
Differential Glycosite Profiling—A Versatile Method to Compare Membrane Glycoproteomes


 and
and
Abstract

1. Introduction
2. Results
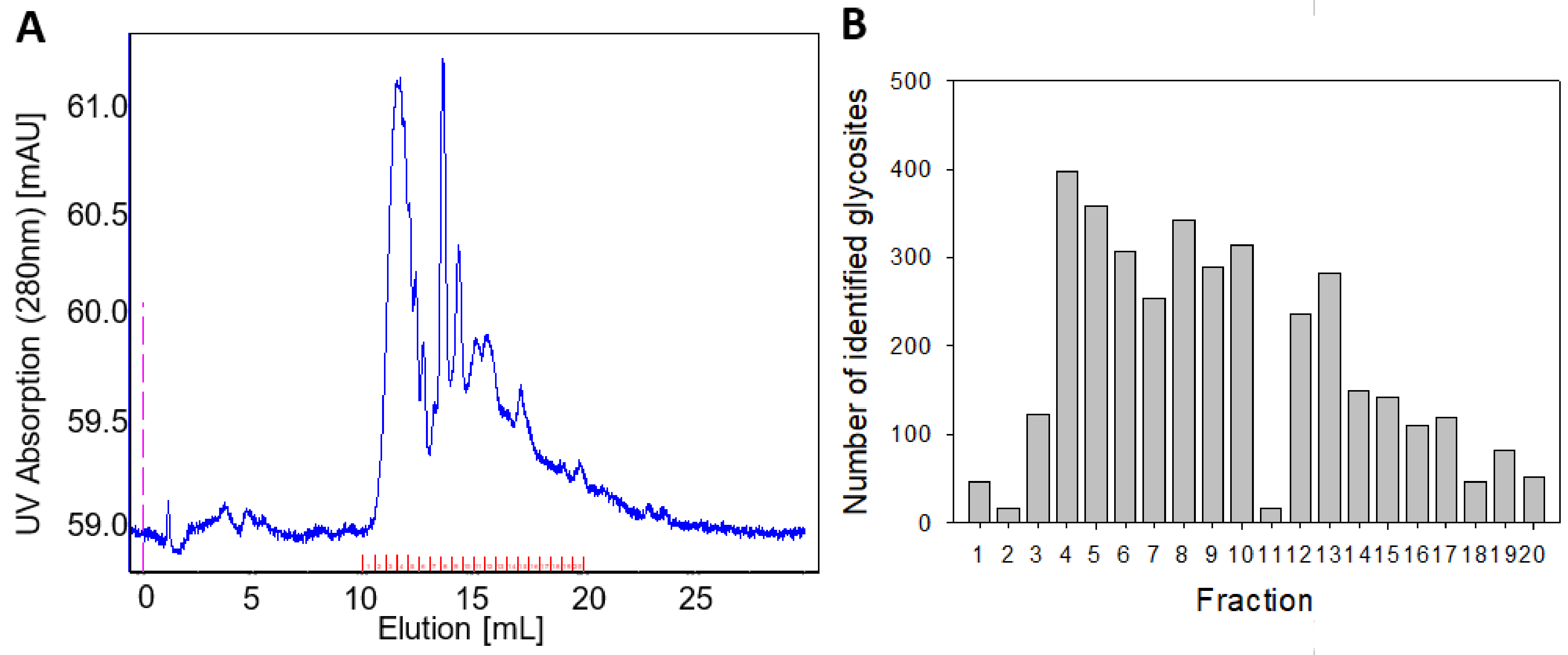
2.1. Optimization of Sample Preparation for LC–MS/MS
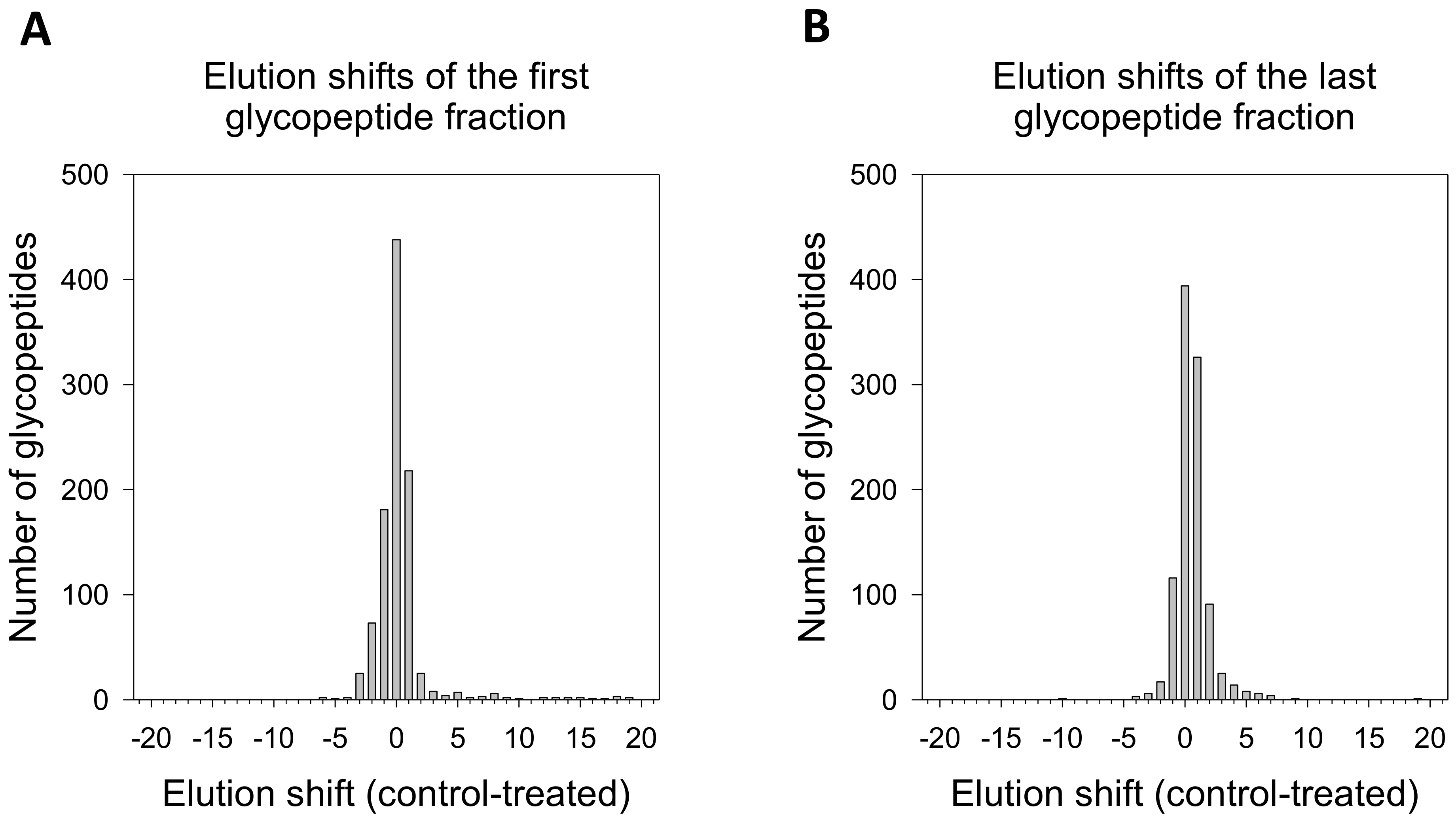
2.2. Method Implementation
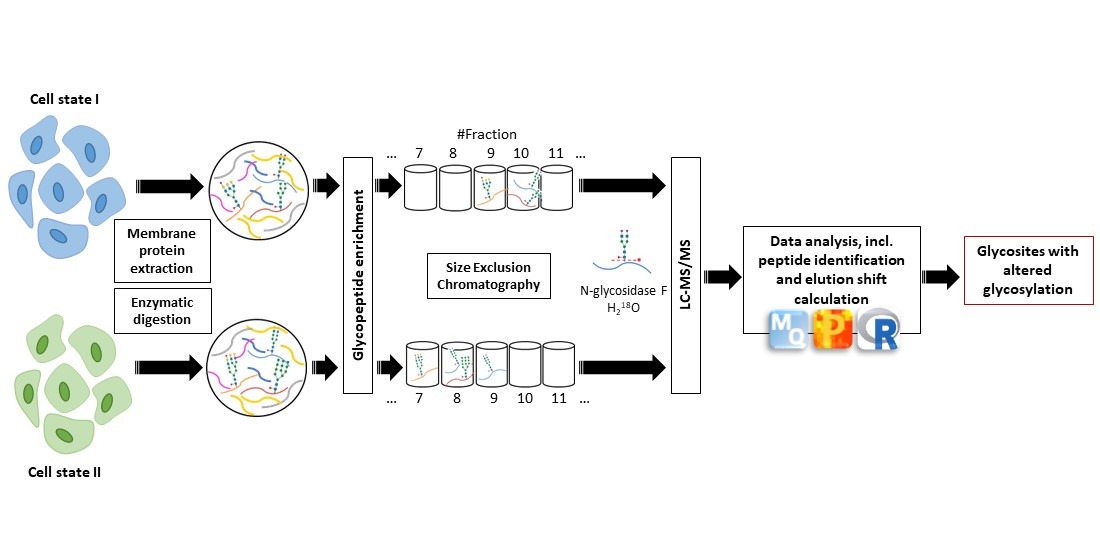
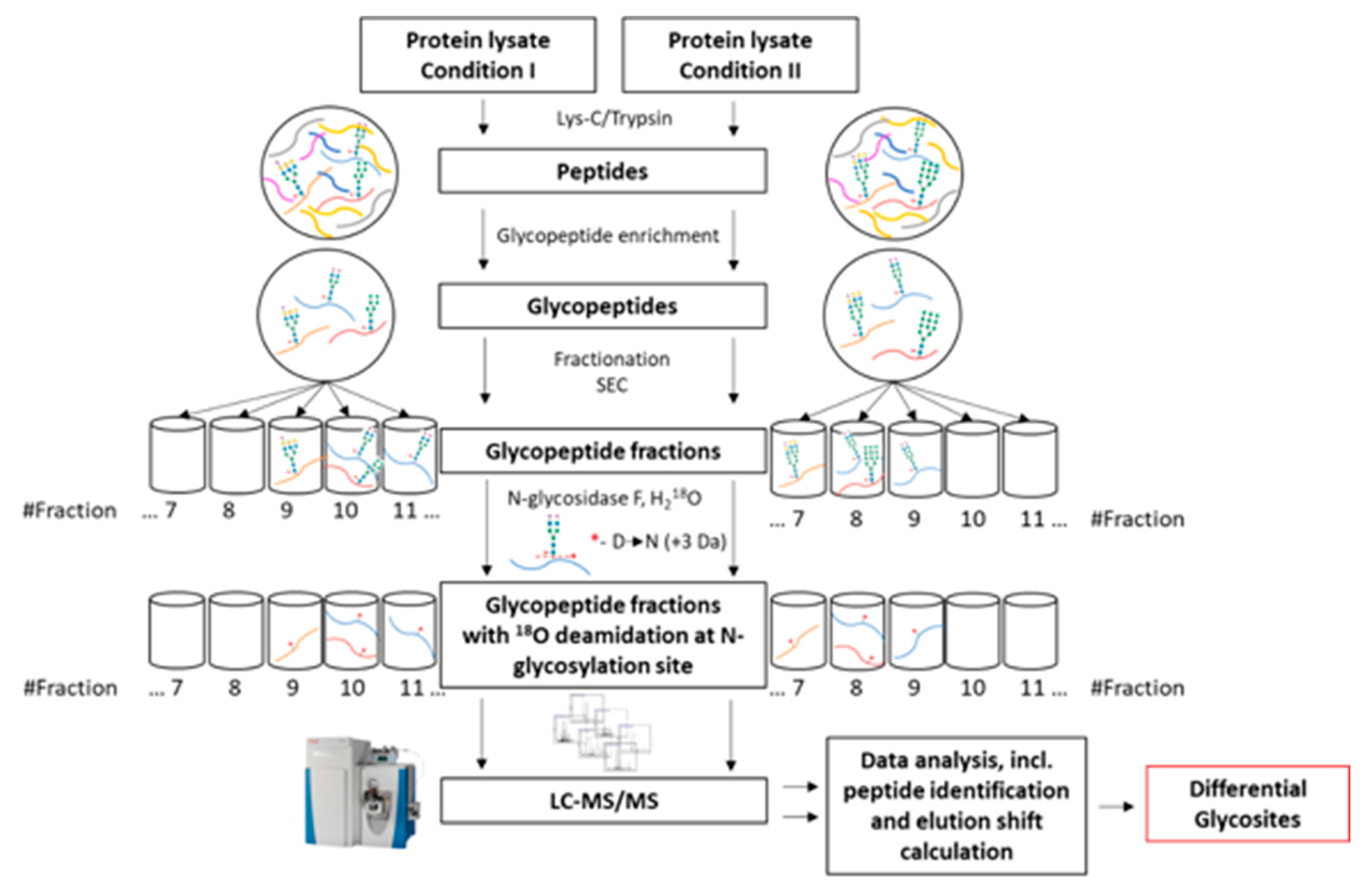
2.3. Overview of the Differential Glycosite Profiling Strategy
- Membrane protein extraction and enzymatic digestion
- Glycopeptide enrichment
- Glycopeptide fractionation
- Glycopeptide deglycosylation
- Mass spectrometric analysis (LC–MS/MS)
- Data analysis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Membrane Extraction
4.2. Membrane Protein In-Solution Lys-C/Tryptic Digestion in NH4HCO3/Urea
4.3. Stage Tipping
4.4. Glycopeptide Enrichment
4.5. Glycopeptide Fractionation
4.6. Glycopeptide Deglycosylation
4.7. LC–MS/MS
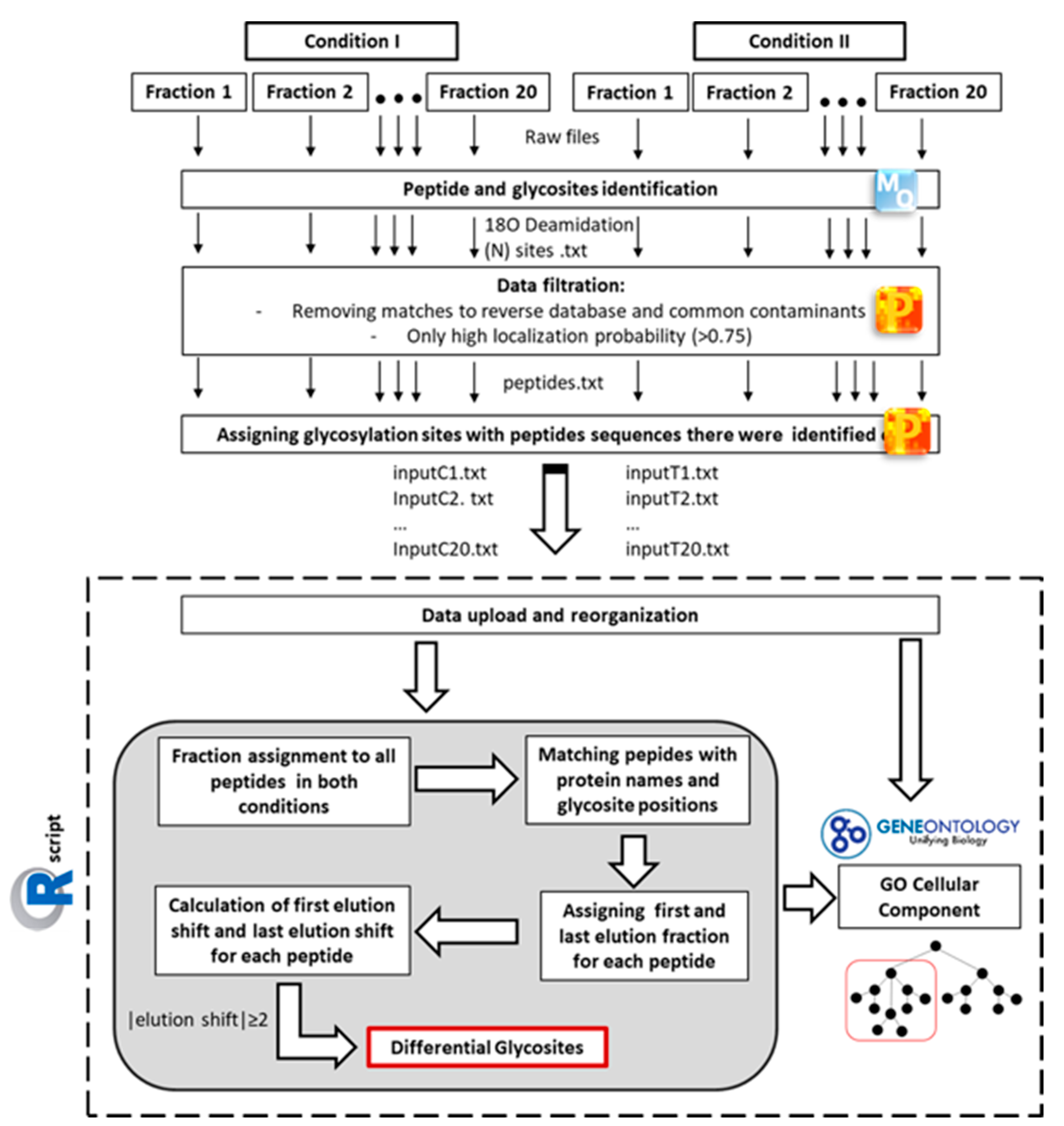
4.8. Peptide and Glycosite Identification
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Zuber, C.; Roth, J. N-Glycosylation. In The Sugar Code; Gabius, H.-J., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 87–110. [Google Scholar]
- Patsos, G.; Corfield, A. O-Glycosylation: Structural diversity and functions. In The Sugar Code; Gabius, H.-J., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 111–138. [Google Scholar]
