
Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins

 , , and
, , and
Abstract
1. Introduction
2. Methods
2.1. Animal Ethics
2.2. Identification of PKAT Family Homologs
2.3. Annotating PKAT Protein Domain Architecture and Intron–Exon Architecture
2.4. Production of Cladogram/Phylograms
2.5. Synteny Analysis
2.6. Functional Domain Alignments
2.7. RPT Domain Identification and Comparison
3. Results and Discussion
3.1. Mutations of PMEL or GPNMB Cause an Overlapping Spectrum of Pathologies but Highlight the Disparate Functional Roles of Each Protein
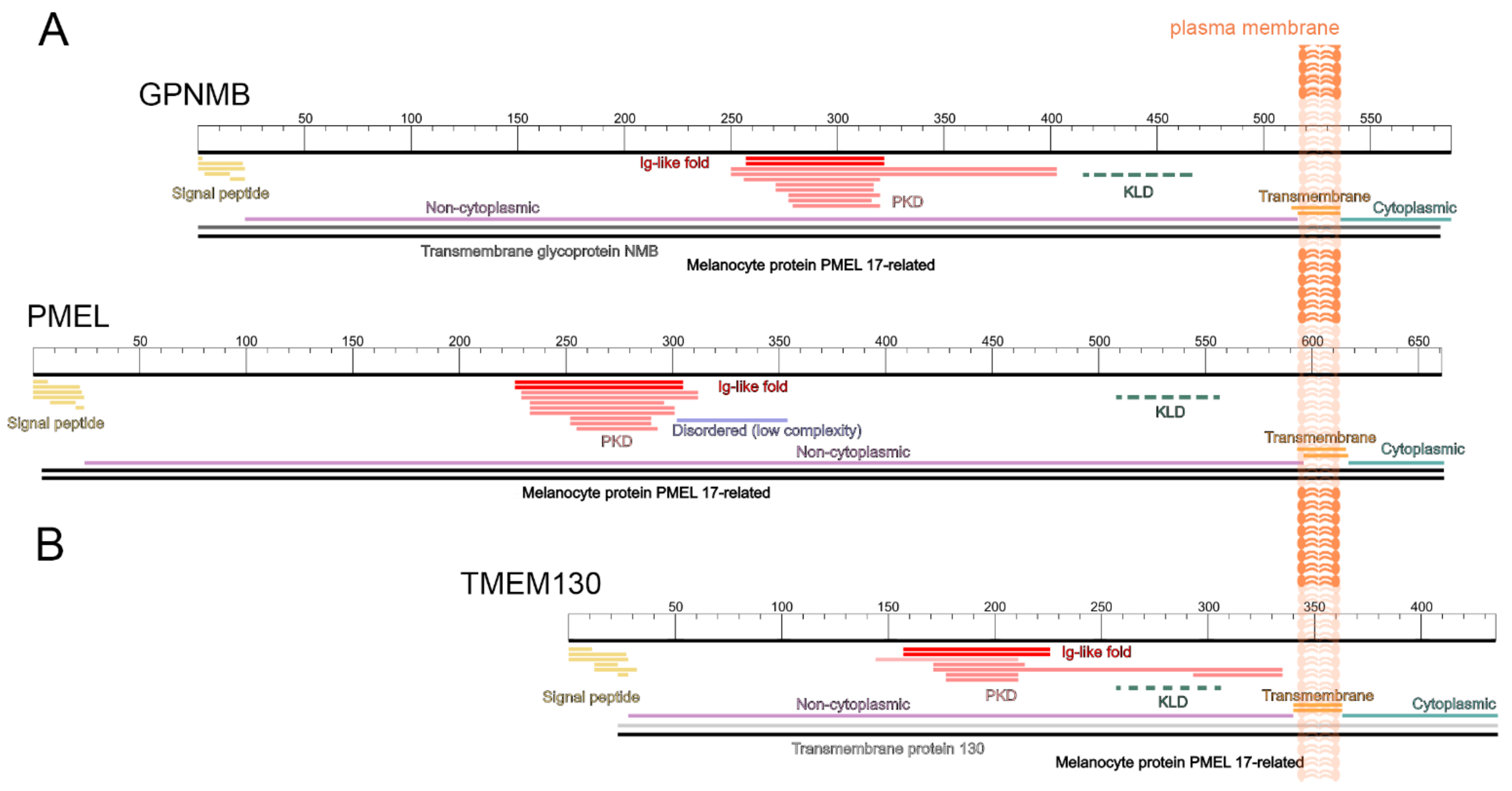
3.2. TMEM130, GPNMB and PMEL Form a Protein Family and Share a Common Protein Domain Architecture
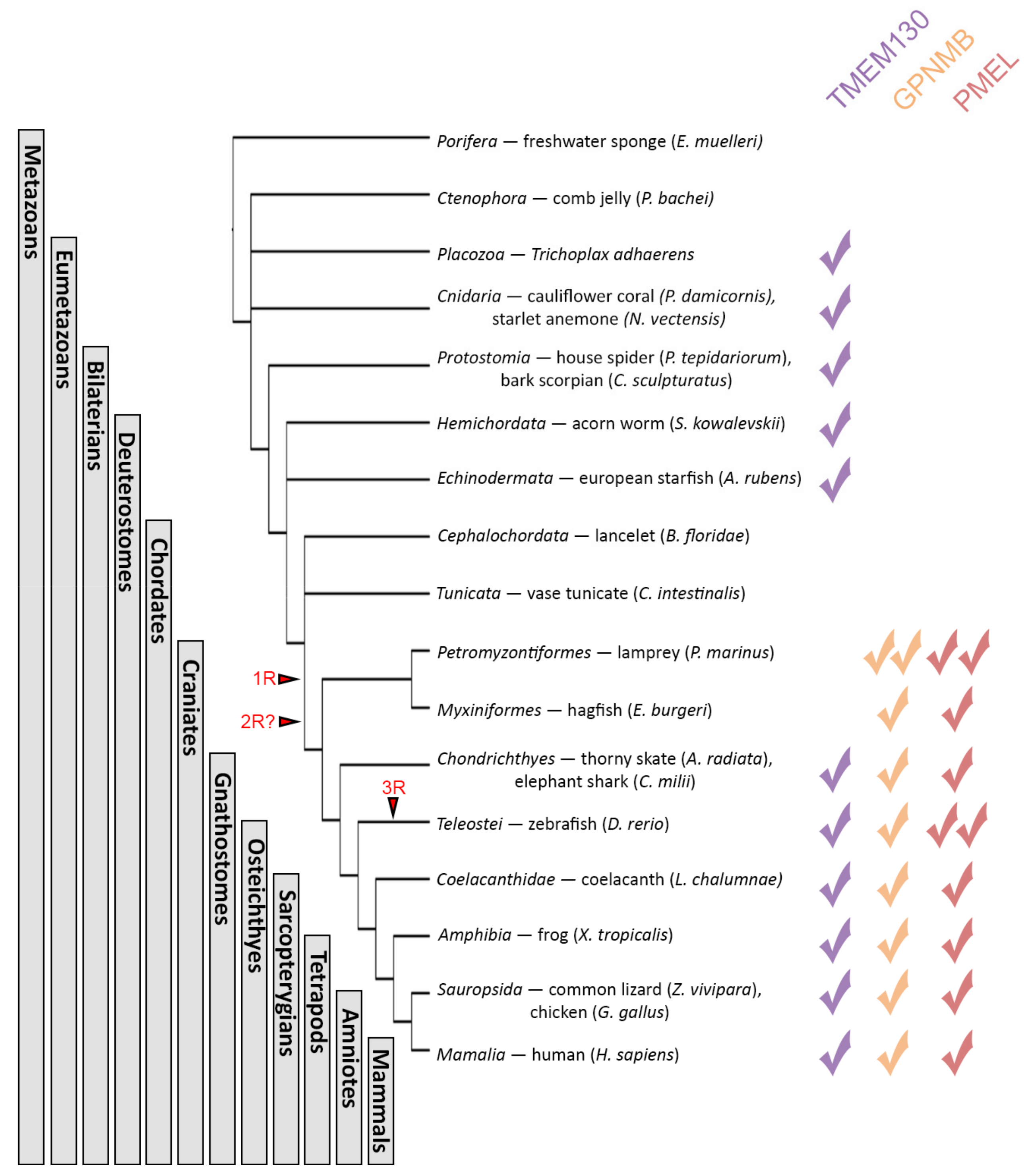
3.3. The PKAT Family Genes Are Distantly Related Paralogs and TMEM130 Represents the Most Ancient Homolog
3.4. TMEM130 Is a Sister Group of Both GPNMB and PMEL
3.5. The Amino-Terminal Region of PKAT Proteins Show Conservation of a Signal Peptide and the Core Amyloid Fragment in PMEL and GPNMB
3.6. Multiple Carboxy-Terminus Domains and Functional Motifs Are Conserved in the PKAT Family
3.7. The PMEL RPT Domain Sequence Is Highly Divergent between Species but Considerably Conserved within Repeat Units of Most Species
3.8. A Strong Evolutionary Pressure for Uniform Repeat Unit Sequence, Length and E/D Ladder Conservation Is Evident in Teleost Fish
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver locus product Pmel17/gp100/Silv/ME20: Controversial in name and in function. Pigment Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef]
- Weterman, M.A.J.; Ajubi, N.; Vandinter, I.M.R.; Degen, W.G.J.; Vanmuijen, G.N.P.; Ruiter, D.J.; Bloemers, H.P.J. NMB, a novel gene, is expressed in low-metastatic human-melanoma cell-lines and xenografts. Int. J. Cancer 1995, 60, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Onaga, M.; Ido, A.; Hasuike, S.; Uto, H.; Moriuchi, A.; Nagata, K.; Hori, T.; Hayash, K.; Tsubouchi, H. Osteoactivin expressed during cirrhosis development in rats fed a choline-deficient, L-amino acid-defined diet, accelerates motility of hepatoma cells. J. Hepatol. 2003, 39, 779–785. [Google Scholar] [CrossRef]
- Rose, A.A.N.; Pepin, F.; Russo, C.; Abou Khalil, J.E.; Hallett, M.; Siegell, P.M. Osteoactivin promotes breast cancer metastasis to bone. Mol. Cancer Res. 2007, 5, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.N.; Wagner, C.; Schultewolter, T.; Goos, M. Analysis of Pmel17/gp100 expression in primary human tissue specimens: Implications for melanoma immuno- and gene-therapy. Cancer Immunol. Immunother. 1997, 44, 239–247. [Google Scholar] [CrossRef]
- Nawrath, M.; Pavlovic, J.; Dummet, R.; Schultz, J.; Strack, B.; Heinrich, J.; Moelling, K. Reduced melanoma tumor formation in mice immunized with DNA expressing the melanoma-specific antigen gp100/pmel17. Leukemia 1999, 13, S48–S51. [Google Scholar] [CrossRef]
- John, S.W.M.; Smith, R.S.; Savinova, O.V.; Hawes, N.L.; Chang, B.; Turnbull, D.; Davisson, M.; Roderick, T.H.; Heckenlively, J.R. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Investig. Ophthalmol. Vis. Sci. 1998, 39, 951–962. [Google Scholar]
- Anderson, M.G.; Smith, R.S.; Hawes, N.L.; Zabaleta, A.; Chang, B.; Wiggs, J.L.; John, S.W.M. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet. 2002, 30, 81–85. [Google Scholar] [CrossRef]
- Lahola-Chomiak, A.A.; Footz, T.; Nguyen-Phuoc, K.; Neil, G.J.; Fan, B.J.; Allen, K.F.; Greenfield, D.S.; Parrish, R.K.; Linkroum, K.; Pasquale, L.R.; et al. Non-Synonymous variants in premelanosome protein (PMEL) cause ocular pigment dispersion and pigmentary glaucoma. Hum. Mol. Genet. 2019, 28, 1298–1311. [Google Scholar] [CrossRef]
- Zhu, S.C.; Wuolikainen, A.; Wu, J.F.; Ohman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643–657. [Google Scholar] [CrossRef]
- Deckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Pihlstrom, L.; Axelsson, G.; Bjornara, K.A.; Dizdar, N.; Fardell, C.; Forsgren, L.; Holmberg, B.; Larsen, J.P.; Linder, J.; Nissbrandt, H.; et al. Supportive evidence for 11 loci from genome-wide association studies in Parkinson’s disease. Neurobiol. Aging 2013, 34, 1708.e7–1708.e13. [Google Scholar] [CrossRef]
- Dean, D.N.; Lee, J.C. Defining an amyloid link Between Parkinson’s disease and melanoma. Proc. Natl. Acad. Sci. USA 2020, 117, 22671–22673. [Google Scholar] [CrossRef]
