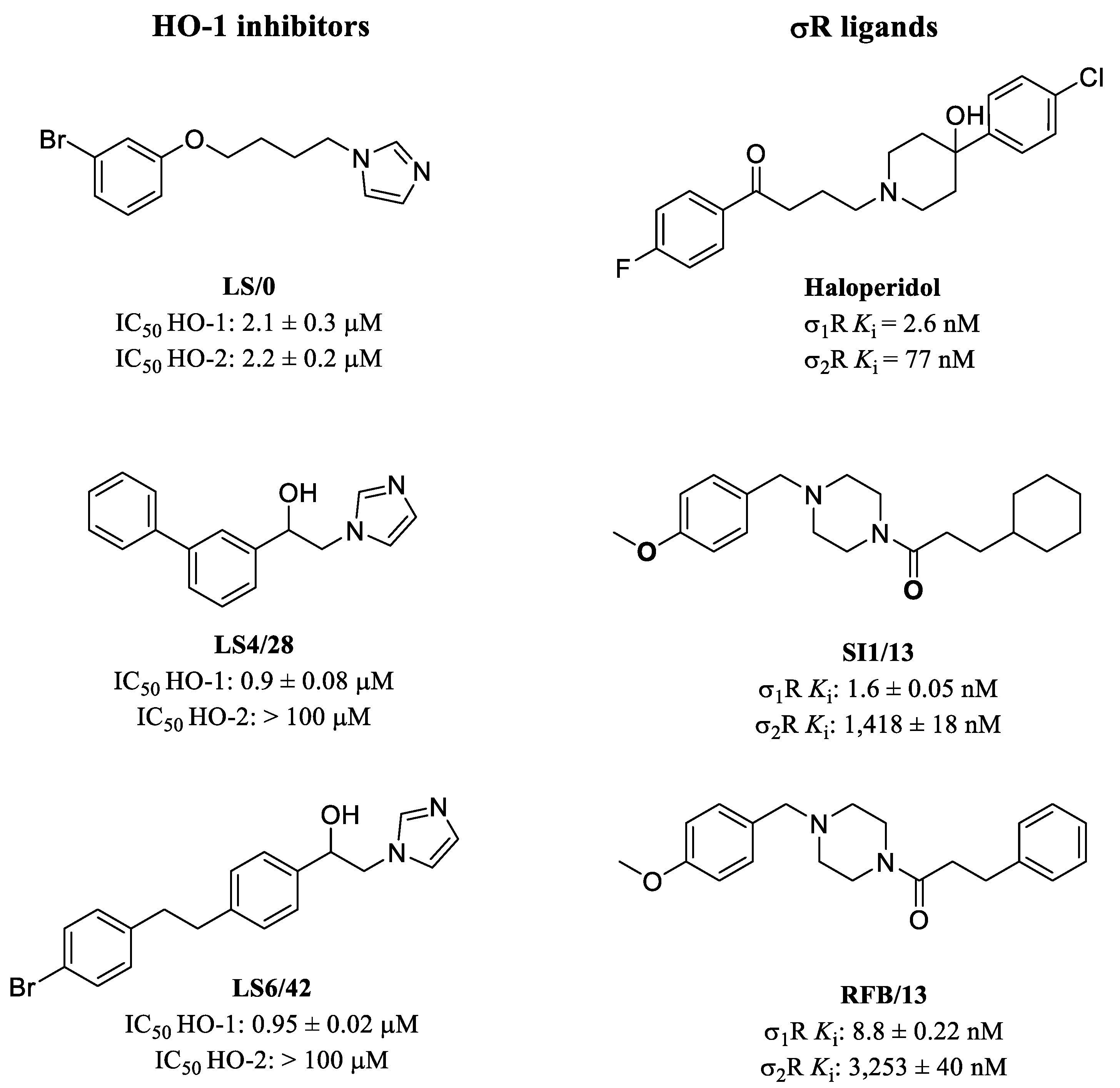
2.2.1. HO-1 Inhibition
2.2.2. σRs Binding Properties
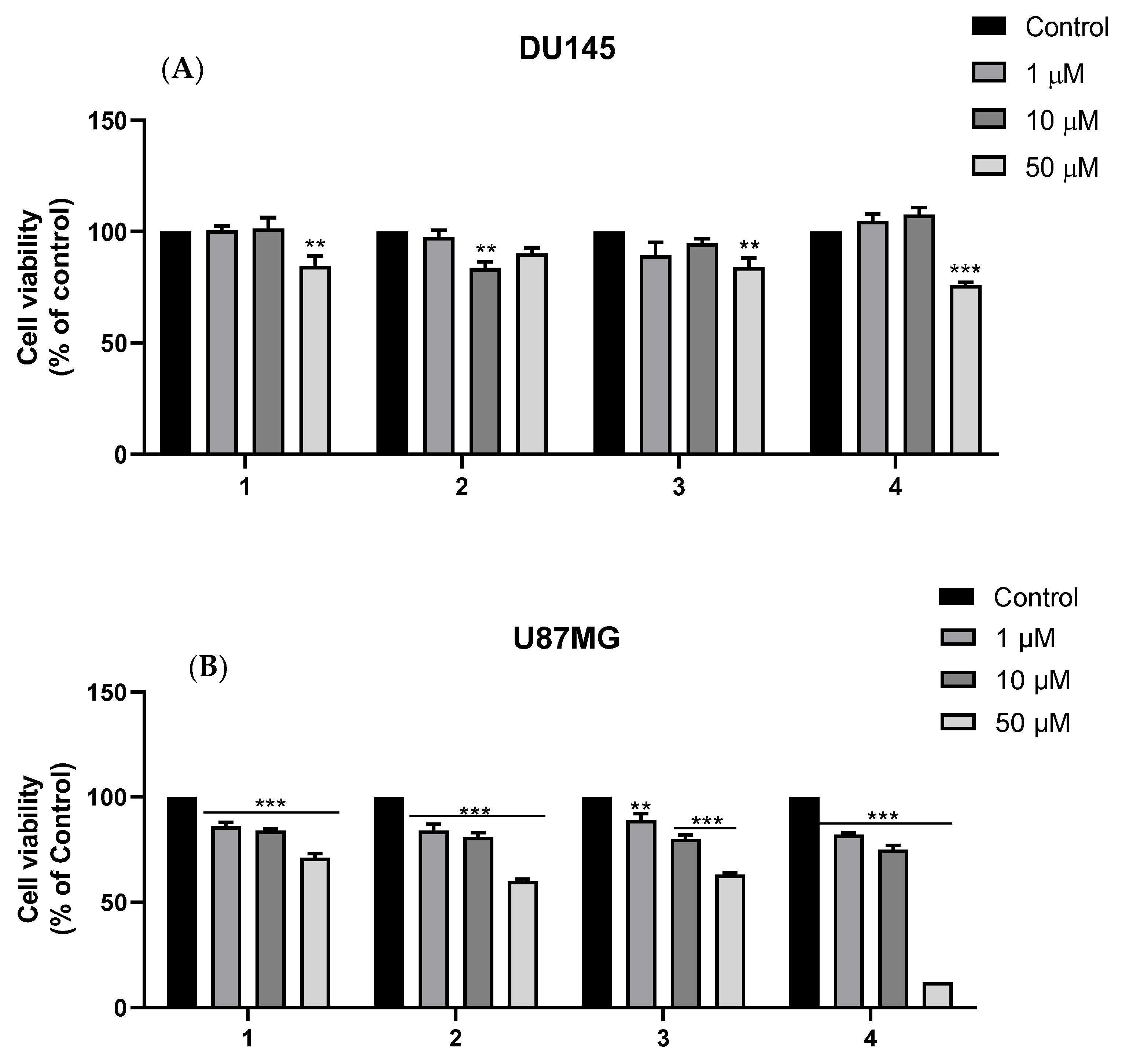
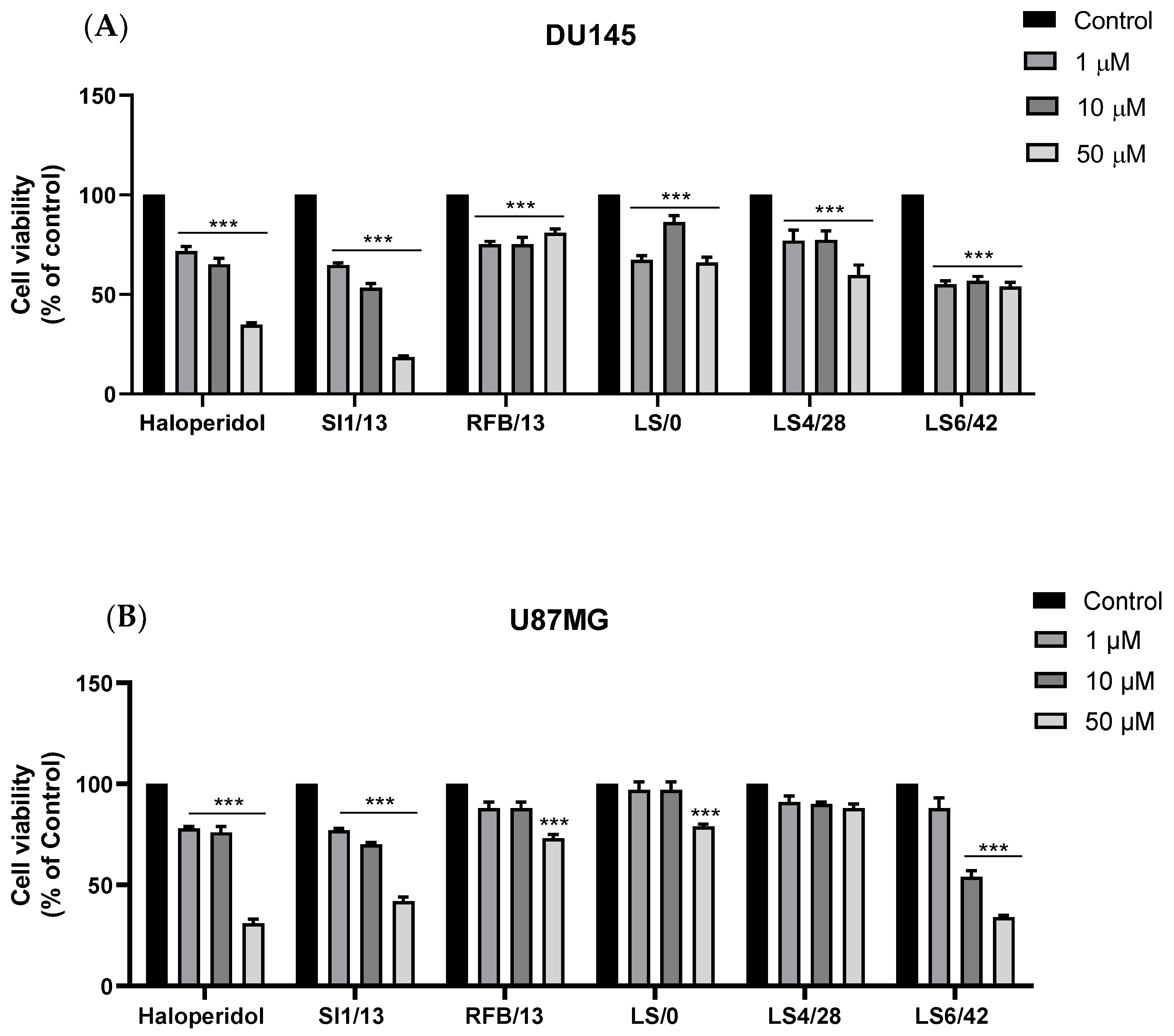
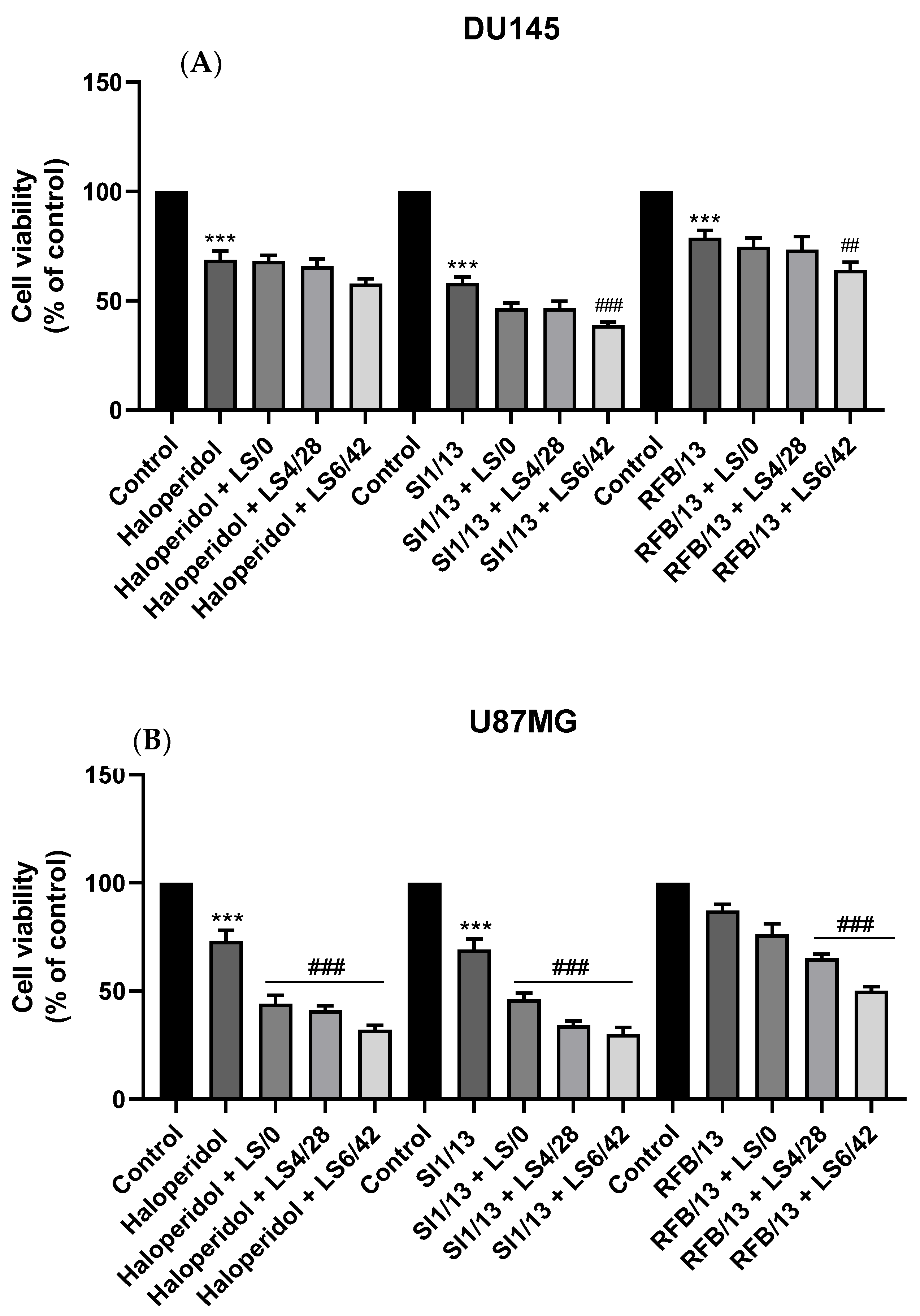
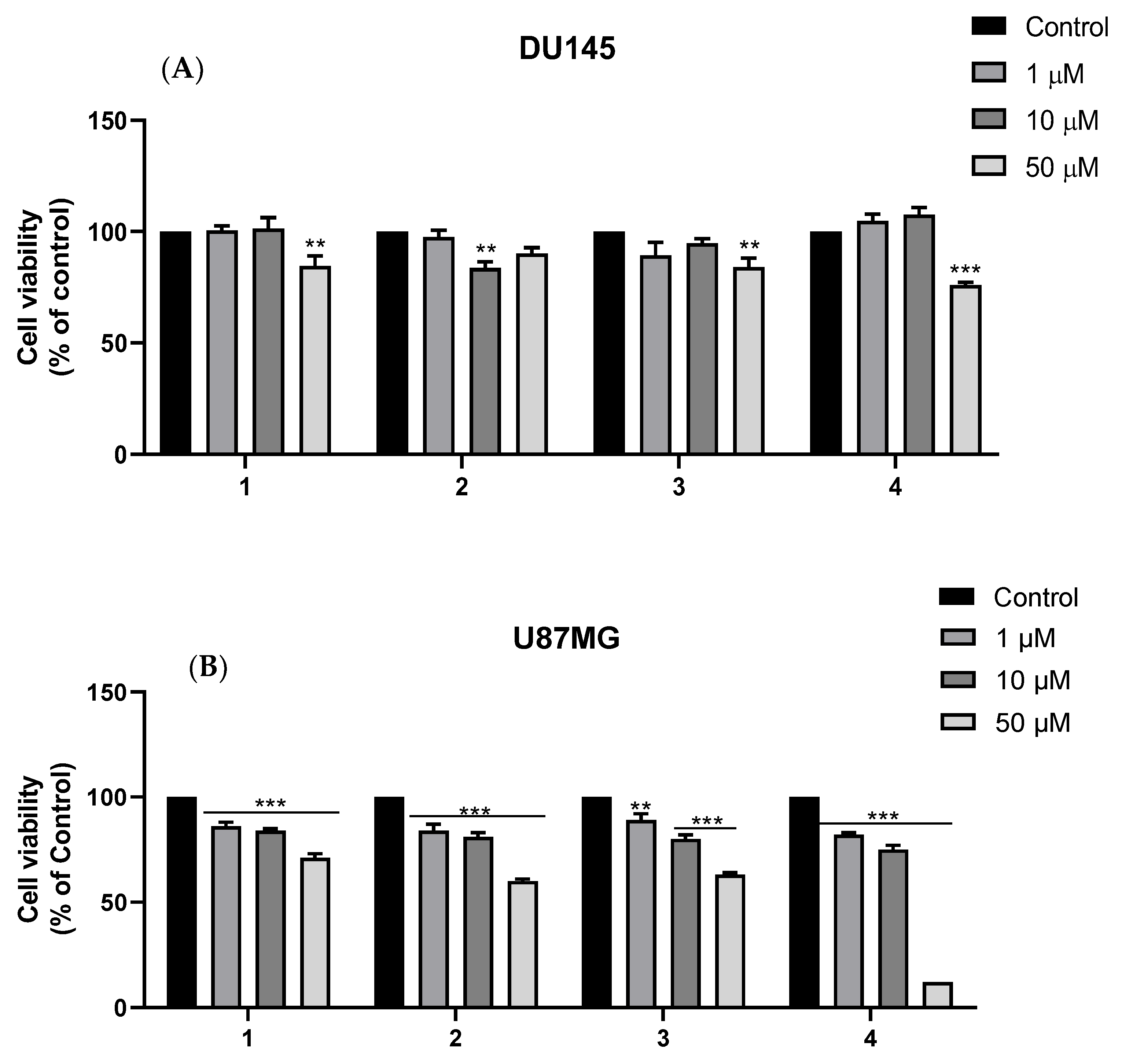
2.2.3. Cytotoxicity against DU145 and U87MG Cell Lines
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. Preparation of Spleen Microsomal Fractions
3.2.2. Preparation of Biliverdin Reductase
3.2.3. Measurement of HO-1 Enzymatic Activity in Microsomal Fraction of Rat Spleen
3.2.4. Radioligand Binding Assay
3.2.5. Cell Cultures
3.2.6. In Vitro Cytotoxicity of HO-1 Inhibitors, σR Ligands, and HO-1/σR Hybrids 1–4 against DU145 and U87MG Cancer Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- World Health Organization. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Antolin, A.A.; Workman, P.; Mestres, J.; Al-Lazikani, B. Polypharmacology in Precision Oncology: Current Applications and Future Prospects. Curr. Pharm. Des. 2016, 22, 6935–6945. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug–Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef]
- Poornima, P.; Kumar, J.D.; Zhao, Q.; Blunder, M.; Efferth, T. Network pharmacology of cancer: From understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol. Res. 2016, 111, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.-Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ferrini, J.-B.; Jbilo, O.; Peleraux, A.; Combes, T.; Vidal, H.; Galiegue, S.; Casellas, P. Transcriptomic Classification of Antitumor Agents: Application to the Analysis of the Antitumoral Effect of SR31747A. Gene Expr. 2003, 11, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef] [Green