
Exploiting Protein N-Terminus for Site-Specific Bioconjugation



Abstract
1. Introduction
2. Chemical Strategies Targeting Protein N-Terminus
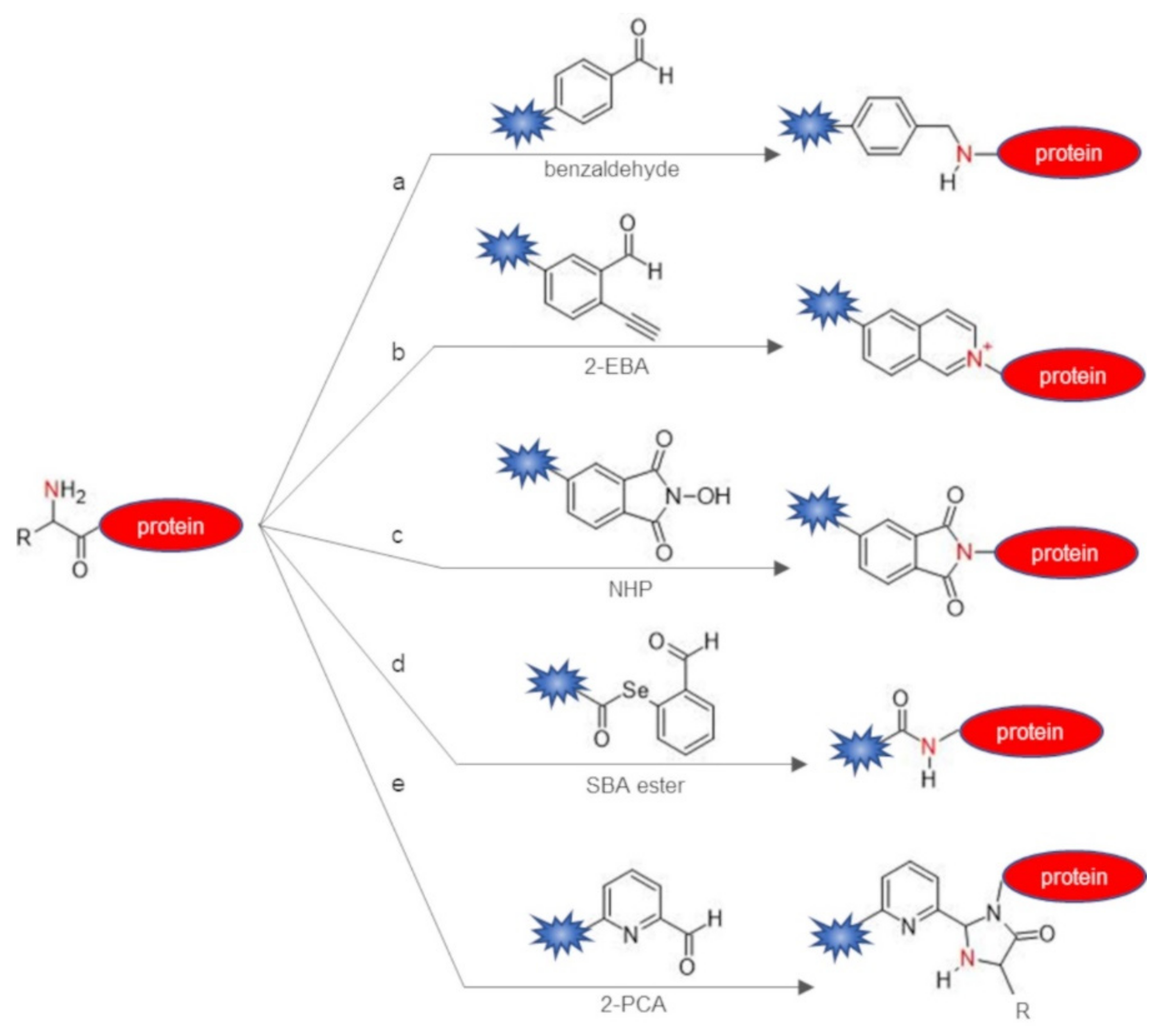
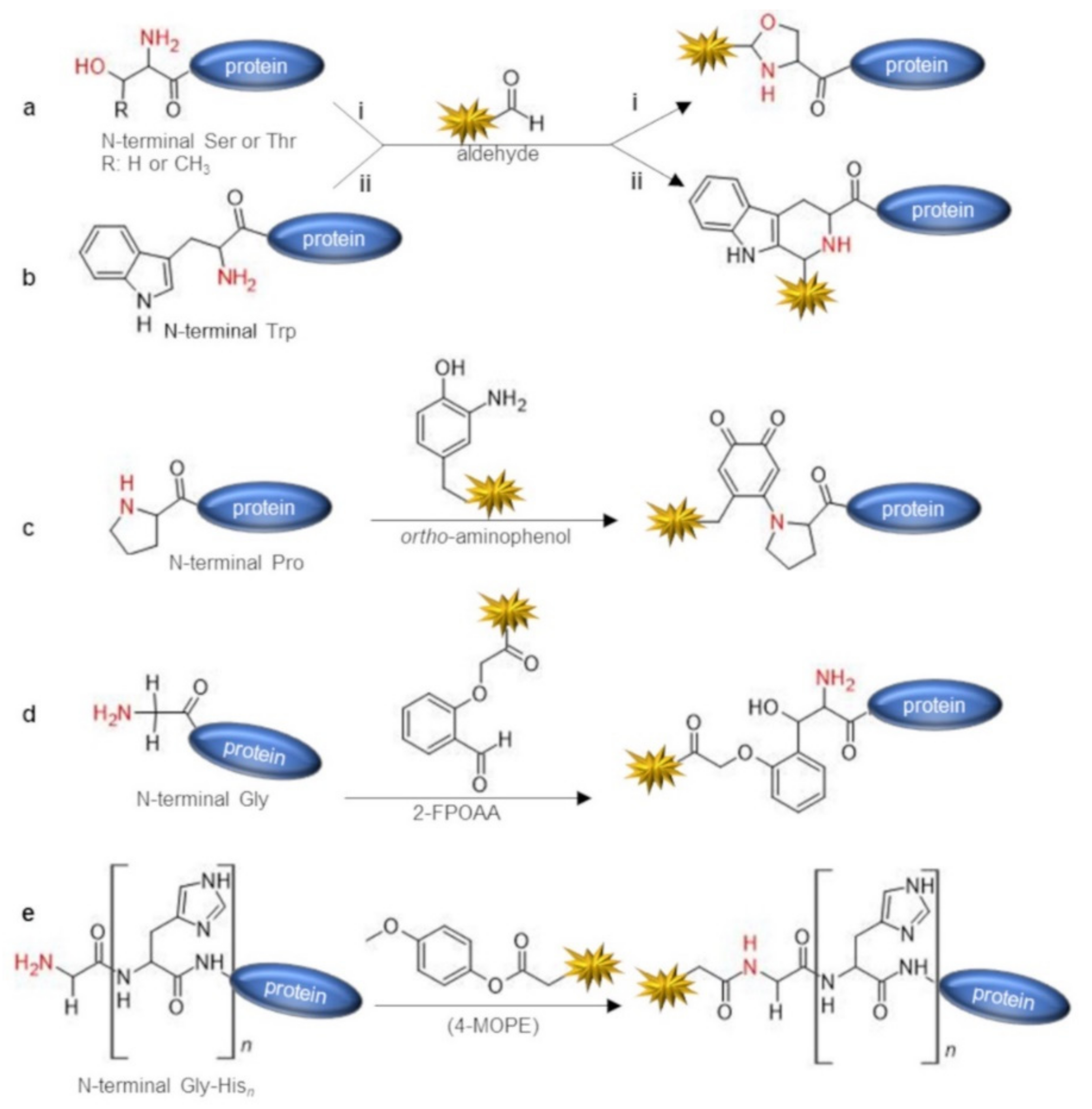
2.1. Direct Labeling of Protein N-Terminus
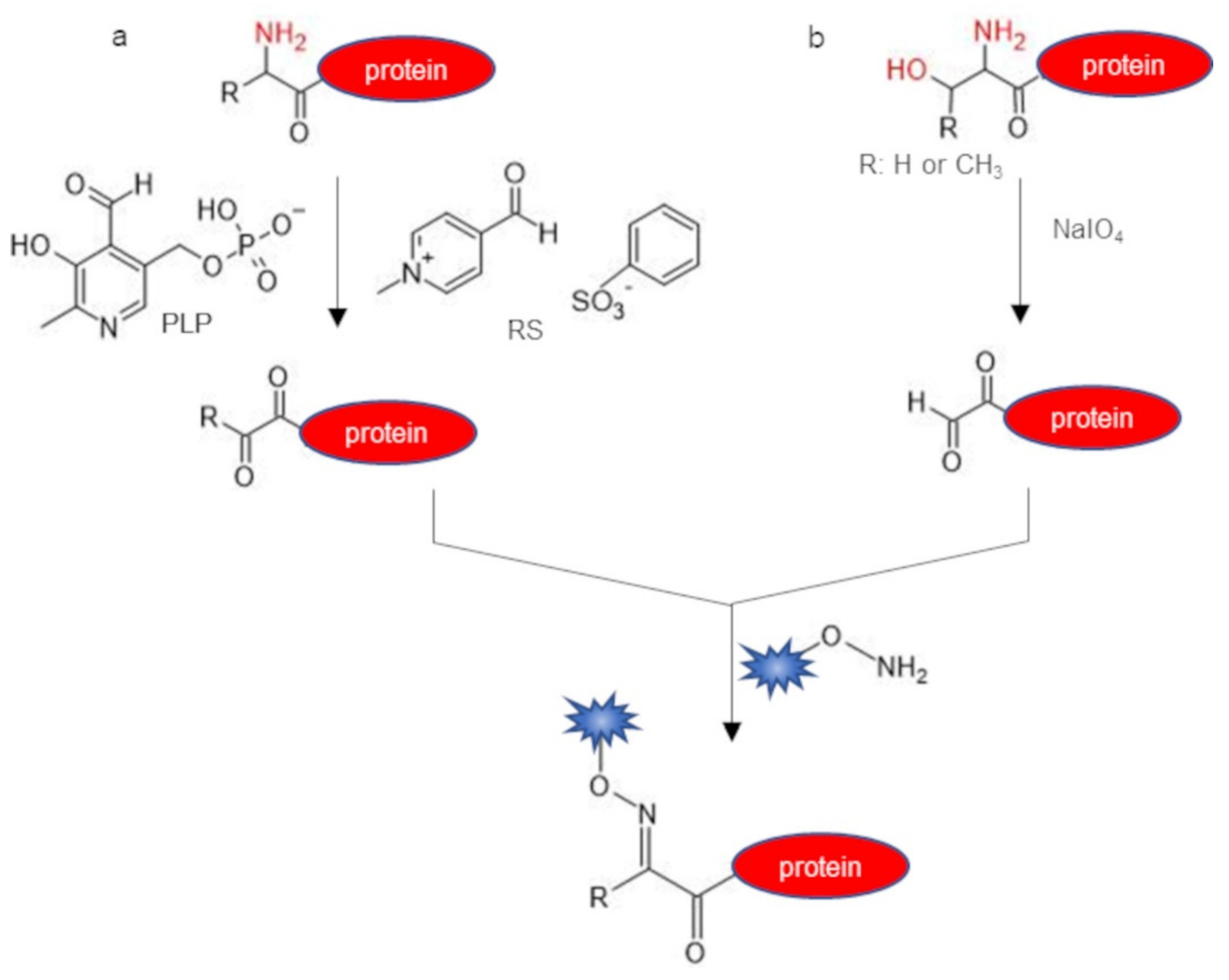
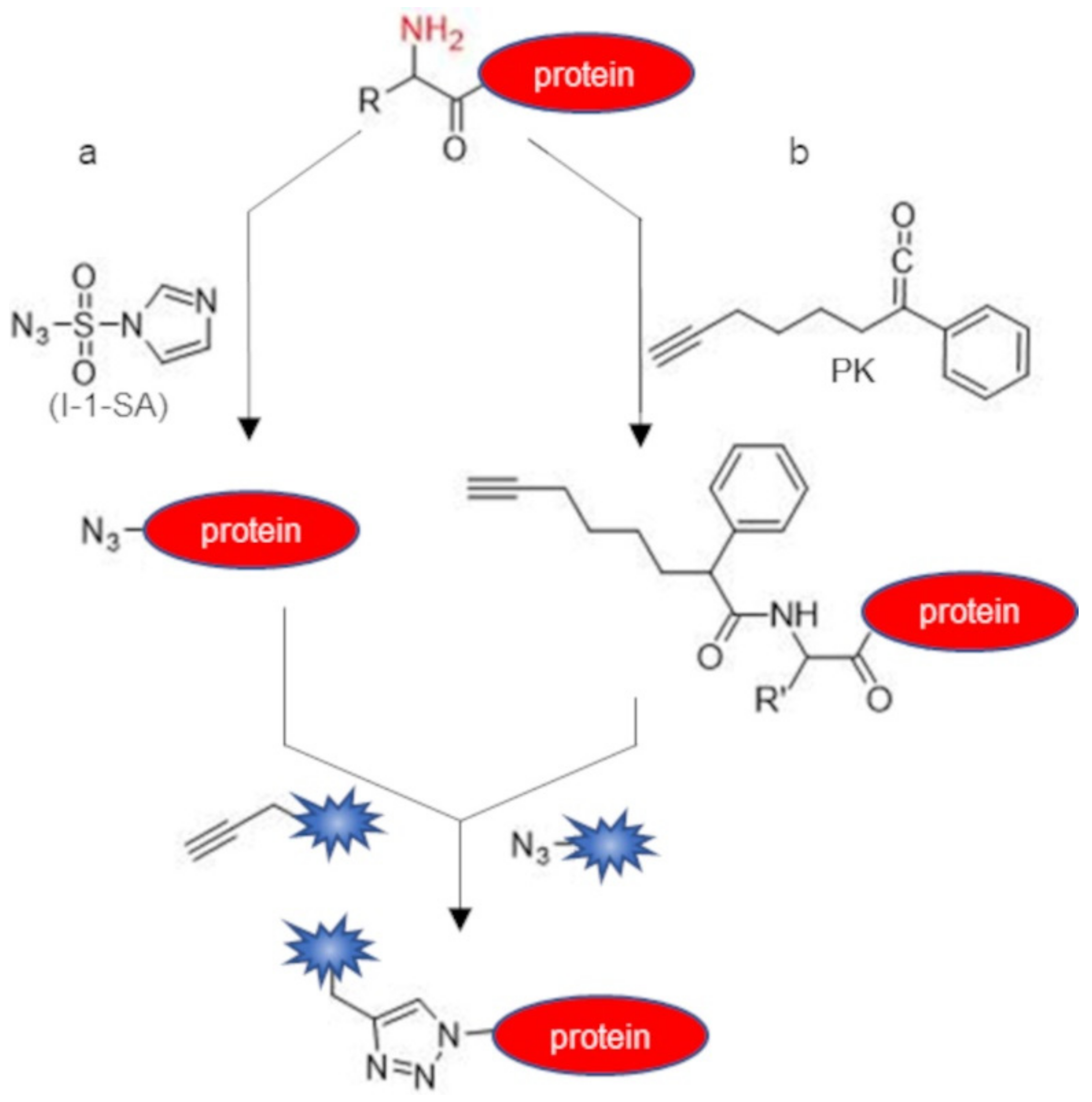
2.2. Indirect Labeling of Protein N-Terminus
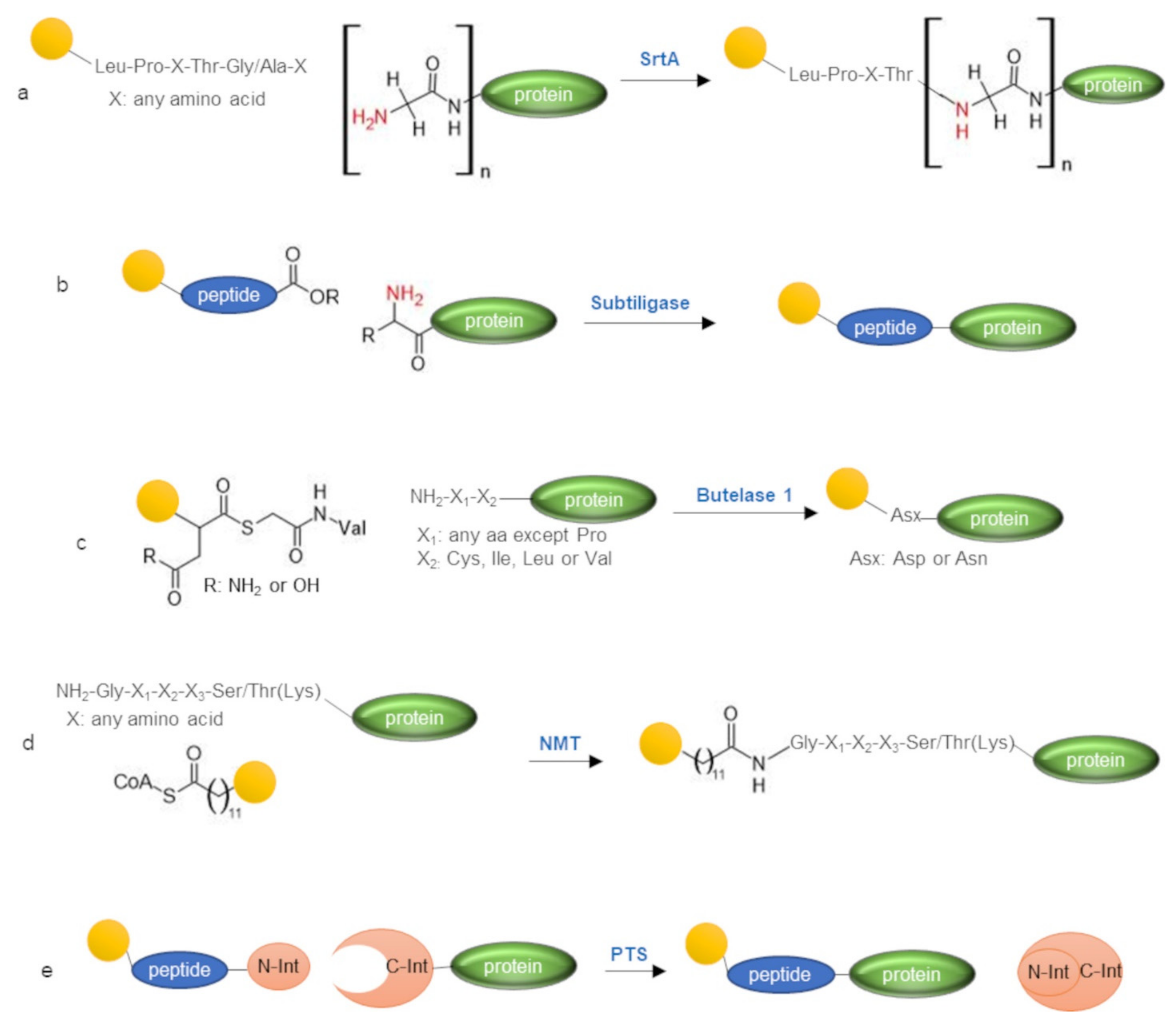
3. Enzymatic Labeling of Protein N-Terminus
3.1. Sortase A
3.2. Subtiligase
3.3. Butelase 1
3.4. N-Myristoyltransferase
3.5. Protein Trans-Splicing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5743. [Google Scholar] [CrossRef] [PubMed]
- Shadish, J.A.; DeForest, C.A. Site-selective protein modification: From functionalized proteins to functional biomaterials. Matter-Us 2020, 2, 50–77. [Google Scholar] [CrossRef]
- Xue, L.; Karpenko, I.A.; Hiblot, J.; Johnsson, K. Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol. 2015, 11, 917–923. [Google Scholar] [CrossRef]
- Stephanopoulos, N.; Francis, M.B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876–884. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of biopharmaceuticals: A review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z.Q. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Xu, L.J.; Kuan, S.L.; Weil, T. Contemporary approaches for site-selective dual functionalization of proteins. Angew. Chem. Int. Ed. 2021, 60, 13757–13777. [Google Scholar] [CrossRef]
- Basle, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef]
- Chalker, J.M.; Bernardes, G.J.L.; Lin, Y.A.; Davis, B.G. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Chen, X.; Muthoosamy, K.; Pfisterer, A.; Neumann, B.; Weil, T. Site-selective lysine modification of native proteins and peptides via kinetically controlled labeling. Bioconjugate Chem. 2012, 23, 500–508. [Google Scholar] [CrossRef]
- Matos, M.J.; Brown, L.; Bernardim, B.; Guerreiro, A.; Jimenez-Oses, G.; Bernardes, G.J.L. Sequential dual site-selective protein labelling enabled by lysine modification. Bioorgan. Med. Chem. 2020, 28, 115783. [Google Scholar] [CrossRef]
- Taylor, M.T.; Nelson, J.E.; Suero, M.G.; Gaunt, M.J. A protein functionalization platform based on selective reactions at methionine residues. Nature 2018, 562, 563–568. [Google Scholar] [CrossRef]
- Xie, L.J.; Liu, L.; Cheng, L. Modifying methionine on proteins. ChemBioChem 2020, 21, 461–463. [Google Scholar] [CrossRef]
- Lin, S.X.; Yang, X.Y.; Jia, S.; Weeks, A.M.; Hornsby, M.; Lee, P.S.; Nichiporuk, R.V.; Iavarone, A.T.; Wells, J.A.; Toste, F.D.; et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. [Google Scholar] [CrossRef]
- Antos, J.M.; McFarland, J.M.; Iavarone, A.T.; Francis, M.B. Chemoselective tryptophan labeling with rhodium carbenoids at mild pH. J. Am. Chem. Soc. 2009, 131, 6301–6308. [Google Scholar] [CrossRef]
- Chen, Z.; Popp, B.V.; Bovet, C.L.; Ball, Z.T. Site-specific protein modification with a dirhodium metallopeptide catalyst. ACS Chem. Biol. 2011, 6, 920–925. [Google Scholar] [CrossRef]
- Joshi, P.N.; Rai, V. Single-site labeling of histidine in proteins, on-demand reversibility, and traceless metal-free protein purification. Chem. Commun. 2019, 55, 1100–1103. [Google Scholar] [CrossRef]
- Sato, S.; Matsumura, M.; Kadonosono, T.; Abe, S.; Ueno, T.; Ueda, H.; Nakamura, H. Site-selective protein chemical modification of exposed tyrosine residues using tyrosine click reaction. Bioconjugate Chem. 2020, 31, 1417–1424. [Google Scholar] [CrossRef]
- Lee, K.J.; Kang, D.; Park, H.S. Site-specific labeling of proteins using unnatural amino acids. Mol. Cells 2019, 42, 386–396. [Google Scholar]