- Shams-Eldin, H.; Debierre-Grockiego, F.; Schwarz, R.T. Glycophosphatidylinositol anchors: Structure, biosynthesis and funtions. In The Sugar Code; Gabius, H.-J., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 155–176. [Google Scholar]
- Lis, H.; Sharon, N. Protein glycosylation. Structural and functional aspects. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef]
- Gu, J.; Isaji, T.; Xu, Q.; Kariya, Y.; Gu, W.; Fukuda, T.; Du, Y. Potential roles of N-glycosylation in cell adhesion. Glycoconj. J. 2012, 29, 599–607. [Google Scholar] [CrossRef]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. [Google Scholar] [CrossRef]
- Zhou, Q.; Qiu, H. The Mechanistic Impact of N-Glycosylation on Stability, Pharmacokinetics, and Immunogenicity of Therapeutic Proteins. J. Pharm. Sci. 2019, 108, 1366–1377. [Google Scholar] [CrossRef]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef]
- Takeuchi, H.; Haltiwanger, R.S. Significance of glycosylation in Notch signaling. Biochem. Biophys. Res. Commun. 2014, 453, 235–242. [Google Scholar] [CrossRef]
- Goth, C.K.; Petäjä-Repo, U.E.; Rosenkilde, M.M. G Protein-Coupled Receptors in the Sweet Spot: Glycosylation and other Post-translational Modifications. ACS Pharmacol. Transl. Sci. 2020, 3, 237–245. [Google Scholar] [CrossRef]
- Mathew, M.P.; Donaldson, J.G. Glycosylation and glycan interactions can serve as extracellular machinery facilitating clathrin-independent endocytosis. Traffic 2019, 20, 295–300. [Google Scholar] [CrossRef]
