
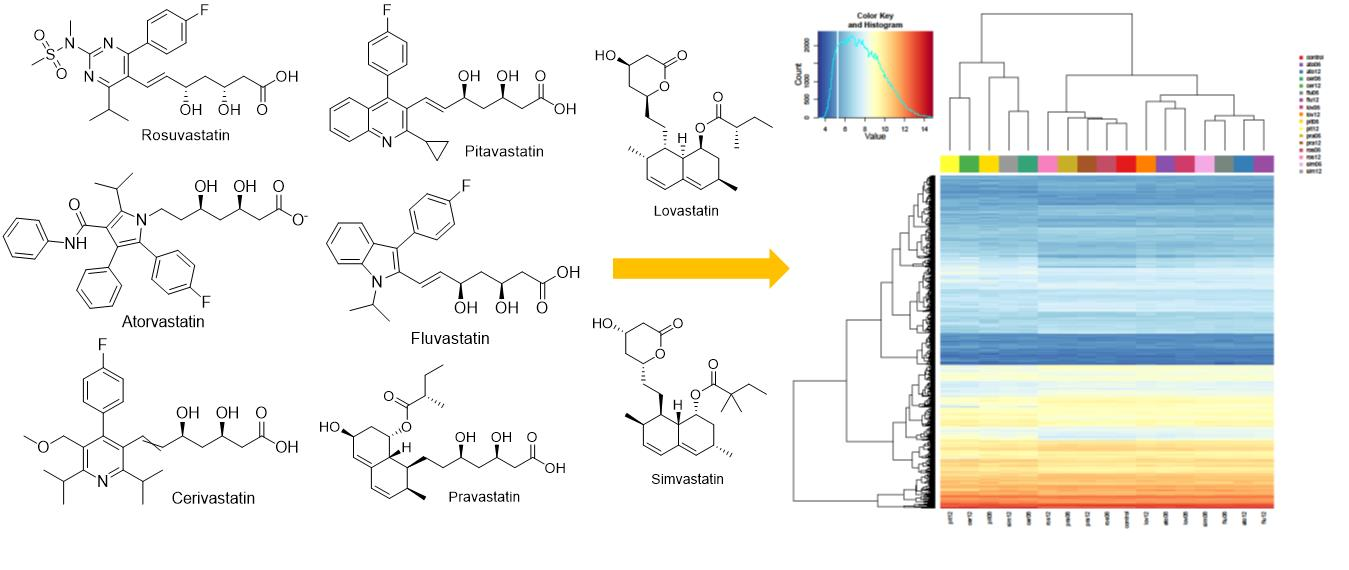
Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins

 ,
,  ,
, 
 ,
,  and
and
Abstract

1. Introduction
2. Results
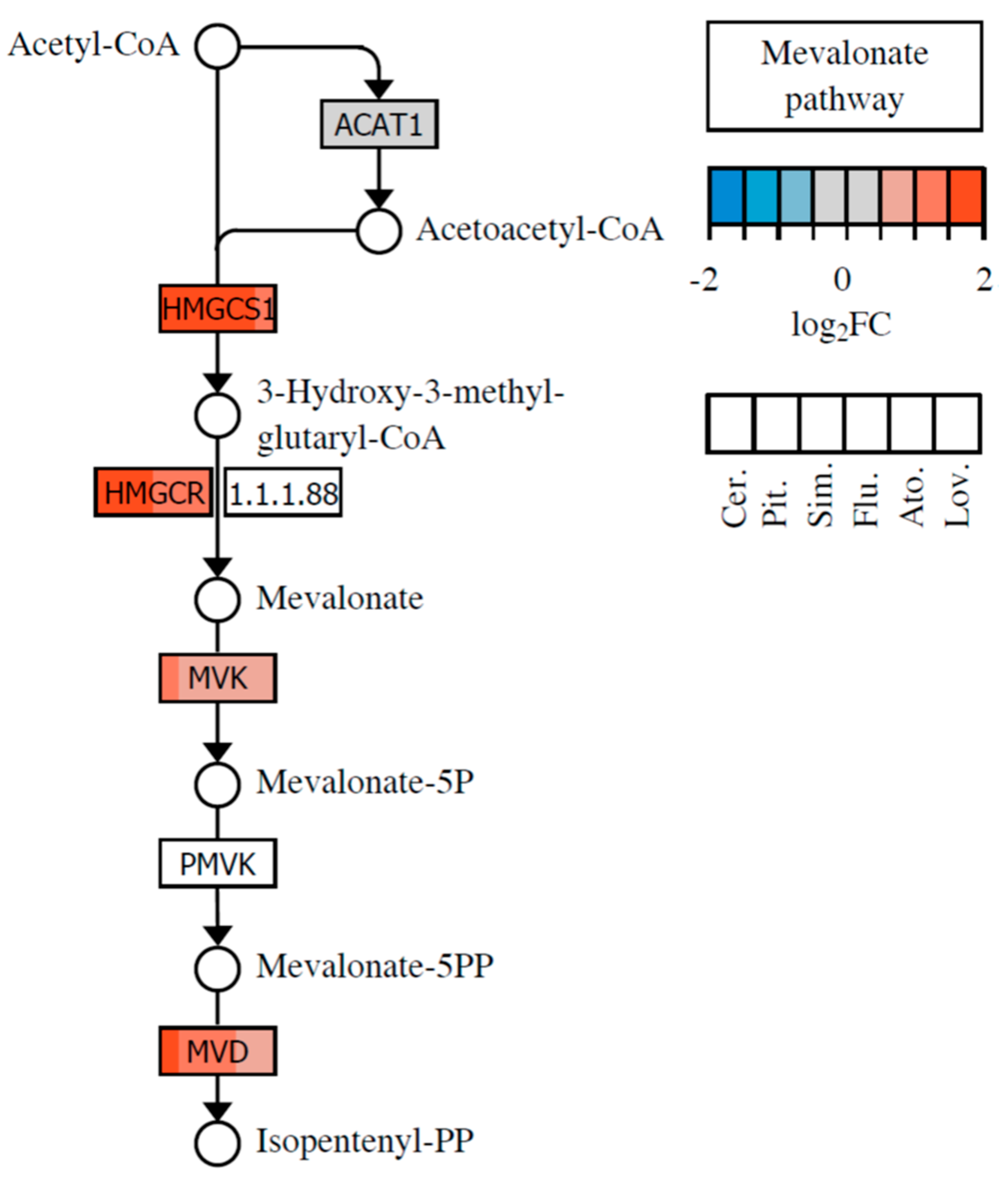
2.1. Effect of Statins on Lipid Metabolism and Synthesis of Steroids
2.2. Statins in the Role of Epigenetic Regulators
2.3. Statins and Their Potential Role as Posttranslational Regulators
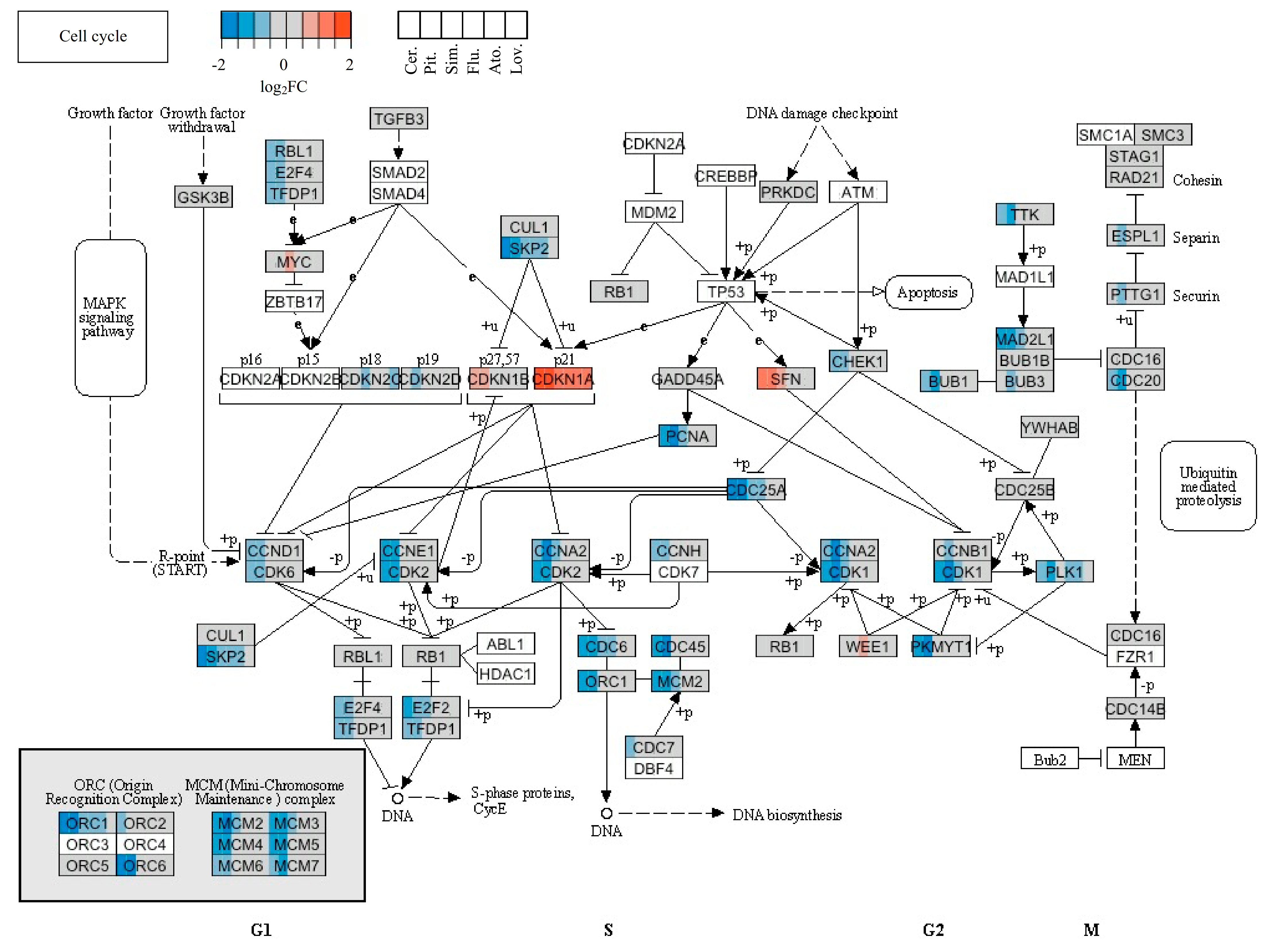
2.4. Statins’ Effect on Cell Cycle and Cell Death
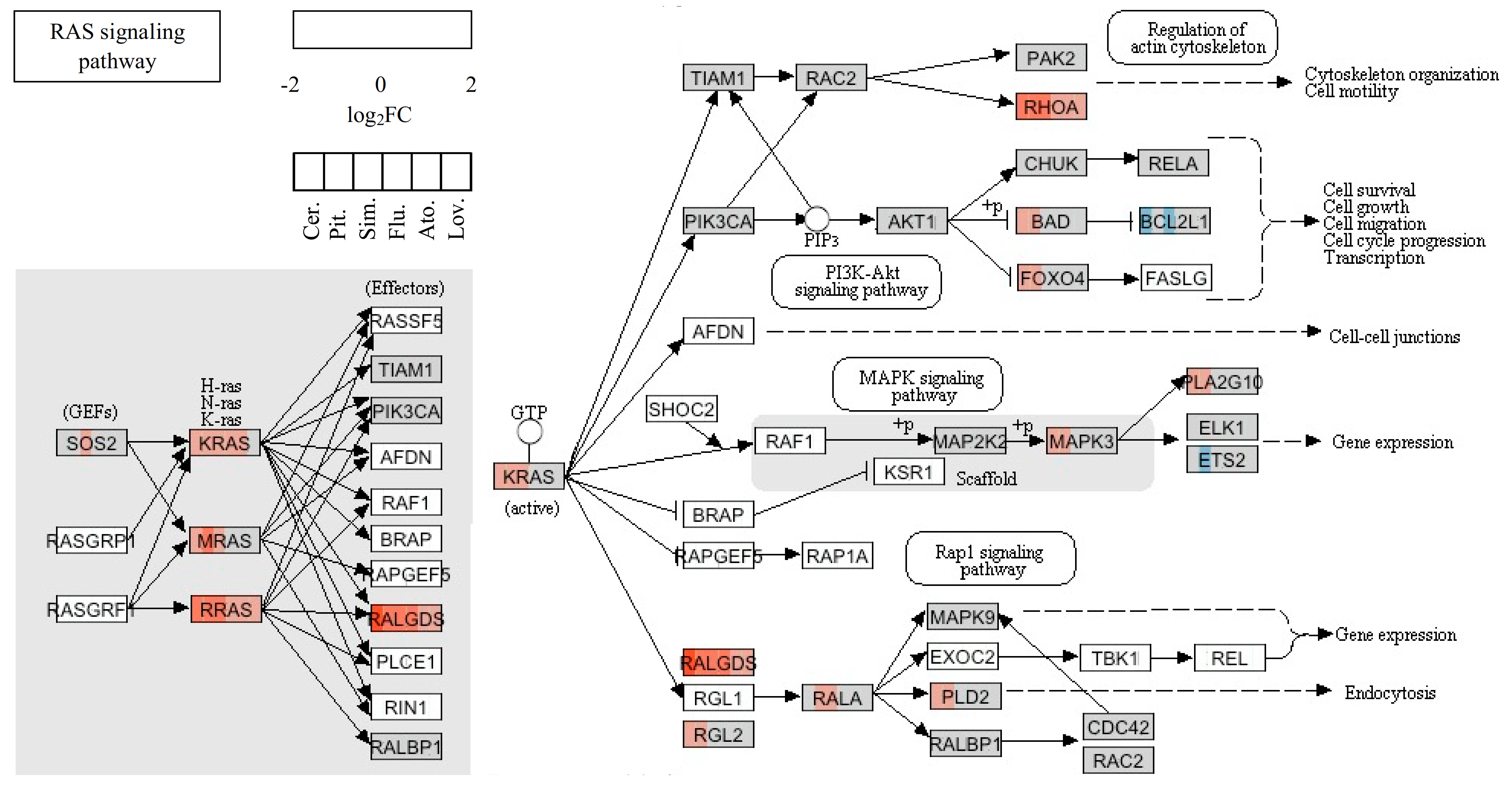
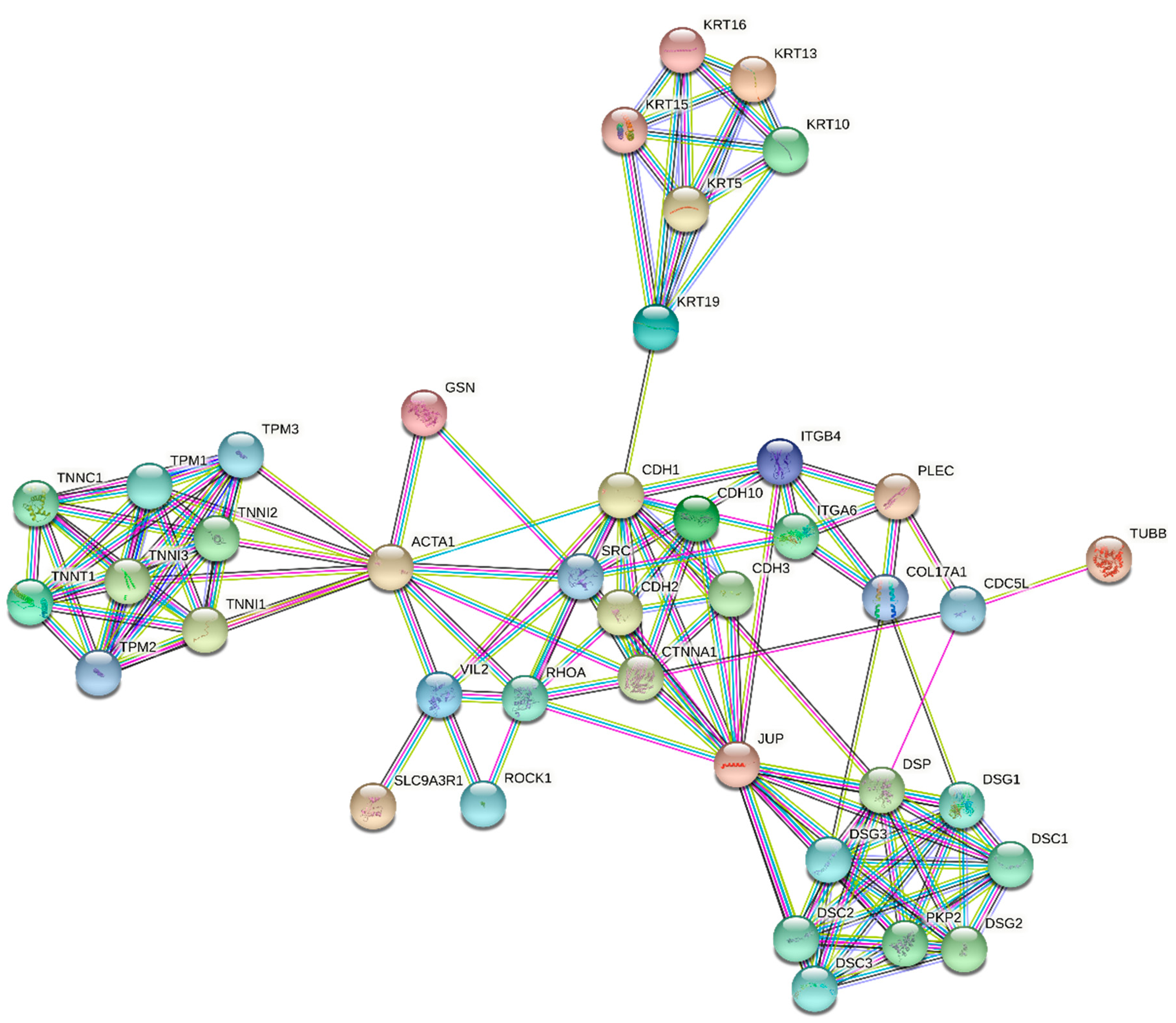
2.5. Statins’ Effect on Migration and Invasion of Cancer Cells
3. Materials and Methods
3.1. DNA Microarray Analysis
3.2. RT-qPCR Analysis
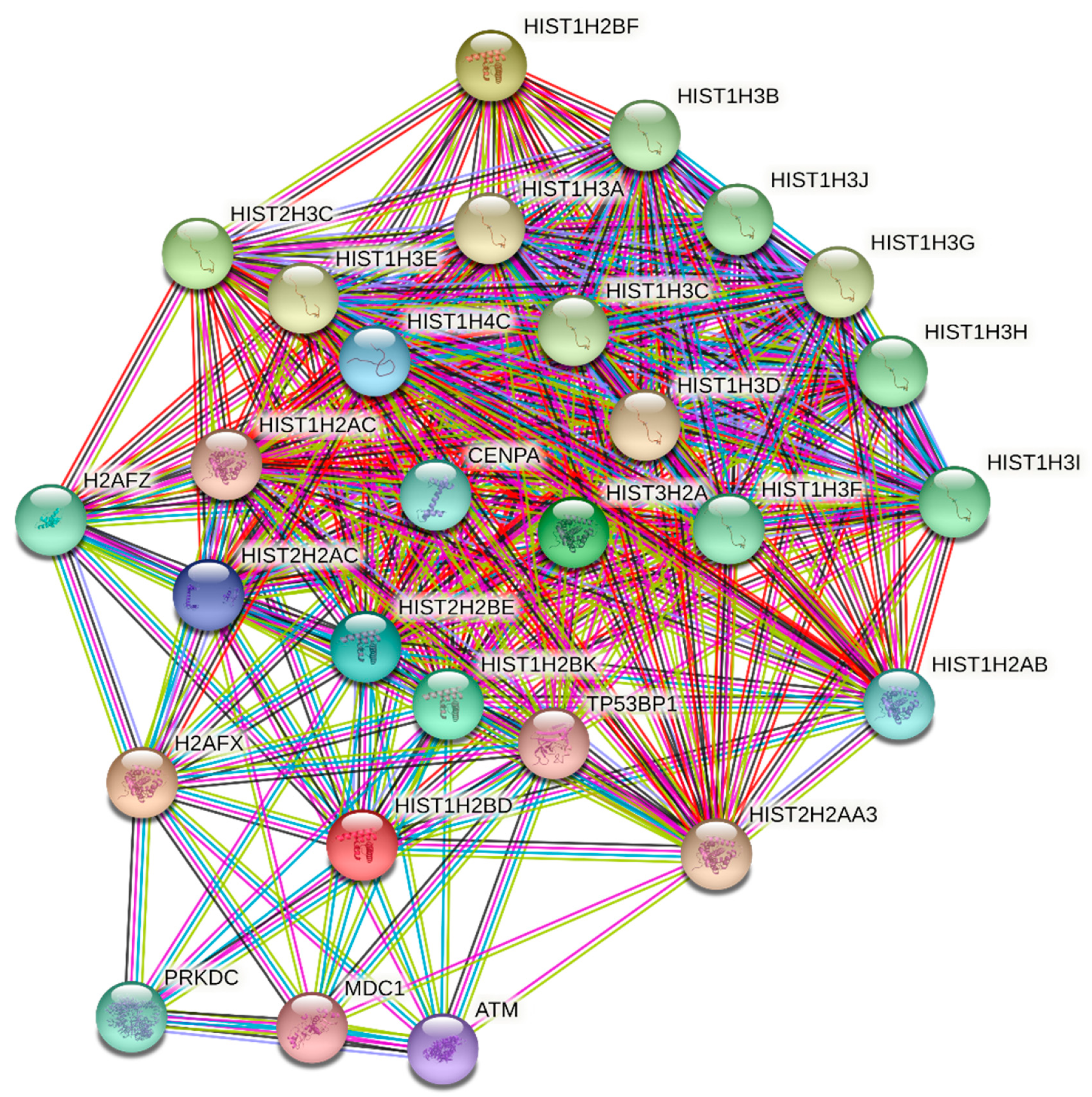
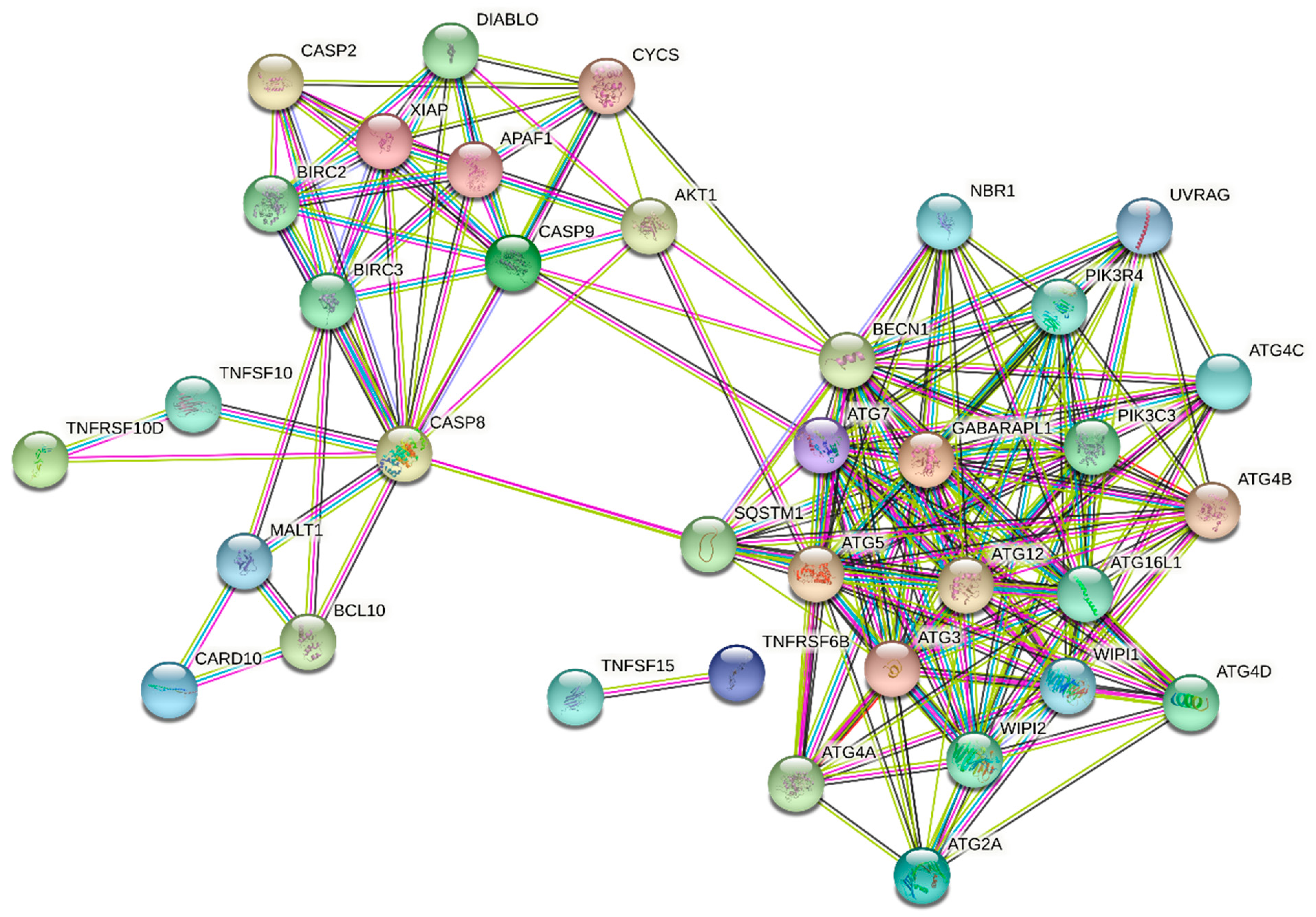
3.3. STRING Analysis
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement:
Informed Consent Statement
Data Availability Statement:
Acknowledgments
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, R.; Dinev, D.; Glavcheva, M.; Danova, J.; Yetik-Anacak, G.; Krasilnikova, J.; Podlipnik, C. Briefly about anticancer properties of statins. Biomed. J. Sci. Tech. Res. 2019, 7, 12655–12659. [Google Scholar] [CrossRef]
- Mohammadkhani, N.; Gharbib, S.; Rajani, H.F.; Farzaneh, A.; Mahjoobe, G.; Hoseinsalari, A.; Korsching, E. Statins: Complex outcomes but increasingly helpful treatment options for patients. European J. Pharmacol. 2019, 863, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tavintharan, S.; Ong, C.N.; Jeyaseelan, K.; Sivakumar, M.; Lim, S.C.; Sum, C.F. Reduced mitochondrial coenzyme Q10 levels in HepG2 cells treated with high-dose simvastatin: A possible role in statin-induced hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 223, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Vyskočil, J.; Nováková, J. Statiny v onkologii. Klin. Farmakol. Farm. 2005, 19, 155–159. [Google Scholar]