Version]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittalà, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Pittalà, V.; Vanella, L.; Platania, C.B.M.; Salerno, L.; Raffaele, M.; Amata, E.; Marrazzo, A.; Floresta, G.; Romeo, G.; Greish, K.; et al. Synthesis, in vitro and in silico studies of HO-1 inducers and lung antifibrotic agents. Futur. Med. Chem. 2019, 11, 1523–1536. [Google Scholar] [CrossRef]
- Carota, G.; Raffaele, M.; Sorrenti, V.; Salerno, L.; Pittalà, V.; Intagliata, S. Ginseng and heme oxygenase-1: The link between an old herb and a new protective system. Fitoterapia 2019, 139, 104370. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxidative Med. Cell. Longev. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Huang, L.-Q.; He, H.-S.; Yan, J.; Huang, C.; Wang, R.; Guan, Y.; Huang, D.-P. Overexpression of heme oxygenase-1 in bone marrow stromal cells promotes multiple myeloma resistance through the JAK2/STAT3 pathway. Life Sci. 2020, 257, 118088. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Song, D.-M.; Niu, Y.-H.; Wang, B.-S. Inhibition of heme oxygenase-1 enhances the chemosensitivity of laryngeal squamous cell cancer Hep-2 cells to cisplatin. Apoptosis 2016, 21, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Białas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [Green Version]
- Raffaele, M.; Pittalà, V.; Zingales, V.; Barbagallo, I.; Salerno, L.; Volti, G.L.; Romeo, G.; Carota, G.; Sorrenti, V.; Vanella, L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. Int. J. Mol. Sci. 2019, 20, 2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, P.E.; Martin, W.R. The effects of morphine and nalorphine-like drugs in the nondependent, morphine-dependent and cyclazocine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 198, 66–82. [Google Scholar]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors: Biology in normal and diseased states. J. Recept. Signal Transduct. 