
σ2 Receptor and Its Role in Cancer with Focus on a MultiTarget Directed Ligand (MTDL) Approach


Abstract

1. Introduction
2. σ Receptors
2.1. σ1 Receptor
2.1.1. σ1 Receptors Involvement in Cancer
2.1.2. σ1/σ2 Receptors MTDLs
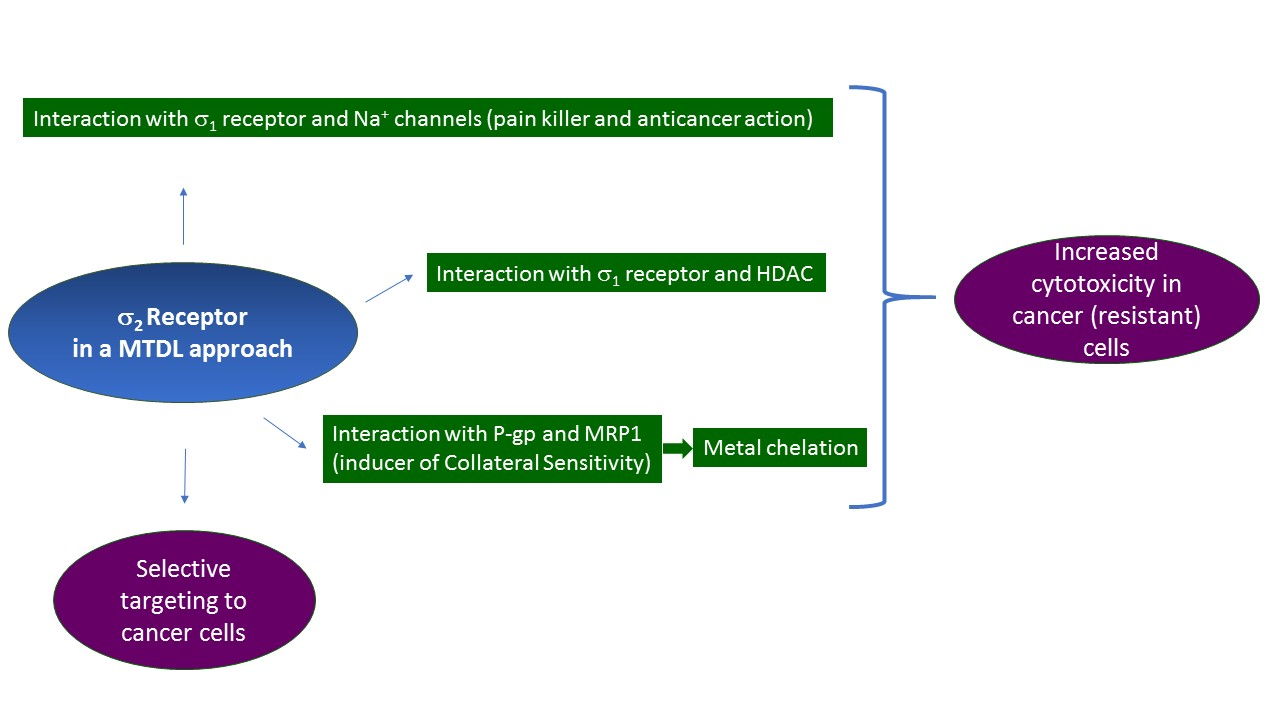
2.2. σ2 Receptor
2.2.1. σ2 Receptor Reference Ligands
- Indoles (Siramesine-like compounds). They were developed starting from indole-3-yl-alkyl-arylpiperazines as 5-HT1A agonists [45]. This class of compounds was generated through replacement of the piperazine with constrained arylpiperidines and introduction of a 4-florophenyl group at the indole N-atom leading to siramesine, that despite its subsequently reported lack of selectivity [46], is still widely used as a reference compound. Binding to phosphatidic acid [47], ROS formation [48], lysosomotropic properties [49], release of cytochrome C by mitochondria [50] were reported as mechanisms of action of siramesine, that eventually lead to tumor cells death.
- Granatanes. This class of ligands was developed from BIMU-1, a 5-HT3/5-HT4 serotonin receptor ligands with nanomolar affinity for σ2 receptors. The bicyclo-octane core of BIMU-1 was replaced by a 9-azabicyclo[3.3.1]nonane (granatane). In addition, the cyclic urea group was opened and replaced by a phenylcarbamate moiety [51,52]. Moreover, the nitrogen atom was functionalized with a benzyl group (WC26 and WC59 [53]) or ω-aminoalkyl chains (SV119 and SW43 [54]) leading to optimal σ2 ligands.