- Haraszti, T.; Trantow, C.M.; Hedberg-Buenz, A.; Grunze, M.; Anderson, M.G. Spectral analysis by XANES reveals that GPNMB influences the chemical composition of intact melanosomes. Pigment Cell Melanoma Res. 2011, 24, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.S.; Chintamaneni, C.; Kozak, C.A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.; Barton, D.; Francke, U.; Kobayashi, Y.; Kim, K.K. A Melanocyte-Specific Gene, Pmel-17, Maps Near The Silver Coat Color Locus On Mouse Chromosome-10 And Is In A Syntenic Region On Human Chromosome-12. Proc. Natl. Acad. Sci. USA 1991, 88, 9228–9232. [Google Scholar] [CrossRef]
- Kerje, S.; Sharma, P.; Gunnarsson, U.; Kim, H.; Bagchi, S.; Fredriksson, R.; Schutz, K.; Jensen, P.; von Heijne, G.; Okimoto, R.; et al. The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 2004, 168, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Brunberg, E.; Andersson, L.; Cothran, G.; Sandberg, K.; Mikko, S.; Lindgren, G. A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet. 2006, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Lin, S.P.; Chiang, C.P.; Wu, Y.H.; H’ng, W.S.; Chang, C.P.; Chen, Y.T.; Wu, J.Y. Loss of GPNMB Causes Autosomal-Recessive Amyloidosis Cutis Dyschromica in Humans. Am. J. Hum. Genet. 2018, 102, 219–232. [Google Scholar] [CrossRef]
- Biswas, K.B.; Takahashi, A.; Mizutani, Y.; Takayama, S.; Ishitsuka, A.; Yang, L.L.; Yang, F.; Iddamalgoda, A.; Katayama, I.; Inoue, S. GPNMB is expressed in human epidermal keratinocytes but disappears in the vitiligo lesional skin. Sci. Rep. 2020, 10, 4930. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Shewmaker, F.; McPhie, P.; Monterroso, B.; Thurber, K.; Wickner, R.B. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 13731–13736. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.; Tzika, A.C.; Mitchell, S.M.; Liu, X.R.; Leonhardt, R.M. Repeat domain-associated O-glycans govern PMEL fibrillar sheet architecture. Sci. Rep. 2019, 9, 6101. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.C.; Rouzaud, F.; Julien, S.; Chen, K.G.; Passeron, T.; Yamaguchi, Y.; Abu-Asab, M.; Tsokos, M.; Costin, G.E.; Yamaguchi, H.; et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J. Biol. Chem. 2007, 282, 11266–11280. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.C.; Theos, A.C.; Herman, K.E.; Tenza, D.; Raposo, G.; Marks, M.S. Premelanosome amyloid-like fibrils are composed of only Golgi-processed forms of Pmel17 that have been proteolytically processed in Endosomes. J. Biol. Chem. 2008, 283, 2307–2322. [Google Scholar] [CrossRef]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Maruyama, H.; Huelsmann, C.; Baches, S.; Weggen, S.; Koo, E.H. Formation of Pmel17 Amyloid Is Regulated by Juxtamembrane Metalloproteinase Cleavage, and the Resulting C-terminal Fragment Is a Substrate for gamma-Secretase. J. Biol. Chem. 2009, 284, 2296–2306. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Hozumi, Y.; Suzuki, T. ADAM protease inhibitors reduce melanogenesis by regulating PMEL17 processing in human melanocytes. J. Dermatol. Sci. 2015, 78, 133–142. [Google Scholar] [CrossRef]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; De Strooper, B.; Raposo, G.; van Niel, G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef]
- Shimshek, D.R.; Jacobson, L.H.; Kolly, C.; Zamurovic, N.; Balavenkatraman, K.K.; Morawiec, L.; Kreutzer, R.; Schelle, J.; Jucker, M.; Bertschi, B.; et al. Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci. Rep. 2016, 6, 21917. [Google Scholar] [CrossRef]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef]
- Kobayashi, T.; Urabe, K.; Orlow, S.J.; Higashi, K.; Imokawa, G.; Kwon, B.S.; Potterf, B.; Hearing, V.J. The Pmel-17 Silver Locus Protein—Characterization And Investigation Of Its Melanogenic Function. J. Biol. Chem. 1994, 269, 29198–29205. [Google Scholar] [CrossRef]
- Lee, Z.H.; Hou, L.; Moellmann, G.; Kuklinska, E.; Antol, K.; Fraser, M.; Halaban, R.; Kwon, B.S. Characterization and subcellular localization of human Pmel 17 silver, a 100-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J. Investig. Dermatol. 1996, 106, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Hurbain, I.; Geerts, W.J.C.; Boudier, T.; Marco, S.; Verkleij, A.J.; Marks, M.S.; Raposo, G. Electron tomography of early melanosomes: Implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl. Acad. Sci. USA 2008, 105, 19726–19731. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef]
- Bissig, C.; Rochin, L.; van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef]
- Silvers, W.K. The Coat Colors of Mice: A Model. for Mammalian Gene Action and Interaction; Springer: New York, NY, USA, 1979. [Google Scholar] [CrossRef]
- Kwon, B.S.; Kim, K.K.; Halaban, R.; Pickard, R.T. Characterization of mouse PMEL-17 gene and silver locus. Pigment Cell Res. 1994, 7, 394–397. [Google Scholar] [CrossRef]