- Johannes, L.; Billet, A. Glycosylation and raft endocytosis in cancer. Cancer Metastasis Rev. 2020, 39, 375–396. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Videira, P.A.Q.; Castro-Caldas, M. Linking Glycation and Glycosylation With Inflammation and Mitochondrial Dysfunction in Parkinson’s Disease. Front. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef]
- Lyons, J.J.; Milner, J.D.; Rosenzweig, S.D. Glycans Instructing Immunity: The Emerging Role of Altered Glycosylation in Clinical Immunology. Front. Pediatrics 2015, 3, 54. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Cavdarli, S.; Uchimura, K.; Allain, F.; Delannoy, P. Glycosylation changes in inflammatory diseases. Adv. Protein Chem. Struct. Biol. 2020, 119, 111–156. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, J.; Lubman, D.M.; Gao, C. Aberrant glycosylation and cancer biomarker discovery: A promising and thorny journey. Clin. Chem. Lab. Med. 2019, 57, 407–416. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta. Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef]
- Roth, Z.; Yehezkel, G.; Khalaila, I. Identification and quantification of protein glycosylation. Int. J. Carbohydr. Chem. 2012, 2012. [Google Scholar] [CrossRef]
- Mulloy, B.; Dell, A.; Stanley, P.; Prestegard, J.H. Structural analysis of glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Eds.; Cold Spring Harbor Press: New York, NY, USA, 2017. [Google Scholar]
