3.1. Synthesis
Starting materials and solvents were purchased from commercial suppliers and used without any additional purification. 1H NMR and 13C NMR spectra were obtained on a Bruker AV-300 nuclear instrument or Bruker AVANCE NEO 400 MHz in CDCl3 or DMSO-d6 using TMS as internal standard, operating at 300 MHz or 400 MHz and 75 MHz, respectively. Chemical shifts (δ) are expressed in ppm and coupling constants J are given in Hz. Analytical thin layer chromatography (TLC) was purchased from Yantai Chemical Industry Research Institute (Cat. no. HSGF254, Yantai, China). All the reactions were monitored by thin layer chromatography in UV absorbance (254 nm). Flash chromatography was performed using silica gel (200–300 or 300–400 mesh). Melting points were measured with an RY-I melting point apparatus. The HPLC analysis was performed on a Shimadzu LC-20AT machine with a BDS Hypersil C18 column, and the column temperature was at 31 °C. Mobile phase B (100% Acetonitrile) and mobile phase A (NaH2PO4 and H3PO4 buffer solution, pH = 7.5) were used in a gradient elution program (0 min: 25% (B), 5 min: 25% (B), 12 min: 75% (B), 20 min: 75% (B), 23 min: 25% (B), 25 min: 25% (B)) with a flow rate of 1.0 mL/min at 254 nM.
3.1.1. 3-Bromo-2-Methylphenol (8)
A mixture of 3-bromo-2-methylaniline and H2SO4 (1M, 65 mL) was stirred for 30 min at room temperature, then 30% NaNO2 (2.25 g, 32.60 mmol) was added to the mixture dropwise at 0–5 °C. After 30 min, toluene (50 mL) was then added to the reaction mixture and allowed to stir at 100 °C for approximately 1 h. The mixture was extracted with ethyl acetate (50 mL × 3) and washed with brine (50 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether/ethyl acetate = 100/1–60/1) to afford compound 8 (white solid, 4.50 g, yield: 89.6%). m.p. 95.0–98.0 °C. 1H NMR (300 MHz, Chloroform-d) δ 7.19 (dd, J = 8.0, 1.2 Hz, 1H, ArH), 6.97 (t, J = 8.0 Hz, 1H, ArH), 6.76 (d, J = 7.9 Hz, 1H, ArH), 4.82 (s, 1H, OH), 2.38 (s, 3H, CH3).
3.1.2. 1-Bromo-3-(3-Bromopropoxy)-2-Methylbenzene (9)
A mixture of 8 (5.00 g, 26.90 mmol), K2CO3 (9.28 g, 67.25 mmol), and 1,3-dibromopropane (10.75 g, 53.80 mmol) in acetone (30 mL) was refluxed for 8 h. The mixture was diluted with water (100 mL), extracted with ethyl acetate (50 mL × 3), and washed with brine (50 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether = 100%) to afford compound 9 (colorless oil, 6.72 g, yield: 81.7%).1H NMR (300 MHz, Chloroform-d) δ 7.16 (dd, J = 8.1, 1.2 Hz, 1H, ArH), 7.00 (td, J = 8.1, 0.6 Hz, 1H, ArH), 6.79 (dd, J = 8.2, 1.2 Hz, 1H, ArH), 4.10 (t, J = 5.7 Hz, 2H, OCH2), 3.62 (t, J = 6.4 Hz, 2H, BrCH2), 2.39–2.32 (m, 2H, CH2CH2CH2), 2.31 (s, 3H, CH3).
3.1.3. 1-(3-(3-Bromo-2-Methylphenoxy)Propyl)Piperidin-4-ol (10)
A mixture of 9 (1.00 g, 3.27 mmol), DIPEA (1.10 mL, 6.54 mmol), and 4-hydroxypiperidine (0.97 g, 3.27 mmol) in MeCN (10 mL) was refluxed for 12 h. The formed precipitates were filtrated and washed with petroleum ether and less water to obtain compound 10 (yellow solid, 0.86 g, yield: 80.8%). m.p.138.0–140.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.20 (dd, J = 8.0, 1.3 Hz, 1H, ArH), 7.13 (t, J = 8.1 Hz, 1H, ArH), 6.99 (d, J = 7.9 Hz, 1H, ArH), 5.02–4.96 (m, 1H, OH), 4.09 (t, J = 5.9 Hz, 2H, OCH2), 4.01–3.92 (m, 1H, CHOH), 3.77–3.40 (m, 2H, CH2N), 3.20–2.86 (m, 4H, NCH2), 2.28 (s, 3H, CH3), 2.23–2.08 (m, 2H, CH2CH2CH2), 2.08–1.69 (m, 4H, CH2CHOH).
