
Novel Substrates for Kinases Involved in the Biosynthesis of Inositol Pyrophosphates and Their Enhancement of ATPase Activity of a Kinase

 , , and
, , and
Abstract
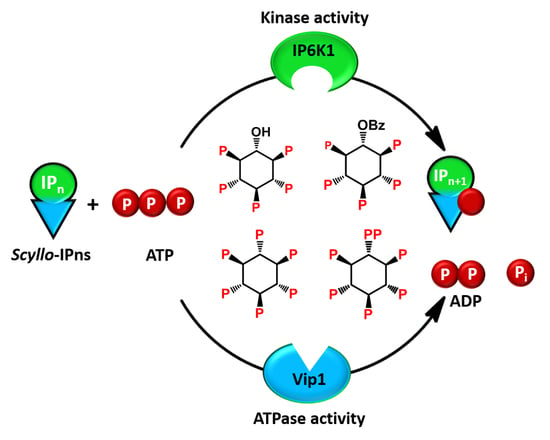
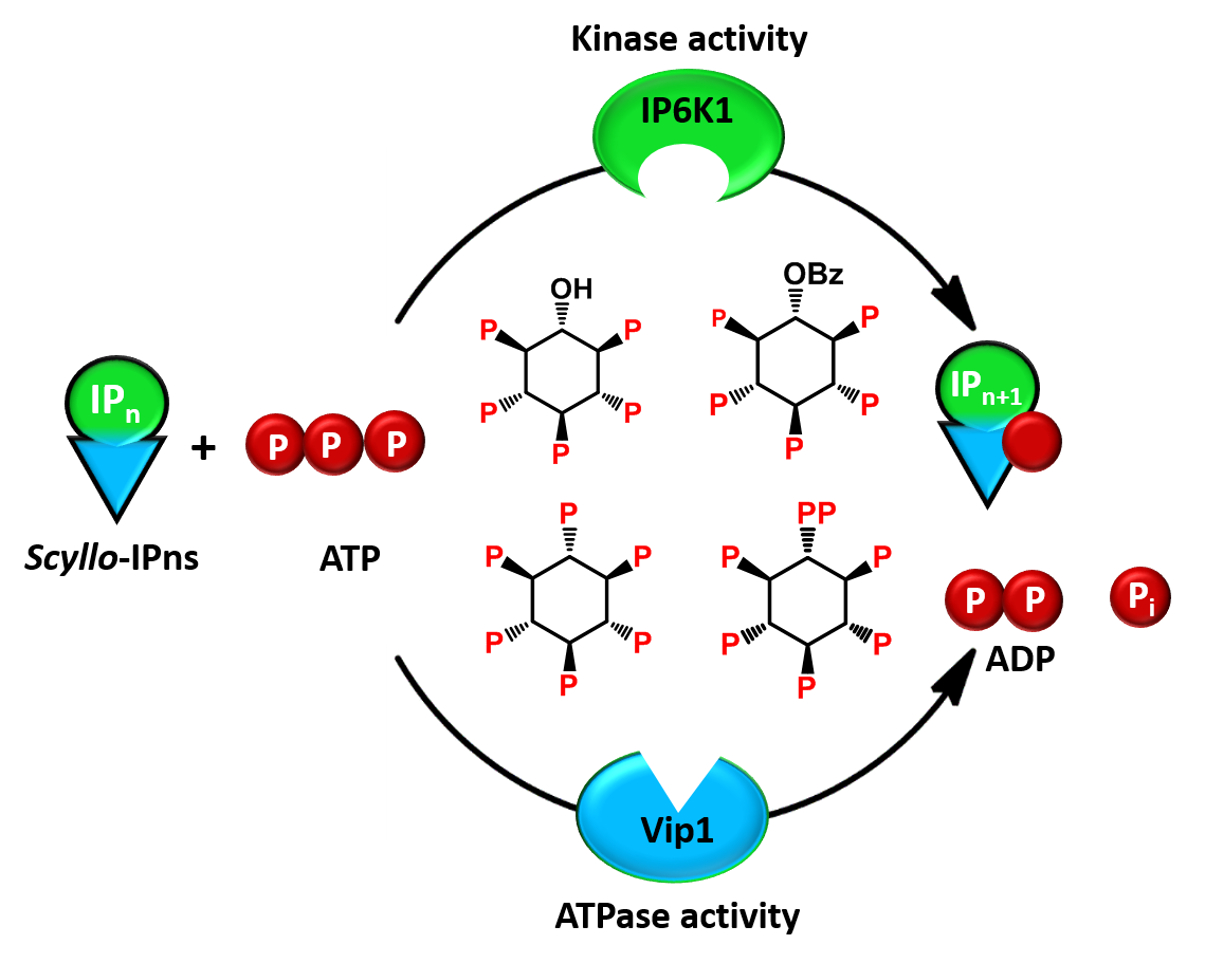{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
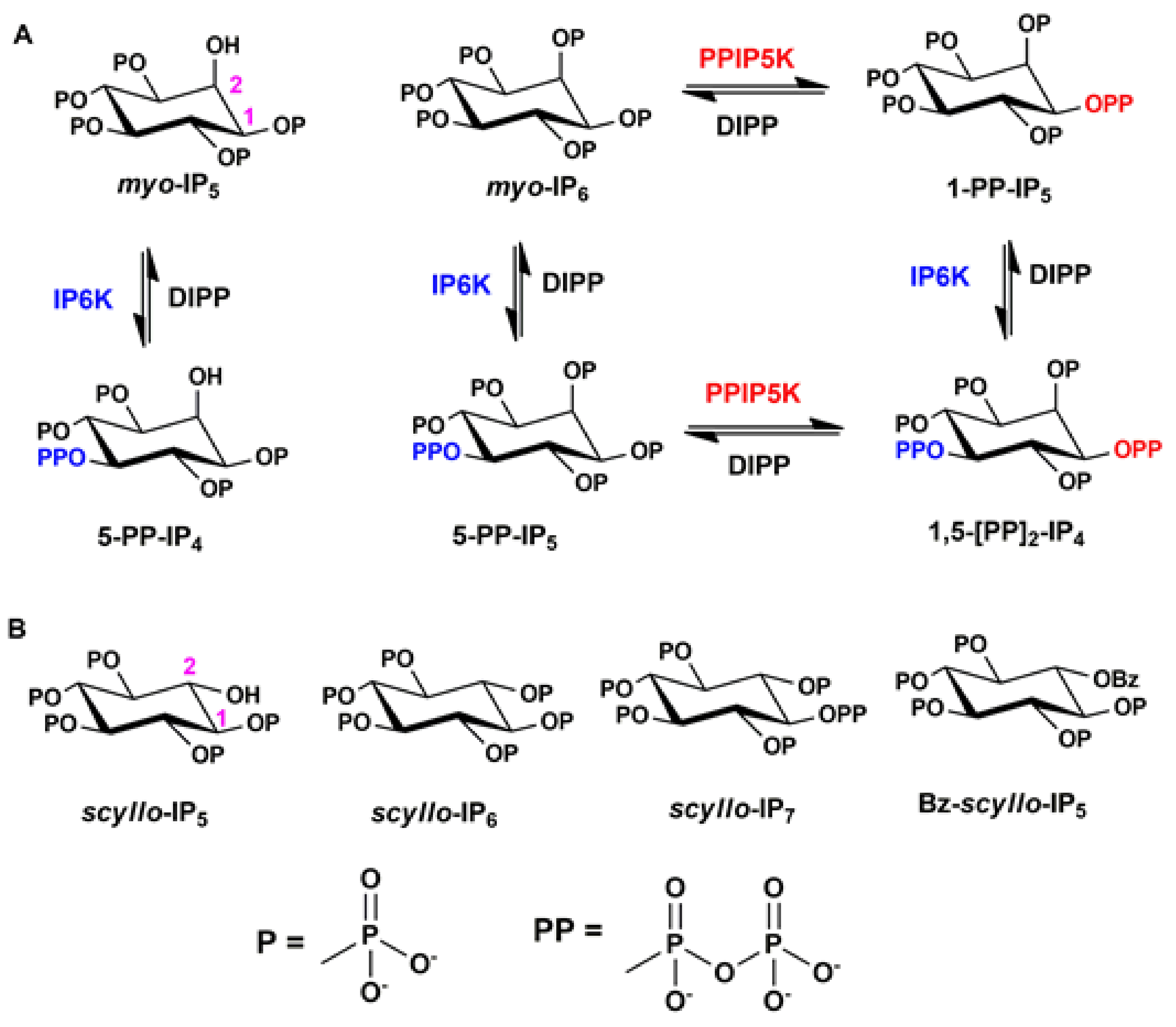
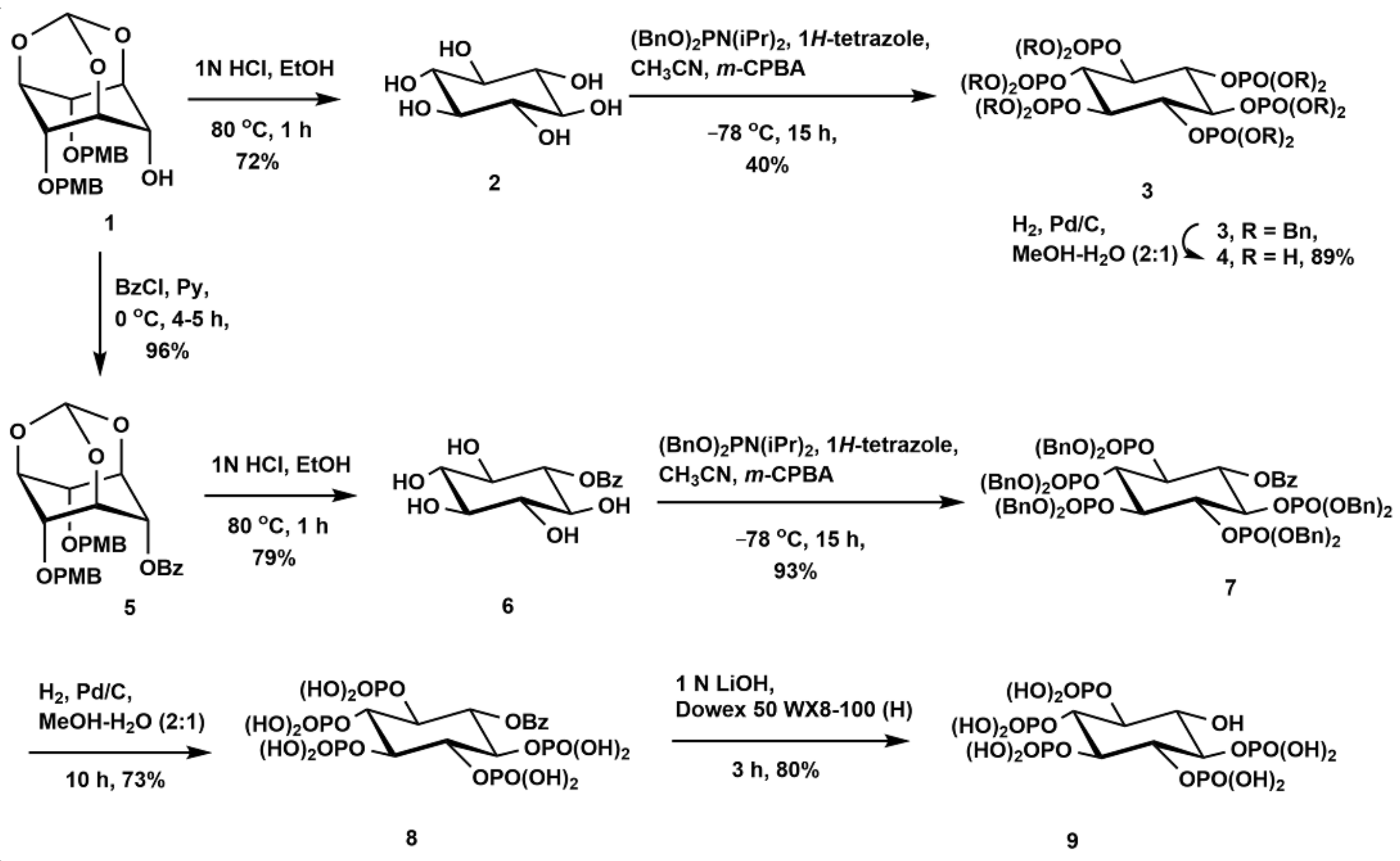
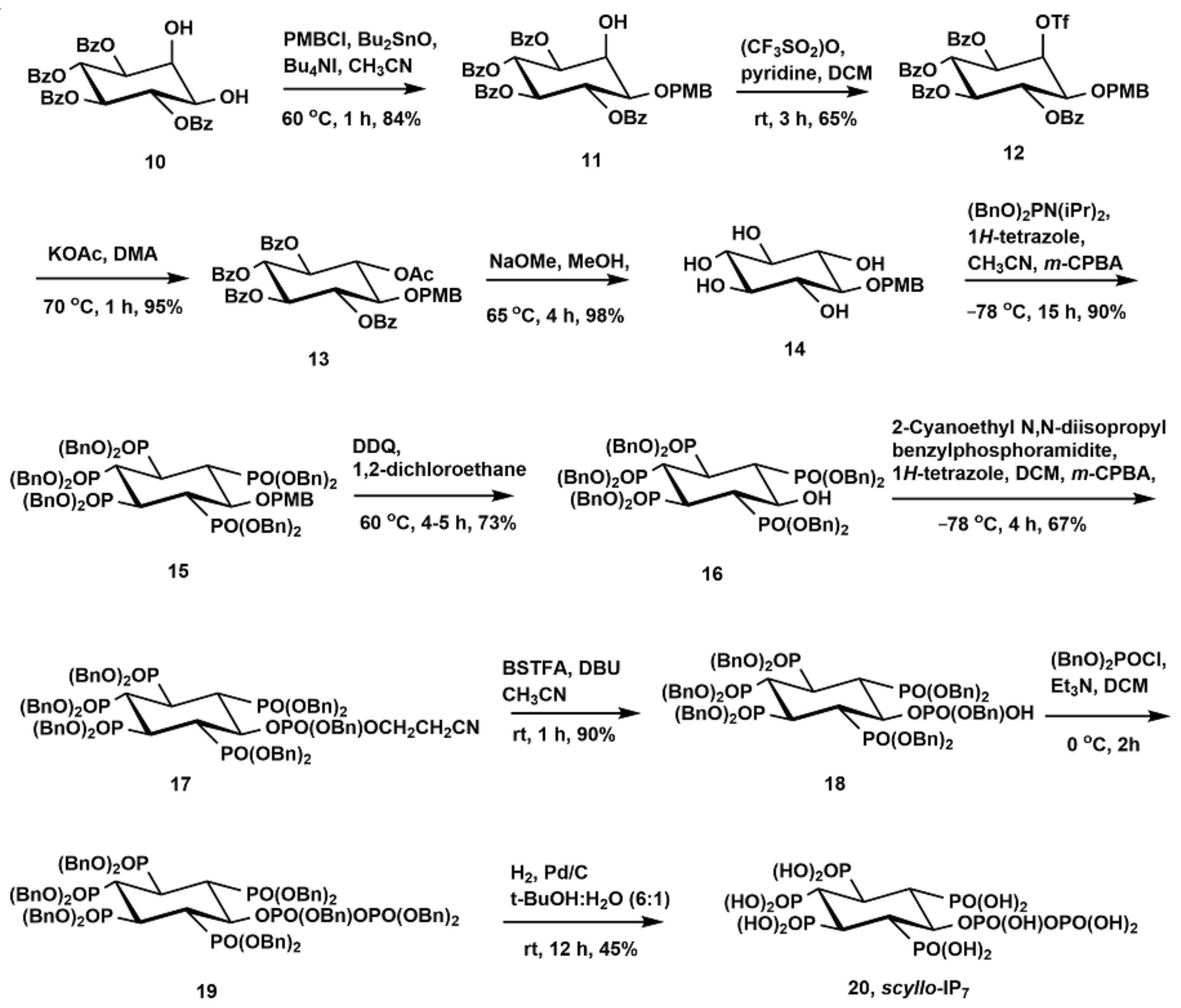
2.1. Synthesis of Scyllo-Inositol Phosphates
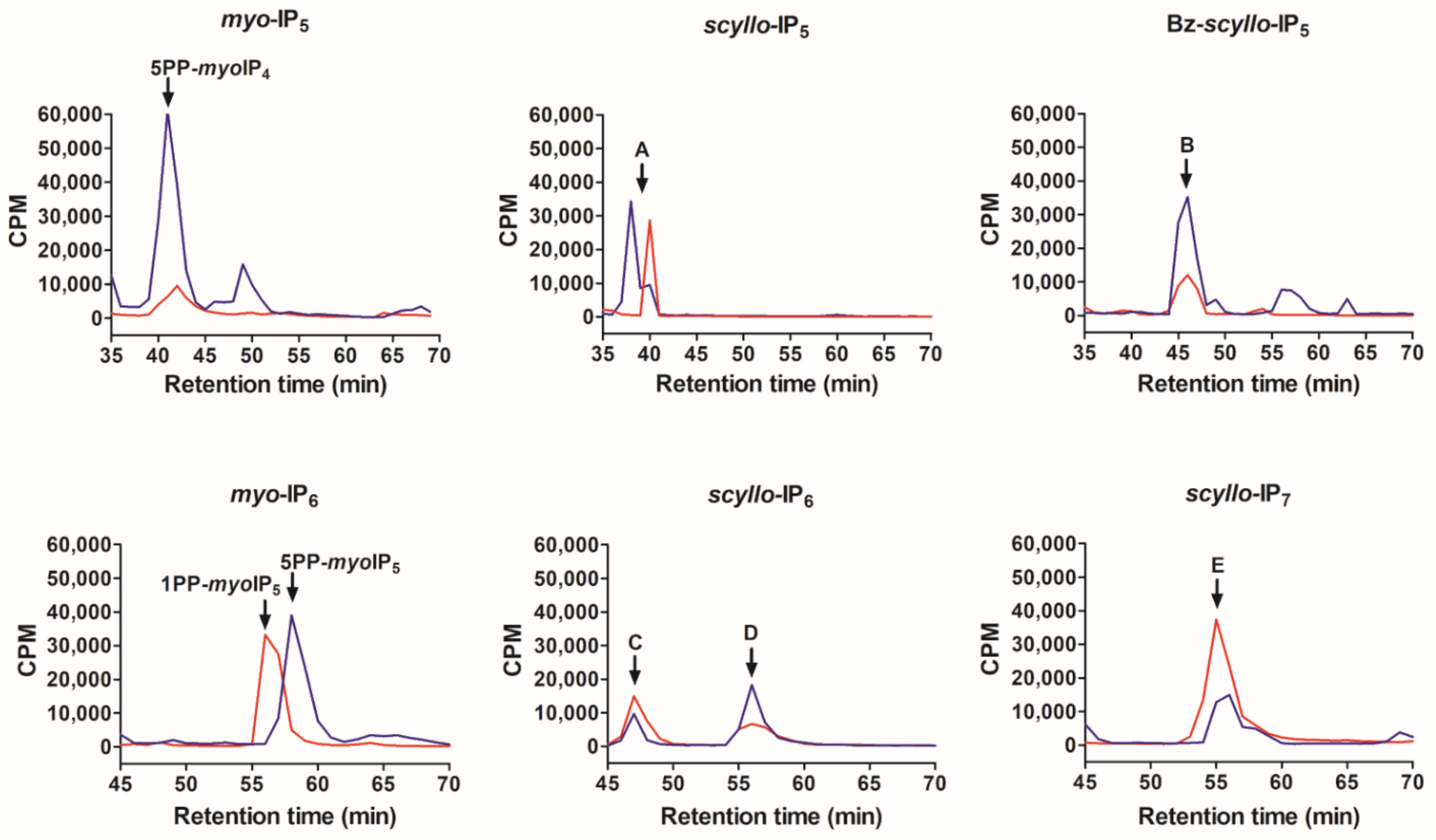
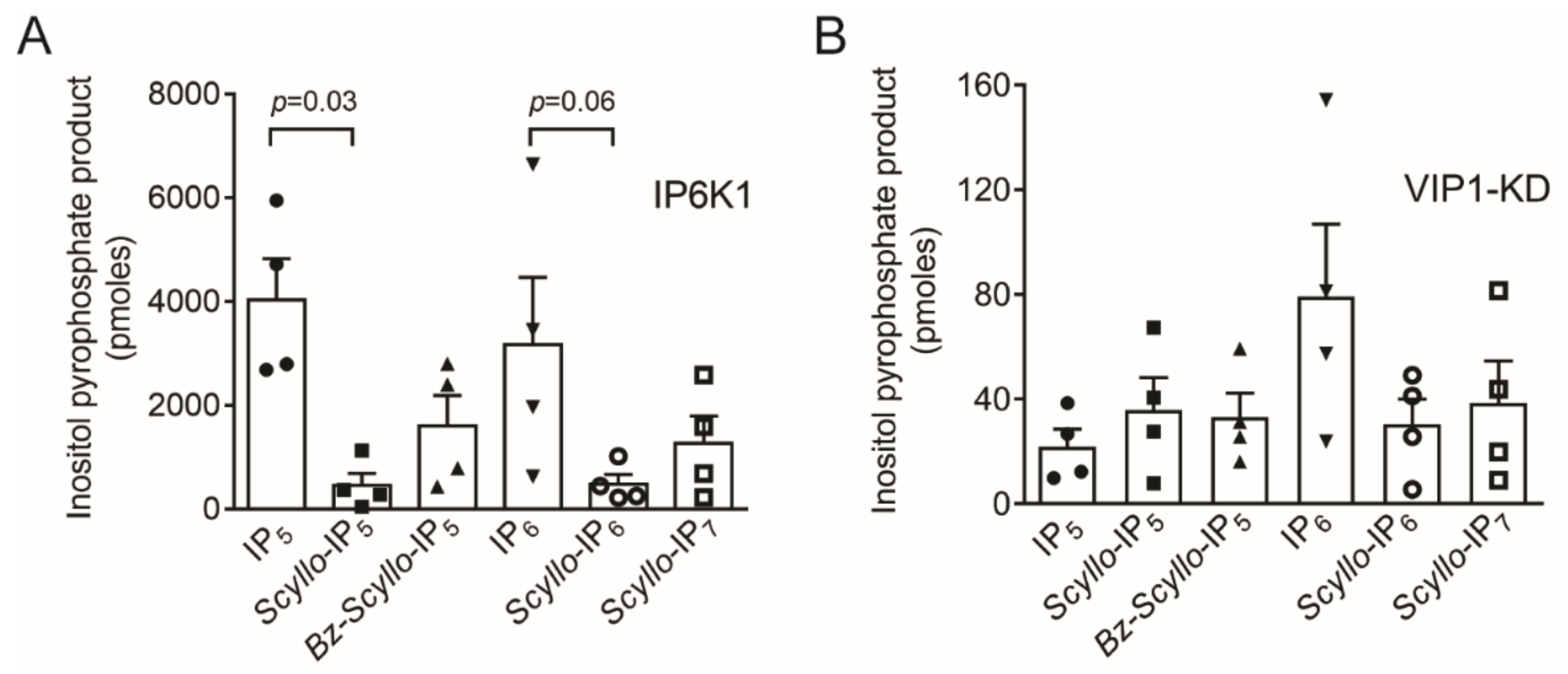
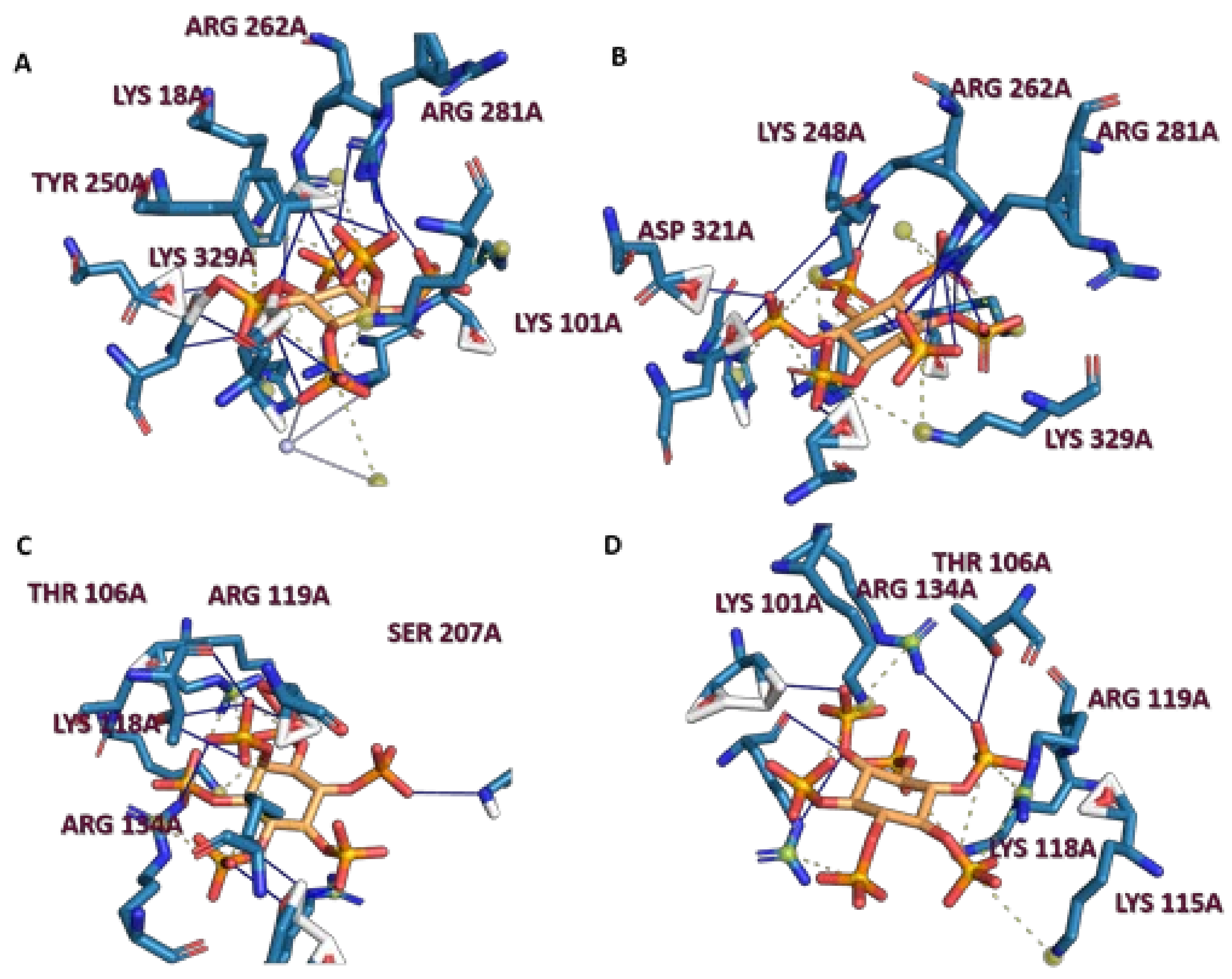
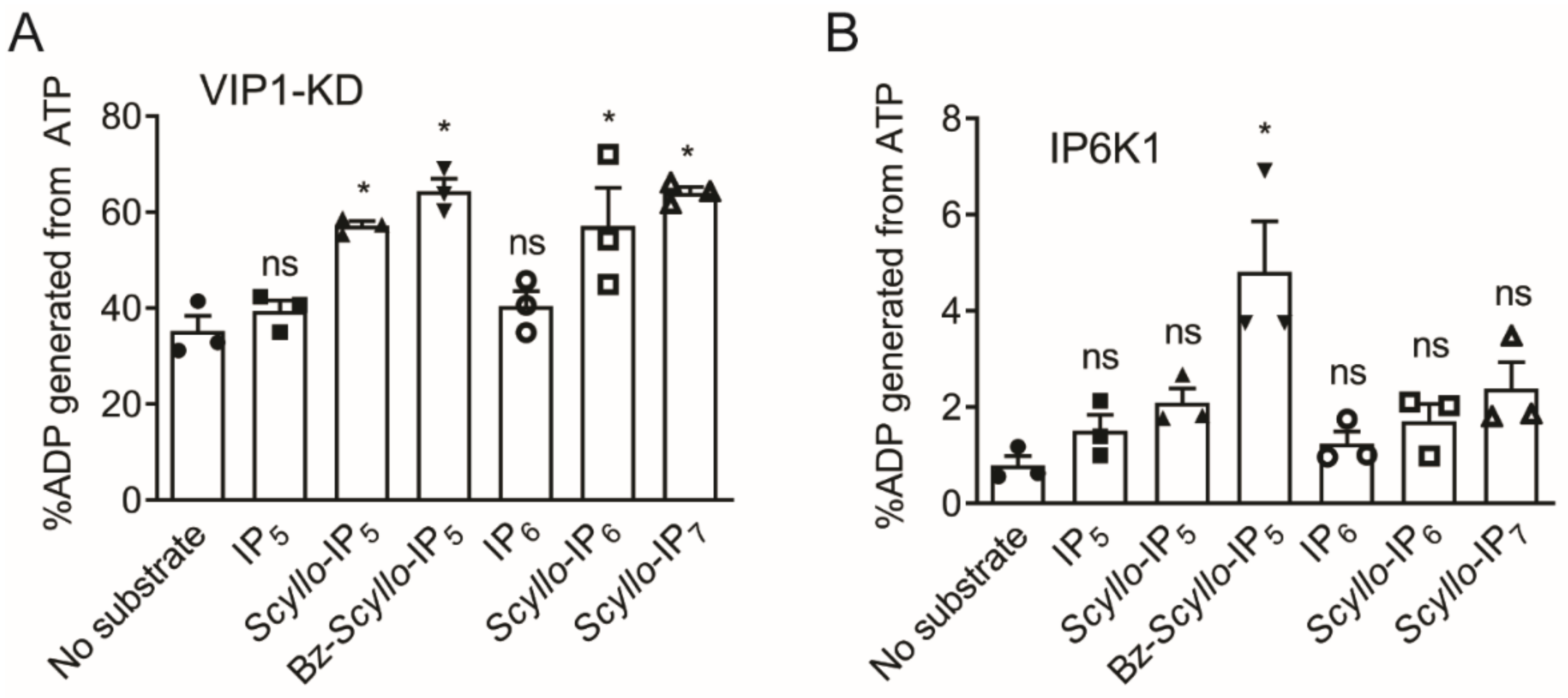
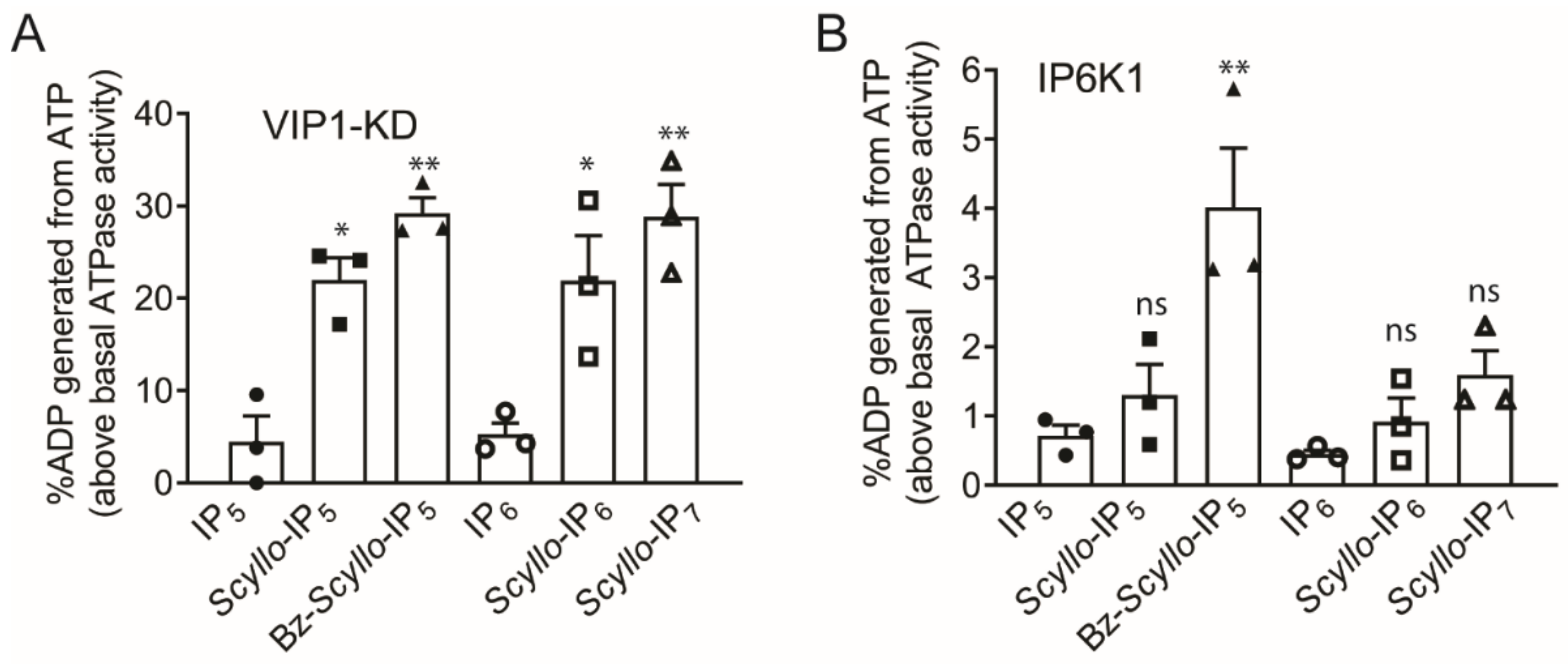
2.2. Analysis of Scyllo-Inositol Phosphates as Substrates for IP6K and PPIP5K
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Synthesis of Scyllo-Inositol (2)
3.2.2. Synthesis of 1,2,3,4,5,6-Hexakis-O-(dibenzyloxyphosphoryl)-Scyllo-Inositol (3)
3.2.3. Synthesis of Scyllo-Inositol Hexakisphosphate (4)
3.2.4. Synthesis of 6-O-Benzoyl-2,4-Di-O-(p-Methoxybenzyl)-1,3,5-O-Methylidyne-Scyllo-Inositol (5)
3.2.5. Synthesis of 1-O-Benzoyl-Scyllo-Inositol (6)
3.2.6. Synthesis of 6-O-Benzoyl-1,2,3,4,5-Pentakis-O-(Dibenzyloxyphosphoryl)-Scyllo-Inositol (7)
3.2.7. Synthesis of 6-O-Benzoyl-Scyllo-Inositol-1,2,3,4,5-Pentakisphosphate (8)
3.2.8. Synthesis of Scyllo-Inositol-1,2,3,4,5-Pentakisphosphate (9)
3.2.9. Synthesis of 1-O-p-Methoxybenzyl-3,4,5,6-Tetra-O-Benzoyl-Myo-Inositol (11)
3.2.10. Synthesis of 1-O-p-Methoxybenzyl-3,4,5,6-Tetra-O-Benzoyl-2-O-Trifluoromethanesulfonyl-Myo-Inositol (12)
3.2.11. Synthesis of 2-O-Acetyl-1-O-p-Methoxybenzyl-3,4,5,6-Tetra-O-Benzoyl-Scyllo-Inositol (13)
3.2.12. Synthesis of 1-O-p-Methoxybenzyl-Scyllo-Inositol (14)
3.2.13. Synthesis of 6-O-p-Methoxybenzyl-1,2,3,4,5-Pentakis-O-(Dibenzyloxyphosphoryl)-Scyllo-Inositol (15)
3.2.14. Synthesis of 1,2,3,4,5-Pentakis-O-(Dibenzyloxyphosphoryl)-Scyllo-Inositol (16)
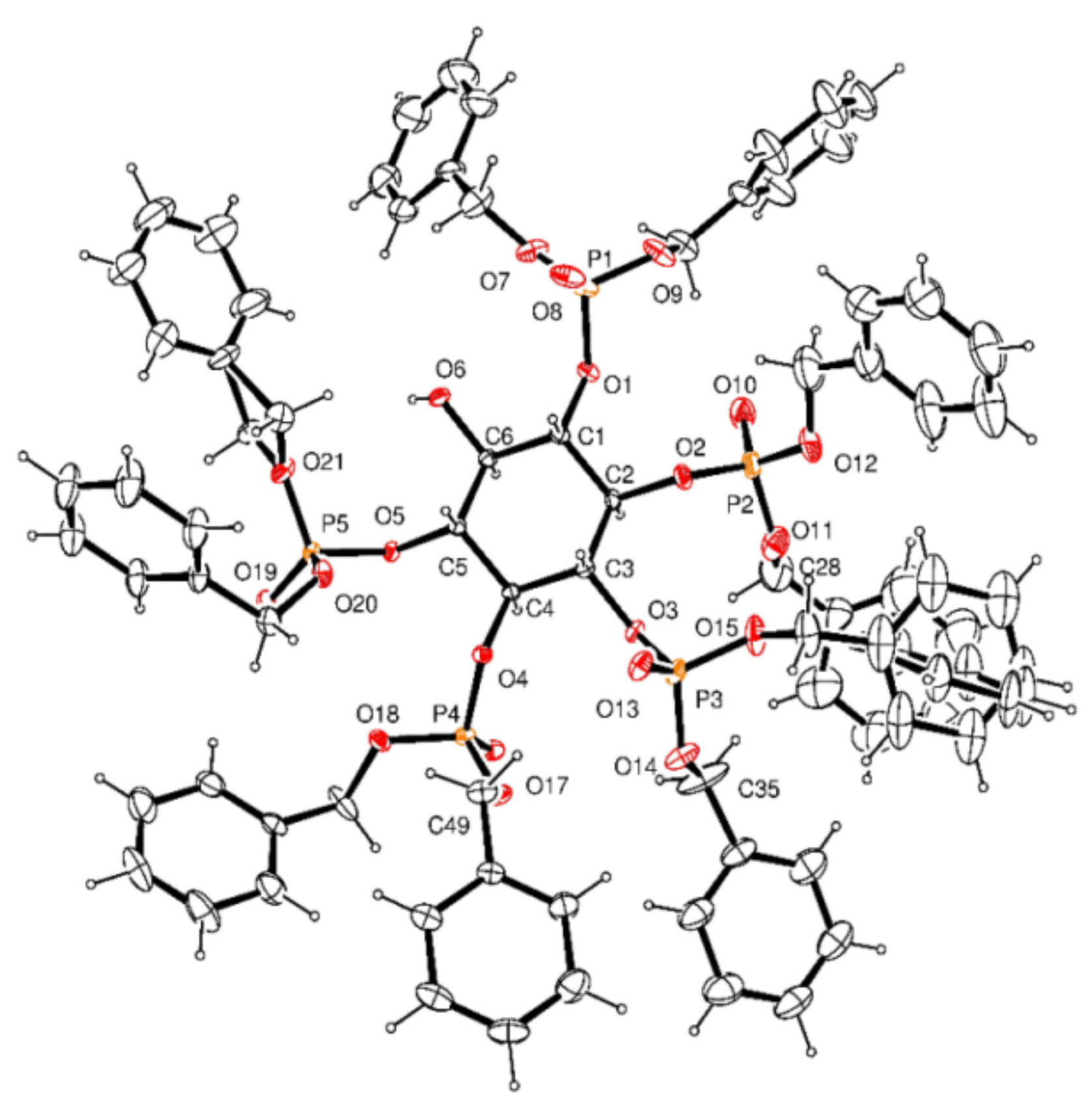
3.2.15. Crystal Data for 16
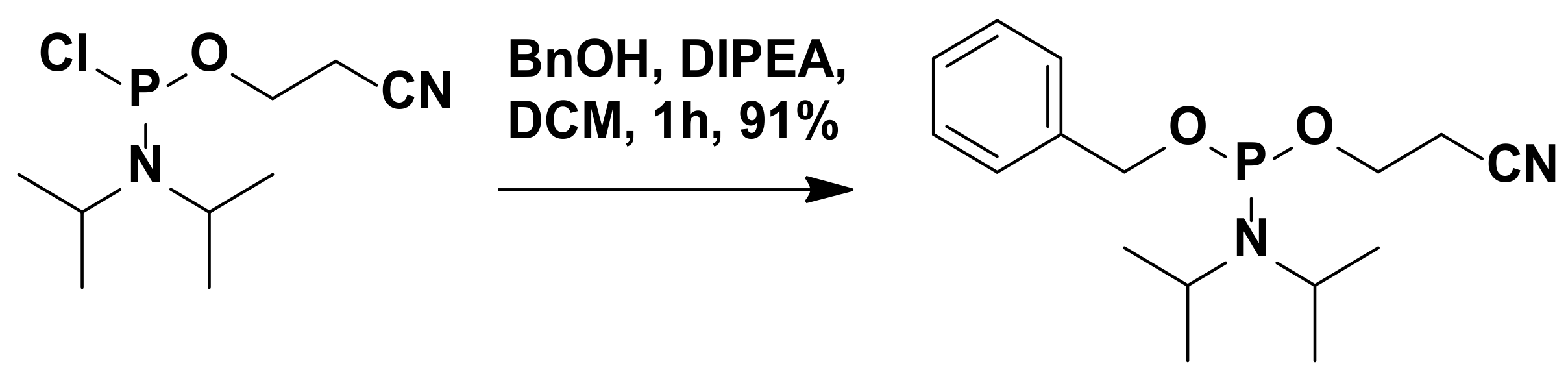
3.2.16. Synthesis of 2-Cyanoethyl-N,N-Diisopropylbenzylphosphoramidite
3.2.17. Synthesis of 6-((Benzyloxy)(2-Cyanoethoxy)Phosphoryl)-1,2,3,4,5-Pentakis-O-(Dibenzyloxyphosphoryl)-Scyllo-Inositol (17)
3.2.18. Synthesis of 6-O-Benzyloxyphosphoryl-1,2,3,4,5-Pentakis-O-(Dibenzyloxyphosphoryl)-Scyllo-Inositol (18)
3.2.19. Synthesis of 6-O-Diphospho-Scyllo-Inositol-1,2,3,4,5-Pentakisphosphate (Scyllo-IP7) (20)