- Benzamides. Structural modifications on D3 receptor ligands led to σ2 receptor high affinity flexible and selective benzamides. The most successful compounds, i.e., RHM-1 [55] and ISO-1 [56], were also produced as radioligands [57] to perform σ2 receptor binding assays or clinical PET studies for the imaging of tumors [58,59,60,61]. An intramolecular H-bond, which forces the flexible benzamides in a bicyclic conformation, was postulated for the interaction of these compounds with the σ2 binding site. Therefore, compound 2 and analogues, in which such H-bond conformation was mimicked, were produced. The nanomolar affinities shown by the rigid bicyclic benzamides validated the hypothesis [62,63]. Importantly, high selectivity rates characterized flexible and rigid benzamides. Corresponding reverse amides were produced (rigid and flexible anilides) as well as the corresponding flexible and rigid anilines, with agreeing results [64].
- Cyclohexyl piperazines. This class, which is based on its lead compound PB28, was developed starting from serotoninergic arylpiperazines [65]. During the last few decades, several analogues were developed and recently reviewed, mostly with the aim of reducing the lipophilicity, as studies on [11C]-radiolabelled PB28 showed high nonspecific binding in mouse brain in vivo [66,67]. However, within the more polar series no ligand showed affinity values comparable to PB28 [68]. Nonetheless, interesting biological profiles in terms of selectivity (compound 3, Table 1) or cytotoxic activity in cancer cells were obtained. Intriguingly, PB28 showed promising anti-SARS-CoV-2 activity in vitro [69], although the effect was later ascribed to the induction of phospholipidosis as an off-target effect [70].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | σ1 R Ki nM | σ2 R Ki nM | Reference |
|---|---|---|---|---|
| Morphans: | - | |||
| (+)-CB-64D |  | 3063 | 16.5 | [43] |
| (+)-CB-184 |  | 7436 | 13.4 | [43] |
| Siramesine |  | 10.5 | 12.6 | [46] |
| Granatane: | ||||
| BIMU-1 |  | 6300 | 32 | [51] |
| WC26 |  | 1436.5 | 2.58 | [53] |
| WC59 |  | 1710.5 | 0.82 | [53] |
| SV119 |  | 1418 | 5.19 | [54] |
| SW43 |  | 134.3 | 7.07 | [54] |
| Benzamides: | ||||
| RHM-1 |  | 3078 | 10.3 | [55] |
| ISO-1 |  | 2150 | 0.26 | [56] |
| 2 |  | 709 | 4.74 | [62] |
| Cyclohexyl piperazines: | ||||
| PB28 |  | 0.38 | 0.68 | [68] |
| 3 |  | 23.3 | 8.52 | [68] |
2.2.2. σ2 Receptor Involvement in Cancer
2.2.3. MTDLs Based on Granatane SV119 and SW43
2.2.4. Collateral Sensitivity (CS) as Multitarget Strategy to Face Cancer
3. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Singh, A.; Beechinor, R.J.; Huynh, J.C.; Li, D.; Dayyani, F.; Valerin, J.B.; Hendifar, A.; Gong, J.; Cho, M. Immunotherapy Updates in Advanced Hepatocellular Carcinoma. Cancers 2021, 13, 2164. [Google Scholar] [CrossRef]
- Teoh, P.J. CAR T-Cell Therapy in Multiple Myeloma: More Room for Improvement. Blood Cancer J. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer Review Douglas. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hellewell, S.B.; Bowen, W.D. A Sigma-like Binding Site in Rat Pheochromocytoma (PC12) Cells: Decreased Affinity for (+)-benzomorphans and Lower Molecular Weight Suggest a Different Sigma Receptor Form from that of Guinea Pig Brain. Brain Res. 1990, 527, 244–253. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal Structure of the Human σ1 Receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the Gene That Codes for the σ2 Receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef]
- Alon, A.; Lyu, J.; Braz, J.M.; Tummino, T.A.; Craik, V.; Matthew, J.; Webb, C.M.; Radchenko, D.S.; Moroz, Y.S.; Huang, X.; et al. Crystal Structures of the σ2 Receptor Template Large-Library Docking for Selective Chemotypes Active in Vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- Abate, C.; Niso, M.; Berardi, F. Sigma-2 Receptor: Past, Present and Perspectives on Multiple Therapeutic Exploitations. Future Med. Chem. 2018, 10, 1997–2018. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Su, T.P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The Sigma-1 Receptor Chaperone as an Inter-Organelle Signaling Modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Anavex® Life Sciences Corp. Available online: https://www.anavex.com/pipeline/ (accessed on 28 April 2021).
- PRidopidine’s Outcome on Function in Huntington Disease, PROOF- HD; ClinicalTrials.Gov Identifier: NCT04556656; ClinicalTrials.gov: Bethesda, MA, USA, 2020.