3.1.4. (3-Bromo-2-Methylphenyl)Methanol (12)
BH3-THF (1 M, 35 mL) was dropwise added to a solution of 3-bromo-2-methylbenzoic acid (5.00 g, 23.20 mmol) in dry THF (25 mL) at 0 °C under N2. The mixture was stirred at an ambient temperature for 12 h. The reaction quenched with 1 M HCl (30 mL), and then extracted with ethyl acetate (20 mL × 3), followed by brine (20 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum to give 12 without any further purification (white solid, 4.5 g, yield, 97.0%). m.p. 103.0–104.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.49 (d, J = 7.9 Hz, 1H, ArH), 7.39 (d, J = 7.8 Hz, 1H, ArH), 7.12 (t, J = 8.1 Hz, 1H, ArH), 5.27 (br, 1H, OH), 4.52 (s, 2H, CH2), 2.30 (s, 3H, ArCH3).
3.1.5. 1-Bromo-3-(Chloromethyl)-2-Methylbenzene (13)
A solution of 12 (4.50 g, 22.50 mmol) in SOCl2 (10 mL) was stirred at 80 °C for 2 h. The solvent was removed in vacuum to obtain compound 13 (colorless oil, 4.80 g, yield, 98.0%). 1H NMR (300 MHz, Chloroform-d) δ 7.47 (d, J = 7.0 Hz, 1H, ArH), 7.25 (d, J = 6.8 Hz, 1H, ArH), 7.04 (t, J = 8.1 Hz, 1H, ArH), 3.90 (s, 2H, CH2), 2.43 (s, 3H, CH3).
3.1.6. 1-(3-Bromo-2-Methylbenzyl)Piperidin-4-One (14)
This compound was prepared following the synthetic procedure similar to that of 10. Compound 14 (colorless oil 0.93 g, yield, 72.1%). 1H NMR (300 MHz, Chloroform-d) δ 7.52 (dd, J = 8.0, 1.6 Hz, 1H, ArH), 7.23 (d, J = 6.4 Hz, 1H, ArH), 7.03 (t, J = 7.8 Hz, 1H, ArH) 3.61 (s, 2H, CH2N), 2.77 (t, J = 6.1 Hz, 4H, 2NCH2), 2.51 (s, 3H, CH3), 2.46 (t, J = 6.2 Hz, 4H, 2CH2CO).
3.1.7. 1′-(3-Bromo-2-Methylbenzyl)-[1,4′-Bipiperidin]-4-ol (15)
A mixture of 14 (400 mg, 1.42 mmol) and 4-hydroxypiperidine (172 mg, 1.71 mmol) in DCM (10 mL) was stirred for 2 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (dichloromethane/methanol = 100/1–30/1) to afford compound 15 (yellow solid, yield, 58.3%). m.p. 102.0–105.0 °C. 1H NMR (300 MHz, Chloroform-d) δ 7.51 (d, J =7.9Hz, 1H, ArH), 7.23 (d, J = 7.4 Hz, 1H, ArH), 7.03 (t, J = 7.7 Hz, 1H, ArH), 4.34–4.27 (m, 1H, CHOH), 3.79–3.66 (m, 1H, CHN), 3.47 (s, 2H, ArCH2), 3.02–2.80 (m, 4H, NCH2), 2.46 (s, 3H, ArCH3), 2.44–2.28 (m, 4H, NCH2), 2.05–1.92 (m, 4H, 2CH2), 1.55–1.70 (m, 4H, 2CH2).
3.1.8. The Synthesis of Compounds 16a–j
Compounds 16a–j were synthesized by the following procedure. To a solution of 3-bromo-2-methylaniline (1.0 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2.8 mmol) in 1,4-dioxane/water (1.0 mL/0.1 mL), Pd(PPh3)4 (0.05 mmol) was added. The mixture was stirred at 80 °C for 8 h under N2 atmosphere. The undissolved solid was filtered off, and the filtrate was concentrated under vacuum. The residue was dissolved in ethyl acetate, and washed with water, followed by brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography (petroleum ether/ethyl acetate = 100/1–80/1) to afford compounds 16a-j.
2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16a)
Brown solid. Yield, 95.6%. m.p. 61.0–62.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.48–7.35 (m, 1H, ArH), 7.34 – 7.22 (m, 3H, ArH), 6.98 (t, J = 7.7 Hz, 1H, ArH), 6.70 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.3 Hz, 1H, ArH), 5.00 (s, 2H, NH2), 1.85 (d, J = 1.8 Hz, 3H, ArCH3).
2,2′-Dimethyl-[1,1′-biphenyl]-3-amine (16b)
Yellow oil. Yield, 90%. m.p. 52.0–55.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.34 – 7.29 (m, 1H, ArH), 7.28–7.22 (m, 2H, ArH), 7.15 (d, J = 7.0 Hz, 1H, ArH), 7.10 (t, J = 7.7 Hz, 1H, ArH), 6.75 (dd, J = 7.9, 1.4 Hz, 1H, ArH), 6.63 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 3.75 (s, 2H, ArNH2), 2.11 (s, 3H, ArCH3), 1.91 (s, 3H, ArCH3).