3.3. Generation and Detection of PPIPn Products
3.4. Detection and Quantification of ATP Consumed and ADP Generated during the Kinase/ATPase Reaction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Irvine, R.F.; Schell, M.J. Back in the water: The return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Best, M.D.; Zhang, H.; Prestwich, G.D. Inositol polyphosphates, diphosphoinositol polyphosphates and phosphatidylinositol polyphosphate lipids: Structure, synthesis, and development of probes for studying biological activity. Nat. Prod. Rep. 2010, 27, 1403–1430. [Google Scholar] [CrossRef]
- Wilson, M.; Livermore, T.M.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369–379. [Google Scholar] [CrossRef]
- Bennett, M.; Onnebo, S.M.N.; Azevedo, C.; Saiardi, A. Inositol pyrophosphates: Metabolism and signaling. Cell. Mol. Life Sci. 2006, 63, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Ganguli, S.; Sen, J.; Bhandari, R. Inositol Pyrophosphates: Energetic, Omnipresent and Versatile Signalling Molecules. J. Indian Inst. Sci. 2017, 97, 23–40. [Google Scholar] [CrossRef]
- Barker, C.J.; Illies, C.; Gaboardi, G.C.; Berggren, P.-O. Inositol pyrophosphates: Structure, enzymology and function. Cell. Mol. Life Sci. 2009, 66, 3851–3871. [Google Scholar] [CrossRef]
- Shears, S.B.; Weaver, J.D.; Wang, H. Structural insight into inositol pyrophosphate turnover. Adv. Biol. Regul. 2013, 53, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gu, C.; Rolfes, R.J.; Jessen, H.J.; Shears, S.B. Structural and biochemical characterization of Siw14: A protein-tyrosine phosphatase fold that metabolizes inositol pyrophosphates. J. Biol. Chem. 2018, 293, 6905–6914. [Google Scholar] [CrossRef]
- Auesukaree, C.; Tochio, H.; Shirakawa, M.; Kaneko, Y.; Harashima, S. Plc1p, Arg82p, and Kcs1p, Enzymes Involved in Inositol Pyrophosphate Synthesis, Are Essential for Phosphate Regulation and Polyphosphate Accumulation in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 25127–25133. [Google Scholar] [CrossRef]
- Azevedo, C.; Szijgyarto, Z.; Saiardi, A. The signaling role of inositol hexakisphosphate kinases (IP6Ks). Adv. Enzym. Regul. 2011, 51, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Chakraborty, A.; Snyder, S.H. Inositol Pyrophosphate Pyrotechnics. Cell Metab. 2007, 5, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.; Hu, X.; Saiardi, A. Are inositol pyrophosphates signalling molecules? J. Cell. Physiol. 2009, 220, 8–15. [Google Scholar] [CrossRef]