- Chin, J.W. Expanding and reprogramming the genetic code of cells and animals. Ann. Rev. Biochem. 2014, 83, 379–408. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Muir, T.W.; Sondhi, D.; Cole, P.A. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 1998, 95, 6705–6710. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; Russomanno, A.; Romanelli, A.; D’Andrea, L.D. Semi-synthesis of labeled proteins for spectroscopic applications. Molecules 2013, 18, 440–465. [Google Scholar] [CrossRef]
- D’Andrea, L.; Romanelli, A. Chemical Ligation: Tools for Biomolecule Synthesis and Modification; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Rosen, C.B.; Francis, M.B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 2017, 13, 697–705. [Google Scholar] [CrossRef]
- Sereda, T.J.; Mant, C.T.; Quinn, A.M.; Hodges, R.S. Effect of alpha-amino group on peptide retention behavior in reversed-phase chromatography—Determination of the Pk(a) values of the alpha-amino group of 19 different N-terminal amino-acid-residues. J. Chromatogr. 1993, 646, 17–30. [Google Scholar] [CrossRef]
- Jacob, E.; Unger, R. A tale of two tails: Why are terminal residues of proteins exposed? Bioinformatics 2007, 23, E225–E230. [Google Scholar] [CrossRef]
- Chen, D.; Disotuar, M.M.; Xiong, X.C.; Wang, Y.X.; Chou, D.H.C. Selective N-terminal functionalization of native peptides and proteins. Chem. Sci. 2017, 8, 2717–2722. [Google Scholar] [CrossRef]
- Deng, J.R.; Lai, N.C.H.; Kung, K.K.Y.; Yang, B.; Chung, S.F.; Leung, A.S.L.; Choi, M.C.; Leung, Y.C.; Wong, M.K. N-Terminal selective modification of peptides and proteins using 2-ethynylbenzaldehydes. Commun. Chem. 2020, 3, 1. [Google Scholar] [CrossRef]
- Singudas, R.; Adusumalli, S.R.; Joshi, P.N.; Rai, V. A phthalimidation protocol that follows protein defined parameters. Chem. Commun. 2015, 51, 473–476. [Google Scholar] [CrossRef]
- Raj, M.; Wu, H.B.; Blosser, S.L.; Vittoria, M.A.; Arora, P.S. Aldehyde capture ligation for synthesis of native peptide bonds. J. Am. Chem. Soc. 2015, 137, 6932–6940. [Google Scholar] [CrossRef]
- Busch, G.K.; Tate, E.W.; Gaffney, P.R.J.; Rosivatz, E.; Woscholski, R.; Leatherbarrow, R.J. Specific N-terminal protein labelling: Use of FMDV 3C(pro) protease and native chemical ligation. Chem. Commun. 2008, 29, 3369–3371. [Google Scholar] [CrossRef][Green Version]
- Ren, H.J.; Xiao, F.; Zhan, K.; Kim, Y.P.; Xie, H.X.; Xia, Z.Y.; Rao, J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. 2009, 48, 9658–9662. [Google Scholar] [CrossRef]
- Casi, G.; Huguenin-Dezot, N.; Zuberbuhler, K.; Scheuermann, J.; Neri, D. Site-specific traceless coupling of potent cytotoxic drugs to recombinant antibodies for pharmacodelivery. J. Am. Chem. Soc. 2012, 134, 5887–5892. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Cambray, S.; Gao, J.M. Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation. Chem. Sci. 2016, 7, 4589–4593. [Google Scholar] [CrossRef]
- Li, X.F.; Zhang, L.S.; Hall, S.E.; Tam, J.P. A new ligation method for N-terminal tryptophan-containing peptides using the Pictet-Spengler reaction. Tetrahedron Lett. 2000, 41, 4069–4073. [Google Scholar] [CrossRef]
- MacDonald, J.I.; Munch, H.K.; Moore, T.; Francis, M.B. One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Biol. 2015, 11, 326–331. [Google Scholar] [CrossRef]
- Obermeyer, A.C.; Jarman, J.B.; Francis, M.B. N-Terminal modification of proteins with o-aminophenols. J. Am. Chem. Soc. 2014, 136, 9572–9579. [Google Scholar] [CrossRef]