- Rudd, P.; Karlsson, N.G.; Khoo, K.H.; Packer, N.H. Glycomics and glycoproteomics. In Essentials of GLycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Eds.; Cold Spring Harbor Press: New York, NY, USA, 2017. [Google Scholar]
- Diaz, S.; Varki, A. Metabolic radiolabeling of animal cell glycoconjugates. Curr. Protoc. Protein Sci. 2009, 26, 1–16. [Google Scholar] [CrossRef]
- Gebert, J.; Schnölzer, M.; Warnken, U.; Kopitz, J. Combining Click Chemistry-Based Proteomics With Dox-Inducible Gene Expression. Methods Enzymol. 2017, 585, 295–327. [Google Scholar] [CrossRef]
- Nilsson, J.; Halim, A.; Grahn, A.; Larson, G. Targeting the glycoproteome. Glycoconj. J. 2013, 30, 119–136. [Google Scholar] [CrossRef]
- Shajahan, A.; Heiss, C.; Ishihara, M.; Azadi, P. Glycomic and glycoproteomic analysis of glycoproteins-a tutorial. Anal. Bioanal. Chem. 2017, 409, 4483–4505. [Google Scholar] [CrossRef]
- Ahadova, A.; Gebert, J.; von Knebel Doeberitz, M.; Kopitz, J.; Kloor, M. Dose-dependent effect of 2-deoxy-D-glucose on glycoprotein mannosylation in cancer cells. IUBMB Life 2015, 67, 218–226. [Google Scholar] [CrossRef]
- Kurtoglu, M.; Maher, J.C.; Lampidis, T.J. Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxid. Redox Signal. 2007, 9, 1383–1390. [Google Scholar] [CrossRef]
- Xi, H.; Kurtoglu, M.; Lampidis, T.J. The wonders of 2-deoxy-D-glucose. IUBMB Life 2014, 66, 110–121. [Google Scholar] [CrossRef]