- Lin, H.; Fridy, P.C.; Ribeiro, A.A.; Choi, J.H.; Barma, D.K.; Vogel, G.; Falck, J.R.; Shears, S.B.; York, J.D.; Mayr, G.W. Structural analysis and detection of biological inositol pyrophosphates reveal that the family of VIP/diphosphoinositol pentakisphosphate kinases are 1/3-kinases. J. Biol. Chem. 2009, 284, 1863–1872. [Google Scholar] [CrossRef]
- Majerus, P.W. A discrete signaling function for an inositol pyrophosphate. Sci. STKE 2007, 2007, pe72. [Google Scholar] [PubMed]
- Mulugu, S.; Bai, W.; Fridy, P.; Bastidas, R.J.; Otto, J.C.; Dollins, D.E.; Haystead, T.A.; Ribeiro, A.A.; York, J.D. A Conserved Family of Enzymes That Phosphorylate Inositol Hexakisphosphate. Science 2007, 316, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Onnebo, S.M.N.; Saiardi, A. Inositol Pyrophosphates Get the Vip1 Treatment. Cell 2007, 129, 647–649. [Google Scholar] [CrossRef]
- Wang, H.; Falck, J.R.; Hall, T.M.T.; Shears, S.B. Structural basis for an inositol pyrophosphate kinase surmounting phosphate crowding. Nat. Chem. Biol. 2011, 8, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wundenberg, T.; Mayr, G.W. Synthesis and biological actions of diphosphoinositol phosphates (inositol pyrophosphates), regulators of cell homeostasis. Biol. Chem. 2012, 393, 979–998. [Google Scholar] [CrossRef]
- Saiardi, A. Cell Signalling by Inositol Pyrophosphates. Prokaryotic Cytoskelet. 2012, 59, 413–443. [Google Scholar] [CrossRef]
- Tusi, M.M.; York, J.D. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv. Enzyme Regul. 2010, 50, 324–337. [Google Scholar]
- Thota, S.G.; Bhandari, R. The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J. Biosci. 2015, 40, 593–605. [Google Scholar] [CrossRef]
- Brown, N.W., Jr.; Marmelstein, A.M.; Fiedler, D. Chemical tools for interrogating inositol pyrophosphate structure and function. Chem. Soc. Rev. 2016, 45, 6311–6326. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Huang, K.; Quiocho, F.A.; O’Shea, E.K. Molecular basis of cyclin-CDK-CKI regulation by reversible binding of an inositol pyrophosphate. Nat. Chem. Biol. 2008, 4, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.-X.; Catimel, B.; Gregory, M.; Condron, M.; Kapp, E.; Holmes, A.B.; Burgess, A.W. Synthesis of an inositol hexakisphosphate (IP6) affinity probe to study the interactome from a colon cancer cell line. Integr. Biol. 2016, 8, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Mulugu, S.; York, J.D.; O’Shea, E.K. Regulation of a Cyclin-CDK-CDK Inhibitor Complex by Inositol Pyrophosphates. Science 2007, 316, 109–112. [Google Scholar] [CrossRef]