- Purushottam, L.; Adusumalli, S.R.; Singh, U.; Unnikrishnan, V.B.; Rawale, D.G.; Gujrati, M.; Mishra, R.K.; Rai, V. Single-site glycine-specific labeling of proteins. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Hjuler, C.T.; Sorensen, K.K.; Thygesen, M.B.; Rasmussen, J.E.; Villadsen, K.; Midtgaard, S.R.; Kol, S.; Schoffelen, S.; Jensen, K.J. Selective N-terminal acylation of peptides and proteins with a Gly-His tag sequence. Nat. Commun. 2018, 9, 1–3307. [Google Scholar] [CrossRef]
- Gilmore, J.M.; Scheck, R.A.; Esser-Kahn, A.P.; Joshi, N.S.; Francis, M.B. N-terminal protein modification through a biomimetic transamination reaction. Angew. Chem. Int. Ed. 2006, 45, 5307–5311. [Google Scholar] [CrossRef]
- Witus, L.S.; Netirojjanakul, C.; Palla, K.S.; Muehl, E.M.; Weng, C.H.; Iavarone, A.T.; Francis, M.B. Site-Specific protein transannination using N-methylpyridinium-4-carboxaldehyde. J. Am. Chem. Soc. 2013, 135, 17223–17229. [Google Scholar] [CrossRef]
- De Rosa, L.; Di Stasi, R.; Longhitano, L.; D’Andrea, L.D. Labeling of VEGFR1D2 through oxime ligation. Bioorg. Chem. 2019, 91, 103160. [Google Scholar] [CrossRef]
- Schoffelen, S.; van Eldijk, M.B.; Rooijakkers, B.; Raijmakers, R.; Heck, A.J.R.; van Hest, J.C.M. Metal-free and pH-controlled introduction of azides in proteins. Chem. Sci. 2011, 2, 701–705. [Google Scholar] [CrossRef]
- Chan, A.O.Y.; Ho, C.M.; Chong, H.C.; Leung, Y.C.; Huang, J.S.; Wong, M.K.; Che, C.M. Modification of N-Terminal alpha-Amino Groups of Peptides and Proteins Using Ketenes. J. Am. Chem. Soc. 2012, 134, 2589–2598. [Google Scholar] [CrossRef]
- Theile, C.S.; Witte, M.D.; Blom, A.E.M.; Kundrat, L.; Ploegh, H.L.; Guimaraes, C.P. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1800–1807. [Google Scholar] [CrossRef]
- Weeks, A.M.; Wells, J.A. N-Terminal modification of proteins with subtiligase specificity variants. Curr. Protoc. Chem. Biol. 2020, 12, e79. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Cao, Y.; Wang, W.; Liu, C.F.; Tam, J.P. Site-specific N-terminal labeling of peptides and proteins using butelase 1 and thiodepsipeptide. Angew. Chem. Int. Ed. 2015, 54, 15694–15698. [Google Scholar] [CrossRef]
- Heal, W.P.; Wickramasinghe, S.R.; Bowyer, P.W.; Holder, A.A.; Smith, D.F.; Leatherbarrow, R.J.; Tate, E.W. Site-specific N-terminal labelling of proteins in vitro and in vivo using N-myristoyl transferase and bioorthogonal ligation chemistry. Chem. Commun. 2008, 4, 480–482. [Google Scholar] [CrossRef]
- Nanda, J.S.; Lorsch, J.R. Labeling a protein with fluorophores using NHS ester derivitization. Lab. Methods Enzymol. Protein A 2014, 536, 87–94. [Google Scholar]
- Selo, I.; Negroni, L.; Creminon, C.; Grassi, J.; Wal, J.M. Preferential labeling of alpha-amino N-terminal groups in peptides by biotin, application to the detection of specific anti-peptide antibodies by enzyme immunoassays. J. Immunol. Methods 1996, 199, 127–138. [Google Scholar] [CrossRef]
- Berezovsky, I.N.; Kilosanidze, G.T.; Tumanyan, V.G.; Kisselev, L.L. Amino acid composition of protein termini are biased in different manners. Protein Eng. 1999, 12, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gentle, I.E.; De Souza, D.P.; Baca, M. Direct production of proteins with N-terminal cysteine for site-specific conjugation. Bioconjugate Chem. 2004, 15, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Villain, M.; Vizzavona, J.; Rose, K. Covalent capture: A new tool for the purification of synthetic and recombinant polypeptides. Chem. Biol. 2001, 8, 673–679. [Google Scholar] [CrossRef][Green Version]