2015, 36, 327–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirino, T.J.; Eans, S.O.; Medina, J.M.; Wilson, L.L.; Mottinelli, M.; Intagliata, S.; McCurdy, C.R.; McLaughlin, J.P. Characterization of Sigma 1 Receptor Antagonist CM-304 and Its Analog, AZ-66: Novel Therapeutics Against Allodynia and Induced Pain. Front. Pharmacol. 2019, 10, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.D.; Longen, C.G.; Oyer, H.M.; Chen, N.; Maher, C.M.; Salvino, J.M.; Kania, B.; Anderson, K.N.; Ostrander, W.F.; Knudsen, K.E.; et al. Sigma1 Targeting to Suppress Aberrant Androgen Receptor Signaling in Prostate Cancer. Cancer Res. 2017, 77, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Mégalizzi, V.; Decaestecker, C.; Debeir, O.; Spiegl-Kreinecker, S.; Berger, W.; Lefranc, F.; Kast, R.E.; Kiss, R. Screening of anti-glioma effects induced by sigma-1 receptor ligands: Potential new use for old anti-psychiatric medicines. Eur. J. Cancer 2009, 45, 2893–2905. [Google Scholar] [CrossRef]
- Vilner, B.J.; John, C.S.; Bowen, W.D. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995, 55, 408–413. [Google Scholar]
- Diaz, J.L.; Cuevas, F.; Oliva, A.I.; Font, D.; Sarmentero, M.A.; Alvarez-Bercedo, P.; Lopez-Valbuena, J.M.; Pericas, M.A.; Enrech, R.; Montero, A.; et al. Tricyclic Triazoles as sigma1 Receptor Antagonists for Treating Pain. J. Med. Chem. 2021, 64, 5157–5170. [Google Scholar] [CrossRef]