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.T.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010, 143, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.B.R.; Huang, Y.E.; Chen, J.M.C.; Saiardi, A.; Iijima, M.; Ye, K.Q.; Huang, Y.F.; Nagata, E.; Devreotes, P.; Snyder, S.H. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell 2003, 114, 559. [Google Scholar] [CrossRef]
- Albert, C.; Safrany, S.; Bembenek, E.M.; Reddy, K.M.; Falck, J.R.; Bröcker, M.; Shears, B.S.; Mayr, W.G. Biological variability in the structures of diphosphoinositol polyphosphates in Dictyostelium discoideum and mammalian cells. Biochem. J. 1997, 327, 553–560. [Google Scholar] [CrossRef]
- Wu, M.; Dul, B.E.; Trevisan, A.J.; Fiedler, D. Synthesis and characterization of non-hydrolysable diphosphoinositol polyphosphate messengers. Chem. Sci. 2012, 4, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Thompson, J.; Prestwich, G.D. A Scalable Synthesis of the IP7 Isomer, 5-PP-Ins(1,2,3,4,6)P5. Org. Lett. 2009, 11, 1551–1554. [Google Scholar] [CrossRef]
- Capolicchio, S.; Thakor, D.T.; Linden, A.; Jessen, H.J. Synthesis of Unsymmetric Diphospho-Inositol Polyphosphates. Angew. Chem. Int. Ed. 2013, 52, 6912–6916. [Google Scholar] [CrossRef]
- Capolicchio, S.; Wang, H.; Thakor, D.T.; Shears, S.B.; Jessen, H.J. Synthesis of Densely Phosphorylated Bis-1,5-Diphospho-myo-Inositol Tetrakisphosphate and its Enantiomer by Bidirectional P-Anhydride Formation. Angew. Chem. Int. Ed. 2014, 53, 9508–9511. [Google Scholar] [CrossRef]
- Pavlovic, I.; Thakor, D.T.; Bigler, L.; Wilson, M.; Laha, D.; Schaaf, G.; Saiardi, A.; Jessen, H.J. Prometabolites of 5-Diphospho-myo-inositol Pentakisphosphate. Angew. Chem. Int. Ed. 2015, 54, 9622–9626. [Google Scholar] [CrossRef]
- Pavlovic, I.; Thakor, D.T.; Vargas, J.R.; McKinlay, C.J.; Hauke, S.; Anstaett, P.; Camuña, R.C.; Bigler, L.; Gasser, G.; Schultz, C.; et al. Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nat. Commun. 2016, 7, 10622. [Google Scholar] [CrossRef]
- Riley, A.M.; Wang, H.; Weaver, J.D.; Shears, S.B.; Potter, B.V.L. First synthetic analogues of diphosphoinositol polyphosphates: Interaction with PP-InsP5 kinase. Chem. Commun. 2012, 48, 11292–11294. [Google Scholar] [CrossRef]