- Abatematteo, F.S.; Niso, M.; Contino, M.; Leopoldo, M.; Abate, C. Multi-Target Directed Ligands (MTDLs) Binding the σ1 Receptor as Promising Therapeutics: State of the Art and Perspectives. Int. J. Mol. Sci. 2021, 22, 6359. [Google Scholar] [CrossRef]
- Sereti, E.; Tsimplouli, C.; Kalaitsidou, E.; Sakellaridis, N.; Dimas, K. Study of the Relationship between Sigma Receptor Expression Levels and Some Common Sigma Ligand Activity in Cancer Using Human Cancer Cell Lines of the Nci-60 Cell Line Panel. Biomedicines 2021, 9, 38. [Google Scholar] [CrossRef]
- Simony-Lafontaine, J.; Esslimani, M.; Bribes, E.; Gourgou, S.; Lequeux, N.; Lavail, R.; Grenier, J.; Kramar, A.; Casellas, P. Immunocytochemical Assessment of Sigma-1 Receptor and Human Sterol Isomerase in Breast Cancer and Their Relationship with a Series of Prognostic Factors. Br. J. Cancer 2000, 82, 1958–1966. [Google Scholar] [CrossRef]
- Wang, B.; Rouzier, R.; Albarracin, C.T.; Sahin, A.; Wagner, P.; Yang, Y.; Smith, T.L.; Bernstam, F.M.; Marcelo, A.C.; Hortobagyi, G.N.; et al. Expression of Sigma 1 Receptor in Human Breast Cancer. Breast Cancer Res. Treat. 2004, 87, 205–214. [Google Scholar] [CrossRef]
- Xu, Q.X.; Li, E.M.; Zhang, Y.F.; Liao, L.D.; Xu, X.E.; Wu, Z.Y.; Shen, J.H.; Xu, L.Y. Overexpression of Sigma1 Receptor and Its Positive Associations With Pathologic TNM Classification in Esophageal Squamous Cell Carcinoma. J. Histochem. Cytochem. 2012, 60, 457–466. [Google Scholar] [CrossRef]
- Xu, D.; Yi, W.; Chen, Y.; Ma, L.; Wang, J.; Yu, G. Overexpression of Sig1R Is Closely Associated with Tumor Progression and Poor Outcome in Patients with Hilar Cholangiocarcinoma. Med. Oncol. 2014, 31, 1–7. [Google Scholar] [CrossRef]
- Aydar, E.; Onganer, P.; Perrett, R.; Djamgoz, M.B.; Palmer, C.P. The Expression and Functional Characterization of Sigma (σ) 1 Receptors in Breast Cancer Cell Lines. Cancer Lett. 2006, 242, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Caveliers, V.; Everaert, H.; Lahoutte, T.; Dierickx, L.O.; John, C.S.; Bossuyt, A. Labelled Sigma Receptor Ligands: Can Their Role in Neurology and Oncology Be Extended? Eur. J. Nucl. Med. 2001, 28, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.J.; Maher, C.M. Sigma1 Pharmacology in the Context of Cancer. Handb. Exp. Pharmacol. 2017, 244, 237–308. [Google Scholar] [CrossRef]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological Profiling of Sigma 1 Receptor Ligands by Novel Receptor Homomer Assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef]
- Brent, P.J.; Pang, G.; Little, G.; Dosen, P.J.; Van Helden, D.F. The Sigma Receptor Ligand, Reduced Haloperidol, Induces Apoptosis and Increases Intracellular-Free Calcium Levels [Ca2+]i in Colon and Mammary Adenocarcinoma Cells. Biochem. Biophys. Res. Commun. 1996, 219, 219–226. [Google Scholar] [CrossRef]
- Sozio, P.; Fiorito, J.; Di Giacomo, V.; Di Stefano, A.; Marinelli, L.; Cacciatore, I.; Cataldi, A.; Pacella, S.; Turkez, H.; Parenti, C.; et al. Haloperidol Metabolite II Prodrug: Asymmetric Synthesis and Biological Evaluation on Rat C6 Glioma Cells. Eur. J. Med. Chem. 2015, 90, 1–9. [Google Scholar] [CrossRef]
- Rui, M.; Rossi, D.; Marra, A.; Paolillo, M.; Schinelli, S.; Curti, D.; Tesei, A.; Cortesi, M.; Zamagni, A.; Laurini, E.; et al. Synthesis and Biological Evaluation of New Aryl-alkyl(alkenyl)-4-benzylpiperidines, Novel Sigma Receptor (SR) Modulators, as Potential Anticancer-Agents. Eur. J. Med. Chem. 2016, 124, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, A.; Fiorito, J.; Zappal, L.; Prezzavento, O.; Ronsisvalle, S.; Pasquinucci, L.; Scoto, G.M.; Bernardini, R.; Ronsisvalle, G. Antiproliferative Activity of Phenylbutyrate Ester of Haloperidol Metabolite II [(±)-MRJF4] in Prostate Cancer Cells. Eur. J. Med. Chem. 2011, 46, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Graciotti, L.; Faronato, M.; Soldovieri, M.V.; Miceli, F.; Amoroso, S.; Annunziato, L.; Procopio, A.; Taglialatela, M. Human Neoplastic Mesothelial Cells Express Voltage-Gated Sodium Channels Involved in Cell Motility. Int. J. Biochem. Cell Biol. 2006, 38, 1146–1159. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Djamgoz, M.B.A.; Isom, L.L. An Emerging Role for Voltage-Gated Na+ Channels in Cellular Migration: Regulation of Central Nervous System Development and Potentiation of Invasive Cancers. Neuroscientist 2008, 14, 571–583. [Google Scholar] [CrossRef]
