
Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins



Abstract
1. Introduction
2. Introduction to Molecular Imaging

| Trade Name | Generic Name | Company | Approval Year | Antibody | Target | Cell Line | Radionuclide | Indications | |
|---|---|---|---|---|---|---|---|---|---|
| EMA | FDA | ||||||||
| OncoScint | Satumomab pendetide | Cytogen | NA | 1992 | B72.3, mouse IgG1 | TAG-72 | Hybridoma | 111In | Colorectal and ovarian carcinoma |
| CEA-Scan | Arcitumomab | Immunomedis | 1996 (withdrawn in 2005) | 1996 | IMMU-4, mouse IgG Fab’ | CEA | Hybridoma | 99mTc | Colorectal cancer |
| Myoscint | Imciromab pentetate | Centocor | NA | 1996 (discontinued) | R11D10, mouse IgG2a Fab’ | Human cardiac myosin | Murine ascites | 111In | Myocardial infarction |
| Verluma | Nofetumomab merpentan | Boehringer Ingelheim, NeoRx | NA | 1996 | NR-LU-10, mouse IgG2b Fab | carcinoma-associated antigen | Hybridoma | 99mTc | Breast, lung, gastrointestinal, ovary, bladder, kidney, cervix, and pancreas carcinomas |
| ProstaScint | Capromab pendetide | Cytogen | NA | 1996 | 7E11-C5.3, mouse IgG1 | PSMA, | Hybridoma | 111In | Prostate carcinoma |
| Zevalin | Ibritumomab tiuxetan | Spectrum Pharms/Biogen | 2004 | 2002 | 2B8, mouse IgG1 | CD20 | CHO | 90Y | Non-Hodgkin lymphoma |
| Bexxar | Tositumomab | Corixa and GSK | NA | 2002 (discontinued in 2014) | B1, mouse IgG2a | CD20 | Hybridoma | 131I | Non-Hodgkin lymphoma |
| NeutroSpec (LeuTech) | Fanolesomab | Palatin Technologies | NA | 2004 | RB5, mouse IgM | CD15 | Hybridoma | 99mTc | Appendicitis |
| Lymphoscan | Bectumomab | Immunomedics | NA | LL2, mouse IgG2a Fab’ | CD22 | 99mTc | Non-Hodgkin lymphoma | ||
| HumaSPECT | Votumumab | KS Biomedix Ltd./Organon Teknika | 1998 (withdrawn in 2003) | NA | 88BV59, human IgG3 | Cytokeratin tumor associated antigen | Human lymphoblastoid cell line transformed with EBV | 99mTc | Carcinoma of the colon and rectum |
| Indimacis-125 | Igovomab | CIS Bio International | 1996 (discontinued) | NA | OC125, mouse IgG1 F(ab’)2 | CA-125 | 111In | Ovarian cancer | |
| LeukoScan | Sulesomab | Immunomedics | 1997 | NA | IMMU MN3, mouse IgG Fab’ | NCA-90 NS0 | 99mTc | Osteomyelitis and appendicitis, including patients with diabetic foot ulcers | |
| Scintimun | Besilesomab | CIS Bio | 2010 | NA | Murine IgG1 | NCA-95 | Hybridoma | 99mTc | Inflammation/infection |
3. Radionuclide and Antibody-Based Tumor Targeting Molecules for Radioimmunoimaging and Therapy
| Radionuclide | Decay | Common Production Process a | Chelator c Labeling Conditions | Properties | ||||
|---|---|---|---|---|---|---|---|---|
| t½ (h) | β+max in KeV (Yield) | β−max in keV (Yield) | γ in keV (Yield) | α in keV (Yield) | ||||
| Alogens | ||||||||
| 18F | 1.83 | 634 (97%) | 140 (41%) | Cyclotron: 18O(p,n)18F (radionuclide/tracers can be transported over short distances) | 18F-labeled prosthetic groups containing click chemistry handles, e.g., azides, [18F]FEA; or alkynes [18F]-FB-DBCO for SPAAC reactions; TCO—and tetrazine for IEDDA reactions | Only suitable for imaging of fast-clearing antibody fragments by PET; Cons: imaging up to 6 h after injection. Defluorination can occur resulting in bone-seeking radionuclide | ||
| 123I | 13.2 | _ | _ | 160 (83%) | Cyclotron 123Te(p,n)123I | Suitable for imaging of non-internalizing antibody fragments by SPECT; cons: dehalogenation can occur resulting in thyroid uptake | ||
| 124I | 100.2 | 2138 (24%) | _ | 0.6 (61%) | Cyclotron: 124Te(p,n)124I (transportation worldwide including RICs) | Ideal for IgG imaging by PET with non-internalizing mAbs; cons: dehalogenation can occur resulting in thyroid uptake | ||
| 131I | 8.03 | _ | 0.63 (90%) | 0.36 (82%) | Nuclear reactor 130Te(n,γ)131m,gT → 131I (transportation worldwide) | Used for RIT; cons: dehalogenation can occur resulting in thyroid uptake. | ||
| Metals | ||||||||
| 44Sc | 3.9 | 1474 (94%) | 1157 (6%) | Sc(p, 2n) 44Ti → 44Sc (Generator) | DOTA 95 °C, 20–30 min, pH 4.0. Lower temperature need to extension of incubation time (hours) | Ideal for RIT with intact IgG and small scaffold proteins. Genuine theranostic. | ||
| 47Sc | 80.4 | 162 | 159 (68.3) | Nuclear reactor 47Ti(n,p)47Sc 46Ca(n,γ)47Ca → 47Sc | ||||
| 64Cu | 12.7 | 653 (18%) | 579 (39%) | Cyclotron: 64Ni(p,n)64Cu (tracers can be transported over short distances) | NOTA/NOTA-type: fast complexation at RT (30–60 min; pH = 5.5–6.5); high kinetic inertness in vivo. Sarcophagine-type Diamsar: quantitative radiolabeling at RT in 2–30 min; pH = 2–9 by using 10−6 M of chelator; compounds have excellent in vivo stability. | Relatively short t½ for imaging antibodies, preferably suitable for imaging of small antibody fragments by PET. Genuine theranostic. | ||
| 67Cu | 61.8 | 576 (20%), 482 (22%), 391 (57%) | 184 (49%) | High energy cyclotron: 68Zn(p,2p)67Cu (not easily available) | Suitable for IgG imaging small antibody fragments by SPECT and RIT. Genuine theranostic. | |||
| 67Ga | 78.3 | 93 (39%), 184 (21%), 300 (17%) | Cyclotron 68Zn(p,2n)67Ga or 67Zn(p,n)67Ga(transportation worldwide) | DOTA: 37 °C, >30 min, pH 4.0–5.5 No optimal | Ideal for imaging with intact IgG by SPECT. | |||
| 68Ga | 1.13 | 1899, 822 (90%) | 108 (3%) | natGa(p,xn)68Ge → 68Ga (Generator) | NOTA: RT, 30–60 min, pH 4.0–5.5. Stable | Only suitable for PET-imaging of fast-clearing antibody fragments. | ||
| 86Y | 14.7 | 3141 (34%) | 1.0(83%) | Cyclotron 86Sr(p,n)86Y | DOTA: 25–100 °C, 15–90 min, pH 4.0–6.0. Stable. NOTA: RT, 5 min, pH 4.0.Stable | Relatively short t½for imaging antibodies, only suitable for imaging with small antibody fragments by PET. Forms an ideal theranostics pair with 90Y. | ||