- Wu, M.; Chong, L.S.; Capolicchio, S.; Jessen, H.J.; Resnick, A.C.; Fiedler, D. Elucidating Diphosphoinositol Polyphosphate Function with Nonhydrolyzable Analogues. Angew. Chem. Int. Ed. 2014, 53, 7192–7197. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chong, L.S.; Perlman, D.H.; Resnick, A.C.; Fiedler, D. Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, E6757–E6765. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.D.; Wang, H.; Shears, S.B. The kinetic properties of a human PPIP5K reveal that its kinase activities are protected against the consequences of a deteriorating cellular bioenergetic environment. Biosci. Rep. 2013, 33, e00022. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Godage, H.Y.; Riley, A.M.; Weaver, J.D.; Shears, S.B.; Potter, B.V.L. Synthetic Inositol Phosphate Analogs Reveal that PPIP5K2 Has a Surface-Mounted Substrate Capture Site that Is a Target for Drug Discovery. Chem. Biol. 2014, 21, 689–699. [Google Scholar] [CrossRef]
- Shears, S.B.; Ali, N.; Craxton, A.; Bembenek, M.E. Synthesis and Metabolism of Bis-diphosphoinositol Tetrakisphosphate in Vitro and in Vivo. J. Biol. Chem. 1995, 270, 10489–10497. [Google Scholar] [CrossRef] [PubMed]
- Craxton, A.; Caffrey, J.J.; Burkhart, W.; Safrany, S.; Shears, B.S. Molecular cloning and expression of a rat hepatic multiple inositol polyphosphate phosphatase. Biochem. J. 1997, 328, 75–81. [Google Scholar] [CrossRef]
- Kilari, R.S.; Weaver, J.D.; Shears, S.B.; Safrany, S. Understanding inositol pyrophosphate metabolism and function: Kinetic characterization of the DIPPs. FEBS Lett. 2013, 587, 3464–3670. [Google Scholar] [CrossRef] [PubMed]
- Wundenberg, T.; Grabinski, N.; Lin, H.; Mayr, G.W. Discovery of InsP6-kinases as InsP6-dephosphorylating enzymes provides a new mechanism of cytosolic InsP6 degradation driven by the cellular ATP/ADP ratio. Biochem. J. 2014, 462, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Dollins, D.E.; Bai, W.; Fridy, P.C.; Otto, J.C.; Neubauer, J.L.; Gattis, S.G.; Mehta, K.P.M.; York, J.D. Vip1 is a kinase and pyrophosphatase switch that regulates inositol diphosphate signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 9356–9364. [Google Scholar] [CrossRef] [PubMed]