- Newman, T.B.; Hulley, S.B. Carcinogenicity of lipid-lowering drugs. JAMA 1996, 275, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Dalen, J.E.; Dalton, W.S. Does lowering cholesterol cause cancer? JAMA 1996, 275, 67–69. [Google Scholar] [CrossRef]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef]
- Downs, J.R.; Clearfield, M.; Weis, S.; Whitney, E.; Shapiro, D.R.; Beere, P.A.; Langendorfer, A.; Stein, E.A.; Kruyer, W.; Gotto, A.M., Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998, 279, 1615–1622. [Google Scholar] [CrossRef]
- Graaf, M.R.; Beiderbeck, A.B.; Egberts, A.C.; Richel, D.J.; Guchelaar, H.J. The risk of cancer in users of statins. Am. J. Clin. Oncol. 2004, 22, 2388–2394. [Google Scholar] [CrossRef]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar] [CrossRef]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; MacFarlane, P.W.; McKillop, J.H.; Packard, C.J. West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995, 333, 1301–1307. [Google Scholar] [CrossRef]
- Lewis, S.J.; Sacks, F.M.; Mitchell, J.S.; East, C.; Glasser, S.; Kell, S.; Letterer, R.; Limacher, M.; Moye, L.A.; Rouleau, J.L.; et al. Effect of pravastatin on cardiovascular events in women after myocardial infarction: The cholesterol and recurrent events (CARE) trial. J. Am. Coll. Cardiol. 1998, 32, 40–46. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.M.; Moye, L.A.; Goldman, S.; Flaker, G.C.; Braunwald, E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CARE) investigators. Circulation 1998, 98, 839–844. [Google Scholar] [CrossRef]
- Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998, 339, 1349–1357. [Google Scholar] [CrossRef]
- Athyros, V.G.; Papageorgiou, A.A.; Mercouris, B.R.; Athyrou, V.V.; Symeonidis, A.N.; Basayannis, E.O.; Demitriadis, D.S.; Kontopoulos, A.G. Treatment with atorvastatin to the National Cholesterol Educational Program goal versus ‘usual’ care in secondary coronary heart disease prevention. The GREek Atorvastatin and Coronary-heart-disease Evaluation (GREACE) study. Curr. Med. Res. Opin. 2002, 18, 220–228. [Google Scholar] [CrossRef]