- Romanelli, A.; Shekhtman, A.; Cowburn, D.; Muir, T.W. Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein-intein junction. Proc. Natl. Acad. Sci. USA 2004, 101, 6397–6402. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, T.J.; Wong, C.H. Conjugation of glycopeptide thioesters to expressed protein fragments: Semisynthesis of glycosylated interleukin-2. Methods Mol. Biol. 2004, 283, 255–266. [Google Scholar] [PubMed]
- Liu, D.S.; Xu, R.; Dutta, K.; Cowburn, D. N-terminal cysteinyl proteins can be prepared using thrombin cleavage. FEBS Lett. 2008, 582, 1163–1167. [Google Scholar] [CrossRef]
- Xu, M.Q.; Evans, T.C. Intein-mediated ligation and cyclization of expressed proteins. Methods 2001, 24, 257–277. [Google Scholar] [CrossRef]
- Wissner, R.F.; Batjargal, S.; Fadzen, C.M.; Petersson, E.J. Labeling proteins with fluorophore/thioamide forster resonant energy transfer pairs by combining unnatural amino acid mutagenesis and native chemical ligation. J. Am. Chem. Soc. 2013, 135, 6529–6540. [Google Scholar] [CrossRef]
- Xiao, J.P.; Tolbert, T.J. Synthesis of N-Terminally linked protein dimers and trimers by a combined native chemical ligation-CuAAC click chemistry strategy. Org. Lett. 2009, 11, 4144–4147. [Google Scholar] [CrossRef]
- Xiao, J.P.; Hamilton, B.S.; Tolbert, T.J. Synthesis of N-terminally linked protein and peptide dimers by native chemical ligation. Bioconjugate Chem. 2010, 21, 1943–1947. [Google Scholar] [CrossRef]
- Xiao, J.P.; Burn, A.; Tolbert, T.J. Increasing solubility of proteins and peptides by site-specific modification with betaine. Bioconjugate Chem. 2008, 19, 1113–1118. [Google Scholar] [CrossRef]
- Hawala, I.; De Rosa, L.; Aime, S.; D’Andrea, L.D. An innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach. Chem. Commun. 2020, 56, 3500–3503. [Google Scholar] [CrossRef]
- Dempsey, D.R.; Jiang, H.J.; Kalin, J.H.; Chen, Z.; Cole, P.A. Site-specific protein labeling with N-hydroxysuccinimide-esters and the analysis of ubiquitin ligase mechanisms. J. Am. Chem. Soc. 2018, 140, 9374–9378. [Google Scholar] [CrossRef]
- De Rosa, L.; Cortajarena, A.L.; Romanelli, A.; Regan, L.; D’Andrea, L.D. Site-specific protein double labeling by expressed protein ligation: Applications to repeat proteins. Org. Biomol. Chem. 2012, 10, 273–280. [Google Scholar] [CrossRef]
- Jiang, H.; Cole, P.A. N-terminal protein labeling with N-hydroxysuccinimide esters and microscale thermophoresis measurements of protein-protein interactions using labeled protein. Curr. Protoc. 2021, 1, e14. [Google Scholar] [CrossRef]
- White, E.H.; McCapra, F.; Field, G.F. The structure and synthesis of firefly luciferin. J. Am. Chem. Soc. 1963, 85, 337–343. [Google Scholar] [CrossRef]
- Liang, G.L.; Ren, H.J.; Rao, J.H. A biocompatible condensation reaction for controlled assembly of nanostructures in living cells. Nat. Chem. 2010, 2, 54–60. [Google Scholar] [CrossRef]
- Jeon, J.; Shen, B.; Xiong, L.Q.; Miao, Z.; Lee, K.H.; Rao, J.; Chin, F.T. Efficient method for site-specific F-18-labeling of biomolecules using the rapid condensation reaction between 2-cyanobenzothiazole and cysteine. Bioconjugate Chem. 2012, 23, 1902–1908. [Google Scholar] [CrossRef]
- Liu, C.F.; Tam, J.P. Chemical ligation approach to form a peptide-bond between unprotected peptide segments—Concept and model study. J. Am. Chem. Soc. 1994, 116, 4149–4153. [Google Scholar] [CrossRef]
- Zhang, L.S.; Tam, J.P. Thiazolidine formation as a general and site-specific conjugation method for synthetic peptides and proteins. Anal. Biochem. 1996, 233, 87–93. [Google Scholar] [CrossRef]
- Liu, C.F.; Tam, J.P. Peptide segment ligation strategy without use of protecting groups. Proc. Natl. Acad. Sci. USA 1994, 91, 6584–6588. [Google Scholar] [CrossRef]