- Xie, X.Y.; Li, Y.Y.; Ma, W.H.; Chen, A.F.; Sun, Y.T.; Lee, J.Y.; Riad, A.; Xu, D.H.; Mach, R.H.; Huang, Y.S. Synthesis, binding, and functional properties of tetrahydroisoquinolino-2-alkyl phenones as selective sigma2R/TMEM97 ligands. Eur. J. Med. Chem. 2021, 209, 112906. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, S.; Agha, H.; Kopajtic, T.A.; Katz, J.L.; Kamble, S.H.; Sharma, A.; Avery, B.A.; McCurdy, C.R. Exploring 1-adamantanamine as an alternative amine moiety for metabolically labile azepane ring in newly synthesized benzo[d]thiazol-2(3H)one sigma receptor ligands. Med. Chem. Res. 2020, 29, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, S.; Sharma, A.; King, T.; Mesangeau, C.; Seminerio, M.; Chin, F.T.; Wilson, L.L.; Matsumoto, R.R.; McLaughlin, J.P.; Avery, B.A.; et al. Discovery of a Highly Selective Sigma-2 Receptor Ligand, 1-(4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-3-methyl-1H-benzo[d]imidazol-2(3H)-one (CM398), with Drug-Like Properties and Antinociceptive Effects In Vivo. AAPS J. 2020, 22, 94. [Google Scholar] [CrossRef]
- Van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R.A. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2703–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, H.E.; Alsharif, W.F.; Comeau, A.B.; Mesangeau, C.; Intagliata, S.; Mottinelli, M.; McCurdy, C.R.; Bowen, W.D. Divergent Cytotoxic and Metabolically Stimulative Functions of Sigma-2 Receptors: Structure-Activity Relationships of 6-Acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (SN79) Derivatives. J. Pharmacol. Exp. Ther. 2019, 368, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, K.-C.; Tsui, K.-H.; Lin, Y.-H.; Hou, C.-P.; Chang, K.-S.; Tsai, H.-H.; Shin, Y.-S.; Chen, C.-C.; Feng, T.-H.; Juang, H.