- Riley, A.M.; Trusselle, M.; Kuad, P.; Borkovec, M.; Cho, J.; Choi, J.H.; Qian, X.; Shears, S.B.; Spiess, B.; Potter, B.V.L. scyllo -Inositol Pentakisphosphate as an Analogue of myo -Inositol 1,3,4,5,6-Pentakisphosphate: Chemical Synthesis, Physicochemistry and Biological Applications. ChemBioChem 2006, 7, 1114–1122. [Google Scholar] [CrossRef]
- Chung, S.-K.; Kwon, Y.-U.; Chang, Y.-T.; Sohn, K.-H.; Shin, J.-H.; Park, K.-H.; Hong, B.-J.; Chung, I.-H. Synthesis of all possible regioisomers of scyllo-Inositol phosphate. Bioorganic Med. Chem. 1999, 7, 2577–2589. [Google Scholar] [CrossRef]
- Kwon, Y.-U.; Im, J.; Choi, G.; Kim, Y.-S.; Choi, K.Y.; Chung, S.-K. Synthesis of three enantiomeric pairs of scyllo-inositol phosphate and molecular interactions between all possible regioisomers of scyllo-inositol phosphate and inositol 1,4,5-trisphosphate 3-kinase. Bioorganic Med. Chem. Lett. 2003, 13, 2981–2984. [Google Scholar] [CrossRef]
- Thomas, M.P.; Mills, S.J.; Potter, B.V.L. The “Other” inositols and their phosphates: Synthesis, biology, and medicine (with recent advances in myo-inositol chemistry). Angew. Chem. Int. 2016, 55, 1614–1650. [Google Scholar] [CrossRef]
- Turner, B.L.; Cheesman, A.W.; Godage, H.Y.; Riley, A.M.; Potter, B.V.L. Determination of neo- and d-chiro-Inositol Hexakisphosphate in Soils by Solution 31P NMR Spectroscopy. Environ. Sci. Technol. 2012, 46, 4994–5002. [Google Scholar] [CrossRef]
- Sureshan, K.M.; Kiyosawa, Y.; Han, F.; Hyodo, S.; Uno, Y.; Watanabe, Y. Resolution of synthetically useful myo-inositol derivatives using the chiral auxiliary O-acetylmandelic acid. Tetrahedron Asymmetry 2005, 16, 231–241. [Google Scholar] [CrossRef]
- Babouri, R.; Traore, L.; Bekro, Y.-A.; Matveeva, V.I.; Sadykova, Y.M.; Voronina, J.K.; Burilov, A.R.; Ayad, T.; Volle, J.-N.; Virieux, D.; et al. Golden Face of Phosphine: Cascade Reaction to Bridgehead Methanophosphocines by Intramolecular Double Hydroarylation. Org. Lett. 2018, 21, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Losito, O.; Szijgyarto, Z.; Resnick, A.C.; Saiardi, A. Inositol Pyrophosphates and Their Unique Metabolic Complexity: Analysis by Gel Electrophoresis. PLoS ONE 2009, 4, e5580. [Google Scholar] [CrossRef]
- Draškovič, P.; Saiardi, A.; Bhandari, R.; Burton, A.; Ilc, G.; Kovačevič, M.; Snyder, S.H.; Podobnik, M. Inositol Hexakisphosphate Kinase Products Contain Diphosphate and Triphosphate Groups. Chem. Biol. 2008, 15, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Derose, E.F.; London, R.E.; Shears, S.B. IP6K Structure and the Molecular Determinants of Catalytic Specificity in an Inositol Phosphate Kinase Family. Nat. Commun. 2014, 5, 4178. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.0 Schrodinger, LLC. Available online: https://pymol.org/2/support.html?#citing (accessed on 9 June 2021).