- Colabufo, N.A.; Berardi, F.; Abate, C.; Contino, M.; Niso, M.; Perrone, R. Is the σ2 Receptor a Histone Binding Protein? J. Med. Chem. 2006, 49, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Abate, C.; Contino, M.; Inglese, C.; Ferorelli, S.; Berardi, F.; Perrone, R. Tritium Radiolabelling of PB28, a Potent Sigma-2 Receptor Ligand: Pharmacokinetic and Pharmacodynamic Characterization. Bioorganic Med. Chem. Lett. 2008, 18, 2183–2187. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Elenewski, J.; Niso, M.; Berardi, F.; Colabufo, N.A.; Azzariti, A.; Perrone, R.; Glennon, R.A. Interaction of the σ2 Receptor Ligand PB28 with the Human Nucleosome: Computational and Experimental Probes of Interaction with the H2A/H2B Dimer. ChemMedChem 2010, 5, 268–273. [Google Scholar] [CrossRef]
- Abate, C.; Hornick, J.R.; Spitzer, D.; Hawkins, W.G.; Niso, M.; Perrone, R.; Berardi, F. Fluorescent Derivatives of σ Receptor Ligand 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) as a Tool for Uptake and Cellular Localization Studies in Pancreatic Tumor Cells. J. Med. Chem. 2011, 54, 5858–5867. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abate, C.; Niso, M.; Marottoli, R.; Riganti, C.; Ghigo, D.; Ferorelli, S.; Ossato, G.; Perrone, R.; Lacivita, E.; Lamb, D.C.; et al. Novel Derivatives of 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) with Improved Fluorescent and σ Receptors Binding Properties. J. Med Chem 2014, 57, 3314–3323. [Google Scholar] [CrossRef]
- Zeng, C.; Vangveravong, S.; Jones, L.A.; Hyrc, K.; Chang, K.C.; Xu, J.; Rothfuss, J.M.; Goldberg, M.P.; Hotchkiss, R.S.; Mach, R.H. Characterization and Evaluation of Two Novel Fluorescent Sigma-2 Receptor Ligands as Proliferation Probes. Mol. Imaging 2011, 10, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zeng, C.; Chu, W.; Pan, F.; Rothfuss, J.M.; Zhang, F.; Tu, Z.; Zhou, D.; Zeng, D.; Vangveravong, S.; et al. Identification of the PGRMC1 Protein Complex as the Putative Sigma-2 Receptor Binding Site. Nat. Commun. 2011, 2, 380–387. [Google Scholar] [CrossRef]
- Chu, U.B.; Mavlyutov, T.A.; Chu, M.L.; Yang, H.; Schulman, A.; Mesangeau, C.; McCurdy, C.R.; Guo, L.W.; Ruoho, A.E. The Sigma-2 Receptor and Progesterone Receptor Membrane Component 1 Are Different Binding Sites Derived From Independent Genes. EBioMedicine 2015, 2, 1806–1813. [Google Scholar] [CrossRef]
- Pati, M.L.; Groza, D.; Riganti, C.; Kopecka, J.; Niso, M.; Berardi, F.; Hager, S.; Heffeter, P.; Hirai, M.; Tsugawa, H.; et al. Sigma-2 receptor and Progesterone Receptor Membrane Component 1 (PGRMC1) are two Different proteins: Proofs by Fluorescent Labeling and Binding of Sigma-2 Receptor Ligands to PGRMC1. Pharmacol Res. 2017, 117, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Niso, M.; Riganti, C.; Pati, M.L.; Ghigo, D.; Berardi, F.; Abate, C. Novel and Selective Fluorescent σ2-Receptor Ligand with a 3,4-Dihydroisoquinolin-1-one Scaffold: A Tool to Study σ2 Receptors in Living Cells. ChemBioChem 2015, 16, 1078–1083. [Google Scholar] [CrossRef]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of Cholesterol-Regulating Genes by Targeted RNAi Screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; Bartz, F.; Werenbeck-Ueding, J.; Praggastis, M.; Zhang, J.; Joggerst-Thomalla, B.; Theiss, S.; Grimm, D.; Ory, D.S.; et al. Reduction of TMEM97 Increases NPC1 Protein Levels and Restores Cholesterol Trafficking in Niemann-Pick Type C1 Disease Cells. Hum. Mol. Genet. 2016, 25, 3588–3599. [Google Scholar] [CrossRef]
- Bowen, W.D.; Bertha, C.M.; Vilner, B.J.; Rice, K.C. CB-64D and CB-184: Ligands with High σ2 Receptor Affinity and Subtype Selectivity. Eur. J. Pharmacol. 1995, 278, 257–260. [Google Scholar] [CrossRef]