| 90Y | 64.1 | _ | 2280 (100%) | 235U(n,f)90Sr → 90Y (Generator)nuclear reactor: 90Zr(n,p)90Y | Only RIT; forms an ideal theranostics pair with 86Y, 90Sr. | |||
| 89Zr | 78.4 | 902 (23%) | _ | 0.9 (99%) | Cyclotron: 89Y(p,n)89Zr (transportation worldwide including RICs) | DFO: 25 °C, 60 min, pH 7–7.3. | Ideal for IgG imaging by PET, also with internalizing mAb; Cons: residualization in organ of mAb catabolism (liver, spleen, kidneys); demetalation, bone-seeking radionuclide. | |
| 99mTc | 6.02 | _ | _ | 142 (89%) | 235U(n,f)99Mo→99mTc (Generator) | N3S- RT, pH 7 >60 min HYNIC- RT, pH 7 > 60 min. Tc(CO)3+- His-Tag RT, pH 7 > 60 min | Only suitable for imaging of fast-clearing antibody fragments by SPECT; Pros: cheap and easily available. | |
| 111In | 67.3 | _ | _ | 172, 245 (100%) | Cyclotron 112Cd(p,2n)111In111Cd(p,n)111In | DOTA: 37–100 °C, 15–60 min, pH 4.0–6.0. Stable | Ideal t½ for IgG imaging by SPECT, Cons: bone-seeking radionuclide | |
| 177Lu | 159.5 | _ | 177 (12%), 385 (9%), 498 (79%) | 112, 208 (100%) | Nuclear reactor 176Lu(n,γ)177Lu | DOTA: 25–100 °C, 15–90 min, pH 4.0–6.0. Stable NOTA: RT, 30–60 min, pH 4.5. Stable | RIT and imaging (SPECT) possible at the same time Genuine theranostic | |
| 225Ac | 240 | 5600–5830 (100%) | 226Ra(p,2n)225Ac 232Th(n)-233U-229Th-225Ac (Generator)b | DOTA: 37–60 °C, 30–120 min, pH 6.0. | RIT | |||
| 213Bi | 0.76 | 5869 (97.8%) | 5549 (2.2%) | 227Ac(n,γ)229Th 228Th(n,γ)229Th-225Ac-213Bi(Generator) b | DOTA: 95–100 °C, 5 min, pH 6.0–8.7 no suitable for proteins. 3p-C-DEPA: RT, 5–10 min, pH 5.5 NOTA: RT, 5 min, pH 4.0. Stable | RIT | ||
 | ||||||||
 |  |  |  |  |  |  |  |  | |
|---|---|---|---|---|---|---|---|---|---|
| IgG | F(ab’)2 | Minibody | Triabody | Diabody | Fab | scFv | Nanobody | Affibody | |
| MW (kDa) | ~150 | ~110 | ~75 | ~75 | ~50 | ~50 | ~25 | ~15–12 | 6 |
| Avidity | bivalent | bivalent | bivalent | bivalent | monovalent | monovalent | monovalent | monovalent | |
| Target specificity |  |  | | | | | | |  / / |
| Tumor uptake |  | ||||||||
| Tumor penetration |  | ||||||||
| Clearance rate |  | ||||||||
| Excretion route | Hepatic | Hepatic/renal | Hepatic | Hepatic | Renal | Renal | Renal | Renal | Renal |
| Blood t1/2 | 1–3 w | 1–7 d | 5–10 h | 3–5 h | 12–20 h | 2–4 h | 30–60 min | 30–60 min | |
| Isotope t1/2 | |  | | | | | | |  |
| Target/non-target | |  | | | | | | | |
| Imaging (p.i) | 4–7 d | 1 d | 1 d | 1 d | 1 d | <1 d | <1 d | <1 d | |
| Radiolabeling process complexity | |  | | | | | | | |
| Complexity development & test | | | | | | | | | |
| Approval process & complexity of clinical translation | | | | | | | | | |
| w = weeks; d = days; h = hours |  | ||||||||
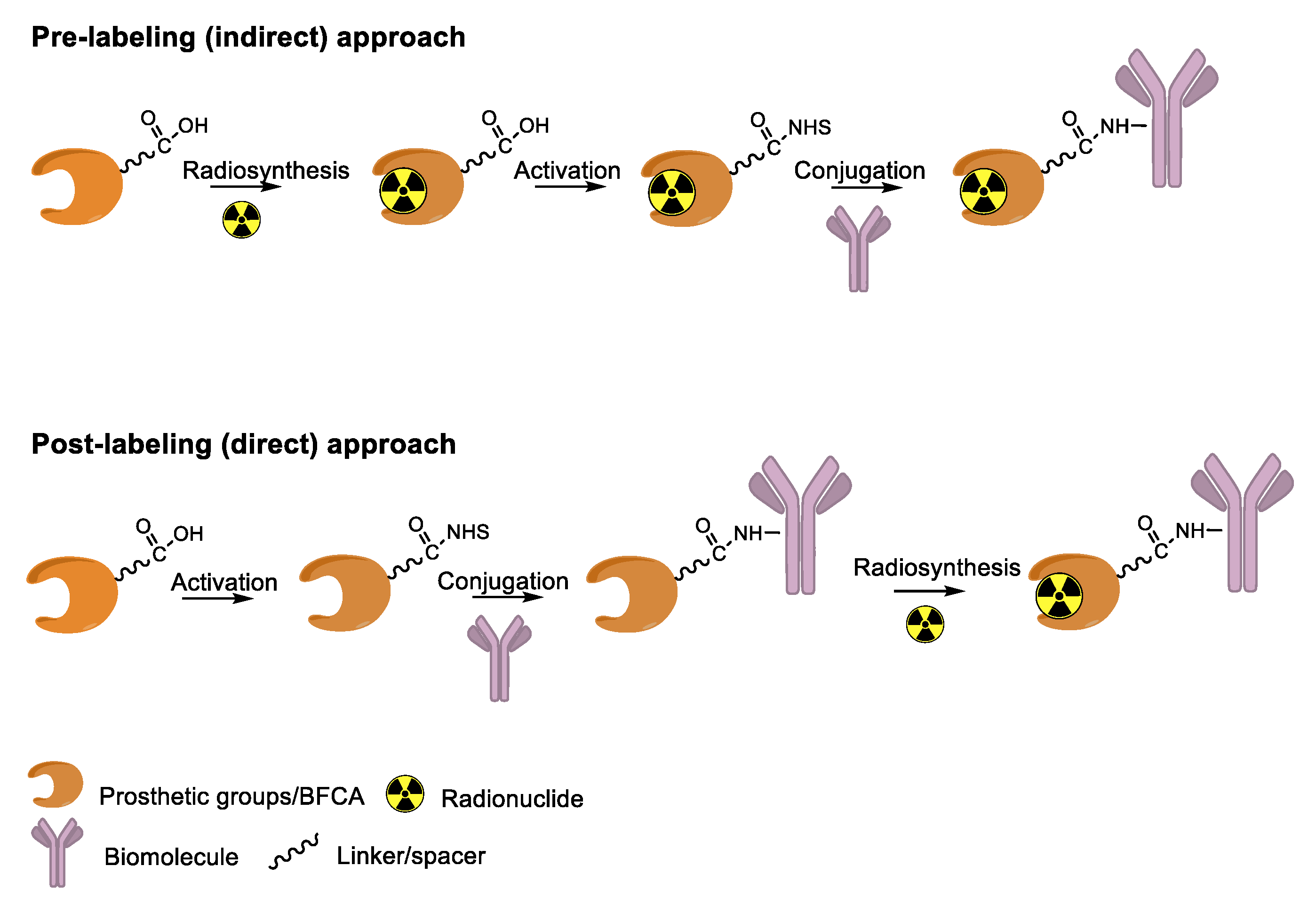
4. Overview of Radiolabeling Strategies for Biomolecules
5. Enzymes Used for the Site-Specific Derivatization of Proteins
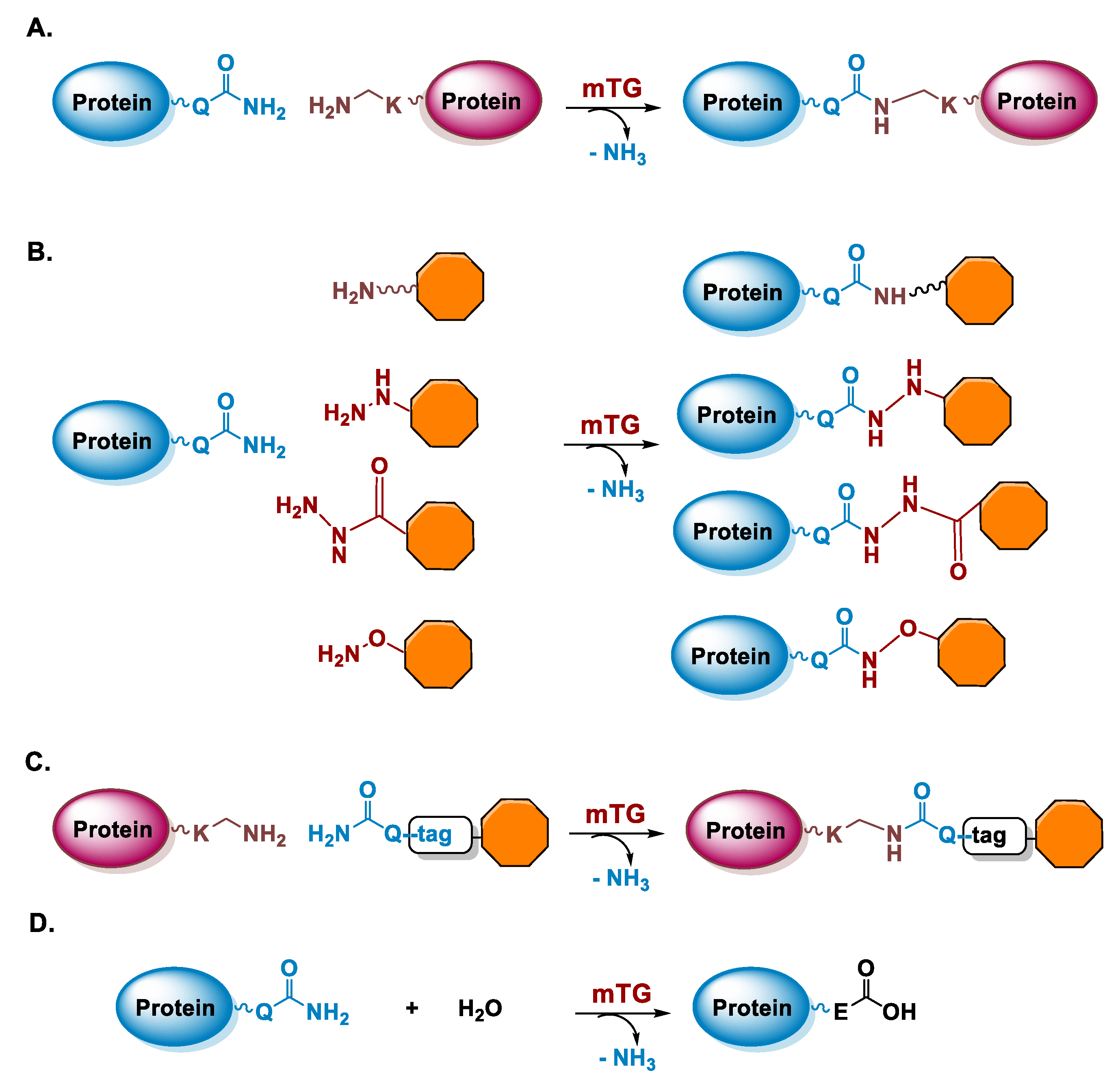
5.1. Microbial Transglutaminase
5.1.1. Determinants for the Site-Specificity of the mTG-Catalyzed Reaction
| Protein (Organism) | N. AA | N. Gln/N. Derivatised Gln | Gln Sequences a | N. Lys/N. Derivatised Lys | Lys Sequences a | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| -5 -1 | +1 +5 | -5 -1 | +1 +5 | |||||||
| Myoglobin, Mb (Equus caballus) | 153 | 6/0 (holoMb) b 6/2 (apoMb) | LKPLA KELGF | Q91 Q152 | SHATK G---- | 19/0 (holoMb) b 19/2 (apoMb) | QSHAT HATKH | K96 K98 | HKIPI IPIKY | [48] |
| α-Lactalbumin, LA (Bos taurus) | 123 | 6/0 (holoLA) c 6/4 (apoLA) | SGYDT TQAIV EYGLF WCKDD | Q39 Q43 Q54 Q65 | AIVQN NNDST INNKI NPHSS | 12/1 (K122; holoLA) c 12/4 (apoLA) | ELKDL FQINN ALCSE QWLCE | K16 K58 K114 K122 | GYGGV IWCKD LDQWL L---- | [48] |
| Avidin (Gallus gallus) | 128 | 4/0 | - | 9/2 | QNTIN RLRTQ | K58 K127 | RTQPT E---- | [49] | ||
| Interferon α-2b (Homo sapiens) | 165 | 12/1 | EACVI | Q101 | GVGVT | 10/2 | LFSCL ESLRS | K31 K164 | DRHDF E---- | [50] |
| Interferon β-1a (Homo sapiens) | 166 | 11/0 | - | 11/2 | LEYCL DFTRG | K33 K115 | DRMNF LMSSL | [51] | ||
| Growth hormone (Homo sapiens) | 191 | 13/2 | YIPKE GQIFK | Q40 Q141 | KYSFL TYSKF | 9/1 | KQTYS | K145 | FDTNS | [53,60] |
| Interleukin-2(Homo sapiens) | 133 | 6/1 | VLNLA | Q74 | SKNFH | 11/ND | - | [61] | ||
| Granulocyte colony-stimulating factor (Homo sapiens) | 174 | 17/1 | ALQPT | Q134 | GAMPA | 4/1 | LCATY | K40 | LCHPE | [53,62] |
| Granulocyte-macrophage colony-stimulating factor (Homo sapiens) | 127 | 8/1 | CWEPV | Q126 | E---- | 6/0 | - | [49] | ||
| Bacteriorhodopsin (Halobacterium salinarum) | 249 | 4/1 | ---QA | Q3 | ITGRP | 7/1 | VGALT | K129 | VYSYR | [63] |
| IgG1 d (Homo sapiens) | HC: 451 LC: 213 | HC:18/1 (degl.) LC: 12/0 | KPREE | Q295 | YDSTY | HC: 36/2 (agl.) LC: 13/0 | EVHNA HNAKT TISKA | K288 K290 K340 | TKPRE PREEQ e GQPRE | [45,56] |
| Notexin (Notechis scutatus scutatus) | 119 | 3/0 | - | 11/6 | KGCFP YCRNI CRNIK RNIKK WNIDT NIDTK | K63 K82 K83 K84 K115 K116 | MSAYD KKCLR KCLRF CLRFV KRCQ- RCQ-- | [52] | ||
| G-actin (Oryctolagus cuniculus) | 375 | 11/1 | GRPRH | Q41 | GVMVG | 19/ND | - | [64] | ||
| Trypsin inhibitor, STI2 (Streptomyces longisporus) | 110 | 1/ND | - | 4/1 | GVICN | K70 | LYDPV | [65] | ||
| Dispase autolysis-inducing protein, DAIP (Streptomyces mobaraensis) | 348 | 5/5 | TTGTL HNDEL AGSDG YGTYF GLEEV | Q39 Q65 Q144 Q298 Q345 | SVSYT RSTDA LYDST AYGTD IHH-- | 10/ND | - | [59] | ||
| Papain inhibitory protein, SPIp (Streptomyces mobaraensis) | 110 | 3/1 | DIPIG | Q6 | KMTGK | 6/ND | - | [66] | ||
| CRM197, mutant of diphtheria toxin (Corynebacterium diphtheriae) | 535 | 16/ND | - | 39/2 | VDSIQ QKGIQ GIQKP | K33 K37 K39 | GIQKP PKSGT SGTQG f | [67] | ||