- Zhao, Z.G.; Im, J.S.; Lam, K.S.; Lake, D.F. Site-specific modification of a single-chain antibody using a novel glyoxylyl-based labeling reagent. Bioconjugate Chem. 1999, 10, 424–430. [Google Scholar] [CrossRef]
- Faustino, H.; Silva, M.J.S.A.; Veiros, L.F.; Bernardes, G.J.L.; Gois, P.M.P. Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalization. Chem. Sci. 2016, 7, 6280. [Google Scholar] [CrossRef]
- Pulka, K.; Slupska, M.; Puszko, A.; Misiak, M.; Wilczek, M.; Kozminski, W.; Misicka, A. Peptides and peptidoaldehydes as substrates for the Pictet-Spengler reaction. J. Pept. Sci. 2013, 19, 433–440. [Google Scholar] [CrossRef]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef]
- Sasaki, T.; Kodama, K.; Suzuki, H.; Fukuzawa, S.; Tachibana, K. N-terminal labeling of proteins by the Pictet-Spengler reaction. Bioorg. Med. Chem. Lett. 2008, 18, 4550–4553. [Google Scholar] [CrossRef]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hirel, P.H.; Schmitter, J.M.; Dessen, P.; Fayat, G.; Blanquet, S. Extent of N-terminal methionine excision from escherichia-coli proteins is governed by the side-chain length of the penultimate amino-acid. Proc. Natl. Acad. Sci. USA 1989, 86, 8247–8251. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, K.F.; Dixon, H.B.F.; Rosner, P.J.; Hoth, L.R.; Lanzetti, A.J.; Borzilleri, K.A.; Marr, E.S.; Pezzullo, L.H.; Martin, L.B.; LeMotte, P.K.; et al. Spontaneous alpha-N-6-phosphogluconoylation of a “His tag” in Escherichia coli: The cause of extra mass of 258 or 178 Da in fusion proteins. Anal. Biochem. 1999, 267, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Scheck, R.A.; Dedeo, M.T.; Lavarone, A.T.; Francis, M.B. Optimization of a biomimetic transamination reaction. J. Am. Chem. Soc. 2008, 130, 11762–11770. [Google Scholar] [CrossRef]
- Scheck, R.A.; Francis, M.B. Regioselective labeling of antibodies through N-terminal transamination. ACS Chem. Biol. 2007, 2, 247–251. [Google Scholar] [CrossRef]
- Buckley, T.F.R.H. Mild and simple biomimetic conversion of amines to carbonyl compounds. J. Am. Chem. Soc. 1982, 104, 4446–4450. [Google Scholar] [CrossRef]
- Palla, K.S.; Witus, L.S.; Mackenzie, K.J.; Netirojjanakul, C.; Francis, M.B. Optimization and expansion of a site-selective N-methylpyridinium-4-carboxaldehyde-mediated transamination for bacterially expressed proteins. J. Am. Chem. Soc. 2015, 137, 1123–1129. [Google Scholar] [CrossRef]
- Geoghegan, K.F.; Stroh, J.G. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate-oxidation of a 2-amino alcohol—Application to modification at N-terminal serine. Bioconjugate Chem. 1992, 3, 138–146. [Google Scholar] [CrossRef]
- El-Mahdi, O.; Melnyk, O. Alpha-oxo aldehyde or glyoxylyl group chemistry in peptide bioconjugation. Bioconjugate Chem. 2013, 24, 735–765. [Google Scholar] [CrossRef]
- Di Stasi, R.; Diana, D.; Capasso, D.; Palumbo, R.; Romanelli, A.; Pedone, C.; Fattorusso, R.; D’Andrea, L.D. VEGFR1(D2) in drug discovery: Expression and molecular characterization. Biopolymers 2010, 94, 800–809. [Google Scholar] [CrossRef]
- Ning, X.; Temming, R.P.; Dommerholt, J.; Guo, J.; Ania, D.B.; Debets, M.F.; Wolfert, M.A.; Boons, G.J.; van Delft, F.L. Protein modification by strain-promoted alkyne-nitrone cycloaddition. Angew. Chem. Int. Ed. Engl. 2010, 49, 3065–3068. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Popp, M.W.L.; Ploegh, H.L. Making and breaking peptide bonds: Protein engineering using sortase. Angew. Chem. Int. Ed. 2011, 50, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Chew, G.L.; Guimaraes, C.P.; Yoder, N.C.; Grotenbreg, G.M.; Popp, M.W.L.; Ploegh, H.L. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J. Am. Chem. Soc. 2009, 131, 10800–10801. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nagamune, T. Expansion of the sortase-mediated labeling method for site-specific N-terminal labeling of cell surface proteins on living cells. Chem. Commun. 2009, 9, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Miller, G.M.; Grotenbreg, G.M.; Ploegh, H.L. Lipid modification of proteins through sortase-catalyzed transpeptidation. J. Am. Chem. Soc. 2008, 130, 16338–16343. [Google Scholar] [CrossRef]