- Bertha, C.M.; Mattson, M.V.; Flippen-anderson, J.L.; Rothman, R.B.; Xu, H.; Cha, X.; Becketts, K.; Rice, K.C. A Marked Change of Receptor Affinity of the 2-Methyl-5-(3-hydroxyphenyl)morphans upon Attachment of an (E)-8-benzylidene Moiety: Synthesis and Evaluation of a New Class of Receptor Ligands. J. Med. Chem. 1994, 37, 3163–3170. [Google Scholar] [CrossRef]
- Perregaard, J.; Moltzen, E.K.; Meier, E.; Sánchez, C. Sigma Ligands with Subnanomolar Affinity and Preference for the Sigma 2 Binding Site. 1. 3-(Omega-aminoalkyl)-1H-indoles. J. Med. Chem. 1995, 38, 1998–2008. [Google Scholar] [CrossRef]
- Niso, M.; Abate, C.; Contino, M.; Ferorelli, S.; Azzariti, A.; Perrone, R.; Colabufo, N.A.; Berardi, F. Sigma-2 Receptor Agonists as Possible Antitumor Agents in Resistant Tumors: Hints for Collateral Sensitivity. ChemMedChem 2013, 8, 2026–2035. [Google Scholar] [CrossRef]
- Parry, M.J.; Alakoskela, J.M.I.; Khandelia, H.; Kumar, S.A.; Jäättelä, M.; Mahalka, A.K.; Kinnunen, P.K.J. High-Affinity Small Molecule-Phospholipid Complex Formation: Binding of Siramesine to Phosphatidic Acid. J. Am. Chem. Soc. 2008, 130, 12953–12960. [Google Scholar] [CrossRef]
- Ostenfeld, M.S.; Fehrenbacher, N.; Hoyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jäättelä, M. Effective Tumor Cell Death by σ-2 Receptor Ligand Siramesine Involves Lysosomal Leakage and Oxidative Stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, M.S.; Høyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sørensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-Cancer Agent Siramesine Is a Lysosomotropic Detergent That Induces Cytoprotective Autophagosome Accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Hafner, Ĉ.M.; Repnik, U.; Turk, V.; Turk, B. Siramesine Triggers Cell Death through Destabilization of Mitochondria, but Not Lysosomes. Cell Death Dis. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Mach, R.H.; Yang, B.; Wu, L.; Kuhner, R.J.; West, T. Synthesis and Sigma Receptor Binding Affinities of 8-azabicyclo[3.2.1]octan-3α-yl and 9-azabicyclo[3.2.1]nonan-3α-yl phenylcarbamates. Med. Chem. Res. 2001, 10, 339–355. [Google Scholar]
- Mach, R.H.; Vangvervong, S.; Huang, Y.; Yang, B.; Blair, J.B.; Wu, L. Synthesis of N-Substituted 9-azabicyclo[3.2.1]nonan-3α-yl phenylcarbamates Analogs as Sigma-2 Receptor Ligands. Med. Chem. Res. 2003, 11, 380–398. [Google Scholar]
- Chu, W.; Xu, J.; Zhou, D.; Zhang, F.; Jones, L.A.; Wheeler, K.T.; Mach, R.H. New N-Substituted 9-azabicyclo[3.3.1]nonan-3α-yl phenylcarbamate Analogs as σ2 Receptor Ligands: Synthesis, in Vitro Characterization, and Evaluation as PET Imaging and Chemosensitization Agents. Bioorganic Med. Chem. 2009, 17, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Vangveravong, S.; Xu, J.; Zeng, C.; Mach, R.H. Synthesis of N-Substituted 9-azabicyclo[3.3.1]nonan-3α-yl carbamate Analogs as σ2 Receptor Ligands. Bioorganic Med. Chem. 2006, 14, 6988–6997. [Google Scholar] [CrossRef]
- Mach, R.H.; Huang, Y.; Freeman, R.A.; Wu, L.; Vangveravong, S.; Luedtke, R.R. Conformationally-Flexible Benzamide Analogues as Dopamine D3 and σ2 Receptor Ligands. Bioorganic Med. Chem. Lett. 2004, 14, 195–202. [Google Scholar] [CrossRef]
- Tu, Z.; Xu, J.; Jones, L.A.; Li, S.; Dumstorff, C.; Vangveravong, S.; Chen, D.L.; Wheeler, K.T.; Welch, M.J.; Mach, R.H. Fluorine-18-Labeled Benzamide Analogues for Imaging the σ2 Receptor Status of Solid Tumors with Positron Emission Tomography. J. Med. Chem. 2007, 50, 3194–3204. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tu, Z.; Jones, L.A.; Vangveravong, S.; Wheeler, K.T.; Mach, R.H. [3H]N-[4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl]-2-methoxy-5-methylbenzamide: A Novel Sigma-2 Receptor Probe. Eur. J. Pharmacol. 2005, 525, 8–17. [Google Scholar] [CrossRef]
- [18F]ISO-1 Positron Emission Tomography (PET/CT) in Primary Breast Cancer; ClinicalTrials.Gov Identifier: NCT02284919; ClinicalTrials.gov: Bethesda, MA, USA, 2014.