2′-Chloro-2-methyl-[1,1′-biphenyl]-3-amine (16c)
Yellow oil. Yield, 93.1%. m.p. 71.5–74.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.58–7.46 (m, 1H, ArH), 7.43–7.30 (m, 2H, ArH), 7.30–7.18 (m, 1H, ArH), 6.94 (t, J = 7.6 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.32 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 4.96 (s, 2H, NH2), 1.76 (s, 3H, ArCH3).
2′-Chloro-2-methyl-[1,1′-biphenyl]-3-amine (16d)
Yellow solid. Yield, 82.1%. m.p. 87.5–90.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79 (dd, J = 7.9, 1.6 Hz, 1H, ArH), 7.67 (td, J = 7.6, 1.7 Hz, 1H, ArH), 7.56 (t, J = 7.6, 1H, ArH), 7.25 (d, J = 7.5 Hz, 1H, ArH), 6.90 (t, J = 7.7 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.31 (d, J = 7.3 Hz, 1H, ArH), 4.98 (s, 2H, NH2), 1.68 (s, 3H, ArCH3).
2-Methyl-[1,1′-biphenyl]-3-amine (16e)
White solid. Yield, 89.6%. 1H NMR (400 MHz, Chloroform-d) δ 7.46–7.40 (m, 2H, ArH), 7.40–7.32 (m, 3H, ArH), 7.11 (t, J = 7.8 Hz, 1H, ArH), 6.75 (d, J = 7.8 Hz, 2H, ArH), 3.82 (s, 2H, NH2), 2.10 (s, 3H, ArCH3).
3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16f)
Yellow oil. Yield, 88.0%.1H NMR (400 MHz, DMSO-d6) δ 7.44 (td, J = 8.1, 6.1 Hz, 1H, ArH), 7.25–7.15 (m, 1H, ArH), 7.12–7.04 (m, 2H, ArH), 6.94 (t, J = 7.8 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.5 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 5.05 (s, 2H, NH2), 1.93 (d, J = 1.9 Hz, 3H, CH3).
4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16g)
White solid. Yield, 88.0%. m.p. 76.5–78.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.32–7.23 (m, 3H, ArH), 7.20 (d, J = 8.8 Hz, 1H, ArH), 6.93 (t, J = 7.7 Hz, 1H, ArH), 6.66 (d, J = 7.9 Hz, 1H, ArH), 6.40 (d, J = 7.5 Hz, 1H, ArH), 4.94 (s, 2H, NH2), 1.92 (s, 3H, ArCH3).
2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-amine (16h)
Yellow solid. Yield, 88.9%. m.p. 81.5–83.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.38–7.27 (m, 2H, ArH), 7.16 (td, J = 8.5, 2.2 Hz, 1H, ArH), 6.97 (t, J = 7.8 Hz, 1H, ArH), 6.70 (dd, J = 8.1, 1.3 Hz, 1H, ArH), 6.40 (d, J = 7.4 Hz, 1H, ArH), 5.01 (s, 2H, NH2), 1.84 (d, J = 1.8 Hz, 3H, ArCH3).
4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16i)
Yellow oil. Yield, 99.2%. 1H NMR (300 MHz, DMSO-d6) δ 7.52 (dd, J = 9.8, 1.9 Hz, 1H, ArH), 7.37 (dd, J = 8.3, 2.0 Hz, 1H, ArH), 7.35–7.28 (m, 1H, ArH), 6.99 (t, J = 7.7 Hz, 1H, ArH), 6.72 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.41 (dd, J = 7.5, 1.3 Hz, 1H, ArH), 5.03(s, 2H, NH2), 1.85 (d, J = 1.8 Hz, 3H, ArCH3).
2. ′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-amine (16j)
Yellow oil. Yield, 80.6%. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (dd, J = 9.8, 2.1 Hz, 1H, ArH), 7.64 (dd, J = 8.0, 2.1 Hz, 1H, ArH), 7.51 (t, J = 7.6 Hz, 1H, ArH), 6.99 (t, J = 7.8 Hz, 1H, ArH), 6.73 (d, J = 8.0 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 5.04 (bs, 2H, NH2), 1.84 (d, J = 1.9 Hz, 3H, ArCH3).
3.1.9. The Synthesis of Compounds 17a–j
Compounds 17a–j were synthesized by the following procedure. HCl (24.84 mL, 3 M) was added to a solution of 16a (4.00 g, 19.9 mmol) in MeOH (40 mL), followed by the addition of H2O (20 mL) at room temperature. The mixture was stirred for 30 min, and NaNO2 (9.92 mL, 2.2 M) was added dropwise very slowly at 0–5 °C. After being stirred for another 30 min, B2pin2 (15.12 g, 59.7 mmol) in MeOH (40 mL) was added to the mixture solution and warmed to room temperature for 2 h. Then the mixture was extracted with DCM (50 mL × 3), washed with brine (50 mL × 2), and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuum and purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 250/1–150/1) to the target compound.