- Khurana, V.; Bejjanki, H.R.; Caldito, G.; Owens, M.W. Statins reduce the risk of lung cancer in humans: A large case-control study of US Veterans. Chest 2007, 131, 1282–1288. [Google Scholar] [CrossRef]
- Kochhar, R.; Khurana, V.; Bejjanki, H.; Caldito, G.; Fort, C. Statins reduce breast cancer risk: A case control study in US female veterans. J. Clin. Oncol. 2005, 23, 514. [Google Scholar] [CrossRef]
- Singal, R.; Khurana, V.; Caldito, G.; Fort, C. Statins and prostate cancer risk. J. Clin. Oncol. 2005, 23, 1004. [Google Scholar] [CrossRef]
- Poynter, J.N.; Gruber, S.B.; Higgins, P.D.; Almog, R.; Bonner, J.D.; Rennert, H.S.; Low, M.; Greenson, J.K.; Rennert, G. Statins and the risk of colorectal cancer. N. Engl. J. Med. 2005, 352, 2184–2192. [Google Scholar] [CrossRef]
- Kawata, S.; Nagase, T.; Yamasaki, E.; Ishiguro, H.; Matsuzawa, Y. Modulation of the mevalonate pathway and cell growth by pravastatin and dlimonene in a human hepatoma cell line (Hep G2). Br. J. Cancer 1994, 69, 1015–1020. [Google Scholar] [CrossRef]
- Hawk, M.A.; Cesen, K.T.; Siglin, J.C.; Stoner, G.D.; Ruch, R.J. Inhibition of lung tumor cell growth in vitro and mouse lung tumor formation by lovastatin. Cancer Lett. 1996, 109, 217–222. [Google Scholar] [CrossRef]
- Feleszko, W.; Jakobisiak, M. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin. Cancer Res. 2000, 6, 1198–1199. [Google Scholar]
- Kawata, S.; Yamasaki, E.; Nagase, T.; Inui, Y.; Ito, N.; Matsuda, Y.; Inada, M.; Tamura, S.; Noda, S.; Imai, Y.; et al. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br. J. Cancer 2001, 84, 886–891. [Google Scholar] [CrossRef]
- Kim, W.S.; Kim, M.M.; Choi, H.J.; Yoon, S.S.; Lee, M.H.; Park, K.; Park, C.H.; Kang, W.K. Phase II study of high-dose lovastatin in patients with advanced Bystric adenocarcinoma. Incest. New Drugs 2001, 19, 81–83. [Google Scholar] [CrossRef]
- Larner, J.; Jane, J.; Laws, E.; Packer, R.; Myers, C.; Shaffrey, M. A phase I-II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am. J. Clin. Oncol. 1998, 21, 579–583. [Google Scholar] [CrossRef]