5.1.2. Conditions of the mTG Reaction
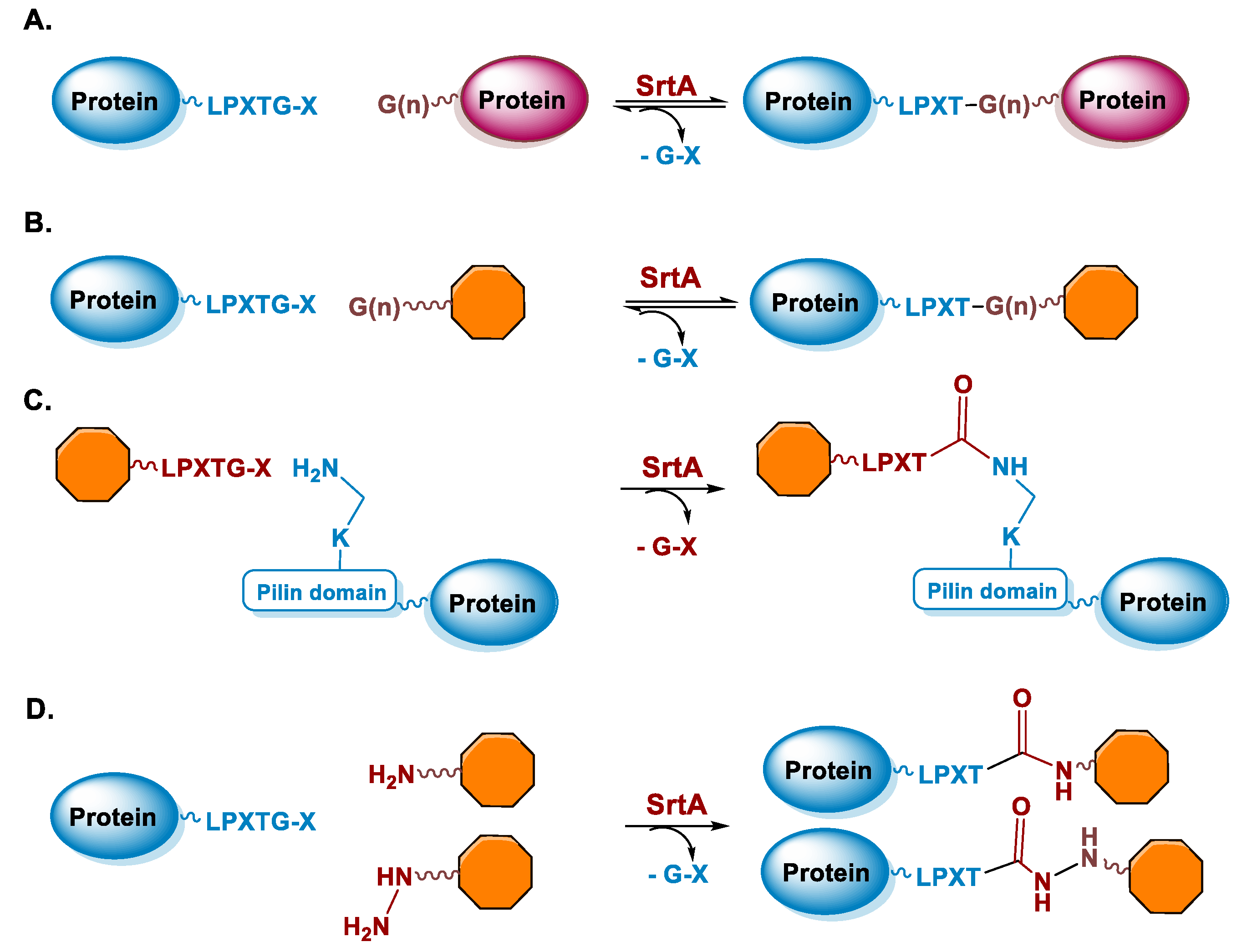
5.2. Sortase
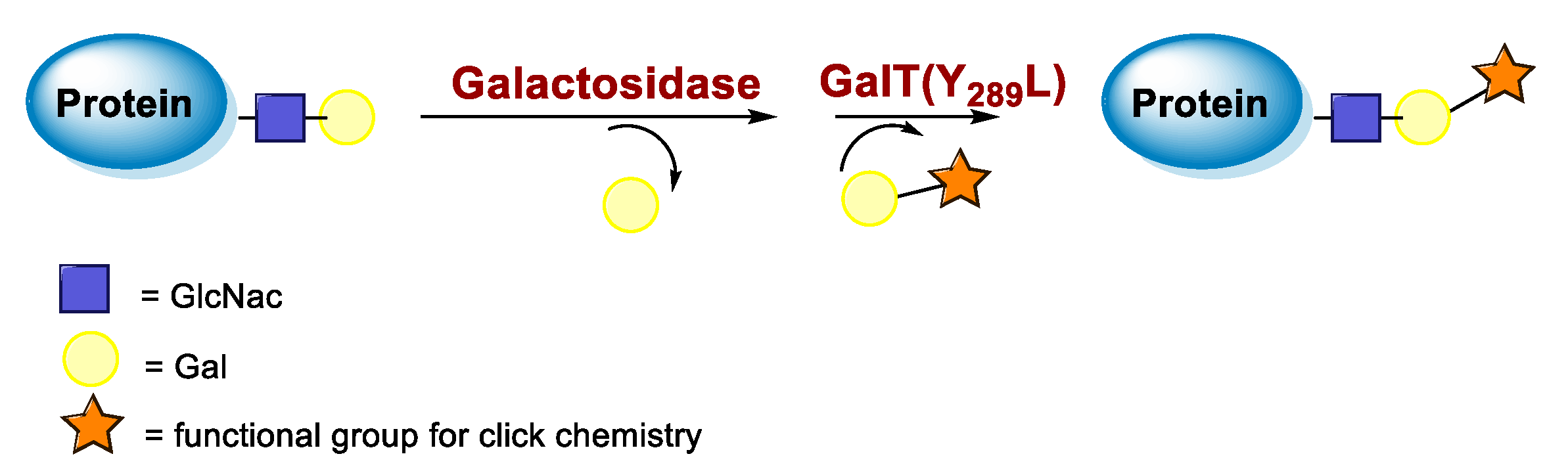
5.3. Galactosyltransferase
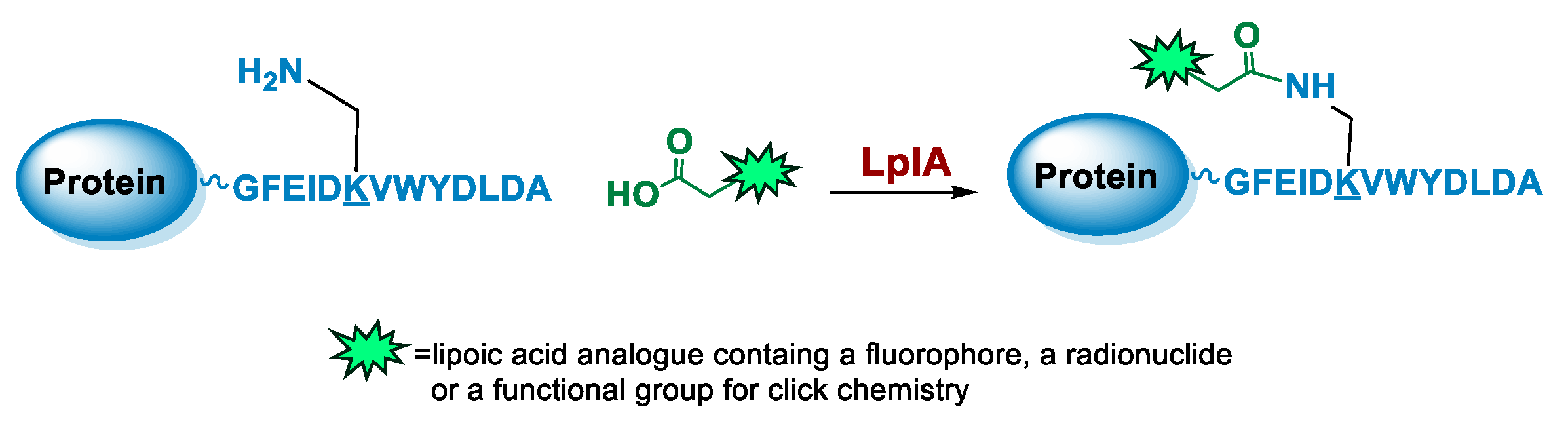
5.4. Lipoic Acid Ligase
6. Enzyme-Mediated Conjugation in Nuclear Molecular Imaging
6.1. Transglutaminase-Mediated Conjugation in MI












6.2. Sortase A-Mediated Conjugation in MI




6.3. Galactosyltransferase-Mediated Conjugation in MI










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6.4. Lipoic Acid Ligase-Mediated Conjugation in MI
| Biomolecules and Strategy | Bi-Functional Substrate | Ref. |
|---|---|---|
 |  | [113] |
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhang, Y.; Park, K.-Y.; Suazo, K.F.; Distefano, M.D. Recent Progress in Enzymatic Protein Labelling Techniques and Their Applications. Chem. Soc. Rev. 2018, 47, 9106–9136. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Fu, R.; Carroll, L.; Yahioglu, G.; Aboagye, E.O.; Miller, P.W. Antibody Fragment and Affibody ImmunoPET Imaging Agents: Radiolabelling Strategies and Applications. ChemMedChem 2018, 13, 2466–2478. [Google Scholar] [CrossRef]
- Bouleau, A.; Lebon, V.; Truillet, C. PET Imaging of Immune Checkpoint Proteins in Oncology. Pharmacol. Ther. 2021, 222, 107786. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.T.; Gambhir, S.S. The Immunoimaging Toolbox. J. Nucl. Med. 2018, 59, 1174–1182. [Google Scholar] [CrossRef]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.-Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef]
- Schneider, H.; Deweid, L.; Avrutina, O.; Kolmar, H. Recent Progress in Transglutaminase-Mediated Assembly of Antibody-Drug Conjugates. Anal. Biochem. 2020, 595, 113615. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.; Kiessling, F.; Pichler, B.J. Quo Vadis, Molecular Imaging? J. Nucl. Med. 2020, 61, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Salsano, M.; Treglia, G. PET Imaging Using Radiolabelled Antibodies: Future Direction in Tumor Diagnosis and Correlate Applications. Res. Rep. Nucl. Med. 2013, 3, 9–17. [Google Scholar] [CrossRef]
- Garousi, J.; Orlova, A.; Frejd, F.Y.; Tolmachev, V. Imaging Using Radiolabelled Targeted Proteins: Radioimmunodetection and Beyond. EJNMMI Radiopharm. Chem. 2020, 5, 1–26. [Google Scholar] [CrossRef]
- Altunay, B.; Morgenroth, A.; Beheshti, M.; Vogg, A.; Wong, N.C.L.; Ting, H.H.; Biersack, H.-J.; Stickeler, E.; Mottaghy, F.M. HER2-Directed Antibodies, Affibodies and Nanobodies as Drug-Delivery Vehicles in Breast Cancer with a Specific Focus on Radioimmunotherapy and Radioimmunoimaging. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–Drug Conjugates for Cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev. 2013, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2018, 119, 902–956. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2018, 119, 870–901. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.M.; Poty, S.; Sharma, S.K.; Lewis, J.S. Preclinical Optimization of Antibody-Based Radiopharmaceuticals for Cancer Imaging and Radionuclide Therapy—Model, Vector, and Radionuclide Selection. J. Label. Compd. Radiopharm. 2018, 61, 611–635. [Google Scholar] [CrossRef]
- Rashidian, M.; Ploegh, H. Nanobodies as Non-Invasive Imaging Tools. Immuno-Oncol. Technol. 2020, 7, 2–14. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A. Affibody Molecules as Targeting Vectors for PET Imaging. Cancers 2020, 12, 651. [Google Scholar] [CrossRef]
- Qaim, S.M. Nuclear Data for Production and Medical Application of Radionuclides: Present Status and Future Needs. Nucl. Med. Biol. 2017, 44, 31–49. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with 225Actinium and 213Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Bolzati, C.; Carta, D.; Salvarese, N.; Refosco, F. Chelating Systems for 99mTc/188Re in the Development of Radiolabeled Peptide Pharmaceuticals. Anti-Cancer Agents Med. Chem. 2012, 12, 428–464. [Google Scholar] [CrossRef]
- Williams, J.D.; Kampmeier, F.; Badar, A.; Howland, K.; Cooper, M.S.; Mullen, G.E.D.; Blower, P.J. Optimal His-Tag Design for Efficient [99mTc(CO)3]+ and [188Re(CO)3]+ Labeling of Proteins for Molecular Imaging and Radionuclide Therapy by Analysis of Peptide Arrays. Bioconj. Chem. 2020. [Google Scholar] [CrossRef]
- Spicer, C.D.; Davis, B.G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. [Google Scholar] [CrossRef]
- Cardinale, J.; Giammei, C.; Jouini, N.; Mindt, T.L. Bioconjugation Methods for Radiopharmaceutical Chemistry. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer: New York, NY, USA, 2019; pp. 449–479. ISBN 978-3-319-98946-4. [Google Scholar]
- Strop, P.; Delaria, K.; Foletti, D.; Witt, J.M.; Hasa-Moreno, A.; Poulsen, K.; Casas, M.G.; Dorywalska, M.; Farias, S.; Pios, A.; et al. Site-Specific Conjugation Improves Therapeutic Index of Antibody Drug Conjugates with High Drug Loading. Nat. Biotechnol. 2015, 33, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation Site Modulates the In Vivo Stability and Therapeutic Activity of Antibody-Drug Conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Lorand, L.; Conrad, S.M. Transglutaminases. Mol. Cell Biochem. 1984, 58, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.; Matte, C.R.; Bizarro, C.V.; Ayub, M.A.Z. Transglutaminases: Part I—Origins, Sources, and Biotechnological Characteristics. World J. Microbiol. Biotechnol. 2020, 36, 1–18. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s Biological Glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Lim, T.S. Bioengineering of Microbial Transglutaminase for Biomedical Applications. Appl. Microbiol. Biotechnol. 2019, 103, 2973–2984. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Caporale, A.; Monti, A.; Sandomenico, A.; Selis, F.; Ruvo, M. A Recent Update on the Use of Microbial Transglutaminase for the Generation of Biotherapeutics. World J. Microbiol. Biotechnol. 2020, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.; Matte, C.R.; Bizarro, C.V.; Ayub, M.A.Z. Review Transglutaminases: Part II—Industrial Applications in Food, Biotechnology, Textiles and Leather Products. World J. Microbiol. Biotechnol. 2020, 36, 1–20. [Google Scholar] [CrossRef]