2-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17a)
Yellow solid. Yield, 60.4%. m.p. 94.0–95.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.70 (dd, J = 5.4, 3.7 Hz, 1H, ArH), 7.53–7.36 (m, 1H, ArH), 7.34–7.25 (m, 5H, ArH), 2.27 (d, J = 1.4 Hz, 3H, ArCH3), 1.31 (s, 12H, CH3).
2-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17b)
Colorless oil. Yield, 40.1%. 1H NMR (300 MHz, DMSO-d6) δ 7.67 (dd, J = 7.4, 1.8 Hz, 1H, ArH), 7.34–7.21 (m, 4H, ArH), 7.15 (dd, J = 7.5, 1.8 Hz, 1H, ArH), 7.10–6.95 (m, 1H, ArH), 2.17 (s, 3H, ArCH3), 1.99 (s, 3H, ArCH3), 1.33 (s, 12H, CCH3).
2-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17c)
Light yellow solid. Yield, 62.9%. m.p. 101.0–103.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.71 (dd, J = 7.3, 1.8 Hz, 1H, ArH), 7.60 (dd, J = 5.8, 3.5 Hz, 1H, ArH), 7.48–7.41 (m, 2H, ArH), 7.33–7.25 (m, 2H, ArH), 7.21 (dd, J = 7.6, 1.8 Hz, 1H, ArH), 2.22 (s, 3H, ArCH3), 1.34 (s, 12H, CH3).
4,4,5,5-Tetramethyl-2-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-1,3,2-dioxaborolane (17d)
Colorless oil. Yield, 68.8%. 1H NMR (300 MHz, DMSO-d6) δ 7.83 (dd, J = 7.9, 1.6 Hz, 1H, ArH), 7.79–7.70 (m, 2H, ArH), 7.68–7.61 (m, 1H, ArH), 7.29 (d, J = 7.5 Hz, 1H), 7.25–7.13 (m, 2H, ArH), 2.12 (s, 3H, ArH), 1.31 (s, 12H, CH3).
4,4,5,5-Tetramethyl-2-(2-methyl-[1,1′-biphenyl]-3-yl)-1,3,2-dioxaborolane (17e)
Colorless oil. Yield, 55.3%. 1H NMR (400 MHz, DMSO-d6) δ 7.69 (d, J = 7.8 Hz, 1H, ArH), 7.57–7.40 (m, 1H, ArH), 7.39–7.20 (m, 4H, ArH), 7.03 (s, 1H, ArH), 6.99 (d, J = 7.9 Hz, 1H, ArH), 2.26 (d, J = 1.3 Hz, 3H, ArCH3), 1.32 (s, 12H, CH3).
2-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17f)
White solid. Yield, 56.8%. 1H NMR (300 MHz, DMSO-d6) δ 7.69 (dd, J = 6.4, 2.5 Hz, 1H, ArH), 7.50 (td, J = 8.1, 6.2 Hz, 1H, ArH), 7.34–7.26 (m, 2H, ArH), 7.26–7.21 (m, 1H, ArH), 7.19–7.16 (m, 2H, ArH), 2.37 (s, 3H, ArCH3), 1.34 (s, 12H, CH3).
2-(4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17g)
Yellow solid. Yield, 60.0%. 1H NMR (300 MHz, DMSO-d6) δ 7.71–7.63 (m, 1H, ArH), 7.38–7.34 (m, 1H, ArH), 7.34–7.29 (m, 2H, ArH), 7.29–7.23 (m, 3H, ArH), 2.35 (s, 3H, ArCH3), 1.33 (s, 12H, CH3).
2-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17h)
Light yellow solid. Yield, 67.5%. m.p. 59.5–61.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79–7.68 (m, 1H, ArH), 7.43–7.32 (m, 2H, ArH), 7.28 (d, J = 4.4 Hz, 2H, ArH), 7.19 (td, J = 8.5, 2.7 Hz, 1H, ArH), 2.28 (d, J = 1.3 Hz, 3H, ArCH3), 1.33 (s, 12H, CH3).
2-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17i)
Light yellow solid. Yield, 63.5%. m.p. 91.5–94.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.72 (q, J = 4.0 Hz, 1H, ArH), 7.57 (dd, J = 9.8, 2.0 Hz, 1H, ArH), 7.41 (dd, J = 8.2, 2.0 Hz, 1H, ArH), 7.37 (d, J = 7.7 Hz, 1H, ArH), 7.30 (d, J = 4.5 Hz, 2H, ArH), 2.28 (s 3H, ArCH3), 1.34 (s, 12H, CH3).
2-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17j)
Brown solid. Yield, 63.5%. m.p. 76.5–78.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.80 (d, J = 9.8Hz, 1H, ArH), 7.74 (d, J = 6.3Hz, 1H, ArH), 7.70–7.66 (m, 1H, ArH), 7.59–7.52 (m, 1H, ArH), 7.33–7.28 (m, 2H, ArH), 2.27 (s, 3H, CH3), 1.31 (s, 12H, CH3).