- Wormald, M.; Liao, G.; Kimos, M.; Barrow, J.; Wei, H. Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS ONE 2017, 12, e0188852. [Google Scholar] [CrossRef] [PubMed]
- Paudel, H.; Carlson, G. The ATPase activity of phosphorylase kinase is regulated in parallel with its protein kinase activity. J. Biol. Chem. 1991, 266, 16524–16529. [Google Scholar] [CrossRef]
- Armstrong, R.N.; Kondo, H.; Kaiser, E.T. Cyclic AMP-dependent ATPase activity of bovine heart protein kinase. Proc. Natl. Acad. Sci. USA 1979, 76, 722–725. [Google Scholar] [CrossRef]
- Ward, N.E.; O’Brian, C.A. The intrinsic ATPase activity of protein kinase C is catalyzed at the active site of the enzyme. Biochemistry 1992, 31, 5905–5911. [Google Scholar] [CrossRef]
- Ahmad, S.; Hughes, M.A.; Johnson, G.L.; Scott, J.E. Development and Validation of a High-Throughput Intrinsic ATPase Activity Assay for the Discovery of MEKK2 Inhibitors. J. Biomol. Screen. 2013, 18, 388–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fox, T.; Fitzgibbon, M.J.; Fleming, M.A.; Hsiao, H.-M.; Brummel, C.L.; Su, M.S.-S. Kinetic mechanism and ATP-binding site reactivity of p38gamma MAP kinase. FEBS Lett. 1999, 461, 323–328. [Google Scholar] [CrossRef]
- Ferreira-Cerca, S.; Sagar, V.; Schaefer, T.; Diop, M.; Wesseling, A.-M.; Lu, H.; Chai, E.; Hurt, E.; LaRonde-LeBlanc, N. ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat. Struct. Mol. Biol. 2012, 19, 1316–1323. [Google Scholar] [CrossRef]
- Trajtenberg, F.; Graña, M.; Ruétalo, N.; Botti, H.; Buschiazzo, A. Structural and Enzymatic Insights into the ATP Binding and Autophosphorylation Mechanism of a Sensor Histidine Kinase. J. Biol. Chem. 2010, 285, 24892–24903. [Google Scholar] [CrossRef] [PubMed]
- Bruker. SADABS (Version 2.05), SMART (Version 5.631), SAINT (Version 6.45) and SHELXTL (Version 6.14); Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Wang, X.J.; Xu, B.; Mullins, A.B.; Neiler, F.K.; Etzkorn, F.A. Conformationally locked isostere of phosphoSer−cis-pro inhibits pin1 23-fold better than phosphoSer−trans-pro isostere. J. Am. Chem. Soc. 2004, 126, 15533–15542. [Google Scholar] [CrossRef]