- Bonovas, S.; Filioussi, K.; Sitaras, N.M. Statins are not associated with a reduced risk of pancreatic cancer at the population level, when taken at low doses for managing hypercholesterolemia: Evidence from a meta-analysis of 12 studies. Am. J. Gastroenterol. 2008, 103, 2646–3651. [Google Scholar] [CrossRef]
- Bonovas, S.; Filioussi, K.; Tsantes, A.; Sitaras, N.M. Use of statins and risk of haematological malignancies: A meta-analysis of six randomized clinical trials and eight observational studies. Br. J. Clin. Pharmacol. 2007, 64, 255–262. [Google Scholar] [CrossRef]
- Bonovas, S.; Sitaras, N.M. Does pravastatin promote cancer in elderly patients? A metaanalysis. CMAJ 2007, 176, 649–654. [Google Scholar] [CrossRef]
- Gbelcová, H.; Rimpelová, S.; Ruml, T.; Fenclová, M.; Kosek, V.; Hajšlová, J.; Strnad, H.; Kolář, M.; Vítek, L. Variability in statin-induced changes in gene expression profiles of pancreatic cancer. Sci. Rep. 2017, 7, 44219. [Google Scholar] [CrossRef]
- Gbelcová, H.; Leníček, M.; Zelenka, J.; Knejzlík, Z.; Dvořáková, G.; Zadinová, M.; Poučková, P.; Kudla, M.; Balaž, P.; Ruml, T.; et al. Differences in antitumor effects of various statins on human pancreatic cancer. Int. J. Cancer 2008, 122, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as anticancer agents in the era of precision medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Di Bello, E.; Zwergel, C.; Mai, A.; Valente, S. The innovative potential of statins in cancer: New targets for new therapies. Front. Chem. 2020, 8, 516. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, M.; Sun, C.; Qu, G.; Shi, T.; Min, M.; Wu, Y.; Sun, Y. Statin use and risk of pancreatic cancer an updated meta-analysis of 26 studies. Pancreas 2019, 48, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Riscal, R.; Skuli, N.; Simon, M.C. Even cancer cells watch their cholesterol! Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef]