3.1.10. 2″-Fluoro-3-Methoxy-2′-Methyl-[1,1′:3′,1″-Terphenyl]-4-Carbaldehyde (19a)
To a solution of 17 (2.50 g, 8.01 mmol), 18a (1.51 g, 8.81 mmol), and K2CO3 (3.10 g, 22.4 mmol) in 1,4-dioxane/water (25 mL/2.5 mL), Pd(PPh3)4 (0.78 g, 0.67 mmol) was added, and the mixture was allowed to stir at 90 °C for 8 h under N2 atmosphere. The undissolved solid was filtered off, and the filtrate was concentrated under vacuum. The residue was dissolved in ethyl acetate (25 mL × 3) and washed with water (25 mL × 2), followed by brine (25 mL × 2). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether/ ethyl acetate = 150/1–80/1) to afford compound 19a (white solid 2.45 g, yield: 95.3%). m.p. 82.0–83.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H, CHO), 7.77 (d, J = 7.9 Hz, 1H, ArH), 7.50–7.43 (m, 1H, ArH), 7.41–7.36 (m, 2H, ArH), 7.36–7.30 (m, 3H, ArH), 7.30–7.24 (m, 1H, ArH), 7.20 (s, 1H, ArH), 7.08 (dt, J = 7.9, 1.0 Hz, 1H, ArH), 3.96 (s, 3H, OCH3), 2.21 (s, 3H, ArCH3).
3.1.11. The Synthesis of Compounds 19b–k
Compounds 19b–k were prepared analogously to compound 16a.
6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19b)
White solid powder. Yield, 72.1%. m.p. 78.5–81.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.32 (s, 1H, CHO), 8.22 (d, J = 7.7 Hz, 1H, ArH), 7.59–7.54 (m, 1H, ArH), 7.51 (d, J = 7.8 Hz, 1H, ArH), 7.46 (d, J = 7.3 Hz, 1H, ArH), 7.42 (d, J = 5.4 Hz, 1H, ArH), 7.40–7.31 (m, 4H, ArH), 4.07 (s, 3H, OCH3), 2.18 (s, 3H, ArCH3).
6-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19c)
Colorless oil. Yield, 74.5%. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.19 (d, J = 7.7 Hz, 1H, ArH), 7.50 (dd, J = 7.8, 1.5 Hz, 1H, ArH), 7.42–7.36 (m, 2H, ArH), 7.35–7.24 (m, 3H, ArH), 7.20 (dd, J = 7.5, 1.5 Hz, 1H, ArH), 7.14 (dd, J = 6.9, 2.0 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.05 (s, 3H, ArCH3), 2.04 (s, 3H, ArCH3).
6-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19d)
Colorless oil, Yield, 61.7%. 1H NMR (300 MHz, DMSO-d6) δ 10.32 (s, 1H, CHO), 8.22 (d, J = 7.7 Hz, 1H, ArH), 7.65–7.61 (m, 1H, ArH), 7.57 (dd, J = 7.8, 1.7 Hz, 1H, ArH), 7.54–7.46 (m, 2H, ArH), 7.46–7.37 (m, 3H, ArH), 7.28 (dd, J = 7.6, 1.5 Hz, 1H, ArH), 4.07 (s, 3H, OCH3), 2.12 (s, 3H, ArCH3).
2-Methoxy-6-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)nicotinaldehyde (19e)
White solid. Yield, 48.4%. 1H NMR (300 MHz, DMSO-d6) δ 10.31 (s, 1H, CHO), 8.20 (d, J = 7.7 Hz, 1H, ArH), 7.89 (d, J = 6.6 Hz, 1H, ArH), 7.77 (t, J = 6.7 Hz, 1H, ArH), 7.66 (t, J = 7.7 Hz, 1H, ArH), 7.56 (dd, J = 7.8, 1.5 Hz, 1H, ArH), 7.52–7.43 (m, 3H, ArH), 7.25 (d, J = 7.8 Hz, 1H, ArH), 4.05 (s, 3H, OCH3), 2.03 (s, 3H, ArCH3).
2-Methoxy-6-(2-methyl-[1,1′-biphenyl]-3-yl)nicotinaldehyde (19f)
White solid. Yield, 56.8%. 1H NMR (300 MHz, DMSO-d6) δ 10.42 (s, 1H, CHO), 7.79 (d, J = 7.8 Hz, 1H, ArH), 7.54–7.45 (m, 1H, ArH), 7.44–7.25 (m, 6H, ArH), 7.23 (s, 1H, ArH), 7.10 (d, J = 7.9 Hz, 1H, ArH), 3.99 (s, 3H, OCH3), 2.04 (s, 3H, ArCH3).
6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19g)
White solid. Yield, 68.8%. m.p. 131.0–132.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.31 (s, 1H, CHO), 8.22 (dd, J = 8.2, 5.4 Hz, 1H, ArH), 7.52 (dq, J = 8.9, 3.7, 3.1 Hz, 2H, ArH), 7.46–7.31 (m, 3H, ArH), 7.26 (t, J = 7.5 Hz, 3H, ArH), 4.06 (s, 3H, OCH3), 2.24 (s, 3H, ArCH3).