-H. Antioxidation and Antiapoptosis Characteristics of Heme Oxygenase-1 Enhance Tumorigenesis of Human Prostate Carcinoma Cells. Transl. Oncol. 2020, 13, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, R.; di Giacomo, C.; Sorrenti, V.; Galvano, F.; Santangelo, R.; Cardile, V.; Gangia, S.; d’Orazio, N.; Abraham, N.G.; Vanella, L. Antiproliferative effect of oleuropein in prostate cell lines. Int. J. Oncol. 2012, 41, 31–38. [Google Scholar]
- John, C.S.; Vilner, B.J.; Geyer, B.C.; Moody, T.; Bowen, W.D. Targeting sigma receptor-binding benzamides as in vivo diagnostic and therapeutic agents for human prostate tumors. Cancer Res. 1999, 59, 4578–4583. [Google Scholar] [PubMed]
- Listro, R.; Stotani, S.; Rossino, G.; Rui, M.; Malacrida, A.; Cavaletti, G.; Cortesi, M.; Arienti, C.; Tesei, A.; Rossi, D.; et al. Exploring the RC-106 Chemical Space: Design and Synthesis of Novel (E)-1-(3-Arylbut-2-en-1-yl)-4-(Substituted) Piperazine Derivatives as Potential Anticancer Agents. Front. Chem. 2020, 8, 495. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Di Chiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Heme Oxygenase Database (HemeOxDB) and QSAR Analysis of Isoform 1 Inhibitors. Chem. Med. Chem. 2017, 12, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Siracusa, M.A.; di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Tibullo, D.; Sorrenti, V. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorganic Med. Chem. 2013, 21, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, M.; Sferrazzo, G.; Pittalà, V.; di Rosa, M.; Vanella, L.; Salerno, L.; Sorrenti, V.; Carota, G.; Parrinello, N.; Raffaele, M.; et al. Non-competitive heme oxygenase-1 activity inhibitor reduces non-small cell lung cancer glutathione content and regulates cell proliferation. Mol. Biol. Rep. 2020, 47, 1949–1964. [Google Scholar] [CrossRef]