- Kwon, B.S.; Halaban, R.; Ponnazhagan, S.; Kim, K.; Chintamaneni, C.; Bennett, D.; Pickard, R.T. Mouse Silver Mutation Is Caused By A Single-Base Insertion In The Putative Cytoplasmic Domain Of Pmel-17. Nucleic Acids Res. 1995, 23, 154–158. [Google Scholar] [CrossRef]
- Theos, A.C.; Berson, J.F.; Theos, S.C.; Herman, K.E.; Harper, D.C.; Tenza, D.; Sviderskaya, E.V.; Lamoreux, M.L.; Bennett, D.C.; Raposo, G.; et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 2006, 17, 3598–3612. [Google Scholar] [CrossRef]
- Hellstrom, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannstrom, P.; Narfstrom, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of Pmel Alters Melanosome Shape But Has Only a Subtle Effect on Visible Pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Lampert, J.M.; von Lintig, J.; Schwarz, H.; Geisler, R.; Neuhauss, S.C.F. A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev. Biol. 2005, 284, 421–436. [Google Scholar] [CrossRef]
- Ishishita, S.; Takahashi, M.; Yamaguchi, K.; Kinoshita, K.; Nakano, M.; Nunome, M.; Kitahara, S.; Tatsumoto, S.; Go, Y.; Shigenobu, S.; et al. Nonsense mutation in PMEL is associated with yellowish plumage colour phenotype in Japanese quail. Sci. Rep. 2018, 8, 16732. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.C.; Kerje, S.; Hallbook, F.; Jensen, P. The Dominant white mutation in the PMEL17 gene does not cause visual impairment in chickens. Vet. Ophthalmol. 2009, 12, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Kuliawat, R.; Santambrogio, L. A mutation within the transmembrane domain of melanosomal protein Silver (Pmel17) changes lumenal fragment interactions. Eur. J. Cell Biol. 2009, 88, 653–667. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the Transmembrane Domain Alter PMEL Amyloid Formation from Functional to Pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef]
- Komaromy, A.M.; Rowlan, J.S.; La Croix, N.C.; Mangan, B.G. Equine Multiple Congenital Ocular Anomalies (MCOA) syndrome in PMEL17 (Silver) mutant ponies: Five cases. Vet. Ophthalmol. 2011, 14, 313–320. [Google Scholar] [CrossRef]
- Andersson, L.S.; Wilbe, M.; Viluma, A.; Cothran, G.; Ekesten, B.; Ewart, S.; Lindgren, G. Equine Multiple Congenital Ocular Anomalies and Silver Coat Colour Result from the Pleiotropic Effects of Mutant PMEL. PLoS ONE 2013, 8, e75639. [Google Scholar] [CrossRef]
- Segard, E.M.; Depecker, M.C.; Lang, J.; Gemperli, A.; Cadore, J.L. Ultrasonographic features of PMEL17 (Silver) mutant gene-associated multiple congenital ocular anomalies (MCOA) in Comtois and Rocky Mountain horses. Vet. Ophthalmol. 2013, 16, 429–435. [Google Scholar] [CrossRef]
- Clark, L.A.; Wahl, J.M.; Rees, C.A.; Murphy, K.E. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. USA 2006, 103, 1376–1381. [Google Scholar] [CrossRef]
- Ballif, B.C.; Ramirez, C.J.; Carl, C.R.; Sundin, K.; Krug, M.; Zahand, A.; Shaffer, L.G.; Flores-Smith, H. The PMEL Gene and Merle in the Domestic Dog: A Continuum of Insertion Lengths Leads to a Spectrum of Coat Color Variations in Australian Shepherds and Related Breeds. Cytogenet. Genome Res. 2018, 156, 22–34. [Google Scholar] [CrossRef]
- Murphy, S.C.; Evans, J.M.; Tsai, K.L.; Clarke, L.A. Length variations within the Merle retrotransposon of canine PMEL: Correlating genotype with phenotype. Mob. DNA 2018, 9, 26. [Google Scholar] [CrossRef]
- Kawahara, K.; Hirata, H.; Ohbuchi, K.; Nishi, K.; Maeda, A.; Kuniyasu, A.; Yamada, D.; Maeda, T.; Tsuji, A.; Sawada, M.; et al. The Novel Monoclonal Antibody 9F5 Reveals Expression of a Fragment of GPNMB/Osteoactivin Processed by Furin-like Protease(s) in a Subpopulation of Microglia in Neonatal Rat Brain. Glia 2016, 64, 1938–1961. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Barbe, M.F.; Rico, M.C.; Salihoglu, S.; Arango-Hisijara, I.; Selim, A.H.; Anderson, M.G.; Owen, T.A.; Popoff, S.N.; Safadi, F.F. Osteoactivin, an anabolic factor that regulates osteoblast differentiation and function. Exp. Cell Res. 2008, 314, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Sato, S.; Yamaguchi, Y.; Passeron, T.; Tamaki, K.; Hearing, V.J. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. Faseb J. 2010, 24, 1616–1629. [Google Scholar] [CrossRef]
- Rose, A.A.N.; Annis, M.G.; Dong, Z.F.; Pepin, F.; Hallett, M.; Park, M.; Siegel, P.M. ADAM10 Releases a Soluble Form of the GPNMB/Osteoactivin Extracellular Domain with Angiogenic Properties. PLoS ONE 2010, 5, e12093. [Google Scholar] [CrossRef]