- Laussel, C.; Léon, S. Cellular toxicity of the metabolic inhibitor 2-deoxyglucose and associated resistance mechanisms. Biochem. Pharmacol. 2020, 182, 114213. [Google Scholar] [CrossRef]
- Colley, K.J.; Varki, A.; Kinoshita, T. Cellular Organization of Glycosylation. In Essentials of Glycobiology; Copyright 2015–2017 by The Consortium of Glycobiology Editors, La Jolla, California; All rights reserved; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015; pp. 41–49. [Google Scholar] [CrossRef]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Brewer, C.F.; Bhattacharyya, L. Specificity of concanavalin A binding to asparagine-linked glycopeptides. A nuclear magnetic relaxation dispersion study. J. Biol. Chem. 1986, 261, 7306–7310. [Google Scholar] [CrossRef]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: A database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteom. 2019, 16, 35. [Google Scholar] [CrossRef]
- Thaysen-Andersen, M.; Packer, N.H.; Schulz, B.L. Maturing Glycoproteomics Technologies Provide Unique Structural Insights into the N-glycoproteome and Its Regulation in Health and Disease. Mol. Cell. Proteom. 2016, 15, 1773–1790. [Google Scholar] [CrossRef]
- Kaltner, H.; Abad-Rodríguez, J.; Corfield, A.P.; Kopitz, J.; Gabius, H.J. The sugar code: Letters and vocabulary, writers, editors and readers and biosignificance of functional glycan-lectin pairing. Biochem. J. 2019, 476, 2623–2655. [Google Scholar] [CrossRef]