- Savoca, M.; Tonoli, E.; Atobatele, A.; Verderio, E. Biocatalysis by Transglutaminases: A Review of Biotechnological Applications. Micromachines 2018, 9, 562. [Google Scholar] [CrossRef]
- Deweid, L.; Avrutina, O.; Kolmar, H. Microbial Transglutaminase for Biotechnological and Biomedical Engineering. Biol. Chem. 2019, 400, 257–274. [Google Scholar] [CrossRef]
- Giordano, D.; Facchiano, A. Classification of Microbial Transglutaminases by Evaluation of Evolution Trees, Sequence Motifs, Secondary Structure Topology and Conservation of Potential Catalytic Residues. Biochem. Biophys. Res. Commun. 2019, 509, 506–513. [Google Scholar] [CrossRef]
- Ando, H.; Adachi, M.; Umeda, K.; Matsuura, A.; Nonaka, M.; Uchio, R.; Tanaka, H.; Motoki, M. Purification and Characteristics of a Novel Transglutaminase Derived from Microorganisms. Agric. Biol. Chem. 1989, 53, 2613–2617. [Google Scholar] [CrossRef]
- Strop, P. Versatility of Microbial Transglutaminase. Bioconj. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Yokoyama, K.; Ishikawa, K.; Ono, K.; Ejima, D.; Matsui, H.; Suzuki, E. Crystal Structure of Microbial Transglutaminase fromStreptoverticillium Mobaraense. J. Biol. Chem. 2002, 277, 44252–44260. [Google Scholar] [CrossRef]
- Takazawa, T.; Kamiya, N.; Ueda, H.; Nagamune, T. Enzymatic Labeling of a Single Chain Variable Fragment of an Antibody with Alkaline Phosphatase by Microbial Transglutaminase. Biotechnol. Bioeng. 2004, 86, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Goto, M.; Kamiya, N. Transglutaminase-Mediated Internal Protein Labeling with a Designed Peptide Loop. Biochem. Biophys. Res. Commun. 2011, 410, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kamiya, N.; Nagamune, T. N-Terminal Glycine-Specific Protein Conjugation Catalyzed by Microbial Transglutaminase. FEBS Lett. 2005, 579, 2092–2096. [Google Scholar] [CrossRef]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef]
- Spidel, J.L.; Vaessen, B.; Albone, E.F.; Cheng, X.; Verdi, A.; Kline, J.B. Site-Specific Conjugation to Native and Engineered Lysines in Human Immunoglobulins by Microbial Transglutaminase. Bioconj. Chem. 2017, 28, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-Specific and Stoichiometric Modification of Antibodies by Bacterial Transglutaminase. Angew. Chem. Int. Ed. 2010, 49, 9995–9997. [Google Scholar] [CrossRef]
- Fontana, A.; Spolaore, B.; Mero, A.; Veronese, F.M. Site-Specific Modification and PEGylation of Pharmaceutical Proteins Mediated by Transglutaminase. Adv. Drug Deliv. Rev. 2008, 60, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Farias, S.E.; Strop, P.; Delaria, K.; Galindo Casas, M.; Dorywalska, M.; Shelton, D.L.; Pons, J.; Rajpal, A. Mass Spectrometric Characterization of Transglutaminase Based Site-Specific Antibody–Drug Conjugates. Bioconj. Chem. 2014, 25, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Spolaore, B.; Raboni, S.; Ramos Molina, A.; Satwekar, A.; Damiano, N.; Fontana, A. Local Unfolding Is Required for the Site-Specific Protein Modification by Transglutaminase. Biochemistry 2012, 51, 8679–8689. [Google Scholar] [CrossRef] [PubMed]
- Spolaore, B.; Damiano, N.; Raboni, S.; Fontana, A. Site-Specific Derivatization of Avidin Using Microbial Transglutaminase. Bioconj. Chem. 2014, 25, 470–480. [Google Scholar] [CrossRef]
- Spolaore, B.; Raboni, S.; Satwekar, A.A.; Grigoletto, A.; Mero, A.; Montagner, I.M.; Rosato, A.; Pasut, G.; Fontana, A. Site-Specific Transglutaminase-Mediated Conjugation of Interferon α-2b at Glutamine or Lysine Residues. Bioconj. Chem. 2016, 27, 2695–2706. [Google Scholar] [CrossRef]
- Spolaore, B.; Forzato, G.; Fontana, A. Site-Specific Derivatization of Human Interferon β-1a at Lysine Residues Using Microbial Transglutaminase. Amino Acids 2018, 50, 923–932. [Google Scholar] [CrossRef]
- Spolaore, B.; Fernández, J.; Lomonte, B.; Massimino, M.L.; Tonello, F. Enzymatic Labelling of Snake Venom Phospholipase A2 Toxins. Toxicon 2019, 170, 99–107. [Google Scholar] [CrossRef]
- Mero, A.; Spolaore, B.; Veronese, F.M.; Fontana, A. Transglutaminase-Mediated PEGylation of Proteins: Direct Identification of the Sites of Protein Modification by Mass Spectrometry Using a Novel Monodisperse PEG. Bioconj. Chem. 2009, 20, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Wright, P.E. Is Apomyoglobin a Molten Globule? Structural Characterization by NMR. J. Mol. Biol. 1996, 263, 531–538. [Google Scholar] [CrossRef]
- Mindt, T.L.; Jungi, V.; Wyss, S.; Friedli, A.; Pla, G.; Novak-Hofer, I.; Grünberg, J.; Schibli, R. Modification of Different IgG1 Antibodies via Glutamine and Lysine Using Bacterial and Human Tissue Transglutaminase. Bioconj. Chem. 2008, 19, 271–278. [Google Scholar] [CrossRef]
- Spycher, P.R.; Amann, C.A.; Wehrmüller, J.E.; Hurwitz, D.R.; Kreis, O.; Messmer, D.; Ritler, A.; Küchler, A.; Blanc, A.; Béhé, M.; et al. Dual, Site-Specific Modification of Antibodies by Using Solid-Phase Immobilized Microbial Transglutaminase. ChemBioChem 2017, 18, 1923–1927. [Google Scholar] [CrossRef]