- Chi, A.; Valencia, J.C.; Hu, Z.Z.; Watabe, H.; Yamaguchi, H.; Mangini, N.J.; Huang, H.Z.; Canfield, V.A.; Cheng, K.C.; Yang, F.; et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J. Proteome Res. 2006, 5, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Tomihari, M.; Hwang, S.H.; Chung, J.S.; Cruz, P.D.; Ariizumi, K. Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp. Dermatol. 2009, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, V.M.; Meadows, N.A.; Raggatt, L.J.; Chang, M.K.; Pettit, A.R.; Cassady, A.I.; Hume, D.A. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene 2008, 413, 32–41. [Google Scholar] [CrossRef]
- Loftus, S.K.; Antonellis, A.; Matera, I.; Renaud, G.; Baxter, L.L.; Reid, D.; Wolfsberg, T.G.; Chen, Y.D.; Wang, C.W.; Prasad, M.K.; et al. Gpnmb is a melanoblast-expressed, MITF-dependent gene. Pigment Cell Melanoma Res. 2009, 22, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, W.; Yuan, X.Y.; Li, D.G.; Gu, W.J.; Gao, T.W. Endothelin-1 enhances the melanogenesis via MITF-GPNMB pathway. Bmb Rep. 2013, 46, 364–369. [Google Scholar] [CrossRef]
- Gutknecht, M.; Geiger, J.; Joas, S.; Dorfel, D.; Salih, H.R.; Muller, M.R.; Grunebach, F.; Rittig, S.M. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun. Signal. 2015, 13, 19. [Google Scholar] [CrossRef]
- Theos, A.C.; Watt, B.; Harper, D.C.; Janczura, K.J.; Theos, S.C.; Herman, K.E.; Marks, M.S. The PKD domain distinguishes the trafficking and amyloidogenic properties of the pigment cell protein PMEL and its homologue GPNMB. Pigment Cell Melanoma Res. 2013, 26, 470–486. [Google Scholar] [CrossRef]
- Rahman, O.U.; Kim, J.; Mahon, C.; Jelani, M.; Kang, C. Two missense mutations in GPNMB cause autosomal recessive amyloidosis cutis dyschromica in the consanguineous pakistani families. Genes Genom. 2021, 43, 471–478. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Hsu, C.K.; Eide, C.R.; Nanda, A.; Orchard, G.E.; Tomita, K.; Sheriff, A.; Scott, W.; Tierney, C.; Lee, J.Y.W.; et al. Semidominant GPNMB Mutations in Amyloidosis Cutis Dyschromica. J. Investig. Dermatol. 2019, 139, 2550. [Google Scholar] [CrossRef] [PubMed]
- Chiu, F.P.C.; Wessagowit, V.; Cakmak, M.F.; Doolan, B.J.; Kootiratrakarn, T.; Chaowalit, P.; Bunnag, T.; Simpson, M.A.; McGrath, J.A.; Onoufriadis, A. Molecular basis and inheritance patterns of amyloidosis cutis dyschromica. Clin. Exp. Dermatol. 2020, 45, 650–653. [Google Scholar] [CrossRef]
- Sha, Y.; Li, L. Amyloidosis cutis dyschromica due to homozygous variants of the GPNMB gene in a Chinese pedigree. Chin. J. Med. Genet. 2021, 38, 123–126. [Google Scholar] [CrossRef]
- Owen, T.A.; Smock, S.L.; Prakash, P.; Pinder, L.; Brees, D.; Krull, D.; Castleberry, T.A.; Clancy, Y.C.; Marks, S.C.; Safadi, F.F.; et al. Identification and characterization of the genes encoding human and mouse osteoactivin. Crit. Rev. Eukaryot. Gene Expr. 2003, 13, 205–220. [Google Scholar] [CrossRef]
- Huang, J.J.; Ma, W.J.; Yokoyama, S. Expression and immunolocalization of Gpnmb, a glioma-associated glycoprotein, in normal and inflamed central nervous systems of adult rats. Brain Behav. 2012, 2, 85–96. [Google Scholar] [CrossRef]
- Shi, F.Y.; Duan, S.Y.; Cui, J.H.; Yan, X.R.; Li, H.M.; Wang, Y.J.; Chen, F.L.; Zhang, L.H.; Liu, J.; Xie, X. Induction of Matrix Metalloproteinase-3 (MMP-3) Expression in the Microglia by Lipopolysaccharide (LPS) via Upregulation of Glycoprotein Nonmetastatic Melanoma B (GPNMB) Expression. J. Mol. Neurosci. 2014, 54, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Yoshino, Y.; Tsuruma, K.; Moriguchi, S.; Oyagi, A.; Tanaka, H.; Ishisaka, M.; Shimazawa, M.; Fukunaga, K.; Hara, H. The extracellular fragment of GPNMB (Glycoprotein nonmelanosoma protein B, osteoactivin) improves memory and increases hippocampal GluA1 levels in mice. J. Neurochem. 2015, 132, 583–594. [Google Scholar] [CrossRef]
- Ripoll, V.M.; Irvine, K.M.; Ravasi, T.; Sweet, M.J.; Hume, D.A. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J. Immunol. 2007, 178, 6557–6566. [Google Scholar] [CrossRef]
- Zhou, L.T.; Zhuo, H.; Ouyang, H.Y.; Liu, Y.X.; Yuan, F.; Sun, L.; Liu, F.Y.; Liu, H. Glycoprotein non-metastatic melanoma protein b (Gpnmb) is highly expressed in macrophages of acute injured kidney and promotes M2 macrophages polarization. Cell. Immunol. 2017, 316, 53–60. [Google Scholar] [CrossRef]
- Mo, J.S.; Anderson, M.G.; Gregory, M.; Smith, R.S.; Savinova, O.V.; Serreze, D.V.; Ksander, B.R.; Streilein, J.W.; John, S.W.M. By altering ocular immune privilege, bone marrow-derived cells pathogenically contribute to DBA/2J pigmentary glaucoma. J. Exp. Med. 2003, 197, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Nair, K.S.; Amonoo, L.A.; Mehalow, A.; Trantow, C.M.; Masli, S.; John, S.W.M. Gpnmb(R150X) allele must be present in bone marrow derived cells to mediate DBA/2J glaucoma. BMC Genet. 2008, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Safadi, F.F.; Xu, J.; Smock, S.L.; Rico, M.C.; Owen, T.A.; Popoff, S.N. Cloning and characterization of osteoactivin, a novel cDNA expressed in osteoblasts. J. Cell. Biochem. 2002, 84, 12–26. [Google Scholar] [CrossRef]