- Gordon, R.E.; Zhang, L.; Peri, S.; Kuo, Y.M.; Du, F.; Egleston, B.L.; Ng, J.M.Y.; Andrews, A.J.; Astsaturov, I.; Curran, T.; et al. Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin. Cancer Res. 2018, 24, 1375–1388. [Google Scholar] [CrossRef]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell. Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The hedgehog-binding proteins GAS1 and CDO cooperate to positively regulate SHH signalling during mouse development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef]
- Martinelli, D.C.; Fan, C.M. Gas1 extends the range of hedgehog action by facilitating its signalling. Genes Dev. 2007, 21, 1231–1243. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. AcetylCoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Rho-GTPases and cancer. Nat. Rev. Cancer 2002, 21, 133–142. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho proteins and cancer. Breast Cancer Res. Treat. 2004, 84, 13–19. [Google Scholar] [CrossRef]
- Mazieres, J.; Tillement, V.; Allal, C.; Clanet, C.; Bobin, L.; Chen, Z.; Sebti, S.M.; Favre, G.; Pradines, A. Geranylgeranylated, but not farnesylated, RhoB suppresses Ras transformation of NIH-3T3 cells. Exp. Cell Res. 2005, 304, 354–364. [Google Scholar] [CrossRef]
- Prendergast, G.C. Actin’ up: RhoB in cancer and apoptosis. Nat. Rev. Cancer 2001, 1, 162–168. [Google Scholar] [CrossRef]
- Furuhjelm, J.; Peränen, J. The C-terminal end of R-Ras contains a focal adhesion targeting signal. J. Cell Sci. 2003, 116, 3729–3738. [Google Scholar] [CrossRef]
- Nowak, J.; Archange, C.; Tardivel-Lacombe, J.; Pontarotti, P.; Pébusque, M.J.; Vaccaro, M.I.; Velasco, G.; Dagorn, J.C.; Iovanna, J.L. The TP53INP2 protein is required for autophagy in mammalian cells. Mol. Biol. Cell 2008, 20, 870–881. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=4998 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=983 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=994 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=6502 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=1870 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=8793 (accessed on 9 February 2021).