- Guimaraes, C.P.; Carette, J.E.; Varadarajan, M.; Antos, J.; Popp, M.W.; Spooner, E.; Brummelkamp, T.R.; Ploegh, H.L. Identification of host cell factors required for intoxication through use of modified cholera toxin. J. Cell Biol. 2011, 195, 751–764. [Google Scholar] [CrossRef]
- Popp, M.W.; Artavanis-Tsakonas, K.; Ploegh, H.L. Substrate filtering by the active site crossover loop in UCHL3 revealed by sortagging and gain-of-function mutations. J. Biol. Chem. 2009, 284, 3593–3602. [Google Scholar] [CrossRef]
- Guimaraes, C.P.; Witte, M.D.; Theile, C.S.; Bozkurt, G.; Kundrat, L.; Blom, A.E.M.; Ploegh, H.L. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1787–1799. [Google Scholar] [CrossRef]
- Popp, M.W.; Dougan, S.K.; Chuang, T.Y.; Spooner, E.; Ploegh, H.L. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174. [Google Scholar] [CrossRef]
- Weeks, A.M.; Wells, J.A. Subtiligase-catalyzed peptide ligation. Chem. Rev. 2020, 120, 3127–3160. [Google Scholar] [CrossRef]
- Abrahmsen, L.; Tom, J.; Burnier, J.; Butcher, K.A.; Kossiakoff, A.; Wells, J.A. Engineering subtilisin and its substrates for efficient ligation of peptide-bonds in aqueous-solution. Biochemistry 1991, 30, 4151–4159. [Google Scholar] [CrossRef]
- Tan, X.H.; Yang, R.L.; Liu, C.F. Facilitating subtiligase-catalyzed peptide ligation reactions by using peptide thioester substrates. Org. Lett. 2018, 20, 6691–6694. [Google Scholar] [CrossRef]
- Weeks, A.M.; Wells, J.A. Engineering peptide ligase specificity by proteomic identification of ligation sites. Nat. Chem. Biol. 2018, 14, 50. [Google Scholar] [CrossRef]
- Chang, T.K.; Jackson, D.Y.; Burnier, J.P.; Wells, J.A. Subtiligase—A tool for semisynthesis of proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 12544–12548. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Wang, S.J.; Qiu, Y.B.; Hemu, X.; Lian, Y.L.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Kam, A.; Loo, S.; Jansson, A.E.; Pan, L.X.; Tam, J.P. Butelase 1: A versatile ligase for peptide and protein macrocyclization. J. Am. Chem. Soc. 2015, 137, 15398–15401. [Google Scholar] [CrossRef]
- Williamson, D.J.; Fascione, M.A.; Webb, M.E.; Turnbull, W.B. Efficient N-terminal labeling of proteins by use of sortase. Angew. Chem. Int. Ed. 2012, 51, 9377–9380. [Google Scholar] [CrossRef]
- Devadas, B.; Lu, T.B.; Katoh, A.; Kishore, N.S.; Wade, A.C.; Mehta, P.P.; Rudnick, D.A.; Bryant, M.L.; Adams, S.P.; Li, Q.; et al. Substrate-specificity of saccharomyces-cerevisiae myristoyl-coa-protein N-myristoyltransferase—Analysis of fatty-acid analogs containing carbonyl groups, nitrogen heteroatoms, and nitrogen-heterocycles in an in vitro enzyme assay and subsequent identification of inhibitors of human immunodeficiency virus-I replication. J. Biol. Chem. 1992, 267, 7224–7239. [Google Scholar]
- Kishore, N.S.; Lu, T.B.; Knoll, L.J.; Katoh, A.; Rudnick, D.A.; Mehta, P.P.; Devadas, B.; Huhn, M.; Atwood, J.L.; Adams, S.P.; et al. The substrate specificity of Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. Analysis of myristic acid analogs containing oxygen, sulfur, double bonds, triple bonds, and/or an aromatic residue. J. Biol. Chem. 1991, 266, 8835–8855. [Google Scholar] [CrossRef]