- Ciaffaglione, V.; Intagliata, S.; Pittalà, V.; Marrazzo, A.; Sorrenti, V.; Vanella, L.; Rescifina, A.; Floresta, G.; Sultan, A.; Greish, K.; et al. New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1923. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Carotti, A.; Ianni, F.; Sorrenti, V.; Intagliata, S.; Rescifina, A.; Salerno, L.; di Michele, A.; Sardella, R.; Pittalà, V. Chromatograpic resolution of phenylethanolic-azole racemic compounds highlighted stereoselective inhibition of heme oxygenase-1 by (R)-enantiomers. Bioorganic Chem. 2020, 99, 103777. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Berardi, F.; Contino, M.; Niso, M.; Abate, C.; Perrone, R.; Tortorella, V. Antiproliferative and cytotoxic effects of some sigma2 agonists and sigma1 antagonists in tumour cell lines. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 370, 106–113. [Google Scholar] [CrossRef]
- Romeo, G.; Bonanno, F.; Wilson, L.L.; Arena, E.; Modica, M.N.; Pittalà, V.; Salerno, L.; Prezzavento, O.; McLaughlin, J.P.; Intagliata, S. Development of New Benzylpiperazine Derivatives as σ1 Receptor Ligands with in Vivo Antinociceptive and Anti-Allodynic Effects. ACS Chem. Neurosci. 2021, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, S.; Alsharif, W.F.; Mesangeau, C.; Fazio, N.; Seminerio, M.; Xu, Y.T.; Matsumoto, R.R.; McCurdy, C.R. Benzimidazolone-based selective sigma2 receptor ligands: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2019, 165, 250–257. [Google Scholar] [CrossRef]