- Juettner, N.E.; Schmelz, S.; Kraemer, A.; Knapp, S.; Becker, B.; Kolmar, H.; Scrima, A.; Fuchsbauer, H. Structure of a Glutamine Donor Mimicking Inhibitory Peptide Shaped by the Catalytic Cleft of Microbial Transglutaminase. FEBS J. 2018, 285, 4684–4694. [Google Scholar] [CrossRef] [PubMed]
- Tagami, U.; Shimba, N.; Nakamura, M.; Yokoyama, K.-I.; Suzuki, E.-I.; Hirokawa, T. Substrate Specificity of Microbial Transglutaminase as Revealed by Three-Dimensional Docking Simulation and Mutagenesis. Protein Eng. Des. Sel. 2009, 22, 747–752. [Google Scholar] [CrossRef]
- Fiebig, D.; Schmelz, S.; Zindel, S.; Ehret, V.; Beck, J.; Ebenig, A.; Ehret, M.; Fröls, S.; Pfeifer, F.; Kolmar, H.; et al. Structure of the Dispase Autolysis-Inducing Protein from Streptomyces Mobaraensis and Glutamine Cross-Linking Sites for Transglutaminase. J. Biol. Chem. 2016, 291, 20417–20426. [Google Scholar] [CrossRef] [PubMed]
- Buchardt, J.; Selvig, H.; Nielsen, P.F.; Johansen, N.L. Transglutaminase-Mediated Methods for Site-Selective Modification of Human Growth Hormone. Biopolymers 2010, 94, 229–235. [Google Scholar] [CrossRef]
- Sato, H.; Hayashi, E.; Yamada, N.; Yatagai, M.; Takahara, Y. Further Studies on the Site-Specific Protein Modification by Microbial Transglutaminase. Bioconj. Chem. 2001, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Grigoletto, A.; Maso, K.; Yoshioka, H.; Rosato, A.; Pasut, G. Site-Selective Enzymatic Chemistry for Polymer Conjugation to Protein Lysine Residues: PEGylation of G-CSF at Lysine-41. Polym. Chem. 2016, 7, 6545–6553. [Google Scholar] [CrossRef]
- Seitz, A.; Schneider, F.; Pasternack, R.; Fuchsbauer, H.-L.; Hampp, N. Enzymatic Cross-Linking of Purple Membranes Catalyzed by Bacterial Transglutaminase. Biomacromolecules 2001, 2, 233–238. [Google Scholar] [CrossRef]
- Kim, E.; Motoki, M.; Seguro, K.; Muhlrad, A.; Reisler, E. Conformational Changes in Subdomain 2 of G-Actin: Fluorescence Probing by Dansyl Ethylenediamine Attached to Gln-41. Biophys. J. 1995, 69, 2024–2032. [Google Scholar] [CrossRef]
- Taguchi, S.; Nishihama, K.-I.; Igi, K.; Ito, K.; Taira, H.; Motoki, M.; Momose, H. Substrate Specificity Analysis of Microbial Transglutaminase Using Proteinaceous Protease Inhibitors as Natural Model Substrates. J. Biochem. 2000, 128, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Juettner, N.E.; Schmelz, S.; Bogen, J.P.; Happel, D.; Fessner, W.-D.; Pfeifer, F.; Fuchsbauer, H.-L.; Scrima, A. Illuminating Structure and Acyl Donor Sites of a Physiological Transglutaminase Substrate from Streptomyces mobaraensis: Transglutaminase Acyl Donor Interactions. Protein Sci. 2018, 27, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Stefanetti, G.; Hu, Q.; Usera, A.; Robinson, Z.; Allan, M.; Singh, A.; Imase, H.; Cobb, J.; Zhai, H.; Quinn, D.; et al. Sugar–Protein Connectivity Impacts on the Immunogenicity of Site-Selective Salmonella O-Antigen Glycoconjugate Vaccines. Angew. Chem. Int. Ed. 2015, 54, 13198–13203. [Google Scholar] [CrossRef]
- Piersma, S.R.; van de Pijpekamp, A.; Wijngaards, G.; Gruppen, H.; Boumans, H. Quantitation and Localisation of (In Vitro) Transglutaminase-Catalysed Glutamine Hydroxylation Using Mass Spectrometry. Enzym. Microb. Technol. 2002, 30, 266–272. [Google Scholar] [CrossRef]
- Malešević, M.; Migge, A.; Hertel, T.C.; Pietzsch, M. A Fluorescence-Based Array Screen for Transglutaminase Substrates. ChemBioChem 2015, 16, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Ota, M.; Nio, N.; Motoki, M. Comparison of Substrate Specificities of Transglutaminases Using Synthetic Peptides as Acyl Donors. Biosci. Biotechnol. Biochem. 2000, 64, 2608–2613. [Google Scholar] [CrossRef]
- Sugimura, Y.; Yokoyama, K.; Nio, N.; Maki, M.; Hitomi, K. Identification of Preferred Substrate Sequences of Microbial Transglutaminase from Streptomyces Mobaraensis Using a Phage-Displayed Peptide Library. Arch. Biochem. Biophys. 2008, 477, 379–383. [Google Scholar] [CrossRef]
- Takahara, M.; Wakabayashi, R.; Minamihata, K.; Goto, M.; Kamiya, N. Design of Lipid–Protein Conjugates Using Amphiphilic Peptide Substrates of Microbial Transglutaminase. ACS Appl. Bio Mater. 2018, 1, 1823–1829. [Google Scholar] [CrossRef]
- Rachel, N.; Pelletier, J. Biotechnological Applications of Transglutaminases. Biomolecules 2013, 3, 870–888. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Sawa, A.; Kawabata, R.; Nio, N.; Motoki, M. Substrate Specificities of Microbial Transglutaminase for Primary Amines. J. Agric. Food Chem. 2000, 48, 6230–6233. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, M.T.; Keillor, J.W.; Pelletier, J.N. Microbial Transglutaminase Displays Broad Acyl-Acceptor Substrate Specificity. Appl. Microbiol. Biotechnol. 2014, 98, 219–230. [Google Scholar] [CrossRef]
- Chio, T.I.; Demestichas, B.R.; Brems, B.M.; Bane, S.L.; Tumey, L.N. Expanding the Versatility of Microbial Transglutaminase Using A-Effect Nucleophiles as Noncanonical Substrates. Angew. Chem. Int. Ed. 2020, 59, 13814–13820. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Schiavon, M.; Veronese, F.M.; Pasut, G. A New Method to Increase Selectivity of Transglutaminase Mediated PEGylation of Salmon Calcitonin and Human Growth Hormone. J. Control. Release 2011, 154, 27–34. [Google Scholar] [CrossRef]