- Lu, T.; Li, Q.; Katoh, A.; Hernandez, J.; Duffin, K.; Jackson-Machelski, E.; Knoll, L.J.; Gokel, G.W.; Gordon, J.I. The substrate specificity of Saccharomyces cerevisiae myristoyl-CoA: Protein N-myristoyltransferase. Polar probes of the enzyme’s myristoyl-CoA recognition site. J. Biol. Chem. 1994, 269, 5346–5357. [Google Scholar] [CrossRef]
- Hang, H.C.; Geutjes, E.J.; Grotenbreg, G.; Pollington, A.M.; Bijlmakers, M.J.; Ploegh, H.L. Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J. Am. Chem. Soc. 2007, 129, 2744–2745. [Google Scholar] [CrossRef]
- Heal, W.P.; Wickramasinghe, S.R.; Leatherbarrow, R.J.; Tate, E.W. N-Myristoyl transferase-mediated protein labelling in vivo. Org. Biomol. Chem. 2008, 6, 2308–2315. [Google Scholar] [CrossRef]
- Kulkarni, C.; Kinzer-Ursem, T.L.; Tirrell, D.A. Selective functionalization of the protein N terminus with N-myristoyl transferase for bioconjugation in cell lysate. ChemBioChem 2013, 14, 1958–1962. [Google Scholar] [CrossRef]
- Heal, W.P.; Wright, M.H.; Thinon, E.; Tate, E.W. Multifunctional protein labeling via enzymatic N-terminal tagging and elaboration by click chemistry. Nat. Protoc. 2011, 7, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.W.; Camarero, J.A. Intein applications: From protein purification and labeling to metabolic control methods. J. Biol. Chem. 2014, 289, 14512–14519. [Google Scholar] [CrossRef]
- Aranko, A.S.; Wlodawer, A.; Iwai, H. Nature’s recipe for splitting inteins. Protein Eng. Des. Sel. 2014, 27, 263–271. [Google Scholar] [CrossRef]
- Ludwig, C.; Pfeiff, M.; Linne, U.; Mootz, H.D. Ligation of a synthetic peptide to the N terminus of a recombinant protein using semisynthetic protein trans-splicing. Angew. Chem. Int. Ed. 2006, 45, 5218–5221. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Reagent | Target Function | Reaction | Junction | Experimental Conditions a | Yield of Conversion (%) a | Ref |
|---|---|---|---|---|---|---|
| benzaldehydes | α-amine | reductive amination | secondary amine | citric acid buffer pH 6.1, 2 eq of benzaldehyde, 5 eq b of NaBH3CN, 6–48 h, r.t. | 30–70 | [29] |
| 2-ethynylbenzaldehydes (2-EBA) | α-amine | imine formation and intramolecular 6-endo-dig cyclization | isoquinolinium | PBS/DMSO 9:1 pH 6.5–7.4, 5–10 eq of 2-EBA, 1–16 h, 37 °C | 10–92 | [30] |
| N-hydroxy-phthalimide (NHP) | α-amine | phthalimidation | phthalimidoamine | phosphate buffer pH 7.0, 1 eq of NHP, 8 h, r.t. | 45–50 | [31] |
| selenobenzaldehyde (SBA) ester | α-amine | acylation through ACL | amide | dimethylformamide/PBS pH 7.0, 5 eq of SBA ester, 40 h, r.t. | 70 | [32] |
| thioesters | 1,2-aminothiol (N-term Cys) | acylation through NCL | amide | phosphate buffer pH 7.4, 20 eq mercaptoethanesulfonate 4 eq of thioester probe, 5 h, r.t. | ~100 | [33] |
| 2-cyanobenzothiazole (CBT) | 1,2-aminothiol (N-term Cys) | condensation | thiazoline | PBS pH 7.4, 2 mM glutathione or TCEP, 5 eq of CBT, 2 h, r.t. | Not reported | [34] |
| aldehydes (e.g., glyoxalaldhehyde) | 1,2-aminothiol (N-term Cys) or 1,2-aminoalcohol (N-term Ser/Thr) | condensation | thiazolidine or oxazolidine | acetate buffer pH 4.5, 1 mM DTT or TCEP 16 h, then 40–200 eq aldehyde, 2.5−4 days, 4 °C | 90 | [35] |
| 2-formyl phenylboronic acid (2-FPBA) | 1,2-aminothiol (N-term Cys) | condensation | thiazolidino-boronate | phosphate buffer pH 7.0, 1 eq of 2-FPBA 30 min, r.t. | ~100 | [36] |
| aldehydes | N-term Trp | Pictet–Spengler condensation | tetrahydro-beta-carboline | Glacial acetic acid, 24 h reagents ratio not reported | ~100 | [37] |