- Sheng, M.H.C.; Wergedal, J.E.; Mohan, S.; Lau, K.H.W. Osteoactivin is a novel osteoclastic protein and plays a key role in osteoclast differentiation and activity. FEBS Lett. 2008, 582, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Selim, A.A.; Abdelmagid, S.M.; Kanaan, R.A.; Smock, S.L.; Owen, T.A.; Popoff, S.N.; Safadi, F.F. Anti-osteoactivin antibody inhibits osteoblast differentiation and function in vitro. Crit. Rev. Eukaryot. Gene Expr. 2003, 13, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Barbe, M.F.; Arango-Hisijara, I.; Owen, T.A.; Popoff, S.N.; Safadi, F.F. Osteoactivin acts as downstream mediator of BMP-2 effects on osteoblast function. J. Cell. Physiol. 2007, 210, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Selim, A.A.; Castaneda, J.L.; Owen, T.A.; Popoff, S.N.; Safadi, F.F. The role of osteoactivin-derived peptides in osteoblast differentiation. Med. Sci. Monit. 2007, 13, BR259–BR270. [Google Scholar]
- Arosarena, O.A.; Del Carpio-Cano, F.E.; Dela Cadena, R.A.; Rico, M.C.; Nwodim, E.; Safadi, F.F. Comparison of Bone Morphogenetic Protein-2 and Osteoactivin for Mesenchymal Cell Differentiation: Effects of Bolus and Continuous Administration. J. Cell. Physiol. 2011, 226, 2943–2952. [Google Scholar] [CrossRef]
- Ogawa, T.; Nikawa, T.; Furochi, H.; Kosyoji, M.; Hirasaka, K.; Suzue, N.; Sairyo, K.; Nakano, S.; Yamaoka, T.; Itakura, M.; et al. Osteoactivin upregulates expression of MMP-3 and MMP-9 in fibroblasts infiltrated into denervated skeletal muscle in mice. Am. J. Physiol. Cell Physiol. 2005, 289, C697–C707. [Google Scholar] [CrossRef]
- Abe, H.; Uto, H.; Takami, Y.; Takahama, Y.; Hasuike, S.; Kodama, M.; Nagata, K.; Moriuchi, A.; Numata, M.; Ido, A.; et al. Transgenic expression of osteoactivin in the liver attenuates hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 2007, 356, 610–615. [Google Scholar] [CrossRef]
- Sheng, M.H.C.; Wergedal, J.E.; Mohan, S.; Amoui, M.; Baylink, D.J.; Lau, K.H.W. Targeted Overexpression of Osteoactivin in Cells of Osteoclastic Lineage Promotes Osteoclastic Resorption and Bone Loss in Mice. PLoS ONE 2012, 7, e35280. [Google Scholar] [CrossRef]
- Tonogai, I.; Takahashi, M.; Yukata, K.; Sato, R.; Nikawa, T.; Yasui, N.; Sairyo, K. Osteoactivin attenuates skeletal muscle fibrosis after distraction osteogenesis by promoting extracellular matrix degradation/remodeling. J. Pediatr. Orthop. Part B 2015, 24, 162–169. [Google Scholar] [CrossRef]
- Arosarena, O.A.; Barr, E.W.; Thorpe, R.; Yankey, H.; Tarr, J.T.; Safadi, F.F. Osteoactivin regulates head and neck squamous cell carcinoma invasion by modulating matrix metalloproteases. J. Cell. Physiol. 2018, 233, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, K.; Owaki, K.; Okumura, Y.; Yano, M.; Oto, T.; Suzuki, E.; Tamura, S.; Abe, T.; Kohno, S.; Ohno, A.; et al. An Intracellular Fragment of Osteoactivin Formed by Ectodomain Shedding Trans located to the Nucleoplasm and Bound to RNA Binding Proteins. Biosci. Biotechnol. Biochem. 2012, 76, 2225–2229. [Google Scholar] [CrossRef]
- Furochi, H.; Tamura, S.; Mameoka, M.; Yamada, C.; Ogawa, T.; Hirasaka, K.; Okumura, Y.; Imagawa, T.; Oguri, S.; Ishidoh, K.; et al. Osteoactivin fragments produced by ectodomain shedding induce MMP-3 expression via ERK pathway in mouse NIH-3T3 fibroblasts. FEBS Lett. 2007, 581, 5743–5750. [Google Scholar] [CrossRef] [PubMed]
- Moussa, F.M.; Hisijara, I.A.; Sondag, G.R.; Scott, E.M.; Frara, N.; Abdelmagid, S.M.; Safadi, F.F. Osteoactivin Promotes Osteoblast Adhesion Through HSPG and alpha v beta 1 Integrin. J. Cell. Biochem. 2014, 115, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Murthy, M.N.; Blauwendraat, C.; Guelfi, S.; Hardy, J.; Lewis, P.A.; Trabzuni, D.; UKBEC. Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson’s disease on chromosome 7p15.3. Neurogenetics 2017, 18, 121–133. [Google Scholar] [CrossRef]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef]
- Huttenrauch, M.; Ogorek, I.; Klafki, H.; Otto, M.; Stadelmann, C.; Weggen, S.; Wiltfang, J.; Wirths, O. Glycoprotein NMB: A novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 2018, 6, 108. [Google Scholar] [CrossRef]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Transgenic Overexpression of GPNMB Protects Against MPTP-Induced Neurodegeneration. Mol. Neurobiol. 2020, 57, 2920–2933. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, M.; Tsuruma, K.; Yamada, M.; Takahashi, H.; Hozumi, I.; Niwa, J.; Iguchi, Y.; et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef]