- Caporale, A.; Monti, A.; Selis, F.; Sandomenico, A.; Tonon, G.; Ruvo, M.; Doti, N. A Comparative Analysis of Catalytic Activity and Stability of Microbial Transglutaminase in Controlled Denaturing Conditions. J. Biotechnol. 2019, 302, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Dickgiesser, S.; Rieker, M.; Mueller-Pompalla, D.; Schröter, C.; Tonillo, J.; Warszawski, S.; Raab-Westphal, S.; Kühn, S.; Knehans, T.; Könning, D.; et al. Site-Specific Conjugation of Native Antibodies Using Engineered Microbial Transglutaminases. Bioconj. Chem. 2020, 31, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Jacobitz, A.W.; Kattke, M.D.; Wereszczynski, J.; Clubb, R.T. Sortase Transpeptidases: Structural Biology and Catalytic Mechanism. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 109, pp. 223–264. ISBN 978-0-12-811876-4. [Google Scholar]
- Bradshaw, W.J.; Davies, A.H.; Chambers, C.J.; Roberts, A.K.; Shone, C.C.; Acharya, K.R. Molecular Features of the Sortase Enzyme Family. FEBS J. 2015, 282, 2097–2114. [Google Scholar] [CrossRef] [PubMed]
- Proft, T. Sortase-Mediated Protein Ligation: An Emerging Biotechnology Tool for Protein Modification and Immobilisation. Biotechnol. Lett. 2010, 32, 1–10. [Google Scholar] [CrossRef]
- Pishesha, N.; Ingram, J.R.; Ploegh, H.L. Sortase A: A Model for Transpeptidation and Its Biological Applications. Annu. Rev. Cell Dev. Biol. 2018, 34, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, L.; Schwarzer, D. Sortase-Mediated Ligations for the Site-Specific Modification of Proteins. Curr. Opin. Chem. Biol. 2014, 22, 122–128. [Google Scholar] [CrossRef]
- Popp, M.W.-L.; Ploegh, H.L. Making and Breaking Peptide Bonds: Protein Engineering Using Sortase. Angew. Chem. Int. Ed. 2011, 50, 5024–5032. [Google Scholar] [CrossRef]
- Mazmanian, S.K. Staphylococcus aureus Sortase, an Enzyme That Anchors Surface Proteins to the Cell Wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Perry, A.M.; Ton-That, H.; Mazmanian, S.K.; Schneewind, O. Anchoring of Surface Proteins to the Cell Wall of Staphylococcus aureus. J. Biol. Chem. 2002, 277, 16241–16248. [Google Scholar] [CrossRef]
- Huang, X.; Aulabaugh, A.; Ding, W.; Kapoor, B.; Alksne, L.; Tabei, K.; Ellestad, G. Kinetic Mechanism of Staphylococcus aureus Sortase SrtA. Biochemistry 2003, 42, 11307–11315. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef] [PubMed]
- Pritz, S.; Wolf, Y.; Kraetke, O.; Klose, J.; Bienert, M.; Beyermann, M. Synthesis of Biologically Active Peptide Nucleic Acid−Peptide Conjugates by Sortase-Mediated Ligation. J. Org. Chem. 2007, 72, 3909–3912. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, T.; Tsukiji, S.; Nagamune, T. Site-Specific Protein Modification on Living Cells Catalyzed by Sortase. ChemBioChem 2008, 9, 802–807. [Google Scholar] [CrossRef]
- Popp, M.W.; Artavanis-Tsakonas, K.; Ploegh, H.L. Substrate Filtering by the Active Site Crossover Loop in UCHL3 Revealed by Sortagging and Gain-of-Function Mutations. J. Biol. Chem. 2009, 284, 3593–3602. [Google Scholar] [CrossRef]
- Bellucci, J.J.; Bhattacharyya, J.; Chilkoti, A. A Noncanonical Function of Sortase Enables Site-Specific Conjugation of Small Molecules to Lysine Residues in Proteins. Angew. Chem. Int. Ed. 2014, 54, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Baer, S.; Nigro, J.; Madej, M.P.; Nisbet, R.M.; Suryadinata, R.; Coia, G.; Hong, L.P.T.; Adams, T.E.; Williams, C.C.; Nuttall, S.D. Comparison of Alternative Nucleophiles for Sortase A-Mediated Bioconjugation and Application in Neuronal Cell Labelling. Org. Biomol. Chem. 2014, 12, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Sadamoto, R.; Naruchi, K.; Togame, H.; Takemoto, H.; Kondo, H.; Nishimura, S.-I. Highly Oriented Recombinant Glycosyltransferases: Site-Specific Immobilization of Unstable Membrane Proteins by Using Staphylococcus aureus Sortase A. Biochemistry 2010, 49, 2604–2614. [Google Scholar] [CrossRef]
- Li, Y.-M.; Li, Y.-T.; Pan, M.; Kong, X.-Q.; Huang, Y.-C.; Hong, Z.-Y.; Liu, L. Irreversible Site-Specific Hydrazinolysis of Proteins by Use of Sortase. Angew. Chem. Int. Ed. 2014, 53, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Truttmann, M.C.; Ploegh, H.L. Recent Advances in Sortase-Catalyzed Ligation Methodology. Curr. Opin. Struct. Biol. 2016, 38, 111–118. [Google Scholar] [CrossRef]
- Policarpo, R.L.; Kang, H.; Liao, X.; Rabideau, A.E.; Simon, M.D.; Pentelute, B.L. Flow-Based Enzymatic Ligation by Sortase A. Angew. Chem. Int. Ed. 2014, 53, 9203–9208. [Google Scholar] [CrossRef]
- Freund, C.; Schwarzer, D. Engineered Sortases in Peptide and Protein Chemistry. ChemBioChem 2021, 22, 1347–1356. [Google Scholar] [CrossRef]
- Hirakawa, H.; Ishikawa, S.; Nagamune, T. Ca2+-Independent Sortase-A Exhibits High Selective Protein Ligation Activity in the Cytoplasm of Escherichia coli. Biotechnol. J. 2015, 10, 1487–1492. [Google Scholar] [CrossRef]
- Glasgow, J.E.; Salit, M.L.; Cochran, J.R. In Vivo Site-Specific Protein Tagging with Diverse Amines Using an Engineered Sortase Variant. J. Am. Chem. Soc. 2016, 138, 7496–7499. [Google Scholar] [CrossRef]
- Qasba, P.K. Glycans of Antibodies as a Specific Site for Drug Conjugation Using Glycosyltransferases. Bioconj. Chem. 2015, 26, 2170–2175. [Google Scholar] [CrossRef]
- Khidekel, N.; Arndt, S.; Lamarre-Vincent, N.; Lippert, A.; Poulin-Kerstien, K.G.; Ramakrishnan, B.; Qasba, P.K.; Hsieh-Wilson, L.C. A Chemoenzymatic Approach toward the Rapid and Sensitive Detection of O-GlcNAc Posttranslational Modifications. J. Am. Chem. Soc. 2003, 125, 16162–16163. [Google Scholar] [CrossRef]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-Specific Antibody-Drug Conjugation through an Engineered Glycotransferase and a Chemically Reactive Sugar. mAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native MAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconj. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Davis, C.B.; Aggeler, R.; Kang, H.C.; Chen, A.; Agnew, B.J.; Lewis, J.S. Enzyme-Mediated Methodology for the Site-Specific Radiolabeling of Antibodies Based on Catalyst-Free Click Chemistry. Bioconj. Chem. 2013, 24, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Davis, C.B.; Abdel-Atti, D.; Carlin, S.D.; Chen, A.; Aggeler, R.; Agnew, B.J.; Lewis, J.S. Chemoenzymatic Strategy for the Synthesis of Site-Specifically Labeled Immunoconjugates for Multimodal PET and Optical Imaging. Bioconj. Chem. 2014, 25, 2123–2128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef]