| 2-pyridinecarbaldehyde (2-PCA) | α-amine | imine condensation | imidazolidinone | phosphate buffer pH 7.5, 25 μM protein substrate, 10 mM 2-PCA, 37 °C, 16 h | 43–95 | [38] |
| ortho-aminophenols | α-amine (especially N-term Pro) | oxidative coupling | secondary or tertiary amine c | phosphate buffer pH 7.5, 20 μM protein substrate, 5 eq of ortho-aminophenol, 5 mM K3Fe(CN)6, 30 min, r.t. | up to ~ 100 (on N-term prolyl-protein) | [39] |
| 2-(2-formylphenoxy)acetic acid (2-FPOAA) | α-amine of N-term Gly | imine condensation + nucleophilic addition | amino-alcohol | bicarbonate buffer pH 7.8, 20 μM protein substrate 10 mM 2-FPOAA, 24–48 h, 20 °C | 40–71 | [40] |
| 4-methoxyphenyl esters (4-MOPE) | α-amine of Gly-Hisn N-term tag | acylation | amide | HEPES buffer pH 7.5, 20 eq of 4-MOPE, 24 h, 4 °C | 45 (on Gly-His6 tagged protein) | [41] |
| pyridoxal-5′-phosphate (PLP) | α-amine | transamination | aldehyde/ketone | phosphate buffer pH 6.5, 10 mM PLP, 18–20 h, 37–55 °C | 30–80 | [42] |
| Rapoport’s salt (RS) | α-amine (especially N-term Glu) | transamination | aldehyde/ketone | phosphate buffer pH 6.5, 100 mM RS 1 h, 37 °C | 67 | [43] |
| sodium periodate (NaIO4) | 1,2 amino-alcohol (N-term Ser or Thr) | oxidation | aldehyde | phosphate buffer/NaCl pH 7.0, 1.5 eq, of NaIO4, 1 h, r.t. | ~100 | [44] |
| imidazole-1-sulfonyl azide (I-1-SA) | α-amine | diazotransfer | azide | diethanolamine buffered solution pH 8.5, 17.5 eq of I-1-SA, overnight, r.t. | ~100 | [45] |
| phenyl ketene (PK) | α-amine | acylation | amide | phosphate buffer pH 6.3 or 9.2, 6–10 eq of PK, 15 min—overnight, 37 °C | 23–38 | [46] |
| Enzyme | Reagent | N-terminal Target Sequence or Function | Reaction Conditions a | Yield a | Ref |
|---|---|---|---|---|---|
| Sortase A (SrtA) | Leu-Pro-X-Thr-Gly/Ala-X peptide (X any a.a.) | α-amine of oligo-Gly | 0.5–1 mM probe-Leu-Pro-X-Thr-Gly/Ala-, 10–50 µM target protein, 20–150 µM SrtA, in 50 mM Tris buffer pH 7.5, 150 mM NaCl, 10 mM CaCl2 15 min—5 h, r.t. or 37 °C | up to 90 | [47] |
| Subtiligase | C-terminal ester peptide | α-amine | 5 mM peptide ester-probe, 10–50 µM target protein, 1 µM subtiligase, in 100 mM tricine pH 8.0, 1–2 h, 4 °C or r.t. | variable b | [48] |
| Butelase 1 | Thiodepsipeptide Asn/Asp-(S) Gly-Val | α-amine of X1-X2 (X1 any a.a. except Pro; X2 Cys, Ile, Leu, Val) | 100 µM target protein, 0.1 µM butelase 1, 400–500 µM thiodepsipeptide (1 eq added every 30 min) 1 mM EDTA, 20 mM phosphate buffer pH 6.5, 100 min—2.5 h, temperature not specified | up to 95 | [49] |
| N-Myristoyltransferase (NMT) | myristoyl-CoA derivatives | α-amine of Gly-X1-X2-X3-Ser/Thr (Lys) (X is any amino acid) | 15 µM target protein, 2 mM DTT 30 µM myristoyl-CoA derivative in 30 mM Tris buffer, 0.5 mM EGTA, 0.1% TRITON® X-100, pH 7.4 18 h—37 °C | not reported | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rosa, L.; Di Stasi, R.; Romanelli, A.; D’Andrea, L.D. Exploiting Protein N-Terminus for Site-Specific Bioconjugation. Molecules 2021, 26, 3521. https://doi.org/10.3390/molecules26123521
De Rosa L, Di Stasi R, Romanelli A, D’Andrea LD. Exploiting Protein N-Terminus for Site-Specific Bioconjugation. Molecules. 2021; 26(12):3521. https://doi.org/10.3390/molecules26123521
Chicago/Turabian StyleDe Rosa, Lucia, Rossella Di Stasi, Alessandra Romanelli, and Luca Domenico D’Andrea. 2021. "Exploiting Protein N-Terminus for Site-Specific Bioconjugation" Molecules 26, no. 12: 3521. https://doi.org/10.3390/molecules26123521
APA StyleDe Rosa, L., Di Stasi, R., Romanelli, A., & D’Andrea, L. D. (2021). Exploiting Protein N-Terminus for Site-Specific Bioconjugation. Molecules, 26(12), 3521. https://doi.org/10.3390/molecules26123521