- Nagahara, Y.; Shimazawa, M.; Ohuchi, K.; Ito, J.; Takahashi, H.; Tsuruma, K.; Kakita, A.; Hara, H. GPNMB ameliorates mutant TDP-43-induced motor neuron cell death. J. Neurosci. Res. 2017, 95, 1647–1665. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Heger, A.; Holm, L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins-Struct. Funct. Genet. 2000, 41, 224–237. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef]
- Ho, T.; Watt, B.; Spruce, L.A.; Seeholzer, S.H.; Marks, M.S. The Kringle-like Domain Facilitates Post-endoplasmic Reticulum Changes to Premelanosome Protein (PMEL) Oligomerization and Disulfide Bond Configuration and Promotes Amyloid Formation. J. Biol. Chem. 2016, 291, 3595–3612. [Google Scholar] [CrossRef]
- Xie, R.; Okita, Y.; Ichikawa, Y.; Fikry, M.A.; Dam, K.T.H.; Tran, S.T.P.; Kato, M. Role of the kringle-like domain in glycoprotein NMB for its tumorigenic potential. Cancer Sci. 2019, 110, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; Hu, K.N.; McPhie, P.; Tycko, R.; Wickner, R.B. Repeat Domains of Melanosome Matrix Protein Pmel17 Orthologs Form Amyloid Fibrils at the Acidic Melanosomal pH. J. Biol. Chem. 2011, 286, 8385–8393. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Muller, J.; Vieira, W.D.; Rouzaud, F.; Kikuchi, K.; Tamaki, K.; Hearing, V.J. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem. 2006, 281, 21198–21208. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal Domains Elicit Formation of Functional Pmel17 Amyloid Fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, R.M.; Vigneron, N.; Hee, J.S.; Graham, M.; Cresswella, P. Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol. Biol. Cell 2013, 24, 964–981. [Google Scholar] [CrossRef]
- Kenny, N.J.; Francis, W.R.; Rivera-Vicens, R.E.; Juravel, K.; de Mendoza, A.; Diez-Vives, C.; Lister, R.; Bezares-Calderon, L.A.; Grombacher, L.; Roller, M.; et al. Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat. Commun. 2020, 11, 3676. [Google Scholar] [CrossRef]
- Moroz, L.L.; Kocot, K.M.; Citarella, M.R.; Dosung, S.; Norekian, T.P.; Povolotskaya, I.S.; Grigorenko, A.P.; Dailey, C.; Berezikov, E.; Buckley, K.M.; et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 2014, 510, 109. [Google Scholar] [CrossRef]
- He, X.L.; Zhang, J.Z. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 2005, 169, 1157–1164. [Google Scholar] [CrossRef]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar] [CrossRef]
- Holland, P.W.H.; Garciafernandez, J.; Williams, N.A.; Sidow, A. Gene Duplications And The Origins Of Vertebrate Development. Development 1994, 1994, 125–133. [Google Scholar] [CrossRef]
- Dehal, P.; Boore, J.L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005, 3, e314. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin, Germany, 1970. [Google Scholar]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.K.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Keinath, M.C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 2015, 25, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Crollius, H.R. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 166. [Google Scholar] [CrossRef] [PubMed]
- Simakov, O.; Marletaz, F.; Yue, J.X.; O’Connell, B.; Jenkins, J.; Brandt, A.; Calef, R.; Tung, C.H.; Huang, T.K.; Schmutz, J.; et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 2020, 4, 820. [Google Scholar] [CrossRef]
- Smith, J.J.; Kuraku, S.; Holt, C.; Sauka-Spengler, T.; Jiang, N.; Campbell, M.S.; Yandell, M.D.; Manousaki, T.; Meyer, A.; Bloom, O.E.; et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 2013, 45, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R.; Canestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Mehta, T.K.; Ravi, V.; Yamasaki, S.; Lee, A.P.; Lian, M.M.; Tay, B.H.; Tohari, S.; Yanai, S.; Tay, A.; Brenner, S.; et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl. Acad. Sci. USA 2013, 110, 16044–16049. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Brunet, F.; Petit, J.L.; Stange-Thomann, N.; Mauceli, E.; Bouneau, L.; Fischer, C.; Ozouf-Costaz, C.; Bernot, A.; et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 2004, 431, 946–957. [Google Scholar] [CrossRef]
- Meyer, A.; Van de Peer, Y. From 2R to 3R: Evidence for a fish-specific genome duplication (FSGD). Bioessays 2005, 27, 937–945. [Google Scholar] [CrossRef]
- Inoue, J.; Sato, Y.; Sinclair, R.; Tsukamoto, K.; Nishida, M. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc. Natl. Acad. Sci. USA 2015, 112, 14918–14923. [Google Scholar] [CrossRef]