- Corpataux, J.M.; Naik, J.; Porter, K.E.; London, N.J. The effect of six different statins on the proliferation, migration, and invasion of human smooth muscle cells. J. Surg. Res. 2005, 129, 52–56. [Google Scholar] [CrossRef]
- Fromigue, O.; Hamidouche, Z.; Vaudin, P.; Lecanda, F.; Patino, A.; Barbry, P.; Mari, B.; Marie, P.J. CYR61 downregulation reduces osteosarcoma cell invasion, migration, and metastasis. J. Bone Miner. Res. 2011, 26, 1533–1542. [Google Scholar] [CrossRef]
- Kidera, Y.; Tsubaki, M.; Yamazoe, Y.; Shoji, K.; Nakamura, H.; Ogaki, M.; Satou, T.; Itoh, T.; Isozaki, M.; Kaneko, J.; et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 2010, 29, 127. [Google Scholar] [CrossRef]
- Brown, M.; Hart, C.; Tawadros, T.; Ramani, V.; Sangar, V.; Lau, M.; Clarke, N. The differential effects of statins on the metastatic behaviour of prostate cancer. Br. J. Cancer 2012, 106, 1689–1696. [Google Scholar] [CrossRef]
- Wang, G.; Cao, R.; Wang, Y.; Qian, G.; Dan, H.C.; Jiang, W.; Ju, J.; Wu, M.; Xiao, Y.; Wang, X. Simvastatin induces cell cycle arrest and inhibits proliferation of bladder cancer cells via PPARγ signalling pathway. Sci. Rep. 2016, 6, 35783. [Google Scholar] [CrossRef]
- Sheng, S.; Barnett, D.H.; Katzenellenbogen, B.S. Differential estradiol and selective estrogen receptor modulator (SERM) regulation of Keratin 13 gene expression and its underlying mechanism in breast cancer cells. Mol. Cell Endocrinol. 2008, 296, 1–9. [Google Scholar] [CrossRef][Green Version]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=7138 (accessed on 9 February 2021).
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 1999, 274, 33179–33182. [Google Scholar] [CrossRef]
- Schnitzer, M.J.; Block, S.M. Kinesin hydrolyses one ATP per 8-nm step. Nature 1997, 388, 386–390. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=3728 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=7430 (accessed on 9 February 2021).
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=1008 (accessed on 9 February 2021).
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.P.; Aken, B.L.; Beal, K.; Ballester, B.; Caccamo, M.; Chen, Y.; Clarke, L.; Coates, G.; Cunningham, F.; Cutts, T.; et al. Ensembl 2007. Nuc. Acids Res. 2007, 35, D610–D617. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing; Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 20 February 2021).
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- STRING. Available online: https://string-db.org/ (accessed on 9 February 2021).
- Stocker, R.; Bowry, V.W.; Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than doe’s α-tocopherol. Med. Sci. 1991, 88, 1646–1650. [Google Scholar] [CrossRef]
- Wong, W.W.; Clendening, J.W.; Martirosyan, A.; Boutros, P.C.; Bros, C.; Khosravi, F.; Jurisica, I.; Stewart, A.K.; Bergsagel, P.L.; Penn, L.Z. Determinants of sensitivity to lovastatin-induced apoptosis in multiple myeloma. Mol. Cancer Ther. 2007, 6, 1886–1897. [Google Scholar] [CrossRef]
- Jacobson, J.R.; Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. [Google Scholar] [CrossRef]
- Jacobson, J.R.; Dudek, S.M.; Birukov, K.G.; Ye, S.Q.; Grigoryev, D.N.; Girgis, R.E.; Garcia, J.G. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am. J. Respir. Cell Mol. Biol. 2004, 30, 662–670. [Google Scholar] [CrossRef]
- Johnson-Anuna, L.N.; Eckert, G.P.; Keller, J.H.; Igbavboa, U.; Franke, C.; Fechner, T.; Schubert-Zsilavecz, M.; Karas, M.; Müller, W.E.; Wood, W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005, 312, 786–793. [Google Scholar] [CrossRef]
- Adamkov, M.; Halasova, E.; Rajcani, J.; Bencat, M.; Vybohova, D.; Rybarova, S.; Galbavy, S. Relation between expression pattern of p53 and survivin in cutaneous basal cell carcinomas. Med. Sci. Monit. 2011, 17, BR74–BR80. [Google Scholar] [CrossRef]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Fujimoto, T.; Ohsaki, Y. Proteasomal and autophagic pathways converge on lipid droplets. Autophagy 2006, 2, 299–301. [Google Scholar] [CrossRef]
- Yang, Y.C.; Huang, W.F.; Chuan, L.M.; Xiao, D.W.; Zeng, Y.L.; Zhou, D.A.; Xu, G.Q.; Liu, W.; Huang, B.; Hu, Q. In vitro and in vivo study of cell growth inhibition of simvastatin on chronic myelogenous leukemia cells. Chemotherapy 2008, 54, 438–446. [Google Scholar] [CrossRef]
- Assmus, B.; Urbich, C.; Aicher, A.; Hofmann, W.K.; Haendeler, J.; Rössig, L.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ. Res. 2003, 92, 1049–1055. [Google Scholar] [CrossRef]
- Denoyelle, C.; Albanese, P.; Uzan, G.; Hong, L.; Vannier, J.P.; Soria, J.; Soria, C. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal. 2003, 15, 327–338. [Google Scholar] [CrossRef]
- Keyomarsi, K.; Sandoval, L.; Band, V.; Pardee, A.B. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991, 51, 3602–3609. [Google Scholar]
- Maltese, W.A.; Sheridan, K.M. Differentiation of neuroblastoma cells induced by an inhibitor of mevalonate synthesis: Relation of neurite outgrowth and acetylcholinesterase activity to changes in cell proliferation and blocked isoprenoid synthesis. J. Cell Physiol. 1985, 125, 540–558. [Google Scholar] [CrossRef]
- Jakóbisiak, M.; Bruno, S.; Skierski, J.S.; Darzynkiewicz, Z. Cell cycle-specific effects of lovastatin. Proc. Natl. Acad. Sci. USA 1991, 88, 3628–3632. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Mott, G.E.; Ghosh-Choudhury, N.; Radnik, R.A.; Stapleton, M.L.; Ghidoni, J.J.; Kreisberg, J.I. Lovastatin induces apoptosis by inhibiting mitotic and post-mitotic events in cultured mesangial cells. Biochim. Biophys. Acta. 1997, 1359, 13–24. [Google Scholar] [CrossRef][Green Version]