- Nitti, M.; Ivaldo, C.; Traverso, N.; Furfaro, A.L. Clinical Significance of Heme Oxygenase 1 in Tumor Progression. Antioxidants 2021, 10, 789. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vukomanovic, D.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. Structural Insights into Azole-based Inhibitors of Heme Oxygenase-1: Development of Selective Compounds for Therapeutic Applications. Curr. Med. Chem. 2018, 25, 5803–5821. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittalà, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.N.; Vlahakis, J.Z.; Vukomanovic, D.; Lee, W.; Szarek, W.A.; Nakatsu, K.; Jia, Z. A novel, “double-clamp” binding mode for human heme oxygenase-1 inhibition. PLoS ONE 2012, 7, e29514. [Google Scholar] [CrossRef] [Green Version]
- Glennon, R.A.; Ablordeppey, S.Y.; Ismaiel, A.M.; El-Ashmawy, M.B.; Fischer, J.B.; Howie, K.B. Structural Features Important for.sigma.1 Receptor Binding. J. Med. Chem. 1994, 37, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Castracani, C.C.; Longhitano, L.; Distefano, A.; di Rosa, M.; Pittalà, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Volti, G.L. Heme Oxygenase-1 and Carbon Monoxide Regulate Growth and Progression in Glioblastoma Cells. Mol. Neurobiol. 2020, 57, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Dichiara, M.; Gentile, D.; Marrazzo, A.; Turnaturi, R.; Arena, E.; la Mantia, A.; Tomasello, B.R.; Acquaviva, R.; di Giacomo, C.; et al. Sigma Receptor Ligands Carrying a Nitric Oxide Donor Nitrate Moiety: Synthesis, In Silico, and Biological Evaluation. ACS Med. Chem. Lett. 2020, 11, 889–894. [Google Scholar] [CrossRef]