6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19h)
White solid. Yield, 72.3%. m.p. 46.0–48.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.18 (s, 1H, ArH), 8.12 (d, J = 7.9 Hz, 1H, ArH), 7.64 (dd, J = 5.7, 3.3 Hz, 1H, ArH), 7.42–7.10 (m, 7H, ArH), 4.01 (s, 3H, OCH3), 2.33 (s, 3H, ArCH3).
6-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19i)
White solid. Yield, 20.8%. m.p. 119.5–222.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.6 Hz, 1H, ArH), 7.54 (dd, J = 7.8, 1.6 Hz, 1H, ArH), 7.48–7.41(m, 2H, ArH), 7.40–7.35 (m, 2H, ArH), 7.33 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.22 (td, J = 8.5, 2.3 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.14 (s, 3H, ArCH3).
6-(4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19j)
White solid. Yield, 29.8%. m.p. 132.0–134.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.8 Hz, 1H, ArH), 7.61–7.53 (m, 2H, ArH), 7.47–7.40 (m, 3H, ArH), 7.37 (d, J = 7.6 Hz, 1H, ArH), 7.34 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.14 (s, 3H, ArCH3).
6-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-2-methoxy nicotinaldehyde (19k)
White solid. Yield, 47.5%. m.p. 62.0–64.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.6 Hz, 1H, ArH), 7.84 (dd, J = 9.9, 1.9 Hz, 1H, ArH), 7.74–7.69 (m, 1H, ArH), 7.65 (t, J = 7.6 Hz, 1H, ArH), 7.58 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.46 (t, J = 7.5 Hz, 1H, ArH), 7.41-7.32 (m, 2H, ArH), 4.04 (s, 3H, OCH3), 2.15 (s, 3H, ArCH3).
3.1.12. The Synthesis of Compounds A1 and A2
Compounds A1 and A2 were prepared analogous to compounds 16a–j.
1-(3-((2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)oxy)propyl)piperidin-4-ol hydrochloride (A1).
White solid. Yield, 82.0%. m.p. 118.0–201.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.27 (dd, J = 6.6, 1.5 Hz, 2H, ArH), 7.26–7.22 (m, 1H, ArH), 7.19 (t, J = 7.8 Hz, 1H, ArH), 7.09–7.00 (m, 1H, ArH), 6.94 (d, J = 8.1 Hz, 1H, ArH), 6.66 (d, J = 7.4 Hz, 1H, ArH), 4.60 (s, 1H, OH), 4.09–3.99 (m, 3H, OCH3), 3.54–3.42 (m, 2H, NCH2), 2.87–2.72 (m, 2H, NCH2-piperidinol), 2.14 (m, 2H, CH2CH2CH2), 1.99 (s, 3H, ArCH3), 1.94 (s, 2H, NCH2-piperidinol), 1.83 (s, 3H, ArCH3), 1.73 (s, 2H, CH2-piperidinol), 1.44 (s, 2H, CH2-piperidinol). 13C NMR (75 MHz, DMSO-d6) δ 156.82, 142.68, 141.40, 135.61, 130.22, 129.45, 127.72, 126.74, 126.13, 124.03, 121.91, 110.52, 65.62, 60.03, 50.72, 47.37, 31.89, 29.75, 19.94(2C), 13.21(2C). ESI-HRMS: m/z [M+H]+calculated for C22H31NO2, 340.2277; Found: 340.2268. LCMS: tR 9.97 min, purity, 95.1%.
1′-((2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)methyl)-[1,4′-bipiperidin]-4-ol (A2).
Light yellow powder. Yield, 89.3%. m.p. 110.0–113.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.52–7.41 (m, 1H, ArH), 7.36–7.25 (m, 4H, ArH), 7.23 (d, J = 7.5 Hz, 1H, ArH), 7.11 (dd, J = 7.5, 1.6 Hz, 1H, ArH), 4.54 (s, 1H, CHOH), 3.46 (s, 3H, ArCH2N/ CHOH), 2.89 (d, J = 11.0 Hz, 2H, NCH2-piperidine), 2.83–2.69 (m, 2H, NCH2-piperidine), 2.36–2.15 (m, 3H, CHN, NCH2-piperidine), 2.12 (s, 3H, CH3), 1.99 (t, J = 10.7 Hz, 2H, NCH2-piperidine), 1.82–1.61 (m, 4H, CH2-piperidine), 1.57–1.18 (m, 4H, CH2-piperidine).13C NMR (75 MHz, DMSO-d6) δ 161.07, 157.84, 137.64, 136.18, 135.79, 132.11, 129.81, 129.14, 125.51, 124.96, 116.01, 115.72, 67.13, 61.95, 61.08, 53.49(2C), 47.13(2C), 35.25(2C), 28.32(2C), 16.09. ESI-HRMS: m/z [M+H]+calculated for C24H32FN2O, 383.2499 ; Found: 383.2495. LCMS: tR 10.69 min, purity, 96.5%.