- Engelke, K.J.; Hacker, M.P. A non-characteristic response of L1210 cells to lovastatin. Biochem. Biophys. Res. Commun. 1994, 203, 400–407. [Google Scholar] [CrossRef]
- Rao, S.; Porter, D.C.; Chen, X.; Herliczek, T.; Lowe, M.; Keyomarsi, K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7797–7802. [Google Scholar] [CrossRef]
- Cooper, S. Reappraisal of G1-phase arrest and synchronization by lovastatin. Cell Biol. Int. 2002, 26, 715–727. [Google Scholar] [CrossRef]
- da Costa, R.F.; Freire, V.N.; Bezerra, E.M.; Cavada, B.S.; Caetano, E.W.; de Lima Filho, J.L.; Albuquerque, E.L. Explaining statin inhibition effectiveness of HMG-CoA reductase by quantum biochemistry computations. Phys. Chem. Chem. Phys. 2012, 14, 1389–1398. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Mück, W.; Ritter, W.; Ochmann, K.; Unger, S.; Ahr, G.; Wingender, W.; Kuhlmann, J. Absolute and relative bioavailability of the HMG-CoA reductase inhibitor cerivastatin. Int. J. Clin. Pharmacol. Ther. 1997, 35, 255–260. [Google Scholar] [PubMed]
- Kajinami, K.; Mabuchi, H.; Saito, Y. NK-104: A novel synthetic HMG-CoA reductase inhibitor. Expert Opin. Investig. Drugs. 2000, 9, 2653–2661. [Google Scholar] [CrossRef]
- Hamelin, B.A.; Turgeon, J. Hydrophilicity/lipophilicity: Relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol. Sci. 1998, 19, 26–37. [Google Scholar] [CrossRef]
- Menter, D.G.; Ramsauer, V.P.; Harirforoosh, S.; Chakraborty, K.; Yang, P.; Hsi, L.; Newman, R.A.; Krishnan, K. Differential effects of pravastatin and simvastatin on the growth of tumor cells from different organ sites. PLoS ONE 2011, 6, e28813. [Google Scholar] [CrossRef] [PubMed]
- Hindler, K.; Cleeland, C.S.; Rivera, E.; Collard, C.D. The role of statins in cancer therapy. Oncologist 2006, 11, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and its metabolites in tumor growth: Therapeutic potential of statins in cancer treatment. Front Endocrinol. (Lausanne) 2019, 9, 807. [Google Scholar] [CrossRef]
- Sopková, J.; Vidomanová, E.; Strnádel, J.; Škovierová, H.; Halašová, E. The role of statins as therapeutic agents in cancer. Gen. Phys. Biophys. 2017, 36, 501–511. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. | Ato. | Lov. |
|---|---|---|---|---|---|---|---|---|
| Fold Change | ||||||||
| HMGCS1 | NM_002130 | HMG-CoA synthase (EC 2.3.3.10) | 5.67 | 2.99 | 4.13 | 2.96 | 2.90 | 2.80 |
| HMGCR | NM_000859 | HMG-CoA reductase (EC 1.1.1.34) | 3.91 | 3.16 | 3.04 | 2.66 | 2.35 | 2.08 |
| MVD | NM_002461 | Mevalonate pyrophosphate decarboxylase (EC 4.1.1.33) | 3.66 | 2.11 | 2.32 | 2.01 | - | - |
| PPAP2A | NM_003711 | Phosphatidic acid phosphatase 2a (EC 3.1.3.4) | 3.41 | 2.78 | 2.15 | - | - | - |
| AGPAT2 | NM_006412 | 1-acyl-glycerol-phosphate acyltransferase 2 (EC 2.3.1.51) | 2.63 | 2.28 | - | - | - | - |
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. |
|---|---|---|---|---|---|---|
| Fold Change | ||||||
| HIST1H4C | NM_003542.3 | Histone H4 | 0.41 | 0.34 | 0.49 | - |
| HIST1H2BF | NM_003522.3 | Histone H2B type 1-K | 0.33 | 0.41 | 0.43 | 0.49 |
| HIST2H3C | NM_021059.2 | Histone H3C type 2 | - | 0.50 | - | - |
| H2AFX | NM_002105.2 | Histone H2A family member X | - | 0.43 | - | - |
| HIST2H2BE | NM_003528.2 | Histone H2B type 2-E | 4.32 | 4.33 | 2.05 | - |
| HIST1H2BD | NM_138720.1 | Histone H2B type 1-D | 4.15 | 4.55 | 2.01 | - |
| HIST1H2BK | NM_080593.1 | Histone H2B type 1-K | 2.10 | 2.67 | - | - |
| HIST3H2A | NM_033445.2 | Histone H2A type 3 | - | 2.02 | - | - |
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. | Ato. | Lov. |
|---|---|---|---|---|---|---|---|---|
| Fold Change | ||||||||
| RHOB | NM_004040.2 | Ras homolog gene family, member B | 12.48 | 11.90 | 10.81 | 7.11 | 5.59 | 4.92 |
| RASL10A | NM_001007279.1 | RAS-like, family 10, member A | 4.14 | 3.58 | 2.65 | 2.17 | 2.01 | - |
| KRAS | NM_033360.2 | Kirsten rat sarcoma viral oncogene homolog | 3.23 | 2.64 | 2.58 | 2.00 | - | - |
| RAB40B | NM_006822.1 | Member RAS oncogene family | 3.14 | 2.89 | 2.16 | - | - | - |
| RRAS | NM_006270.3 | Related RAS viral (r-ras) oncogene homolog | 2.58 | 2.53 | 2.04 | - | - | - |
| RAB5B | NM_002868.2 | RAS oncogene family member | 2.46 | 2.04 | - | - | - | - |
| RAB6B | NM_016577.3 | RAS oncogene family member | 2.32 | 2.31 | - | - | - | - |
| RHOA | NM_001664.2 | Ras homolog gene family, member A | 2.21 | 2.40 | 2.03 | - | - | - |
| ARHGEF3 | NM_019555.1 | Rho guanine nucleotide exchange factor (GEF) 3 | 2.21 | 2.09 | - | - | - | - |
| RHOQ | NM_012249.3 | Ras homolog gene family, member Q | 2.17 | - | - | - | - | - |
| RRAGC | NM_022157.2 | Ras-related GTP binding C | 2.06 | - | - | - | - | - |
| RAB38 | NM_022337.1 | Member RAS oncogene family | 0.41 | - | - | - | - | - |
| ARHGAP19 | NM_032900.4 | Rho GTPase activating protein 19 | 0.47 | 0.38 | - | - | - | - |
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. | Ato. | Lov. |
|---|---|---|---|---|---|---|---|---|
| Fold Change | ||||||||
| CDKN1A | NM_000389.2 | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 3.64 | 4.21 | 2.74 | 2.59 | 2.33 | 2.40 |
| SFN | NM_006142.3 | Stratifin | 2.40 | 2.57 | - | - | - | - |
| CDKN1C | NM_057735.1 | Cyclin-dependent kinase inhibitor 1C (p57, Kip2) | - | 2.37 | - | - | - | - |
| CCNE2 | NM_057735.1 | Cyclin E2 | 0.30 | 0.29 | - | - | - | - |
| CDC25A | NM_001789.2 | Cell division cycle 25 homolog A | 0.30 | 0.33 | - | - | - | - |
| ORC1L | NM_004153.2 | Origin recognition complex, subunit 1-like | 0.30 | 0.22 | - | - | - | - |
| ORC6L | NM_014321.2 | Origin recognition complex, subunit 6 like | 0.31 | 0.30 | - | - | - | - |