- Cronan, J.E.; Zhao, X.; Jiang, Y. Function, Attachment and Synthesis of Lipoic Acid in Escherichia coli. Adv. Microb. Physiol. 2005, 50, 103–146. [Google Scholar] [CrossRef]
- Puthenveetil, S.; Liu, D.S.; White, K.A.; Thompson, S.; Ting, A.Y. Yeast Display Evolution of a Kinetically Efficient 13-Amino Acid Substrate for Lipoic Acid Ligase. J. Am. Chem. Soc. 2009, 131, 16430–16438. [Google Scholar] [CrossRef] [PubMed]
- Plaks, J.G.; Falatach, R.; Kastantin, M.; Berberich, J.A.; Kaar, J.L. Multisite Clickable Modification of Proteins Using Lipoic Acid Ligase. Bioconj. Chem. 2015, 26, 1104–1112. [Google Scholar] [CrossRef]
- Uttamapinant, C.; Sanchez, M.I.; Liu, D.S.; Yao, J.Z.; White, K.A.; Grecian, S.; Clark, S.; Gee, K.R.; Ting, A.Y. Site-Specific Protein Labeling Using PRIME and Chelation-Assisted Click Chemistry. Nat. Protoc. 2013, 8, 1620–1634. [Google Scholar] [CrossRef]
- Drake, C.R.; Sevillano, N.; Truillet, C.; Craik, C.S.; VanBrocklin, H.F.; Evans, M.J. Site-Specific Radiofluorination of Biomolecules with 8-[18F]-Fluorooctanoic Acid Catalyzed by Lipoic Acid Ligase. ACS Chem. Biol. 2016, 11, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Baalmann, M.; Ziegler, M.J.; Werther, P.; Wilhelm, J.; Wombacher, R. Enzymatic and Site-Specific Ligation of Minimal-Size Tetrazines and Triazines to Proteins for Bioconjugation and Live-Cell Imaging. Bioconj. Chem. 2019, 30, 1405–1414. [Google Scholar] [CrossRef]
- Cohen, J.D.; Zou, P.; Ting, A.Y. Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. ChemBioChem 2012, 13, 888–894. [Google Scholar] [CrossRef]
- Fernández-Suárez, M.; Baruah, H.; Martínez-Hernández, L.; Xie, K.T.; Baskin, J.M.; Bertozzi, C.R.; Ting, A.Y. Redirecting Lipoic Acid Ligase for Cell Surface Protein Labeling with Small-Molecule Probes. Nat. Biotechnol. 2007, 25, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; White, K.A.; Baruah, H.; Thompson, S.; Fernandez-Suarez, M.; Puthenveetil, S.; Ting, A.Y. A Fluorophore Ligase for Site-Specific Protein Labeling inside Living Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10914–10919. [Google Scholar] [CrossRef] [PubMed]
- Baalmann, M.; Neises, L.; Bitsch, S.; Schneider, H.; Deweid, L.; Werther, P.; Ilkenhans, N.; Wolfring, M.; Ziegler, M.J.; Wilhelm, J.; et al. A Bioorthogonal Click Chemistry Toolbox for Targeted Synthesis of Branched and Well-Defined Protein–Protein Conjugates. Angew. Chem. Int. Ed. 2020, 59, 12885–12893. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Schibli, R.; Fischer, E. Enzymatic Antibody Modification by Bacterial Transglutaminase. Antib. Drug Conj. 2013, 205–215. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef]
- Grünberg, J.; Jeger, S.; Sarko, D.; Dennler, P.; Zimmermann, K.; Mier, W.; Schibli, R. DOTA-Functionalized Polylysine: A High Number of DOTA Chelates Positively Influences the Biodistribution of Enzymatic Conjugated Anti-Tumor Antibody ChCE7agl. PLoS ONE 2013, 8, e60350. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Bailey, L.K.; Spycher, P.R.; Schibli, R.; Fischer, E. Microbial Transglutaminase and C-Myc-Tag: A Strong Couple for the Functionalization of Antibody-Like Protein Scaffolds from Discovery Platforms. ChemBioChem 2015, 16, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Salvarese, N.; Spolaore, B.; Marangoni, S.; Pasin, A.; Galenda, A.; Tamburini, S.; Cicoria, G.; Refosco, F.; Bolzati, C. Transglutaminase-Mediated Conjugation and Nitride-Technetium-99m Labelling of a Bis(Thiosemicarbazone) Bifunctional Chelator. J. Inorg. Biochem. 2018, 183, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Bolzati, C.; Ghiani, S.; Maiocchi, A.; Refosco, F.; Salvarese, N.; Spolaore, B. Method for Labeling of Sensitive and Thermosensitive Targeting Biomolecules with Technetium Based Compounds. Patent Application No. WO2017EP83024, 15 December 2017. [Google Scholar]
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A Highly Functional Synthetic Phage Display Library Containing over 40 Billion Human Antibody Clones. PLoS ONE 2014, 9, e100000. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Esposito, C.; Metafora, S.; Pucci, P.; Malorni, A.; Marino, G. Substance P as a Transglutaminase Substrate: Identification of the Reaction Products by Fast Atom Bombardment Mass Spectrometry. Anal. Biochem. 1988, 172, 499–503. [Google Scholar] [CrossRef]
- Ferrandiz, C.; Perezpaya, E.; Braco, L.; Abad, C. Gln5 Selectively Monodansylated Substance P as a Sensitive Tool for Interaction Studies with Membranes. Biochem. Biophys. Res. Commun. 1994, 203, 359–365. [Google Scholar] [CrossRef]
- Mancuso, F.; Costa, C.; Calignano, A.; Mariniello, L.; Rossi, F.; Porta, R.; Esposito, C. Transglutaminase-Synthesized γ-(Glutamyl5) Spermidine Derivative of Substance P Is a Selective Tool for Neurokinin-2 Receptors Characterization. Peptides 1998, 19, 683–690. [Google Scholar] [CrossRef]
- Marculescu, C.; Lakshminarayanan, A.; Gault, J.; Knight, J.C.; Folkes, L.K.; Spink, T.; Robinson, C.V.; Vallis, K.; Davis, B.G.; Cornelissen, B. Probing the Limits of Q-Tag Bioconjugation of Antibodies. Chem. Commun. 2019, 55, 11342–11345. [Google Scholar] [CrossRef]