- Laine, R.A. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide: The Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994, 4, 759–767. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Kim, J.Y.; Yoo, J.S. Quantitative mass spectrometric analysis of glycoproteins combined with enrichment methods. Mass Spectrom. Rev. 2015, 34, 148–165. [Google Scholar] [CrossRef]
- Thaysen-Andersen, M.; Kolarich, D.; Packer, N.H. Glycomics & Glycoproteomics: From Analytics to Function. Mol. Omics 2021, 17, 8–10. [Google Scholar] [CrossRef]
- Tabb, D.L.; Vega-Montoto, L.; Rudnick, P.A.; Variyath, A.M.; Ham, A.J.; Bunk, D.M.; Kilpatrick, L.E.; Billheimer, D.D.; Blackman, R.K.; Cardasis, H.L.; et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010, 9, 761–776. [Google Scholar] [CrossRef]
- Hendrickson, O.D.; Zherdev, A.V. Analytical Application of Lectins. Crit. Rev. Anal. Chem. 2018, 48, 279–292. [Google Scholar] [CrossRef]
- Tikhonov, A.; Smoldovskaya, O.; Feyzkhanova, G.; Kushlinskii, N.; Rubina, A. Glycan-specific antibodies as potential cancer biomarkers: A focus on microarray applications. Clin. Chem. Lab. Med. 2020, 58, 1611–1622. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Consortium, T.G.O. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Chen, S.H. Stable isotope dimethyl labelling for quantitative proteomics and beyond. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374. [Google Scholar] [CrossRef]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, M.H.; Clauser, K.R.; Mertins, P.; Carr, S.A.; Kuster, B. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol. Cell. Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. Crit. Rev. Biochem. 1984, 16, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Valverde, P.; Ardá, A.; Reichardt, N.C.; Jiménez-Barbero, J.; Gimeno, A. Glycans in drug discovery. MedChemComm 2019, 10, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flügge, U.-I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Michalak, M.; Warnken, U.; Schnölzer, M.; Gabius, H.J.; Kopitz, J. Detection of malignancy-associated phosphoproteome changes in human colorectal cancer induced by cell surface binding of growth-inhibitory galectin-4. IUBMB Life 2019, 71, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Block, G.; Chen, H.; Folch-Puy, E.; Foronjy, R.; Jalili, R.; Jendresen, C.B.; Kimura, M.; Kraft, E.; Lindemose, S.; et al. One-step isolation of plasma membrane proteins using magnetic beads with immobilized concanavalin A. Protein Expr Purif 2008, 62, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- R Team. RStudio: Integrated Development Environment for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Wickham, H.; Hester, J.; Francois, R. Readr: Read Rectangular Text Data. R Package Version: 2018. Available online: https://cran.r-project.org/package=readr (accessed on 25 February 2021).