| SKP2 | NM_005983.2 | S-phase kinase-associated protein 2 (p45) | 0.34 | 0.37 | - | - | - | - |
| MCM7 | NM_005916.3 | Minichromosome maintenance complex component 7 | 0.38 | 0.45 | - | - | - | - |
| E2F2 | NM_004091.2 | E2F transcription factor 2 | 0.39 | 0.40 | - | - | - | - |
| CDC45L | NM_003504.3 | CDC45 cell division cycle 45-like | 0.39 | 0.27 | - | - | - | - |
| MCM2 | NM_004526.2 | Minichromosome maintenance complex component 2 | 0.40 | 0.30 | - | - | - | - |
| MCM3 | NM_002388.3 | Minichromosome maintenance complex component 3 | 0.41 | 0.33 | - | - | - | - |
| CDC6 | NM_001254.3 | Cell division cycle 6 homolog | 0.41 | 0.47 | - | - | - | - |
| CDC2 | NM_001786.2 | Cell division cycle 2, G1 to S and G2 to M | 0.41 | 0.32 | - | - | - | - |
| CCND1 | NM_053056.2 | Cyclin D1 | 0.43 | - | - | - | - | - |
| MCM4 | NM_005914.2 | Minichromosome maintenance complex component 4 | 0.43 | 0.36 | - | - | - | - |
| MCM5 | NM_006739.3 | Minichromosome maintenance complex component 5 | 0.45 | 0.35 | - | - | - | - |
| PCNA | NM_182649.1 | Proliferating cell nuclear antigen (PCNA), transcript variant 2 | 0.46 | 0.45 | - | - | - | - |
| CDK2 | NM_001798.2 | Cyclin-dependent kinase 2 | 0.48 | 0.44 | - | - | - | - |
| PKMYT1 | NM_182687.1 | Protein kinase, membrane associated tyrosine/threonine 1 | 0.49 | 0.30 | - | - | - | - |
| MAD2L1 | NM_002358.2 | MAD2 mitotic arrest deficient-like 1 | 0.49 | 0.36 | - | - | - | - |
| CCNA2 | NM_001237.2 | Cyclin A2 | - | 0.31 | - | - | - | - |
| TTK | NM_003318.3 | TTK protein kinase | - | 0.41 | - | - | - | - |
| CDC20 | NM_001255.2 | Cell division cycle 20 homolog | - | 0.46 | - | - | - | - |
| CDC25C | NM_001790.3 | Cell division cycle 25 homolog C | - | 0.46 | - | - | - | - |
| PLK1 | NM_005030.3 | Polo-like kinase 1 | - | 0.47 | - | - | - | - |
| BUB1 | NM_004336.2 | BUB1 budding uninhibited by benzimidazoles 1 homolog | - | 0.48 | - | - | - | - |
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. | Ato. | Lov. |
|---|---|---|---|---|---|---|---|---|
| Fold Change | ||||||||
| TNFRSF10D | NM_003840.3 | Tumor necrosis factor receptor superfamily, member 10d | 3.71 | 3.17 | 4.68 | 3.06 | 2.76 | 2.60 |
| SLC6A12 | NM_003044.2 | Solute carrier family 6 (betaine/GABA), member 12 | 2.45 | 2.16 | 2.84 | 2.45 | - | - |
| DRAM | NM_018370.2 | Damage-regulated autophagy modulator | 2.18 | 2.41 | - | - | - | - |
| CASP9 | NM_032996.1 | Caspase 9, apoptosis-related cysteine peptidase | 2.16 | - | - | - | - | - |
| GABARAPL1 | NM_031412.2 | GABA(A) receptor-associated protein like 1 | 2.13 | 3.76 | 2.49 | 2.13 | - | - |
| ATG2A | NM_015104.1 | ATG2 autophagy related 2 homolog A | 2.08 | - | - | - | - | - |
| TNFAIP1 | NM_021137.3 | Tumor necrosis factor, alpha-induced protein 1 | 2.01 | - | - | - | - | - |
| CARD10 | NM_014550.3 | Caspase recruitment domain family, member 10 | 0.43 | 0.49 | - | - | - | - |
| TNFRSF6B | NM_032945.2 | Tumor necrosis factor receptor superfamily, member 6b | 0.48 | - | - | - | - | - |
| Gene Symbol | Ref. ID | Product Name | Cer. | Pit. | Sim. | Flu. | Ato. | Lov. |
|---|---|---|---|---|---|---|---|---|
| Fold Change | ||||||||
| LOC400578 | XR_017543.1 | Similar to keratin, type I cytoskeletal 14 (Cytokeratin-14) | 15.80 | 12.21 | 5.44 | 3.62 | 3.32 | 2.59 |
| KRT16 | NM_005557.2 | Keratin 16 | 13.85 | 12.21 | 4.60 | 3.52 | 3.28 | 2.23 |
| MGC102966 | XR_015970.1 | Similar to keratin, type I cytoskeletal 16 (Cytokeratin-16) | 13.54 | 11.95 | 4.84 | 3.53 | 3.18 | 2.31 |
| KRT15 | NM_002275.2 | Homo sapiens keratin 15 | 5.97 | 5.86 | 3.85 | 3.43 | 3.02 | 3.25 |
| KLHL24 | NM_017644.3 | Kelch-like 24 | 5.78 | 4.56 | 2.89 | 2.21 | 2.21 | - |
| JUP | NM_002230.1 | Junction plakoglobin | 5.33 | 4.09 | 2.78 | 2.51 | 2.00 | - |
| TUBB1 | NM_030773.2 | Tubulin, beta 1 | 4.02 | - | - | 2.23 | - | - |
| KRT13 | NM_002274.3 | Keratin 13 | 3.16 | 4.35 | 3.38 | 2.84 | 2.68 | 3.05 |
| KRT19 | NM_002276.3 | Keratin 19 | 3.13 | 2.88 | 2.50 | 2.20 | 2.11 | - |
| GSN | NM_198252.2 | Gelsolin (amyloidosis, Finnish type) | 2.97 | 2.32 | 2.45 | 2.00 | - | - |
| KIF1A | NM_004321.4 | Kinesin family member 1A | 2.73 | 3.05 | 2.26 | - | - | - |
| TNNT1 | NM_003283.3 | Troponin T, slow skeletal muscle | 2.70 | 3.57 | 2.10 | - | - | - |
| ITGB4 | NM_001005619.1 | Homo sapiens integrin, beta 4 | 2.45 | 2.49 | 2.18 | 2.03 | - | 2.20 |
| VIL2 | NM_003379.3 | Villin 2 (ezrin) | 2.16 | 2.18 | 2.01 | - | - | - |
| NAV1 | NM_020443.2 | Neuron navigator 1 | 2.14 | 2.38 | 2.13 | - | - | - |
| GSN | NM_198252.2 | Gelsolin (amyloidosis, Finnish type) | - | - | - | - | - | - |
| CDH10 | NM_006727.2 | Cadherin 10, type 2 (T2-cadherin) | 0.31 | 0.28 | 0.33 | 0.48 | 0.45 | - |
| SYNM | NM_015286.5 | Synemin, intermediate filament protein | 0.48 | 0.50 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimpelová, S.; Kolář, M.; Strnad, H.; Ruml, T.; Vítek, L.; Gbelcová, H. Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins. Molecules 2021, 26, 3528. https://doi.org/10.3390/molecules26123528
Rimpelová S, Kolář M, Strnad H, Ruml T, Vítek L, Gbelcová H. Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins. Molecules. 2021; 26(12):3528. https://doi.org/10.3390/molecules26123528
Chicago/Turabian StyleRimpelová, Silvie, Michal Kolář, Hynek Strnad, Tomáš Ruml, Libor Vítek, and Helena Gbelcová. 2021. "Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins" Molecules 26, no. 12: 3528. https://doi.org/10.3390/molecules26123528
APA StyleRimpelová, S., Kolář, M., Strnad, H., Ruml, T., Vítek, L., & Gbelcová, H. (2021). Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins. Molecules, 26(12), 3528. https://doi.org/10.3390/molecules26123528