- Paterson, B.M.; Alt, K.; Jeffery, C.M.; Price, R.I.; Jagdale, S.; Rigby, S.; Williams, C.C.; Peter, K.; Hagemeyer, C.E.; Donnelly, P.S. Enzyme-Mediated Site-Specific Bioconjugation of Metal Complexes to Proteins: Sortase-Mediated Coupling of Copper-64 to a Single-Chain Antibody. Angew. Chem. Int. Ed. 2014, 53, 6115–6119. [Google Scholar] [CrossRef]
- Alt, K.; Paterson, B.M.; Westein, E.; Rudd, S.E.; Poniger, S.S.; Jagdale, S.; Ardipradja, K.; Connell, T.U.; Krippner, G.Y.; Nair, A.K.N.; et al. A Versatile Approach for the Site-Specific Modification of Recombinant Antibodies Using a Combination of Enzyme-Mediated Bioconjugation and Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 7515–7519. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-Fluorodeoxyglucose to Label Antibody Fragments for Immuno-Positron Emission Tomography of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Bilate, A.M.; Duarte, J.N.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Cragnolini, J.; Swee, L.K.; Victora, G.D.; Weissleder, R.; et al. Noninvasive Imaging of Immune Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6146–6151. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Wang, L.; Edens, J.G.; Jacobsen, J.T.; Hossain, I.; Wang, Q.; Victora, G.D.; Vasdev, N.; Ploegh, H.; Liang, S.H. Enzyme-Mediated Modification of Single-Domain Antibodies for Imaging Modalities with Different Characteristics. Angew. Chem. Int. Ed. 2016, 55, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Vikani, N.; Betti, C.; Ballet, S.; Vanderhaegen, S.; Steyaert, J.; Descamps, B.; Vanhove, C.; Bunschoten, A.; van Leeuwen, F.W.B.; et al. Sortase A-Mediated Site-Specific Labeling of Camelid Single-Domain Antibody-Fragments: A Versatile Strategy for Multiple Molecular Imaging Modalities. Contrast Media Mol. Imaging 2016, 11, 328–339. [Google Scholar] [CrossRef]
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef]
- Crauwels, M.; Massa, S.; Martin, C.; Betti, C.; Ballet, S.; Devoogdt, N.; Xavier, C.; Muyldermans, S. Site-Specific Radioactive Labeling of Nanobodies. In Antibody Engineering: Methods and Protocols; Nevoltris, D., Chames, P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 505–540. ISBN 978-1-4939-8648-4. [Google Scholar]
- Zeglis, B.M.; Brand, C.; Abdel-Atti, D.; Carnazza, K.E.; Cook, B.E.; Carlin, S.; Reiner, T.; Lewis, J.S. Optimization of a Pretargeted Strategy for the PET Imaging of Colorectal Carcinoma via the Modulation of Radioligand Pharmacokinetics. Mol. Pharm. 2015, 12, 3575–3587. [Google Scholar] [CrossRef]
- Cook, B.E.; Adumeau, P.; Membreno, R.; Carnazza, K.E.; Brand, C.; Reiner, T.; Agnew, B.J.; Lewis, J.S.; Zeglis, B.M. Pretargeted PET Imaging Using a Site-Specifically Labeled Immunoconjugate. Bioconj. Chem. 2016, 27, 1789–1795. [Google Scholar] [CrossRef]
- Keinänen, O.; Brennan, J.M.; Membreno, R.; Fung, K.; Gangangari, K.; Dayts, E.J.; Williams, C.J.; Zeglis, B.M. Dual Radionuclide Theranostic Pretargeting. Mol. Pharm. 2019, 16, 4416–4421. [Google Scholar] [CrossRef]
- Houghton, J.L.; Zeglis, B.M.; Abdel-Atti, D.; Aggeler, R.; Sawada, R.; Agnew, B.J.; Scholz, W.W.; Lewis, J.S. Site-Specifically Labeled CA19.9-Targeted Immunoconjugates for the PET, NIRF, and Multimodal PET/NIRF Imaging of Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15850–15855. [Google Scholar] [CrossRef] [PubMed]
- Adumeau, P.; Vivier, D.; Sharma, S.K.; Wang, J.; Zhang, T.; Chen, A.; Agnew, B.J.; Zeglis, B.M. Site-Specifically Labeled Antibody–Drug Conjugate for Simultaneous Therapy and ImmunoPET. Mol. Pharm. 2018, 15, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.K.; Christensen, C.; Jensen, M.M.; Agnew, B.J.; Schjöth-Frydendahl, C.; Kjaer, A.; Nielsen, C.H. Site-Specifically Labeled 89Zr-DFO-Trastuzumab Improves Immuno-Reactivity and Tumor Uptake for Immuno-PET in a Subcutaneous HER2-Positive Xenograft Mouse Model. Theranostics 2019, 9, 4409–4420. [Google Scholar] [CrossRef]
- Vivier, D.; Fung, K.; Rodriguez, C.; Adumeau, P.; Ulaner, G.A.; Lewis, J.S.; Sharma, S.K.; Zeglis, B.M. The Influence of Glycans-Specific Bioconjugation on the FcγRI Binding and In Vivo Performance of 89Zr-DFO-Pertuzumab. Theranostics 2020, 10, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Vivier, D.; Sharma, S.K.; Adumeau, P.; Rodriguez, C.; Fung, K.; Zeglis, B.M. The Impact of FcγRI Binding on Immuno-PET. J. Nucl. Med. 2019, 60, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 1: Cysteine Residues and Glycans. Mol. Imaging Biol. 2016, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]





| Enzyme | Substrates | Biomolecules on MI | Pros | Cons |
|---|---|---|---|---|
| mTG |
| Wild type molecules ranging from whole mAbs to peptides |
|
|
| SortA |
| Full-length mAbs and their fragments |
|
|
| Galactosidase and GalT(Y289L) |
| Full-length mAbs with pendant sugar chains |
|
|
| LplA |
| Full-length mAbs and their fragments |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolzati, C.; Spolaore, B. Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins. Molecules 2021, 26, 3492. https://doi.org/10.3390/molecules26123492
Bolzati C, Spolaore B. Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins. Molecules. 2021; 26(12):3492. https://doi.org/10.3390/molecules26123492
Chicago/Turabian StyleBolzati, Cristina, and Barbara Spolaore. 2021. "Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins" Molecules 26, no. 12: 3492. https://doi.org/10.3390/molecules26123492
APA StyleBolzati, C., & Spolaore, B. (2021). Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins. Molecules, 26(12), 3492. https://doi.org/10.3390/molecules26123492