- Pasquier, J.; Braasch, I.; Batzel, P.; Cabau, C.; Montfort, J.; Nguyen, T.; Jouanno, E.; Berthelot, C.; Klopp, C.; Journot, L.; et al. Evolution of gene expression after whole-genome duplication: New insights from the spotted gar genome. J. Exp. Zool. Part. B Mol. Dev. Evol. 2017, 328, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Vonheijne, G. The Signal Peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Duffaud, G.D.; Lehnhardt, S.K.; March, P.E.; Inouye, M. Structure And Function Of The Signal Peptide. Curr. Top. Membr. Transport. 1985, 24, 65–104. [Google Scholar]
- Hee, J.S.; Mitchell, S.M.; Liu, X.R.; Leonhardt, R.M. Melanosomal formation of PMEL core amyloid is driven by aromatic residues. Sci. Rep. 2017, 7, 44064. [Google Scholar] [CrossRef]
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; Sanmillan, J.L.; Gamble, V.; Harris, P.C. The Polycystic Kidney-Disease-1 (Pkd1) Gene Encodes A Novel Protein With Multiple Cell Recognition Domains. Nat. Genet. 1995, 10, 151–160. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Bukanov, N.O.; Donohue, L.C.; Dackowski, W.R.; Klinger, K.W.; Landes, G.M. Strong homophilic interactions of the Ig-like domains of polycystin-1,the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum. Mol. Genet. 2000, 9, 1641–1649. [Google Scholar] [CrossRef]
- Bycroft, M.; Bateman, A.; Clarke, J.; Hamill, S.J.; Sandford, R.; Thomas, R.L.; Chothia, C. The structure of a PKD domain from polycystin-1: Implications for polycystic kidney disease. EMBO J. 1999, 18, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef]
- Le Borgne, R.; Planque, N.; Martin, P.; Dewitte, F.; Saule, S.; Hoflack, B. The AP-3-dependent targeting of the melanosomal glycoprotein QNR-71 requires a di-leucine-based sorting signal. J. Cell Sci. 2001, 114, 2831–2841. [Google Scholar] [CrossRef]
- Hoashi, T.; Muller, J.; Tamaki, K.; Ohara, K.; Hearing, V.J. The internal repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for fibrillogenesis but not for intracellular trafficking. Pigment Cell Melanoma Res. 2008, 21, 303. [Google Scholar]
- Goujon, M.; McWilliam, H.; Li, W.Z.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Animal | HGVS cDNA | HGVS Protein | Inheritance | Phenotype | Notes | Reference |
|---|---|---|---|---|---|---|---|
| SP | Cattle | NM_001080215.2: c.50_52del | NP_001073684.2: p.(Leu18del) | dominant | dilute coat | Highland/Galloway (c.64G > A) allele, interacts with MC1R (e) allele | Schmutz 2013 |
| SP | Cattle | NM_001080215.2: c.64G > A | NP_001073684.2: p.(Gly22Arg) | dominant | dilute coat | Charolais (Dc) allele | Kühn 2007 |
| NTR | Mouse | NM_021882.4: c.74_331del | NP_068682.2: p.(Gly25_Asn110del) | recessive | dilute coat and tail, loss of rod-shape in melanosomes (including uveal melanocytes and RPE cells) | null allele | Hellström 2011 |
| NTR | Quail | XM_032441057.1: c.271T > G, 353G > A, 446G > A | XP_032296948.1: p.(Ser91Ala), (Arg118His), (Trp149Ter) | recessive | yellowish plumage | “L” strain | Ishishita 2018 |
| PKD | Chicken | - | “del.280-284PTVT” relative to AY636124.1 | dominant | grayish plumage | Smoky allele, modifies Dominant White mutation | Kerje 2004 |
| RPT | Zebrafish | NM_001045330.1: c.1474G > T | NP_001038795.1: p.(Glu492Ter) | n/a | hypopigmented body and RPE, vision defects | “fading vision” (fdv) mutant | Schonthaler 2005 |
| TM | Chicken | - | “ins.723-725WAP” relative to AY636124.1 | dominant | white plumage | Dominant white allele, coincident with N399D variant upstream of RPT | Kerje 2004 |
| TM | Chicken | - | “del.731-7135LGTAA” relative to AY636124.1 | dominant | brown/khaki plumage | Dun allele, coincident with A35V, G105S, and R740C variants in NTR or Cyt domains | Kerje 2004 |
| TM | Horse | NM_001163889.1: c.1849C > T | NP_001157361.1: p.(Arg617Cys) | dominant | silver mane and tail, body dapples, MCOA syndrome | Multiple Congenital Ocular Anomalies | Andersson 2013 |
| Cyt | Dog | “SINE insertion in final intron” | (predicted to affect splicing) | dominant | merle (diluted) coat pattern, various auditory and ocular defects | variable oligo(dA) length affects phenotype | Clark 2006; Murphy 2018 |
| Cyt | Mouse | NM_021882.4: c.1805insA | NP_068682.2: p.(Trp602Ter) | recessive | silver coat | reduction in melanocyte density | Martínez-Esparza 1999 |
| Cyt | Zebrafish | NM_001045330.1: c.2426_2436del | NP_001038795.1: p.(Arg810fs) | recessive | global hypopigmentation, ocular defects | pmelaua5022 allele | Lahola-Chomiak 2018 |
| Domain | HGVS cDNA | HGVS Protein | Inheritance | Phenotype | Notes | Reference |
|---|---|---|---|---|---|---|