- Assessment of Cellular Proliferation in Tumors by Positron Emission Tomography (PET) Using [18F]ISO-1; ClinicalTrials.Gov Identifier: NCT00968656; ClinicalTrials.gov: Bethesda, MA, USA, 2009.
- Imaging of in Vivo Sigma-2 Receptor Expression With 18F-ISO-1 Positron Emission Tomography in Metastatic Breast Cancer; ClinicalTrials.Gov Identifier: NCT03057743; ClinicalTrials.gov: Bethesda, MA, USA, 2021.
- PET Assessment of Acute Lung Transplant Rejection; ClinicalTrials.Gov Identifier: NCT02204202; ClinicalTrials.gov: Bethesda, MA, USA, 2014.
- Abate, C.; Ferorelli, S.; Contino, M.; Marottoli, R.; Colabufo, N.A.; Perrone, R.; Berardi, F. Arylamides Hybrids of Two High-Affinity σ 2 Receptor Ligands as Tools for the Development of PET Radiotracers. Eur. J. Med. Chem. 2011, 46, 4733–4741. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Selivanova, S.V.; Müller, A.; Krämer, S.D.; Schibli, R.; Marottoli, R.; Perrone, R.; Berardi, F.; Niso, M.; Ametamey, S.M. Development of 3,4-dihydroisoquinolin-1(2H)-one Derivatives for the Positron Emission Tomography (PET) Imaging of σ2 Receptors. Eur. J. Med. Chem. 2013, 69, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Niso, M.; Pati, M.L.; Berardi, F.; Abate, C. Rigid: Versus Flexible Anilines or Anilides Confirm the Bicyclic Ring as the Hydrophobic Portion for Optimal σ2 Receptor Binding and Provide Novel Tools for the Development of Future σ2 Receptor PET Radiotracers. RSC Adv. 2016, 6, 88508–88518. [Google Scholar] [CrossRef]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Leopoldo, M.; Abate, C.; Tortorella, V. N-aryl or N-alkylpiperazine Derivatives: The Role of N-Substituent on σ1, σ2, 5-HT1A and D2 Receptor Affinity. Med. Chem. Res. 2000, 10, 201–207. [Google Scholar]
- Abate, C.; Niso, M.; Abatematteo, F.S.; Contino, M.; Colabufo, N.A.; Berardi, F. PB28, the Sigma-1 and Sigma-2 Receptors Modulator With Potent Anti–SARS-CoV-2 Activity: A Review About Its Pharmacological Properties and Structure Affinity Relationships. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Kassiou, M.; Dannals, R.F.; Liu, X.; Wong, D.F.; Ravert, H.T.; Scheffel, U.A. Synthesis and in Vivo Evaluation of a New PET Radioligand for Studying Sigma-2 Receptors. Bioorganic Med. Chem. 2005, 13, 3623–3626. [Google Scholar] [CrossRef]
- Abate, C.; Niso, M.; Lacivita, E.; Mosier, P.D.; Toscano, A.; Perrone, R. Analogues of σ Receptor Ligand 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) with Added Polar Functionality and Reduced Lipophilicity for Potential Use as Positron Emission Tomography Radiotracers. J. Med. Chem. 2011, 54, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; Meara, M.J.; Monel, B.; Vallet, T.; Zhang, Z.; Alon, A.; Donnell, H.R.; et al. Phospholipidosis Is a Shared Mechanism Underlying the in Vitro Antiviral Activity of Many Repurposed Drugs against SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Izzo, N.J.; Colom-Cadena, M.; Riad, A.A.; Xu, J.; Singh, M.; Abate, C.; Cahill, M.A.; Spires-Jones, T.L.; Bowen, W.D.; Mach, R.H.; et al. Proceedings from the Fourth International Symposium on σ-2 Receptors: Role in Health and Disease. eNeuro 2020, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=CT1812&cntry=&state=&city=&dist= (accessed on 28 April 2021).