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. R Package Version: 2019. Available online: https://cran.r-project.org/package=stringr (accessed on 25 February 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H.; Henry, L. tidyr: Tidy Messy Data. R Package Version: 2020. Available online: https://cran.r-project.org/package=tidyr (accessed on 25 February 2021).
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R Package Version: 2019. Available online: https://cran.r-project.org/package=dplyr (accessed on 25 February 2021).
- Henry, L.; Wickham, H. purrr: Functional Programming Tools. R Package Version: 2020. Available online: https://cran.r-project.org/package=purrr (accessed on 25 February 2021).
- Müller, K.; Wickham, H. tibble: Simple Data Frames. R Package Version: 2020. Available online: https://cran.r-project.org/package=tibble (accessed on 25 February 2021).
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. R Package Version: 2019. Available online: http://bioconductor.org/packages/org.Hs.eg.db/ (accessed on 25 February 2021).
- Carlson, M. GO.db: A Set of Annotation Maps Describing the Entire Gene Ontology. R Package Version: 2019. Available online: http://bioconductor.org/packages/GO.db/ (accessed on 25 February 2021).
- Cheng, K.; Zhou, Y.; Neelamegham, S. DrawGlycan-SNFG: A robust tool to render glycans and glycopeptides with fragmentation information. Glycobiology 2017, 27, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # Total | |
|---|---|
| Identified 18O deamidated sites | 1879 |
| Known glycosites 1 | 1331 |
| Identified individual glycopeptides in all fractions | 1820 |
| Glycopeptides in both conditions | 1066 |
| Differential glycopeptides | 320 |
| Differential plasma membrane glycopeptides 2 | 136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalak, M.; Kalteis, M.S.; Ahadova, A.; Kloor, M.; Kriegsmann, M.; Kriegsmann, K.; Warnken, U.; Helm, D.; Kopitz, J. Differential Glycosite Profiling—A Versatile Method to Compare Membrane Glycoproteomes. Molecules 2021, 26, 3564. https://doi.org/10.3390/molecules26123564
Michalak M, Kalteis MS, Ahadova A, Kloor M, Kriegsmann M, Kriegsmann K, Warnken U, Helm D, Kopitz J. Differential Glycosite Profiling—A Versatile Method to Compare Membrane Glycoproteomes. Molecules. 2021; 26(12):3564. https://doi.org/10.3390/molecules26123564
Chicago/Turabian StyleMichalak, Malwina, Martin Simon Kalteis, Aysel Ahadova, Matthias Kloor, Mark Kriegsmann, Katharina Kriegsmann, Uwe Warnken, Dominic Helm, and Jürgen Kopitz. 2021. "Differential Glycosite Profiling—A Versatile Method to Compare Membrane Glycoproteomes" Molecules 26, no. 12: 3564. https://doi.org/10.3390/molecules26123564
APA StyleMichalak, M., Kalteis, M. S., Ahadova, A., Kloor, M., Kriegsmann, M., Kriegsmann, K., Warnken, U., Helm, D., & Kopitz, J. (2021). Differential Glycosite Profiling—A Versatile Method to Compare Membrane Glycoproteomes. Molecules, 26(12), 3564. https://doi.org/10.3390/molecules26123564