| NTR | NM 001200054.1 c.332A | NP 001186983.1: p.(Asn111Ser) | putative dominant | PDS | singleton | Lahola-Chomiak 2018 |
| CAF | NM 001200054.1 c.523G > A | NP 001186983.1: p.(Gly175Ser) | dominant | PDS / PG | 13 member family | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.974G > T | NP 001186983.1: p.(Gly325Val) | putative dominant | PDS / PG | singleton | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.994G > A | NP 001186983.1: p.(Val332Ile) | putative dominant | PDS | singleton | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.1019C > T | NP 001186983.1: p.(Ala340Val) | putative dominant | PDS / PG | 2 member family | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.1110G > C | NP 001186983.1: p.(Glu370Asp) | putative dominant | PDS / PG | singleton x3 | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.1112G > C | NP 001186983.1: p.(Ser371Thr) | putative dominant | PDS | singleton | Lahola-Chomiak 2018 |
| RPT | NM 001200054.1 c.1166T > C | NP 001186983.1: p.(Leu389Pro) | putative dominant | PDS | singleton x3 | Lahola-Chomiak 2018 |
| Cyt | NM 001200054.1 c.1921_1926del | NP 001186983.1: p.(Ser641_Ser642del) | putative dominant | PDS / PG | singleton | Lahola-Chomiak 2018 |
| Domain | HGVS cDNA | HGVS Protein | Inheritance | Phenotype | Notes | Reference |
|---|---|---|---|---|---|---|
| NTR/CAF | NM_001005340.2: c.296del, c.565C > T | NP_001005340.1: p.(Asn99ThrfsTer2) p.(Arg189Ter) | homozygous; putative recessive | ACD | Han Chinese | Yang 2018 |
| CAF | NM_001005340.2: c.522C > G | NP_001005340.1: p.(Ile174Met) | homozygous; recessive | ACD | consanguineous; Pakistani | Rahman 2021 |
| CAF | NM_001005340.2: c.565C > T | NP_001005340.1: p.(Arg189Ter) | homozygous; putative recessive | ACD | Han Chinese | Yang 2018 |
| CAF | NM_001005340.2: c.565C > T | NP_001005340.1: p.(Arg189Ter) | homozygous; recessive | ACD | Chinese | Sha 2021 |
| CAF/CAF | NM_001005340.2: c.565C > T, c.660T > G | NP_001005340.1: p.(Arg189Ter) p.(Tyr220Ter) | compound heterozygous; recessive | ACD, skin blisters | Han Chinese | Yang 2018 |
| CAF/btw PKD + KLD | NM_001005340.2: c.565C > T, c.1092del | NP_001005340.1: p.(Arg189Ter) p.(Pro365LeufsTer20) | compound heterozygous; semi-dominant | ACD | Taiwanese; Han Chinese | Onoufriadis 2019; Yang 2018 |
| CAF/KLD | NM_001005340.2: c565C > T, c.1273T > C | NP_001005340.1: p.(Arg189Ter) p.(Cys425Arg) | compound heterozygous; recessive | ACD | Thai | Chiu 2021 |
| CAF | NM_001005340.2: c.700 + 5G > T | NP_001005340.1: p.(Asp234GlyfsTer7) | homo- / heterozygous; semi-dominant | ACD | consanguineous; Kuwaiti Bedouin | Onoufriadis 2019 |
| CAF/PKD | NM_001005340.2: c.719_720del, c.877_880del | NP_001005340.1: p.(Val240AspfsTer24) p.(Val293ProfsTer6) | compound heterozygous; putative recessive | ACD | Han Chinese | Yang 2018 |
| btw PKD + KLD | NM_001005340.2: c.1124G > T | NP_001005340.1: p.(Gly375Val) | homozygous; recessive | ACD | consanguineous; Pakistani | Rahman 2021 |
| KLD | NM_001005340.2 c.1274G > C | NP_001005340.1: p.(Cys425Ser) | homo- / heterozygous; semi-dominant | ACD | Filipino | Onoufriadis 2019 |
| Domain | Animal | HGVS cDNA | HGVS Protein | Inheritance | Phenotype | Notes | Reference |
|---|---|---|---|---|---|---|---|
| NTR | Mouse | NM_053110.4: c.653C > T | NP_444340.3: p.(Arg150Ter) | recessive | PDS, PG | DBA/2J (D2) | Anderson 2002 |
| btw CAF + PKD | Zebrafish | ENSDART00000090883.6: c.799C > T | ENSDARP00000085316.5 P.(Gln267Ter) | N/A | none reported | ZFIN ID: ZDB-ALT-130411-2760 | Dooley 2019 |
| PKD | Zebrafish | ENSDART00000090883.6: c.854G > A | ENSDARP00000085316.5 p.(Trp285Ter) | N/A | none reported | ZFIN ID: ZDB-ALT-130411-2835 | Dooley 2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrystal, P.W.; Footz, T.; Hodges, E.D.; Jensen, J.A.; Walter, M.A.; Allison, W.T. Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins. Molecules 2021, 26, 3529. https://doi.org/10.3390/molecules26123529
Chrystal PW, Footz T, Hodges ED, Jensen JA, Walter MA, Allison WT. Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins. Molecules. 2021; 26(12):3529. https://doi.org/10.3390/molecules26123529
Chicago/Turabian StyleChrystal, Paul W., Tim Footz, Elizabeth D. Hodges, Justin A. Jensen, Michael A. Walter, and W. Ted Allison. 2021. "Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins" Molecules 26, no. 12: 3529. https://doi.org/10.3390/molecules26123529
APA StyleChrystal, P. W., Footz, T., Hodges, E. D., Jensen, J. A., Walter, M. A., & Allison, W. T. (2021). Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins. Molecules, 26(12), 3529. https://doi.org/10.3390/molecules26123529