- Hornick, J.R.; Vangveravong, S.; Spitzer, D.; Abate, C.; Berardi, F.; Goedegebuure, P.; MacH, R.H.; Hawkins, W.G. Lysosomal Membrane Permeabilization Is an Early Event in Sigma-2 Receptor Ligand Mediated Cell Death in Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2012, 31, 1–11. [Google Scholar] [CrossRef]
- Colgan, S.M.; Al-Hashimi, A.A.; Austin, R.C. Endoplasmic Reticulum Stress and Lipid Dysregulation. Expert Rev. Mol. Med. 2011, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mach, R.H.; Zeng, C.; Hawkins, W.G. The σ2 Receptor: A Novel Protein for the Imaging and Treatment of Cancer. J. Med. Chem. 2013, 56, 7137–7160. [Google Scholar] [CrossRef]
- Cantonero, C.; Camello, P.J.; Abate, C.; Berardi, F.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines. Cancers 2020, 12, 257. [Google Scholar] [CrossRef]
- Pati, M.L.; Hornick, J.R.; Niso, M.; Berardi, F.; Spitzer, D.; Abate, C.; Hawkins, W. Sigma-2 Receptor Agonist Derivatives of 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) Induce Cell Death via Mitochondrial Superoxide Production and Caspase Activation in Pancreatic Cancer. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Crawford, K.W.; Bowen, W.D. Sigma-2 Receptor Agonists Activate a Novel Apoptotic Pathway and Potentiate Antineoplastic Drugs in Breast Tumor Cell Lines. Cancer Res. 2002, 62, 313–322. [Google Scholar] [PubMed]
- Zeng, C.; Weng, C.C.; Schneider, M.E.; Puentes, L.; Riad, A.; Xu, K.; Makvandi, M.; Jin, L.; Hawkins, W.G.; Mach, R.H. TMEM97 and PGRMC1 Do Not Mediate Sigma-2 Ligand-Induced Cell Death. Cell Death Discov. 2019, 5. [Google Scholar] [CrossRef]
- Spitzer, D.; Simon, P.O.; Kashiwagi, H.; Xu, J.; Zeng, C.; Vangveravong, S.; Zhou, D.; Chang, K.; McDunn, J.E.; Hornick, J.R.; et al. Use of Multifunctional Sigma-2 Receptor Ligand Conjugates to Trigger Cancer-Selective Cell Death Signaling. Cancer Res. 2012, 72, 201–209. [Google Scholar] [CrossRef]
- Ohman, K.A.; Hashim, Y.M.; Vangveravong, S.; Nywening, T.M.; Cullinan, D.R.; Goedegebuure, S.P.; Liu, J.; Van Tine, B.A.; Tiriac, H.; Tuveson, D.A.; et al. Conjugation to the Sigma-2 Ligand SV119 Overcomes Uptake Blockade and Converts Dm-Erastin into a Potent Pancreatic Cancer Therapeutic. Oncotarget 2016, 7, 33529–33541. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Vangveravong, S.; Mcdunn, J.E.; Hawkins, W.G.; Mach, R.H. Sigma-2 Receptor Ligand as a Novel Method for Delivering a SMAC Mimetic Drug for Treating Ovarian Cancer. Br. J. Cancer 2013, 109, 2368–2377. [Google Scholar] [CrossRef]
- Hashim, Y.M.; Spitzer, D.; Vangveravong, S.; Hornick, M.C.; Garg, G.; Hornick, J.R.; Goedegebuure, P.; Mach, R.H.; Hawkins, W.G. Targeted Pancreatic Cancer Therapy with the Small Molecule Drug Conjugate SW IV-134. Mol. Oncol. 2014, 8, 956–967. [Google Scholar] [CrossRef]
- Garg, G.; Vangveravong, S.; Zeng, C.; Collins, L.; Hornick, M.; Hashim, Y.; Piwnica-Worms, D.; Powell, M.A.; Mutch, D.G.; Mach, R.H.; et al. Conjugation to a SMAC Mimetic Potentiates Sigma-2 Ligand Induced Tumor Cell Death in Ovarian Cancer. Mol. Cancer 2014, 13, 1–13. [Google Scholar] [CrossRef]
- Makvandi, M.; Tilahun, E.D.; Lieberman, B.P.; Anderson, R.C.; Zeng, C.; Xu, K.; Hou, C.; McDonald, E.S.; Pryma, D.A.; Mach, R.H. The Sigma-2 Receptor as a Therapeutic Target for Drug Delivery in Triple Negative Breast Cancer. Biochem. Biophys. Res. Commun. 2015, 467, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Szybalski, W.; Bryson, V. Genetic Studies on Microbial Cross Resistance To Toxic Agents, I: Cross resistance of Escherichia coli to fifteen antibiotics1, 2. J. Bacteriol. 1952, 64, 489–499. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP-Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral Sensitivity as a Strategy against Cancer Multidrug Resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef]