- Bhandari, R.; Saiardi, A.; Ahmadibeni, Y.; Snowman, A.M.; Resnick, A.C.; Kristiansen, T.Z.; Molina, H.; Pandey, A.; Werner, J.K.; Juluri, K.R.; et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc. Natl. Acad. Sci. USA 2007, 104, 15305–15310. [Google Scholar] [CrossRef]
- Saiardi, A.; Erdjument-Bromage, H.; Snowman, A.M.; Tempst, P.; Snyder, S.H. Synthesis of diphosphoinositolpentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 1999, 9, 1323–1326. [Google Scholar] [CrossRef]
- Azevedo, C.; Saiardi, A. Extraction and analysis of soluble inositol polyphosphates from yeast. Nat. Protoc. 2006, 1, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Onnebo, S.M.N.; Saiardi, A. Inositol pyrophosphates modulate hydrogen peroxide signalling. Biochem. J. 2009, 423, 109–118. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.1. Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Version 1.2.0. Available online: http://avogadro.cc/ (accessed on 11 June 2021).
- Riley, A.M.; Unterlass, J.; Konieczny, V.; Taylor, C.W.; Helleday, T.; Potter, B.V.L. A synthetic diphosphoinositol phosphate analogue of inositol trisphosphate. MedChemComm 2018, 9, 1105–1113. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanrao, R.; Manorama, R.; Ganguli, S.; Madhusudhanan, M.C.; Bhandari, R.; Sureshan, K.M. Novel Substrates for Kinases Involved in the Biosynthesis of Inositol Pyrophosphates and Their Enhancement of ATPase Activity of a Kinase. Molecules 2021, 26, 3601. https://doi.org/10.3390/molecules26123601
Mohanrao R, Manorama R, Ganguli S, Madhusudhanan MC, Bhandari R, Sureshan KM. Novel Substrates for Kinases Involved in the Biosynthesis of Inositol Pyrophosphates and Their Enhancement of ATPase Activity of a Kinase. Molecules. 2021; 26(12):3601. https://doi.org/10.3390/molecules26123601
Chicago/Turabian StyleMohanrao, Raja, Ruth Manorama, Shubhra Ganguli, Mithun C. Madhusudhanan, Rashna Bhandari, and Kana M. Sureshan. 2021. "Novel Substrates for Kinases Involved in the Biosynthesis of Inositol Pyrophosphates and Their Enhancement of ATPase Activity of a Kinase" Molecules 26, no. 12: 3601. https://doi.org/10.3390/molecules26123601
APA StyleMohanrao, R., Manorama, R., Ganguli, S., Madhusudhanan, M. C., Bhandari, R., & Sureshan, K. M. (2021). Novel Substrates for Kinases Involved in the Biosynthesis of Inositol Pyrophosphates and Their Enhancement of ATPase Activity of a Kinase. Molecules, 26(12), 3601. https://doi.org/10.3390/molecules26123601