3.1.13. The Synthesis of Compounds A3–A8
Compounds A3–A8 were synthesized by the following procedure. A mixture of 19a or 19b (0.92 mmol), amide (1.11 mmol), and HAc (0.11 mL, 1.84 mmol) in DCM/MeOH (5 mL/1 mL) was stirred for 4 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography to afford the product.
1-((2″-Fluoro-3-methoxy-2′-methyl-[1,1′:3′,1″-terphenyl]-4-yl)methyl)piperidin-4-ol (A3).
White solid. Yield, 66.4%. m.p. 61.5–63.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.52–7.43 (m, 1H, ArH), 7.41–7.33 (m, 3H, ArH), 7.32–7.26 (m, 3H, ArH), 7.21 (dd, J = 7.3, 1.9 Hz, 1H, ArH), 6.92 (d, J = 8.4 Hz, 2H, ArH), 4.57 (s, 1H, OH), 3.81 (s, 3H, OCH3), 3.47 (br, 3H, ArCH2/piperidine-CH), 2.75 (b r, 2H, piperidine-CH2), 2.11 (b r, 2H, piperidine-CH2), 2.01 (s, 3H, ArCH3), 1.79–1.63 (m, 2H, piperidine-CH2), 1.50–1.36 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 161.08, 157.85(F-C), 157.45(F-C), 142.66, 141.71, 136.56, 132.13(d), 132.08, 130.12(d), 129.93, 129.51, 129.29, 126.04, 125.25, 125.02(d), 121.37, 116.12, 115.83, 112.12, 66.71, 55.85 (2C), 55.73, 51.59, 34.82, 18.35 (2C). ESI-HRMS: m/z [M+H]+ calculated for C26H28FNO2, 406.2104; Found: 406.2177. LCMS: tR 10.48 min, purity, 98.4%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidin-4-ol (A4).
White solid powder. Yield, 56.4%. m.p. 84.0–86.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.76 (d, J = 7.5 Hz, 1H, ArH), 7.52 (dd, J = 7.6, 1.7 Hz, 1H, ArH), 7.42–7.28 (m, 5H, ArH), 7.24–7.16 (m, 1H, ArH), 7.09 (d, J = 7.3 Hz, 1H, ArH), 4.02 (s, 3H, OCH3), 3.77 (dt, J = 8.8, 4.5 Hz, 1H, CHOH), 3.61 (s, 2H, ArCH2N), 2.90 (dt, J = 10.8, 4.7 Hz, 2H, CH2-piperidine), 2.37–2.28 (m, 2H, CH2-piperidine), 2.27 (s, 3H, ArCH3), 2.05–1.96 (m, 2H, CH2-piperidine), 1.84 (br, 1H, OH) 1.78–1.68 (m, 2H, CH2-piperidine), 1.47–1.36 (m, 2H, CH2-piperidine). 13C NMR (75 MHz, Chloroform-d) δ 161.16, 155.99, 140.94, 138.50, 136.90, 134.92, 131.75, 130.08, 129.69, 129.58, 129.17, 129.06, 125.43, 124.00, 118.64, 117.24, 115.70, 115.40, 67.97, 55.53, 53.49 (2C), 51.19, 34.51 (2C),18.05. ESI-HRMS: m/z [M+H]+ calculated for C26H29FNO2, 407.2182; Found: 407.2137. LCMS: tR 11.76 min, purity, 96.1%.
4-(((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)cyclohexan-1-ol (A5).
White solid. Yield, 51.3%. m.p. 80.0–82.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 7.5 Hz, 1H, ArH), 7.54–7.44 (m, 2H, ArH), 7.43–7.33 (m, 4H, ArH), 7.32–7.25 (m, 1H, ArH), 7.14 (d, J = 7.3 Hz, 1H, ArH), 4.52 (d, J = 4.5 Hz, 1H, OH), 3.90 (s, 3H, OCH3), 3.72 (s, 2H, CH2NH), 3.45–3.40 (m,1H, CHOH), 2.43–2.32 (m, 1H, NHCH), 2.13 (s, 3H, ArCH3), 1.94–1.78 (m, 4H, CH2-cyclohexane), 1.21–1.01 (m, 4H, CH2-cyclohexane). 13C NMR (75 MHz, DMSO-d6) δ 160.55, 157.88(F-C), 155.05(F-C), 141.03, 138.01, 136.75, 134.40, 132.12,130.12, 130.23, 129.95, 129.3(d), 126.05, 125.04, 122.11, 117.48, 116.11, 115.82, 69.29, 55.91, 53.59, 44.53, 34.32(2C), 31.26(2C), 18.17. ESI-HRMS: m/z [M+H]+ calculated for C26H30FN2O2, 421.2291; Found: 421.2284. LCMS: tR 9.46 min, purity, 95.1%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-3-carboxamide (A6).