- Abate, C.; Pati, M.L.; Contino, M.; Colabufo, N.A.; Perrone, R.; Niso, M.; Berardi, F. From Mixed Sigma-2 Receptor/P-Glycoprotein Targeting Agents to Selective P-Glycoprotein Modulators: Small Structural Changes Address the Mechanism of Interaction at the Efflux Pump. Eur. J. Med. Chem. 2015, 89, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Pati, M.L.; Abate, C.; Contino, M.; Ferorelli, S.; Luisi, R.; Carroccia, L.; Niso, M.; Berardi, F. Deconstruction of 6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline Moiety to Separate P-Glycoprotein (P-gp) Activity from Sigma-2 (σ2) Receptor Affinity in Mixed P-gp/σ2 Receptor Agents. Eur. J. Med. Chem. 2015, 89, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Niso, M.; Riganti, C.; Abate, C. Collateral Sensitivity of Sigma-2 Receptor Ligands: Potentials in the Treatment of Multidrug Resistant Tumors. Recept. Clin. Investig. 2014, 1–8. [Google Scholar] [CrossRef]
- Riganti, C.; Giampietro, R.; Kopecka, J.; Costamagna, C.; Abatematteo, F.S.; Contino, M.; Abate, C. MRP1-Collateral Sensitizers as a Novel Therapeutic Approach in Resistant Cancer Therapy: An in Vitro and in Vivo Study in Lung Resistant Tumor. Int. J. Mol. Sci. 2020, 21, 3333. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Salam, N.K.; Hellawell, J.L.; Fales, H.M.; Kensler, C.B.; Ludwig, J.A.; Szakács, G.; Hibbs, D.E.; Gottesman, M.M. Synthesis, Activity, and Pharmacophore Development for Isatin-β- Thiosemicarbazones with Selective Activity toward Multidrug-Resistant Cells. J. Med. Chem. 2009, 52, 3191–3204. [Google Scholar] [CrossRef]
- Pati, M.L.; Niso, M.; Ferorelli, S.; Abate, C.; Berardi, F. Novel Metal Chelators Thiosemicarbazones with Activity at the σ2 Receptors and P-Glycoprotein: An Innovative Strategy for Resistant Tumor Treatment. RSC Adv. 2015, 5, 103131–103146. [Google Scholar] [CrossRef]
- Pati, M.L.; Niso, M.; Spitzer, D.; Berardi, F.; Contino, M.; Riganti, C.; Hawkins, W.G.; Abate, C. Multifunctional Thiosemicarbazones and Deconstructed Analogues as a Strategy to Study the Involvement of Metal Chelation, Sigma-2 (σ2) Receptor and P-gp Protein in the Cytotoxic Action: In Vitro and in Vivo Activity in Pancreatic Tumors. Eur. J. Med. Chem. 2018, 144, 359–371. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abatematteo, F.S.; Niso, M.; Lacivita, E.; Abate, C. σ2 Receptor and Its Role in Cancer with Focus on a MultiTarget Directed Ligand (MTDL) Approach. Molecules 2021, 26, 3743. https://doi.org/10.3390/molecules26123743
Abatematteo FS, Niso M, Lacivita E, Abate C. σ2 Receptor and Its Role in Cancer with Focus on a MultiTarget Directed Ligand (MTDL) Approach. Molecules. 2021; 26(12):3743. https://doi.org/10.3390/molecules26123743
Chicago/Turabian StyleAbatematteo, Francesca Serena, Mauro Niso, Enza Lacivita, and Carmen Abate. 2021. "σ2 Receptor and Its Role in Cancer with Focus on a MultiTarget Directed Ligand (MTDL) Approach" Molecules 26, no. 12: 3743. https://doi.org/10.3390/molecules26123743
APA StyleAbatematteo, F. S., Niso, M., Lacivita, E., & Abate, C. (2021). σ2 Receptor and Its Role in Cancer with Focus on a MultiTarget Directed Ligand (MTDL) Approach. Molecules, 26(12), 3743. https://doi.org/10.3390/molecules26123743