- Amata, E.; Rescifina, A.; Prezzavento, O.; Arena, E.; Dichiara, M.; Pittala, V.; Montilla-Garcia, A.; Punzo, F.; Merino, P.; Cobos, E.J.; et al. (+)-Methyl (1R,2S)-2-{(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)methyl}-1-phenylcyclopropa necarboxylate ((+)-MR200) Derivatives as Potent and Selective Sigma Receptor Ligands: Stereochemistry and Pharmacological Properties. J. Med. Chem. 2018, 61, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Di Chiara, M.; Arena, E.; Pittalà, V.; Pistarà, V.; Cardile, V.; Graziano, A.C.E.; Fraix, A.; Marrazzo, A.; Sortino, S.; et al. Novel Sigma Receptor Ligand–Nitric Oxide Photodonors: Molecular Hybrids for Double-Targeted Antiproliferative Effect. J. Med. Chem. 2017, 60, 9531–9544. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, G.; Ciaffaglione, V.; Amata, E.; Dichiara, M.; Calabrese, L.; Vanella, L.; Sorrenti, V.; Grosso, S.; D’Amico, A.G.; D’Agata, V.; et al. Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity. Molecules 2021, 26, 3860. https://doi.org/10.3390/molecules26133860
Romeo G, Ciaffaglione V, Amata E, Dichiara M, Calabrese L, Vanella L, Sorrenti V, Grosso S, D’Amico AG, D’Agata V, et al. Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity. Molecules. 2021; 26(13):3860. https://doi.org/10.3390/molecules26133860
Chicago/Turabian StyleRomeo, Giuseppe, Valeria Ciaffaglione, Emanuele Amata, Maria Dichiara, Loredana Calabrese, Luca Vanella, Valeria Sorrenti, Salvo Grosso, Agata Grazia D’Amico, Velia D’Agata, and et al. 2021. "Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity" Molecules 26, no. 13: 3860. https://doi.org/10.3390/molecules26133860
APA StyleRomeo, G., Ciaffaglione, V., Amata, E., Dichiara, M., Calabrese, L., Vanella, L., Sorrenti, V., Grosso, S., D’Amico, A. G., D’Agata, V., Intagliata, S., & Salerno, L. (2021). Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity. Molecules, 26(13), 3860. https://doi.org/10.3390/molecules26133860