White solid powder. Yield, 41.0%. m.p. 88.0–91.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 7.5 Hz, 1H, ArH), 7.55–7.44 (m, 2H, ArH), 7.42–7.33 (m, 4H, ArH, 1/2CONH2), 7.32–7.26 (m, 2H, ArH), 7.16 (d, J = 7.5 Hz, 1H), 6.82 (br, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.50 (s, 2H, ArCH2), 2.89–2.71 (m, 2H, piperidine-CH2), 2.45–2.33 (m, 1H, piperidine-CHCO), 2.23–2.00 (m, 5H, ArCH3, piperidine-CH2), 1.84–1.62 (m, 2H, piperidine-CH2), 1.56–1.36 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 176.04, 161.00, 157.8(C-F), 155.51(C-F), 140.95, 139.15, 136.76, 134.41, 132.14, 130.28, 129.97, 129.39, 129.16, 126.07, 125.09, 119.21, 117.53, 115.9(d), 56.57, 56.05, 53.86, 53.65, 42.77, 27.54, 24.76, 18.19. ESI-HRMS: m/z [M+H]+ calculated for C26H29FN3O2, 434.2244; Found: 434.2238. LCMS: tR 12.04 min, purity, 95.6%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-4-carboxamide (A7).
White solid powder. Yield, 44.7%. m.p. 194.0–196.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.53–7.45 (m, 2H, ArH), 7.43–7.34 (m, 3H, ArH), 7.33–7.26 (m, 2H, ArH), 7.25 (s, 1H, 1/2CONH2), 7.17 (d, J = 7.5 Hz, 1H, ArH), 6.75 (s, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.54 (s, 2H, ArCH2), 2.91 (m, 2H, piperidine-CH2), 2.18–2.12 (s, 3H, ArCH3), 2.11–1.99 (m, 2H, piperidine-CH2), 1.78–1.54 (m, 4H, piperidine-CH2). 13C NMR (75 MHz, Chloroform-d) δ 177.63, 161.37, 161.16,158.16(C-F) 156.27(C-F), 140.83, 138.79, 136.90, 134.90, 131.76, 130.12, 129.74, 129.66, 129.52, 129.17(d), 125.43, 124.01, 117.29, 115.53(d), 55.64, 53.51(2C), 53.03, 42.38, 28.70(2C), 18.04. ESI-HRMS: m/z [M+H]+ calculated for C26H29FN3O2, 434.2244; Found: 434.2243. LCMS: tR 10.05 min, purity, 96.3%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-4-carboxylic acid (A8).
White solid powder. Yield, 19.9%. m.p. 218.0–220.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 12.27 (Br, 1H, COOH), 7.78 (d, J = 7.3 Hz, 1H, ArH), 7.53–7.44 (m, 2H, ArH), 7.43–7.32 (m, 4H, ArH), 7.32–7.24 (m, 1H, ArH), 7.15 (d, J = 7.3 Hz, 1H, ArH), 3.90 (s, 3H, OCH3), 3.49 (s, 2H, ArCH2N), 2.83 (d, J = 12.0 Hz, 2H, piperidine-CH2), 2.32–2.16 (m, 1H, CHCOOH), 2.17–2.03 (m, 5H, ArCH3/ piperidine-CH2), 1.83 (d, J = 13.0 Hz, 2H, piperidine-CH2), 1.68–1.54 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 176.78, 160.90, 157.85(C-F), 155.41(C-F), 140.94, 138.98, 136.75, 134.40, 132.16, 130.28, 130.16, 129.96, 129.35, 129.14, 126.08, 125.07(d), 119.20, 117.59, 115.97(d), 55.66, 53.65(2C), 53.16, 40.76, 28.62(2C),18.19. ESI-HRMS: m/z [M+H]+ calculated for C26H28FN2O3, 435.2084; Found: 435.2806. LCMS: tR 6.52 min, purity, 99.1%.
3.1.14. The Synthesis of Compounds A9–A18
Compounds A9–A18 were synthesized by the following procedure. A mixture of aldehyde (0.92 mmol), β-alanine amide hydrochloride (1.11 mmol), and Et3N (1.84 mmol) in DCM/MeOH (5 mL/1 mL) was stirred for 12 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography to afford the product.
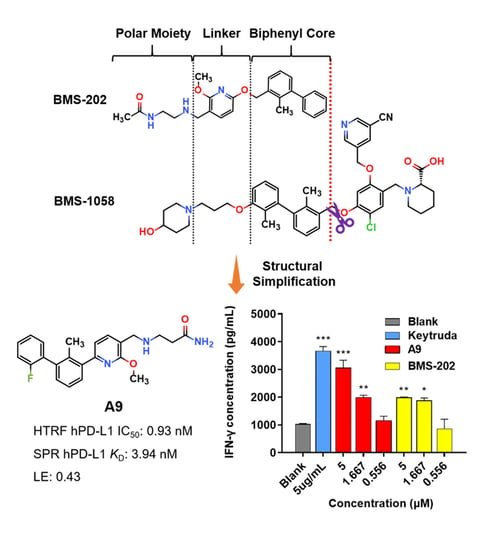
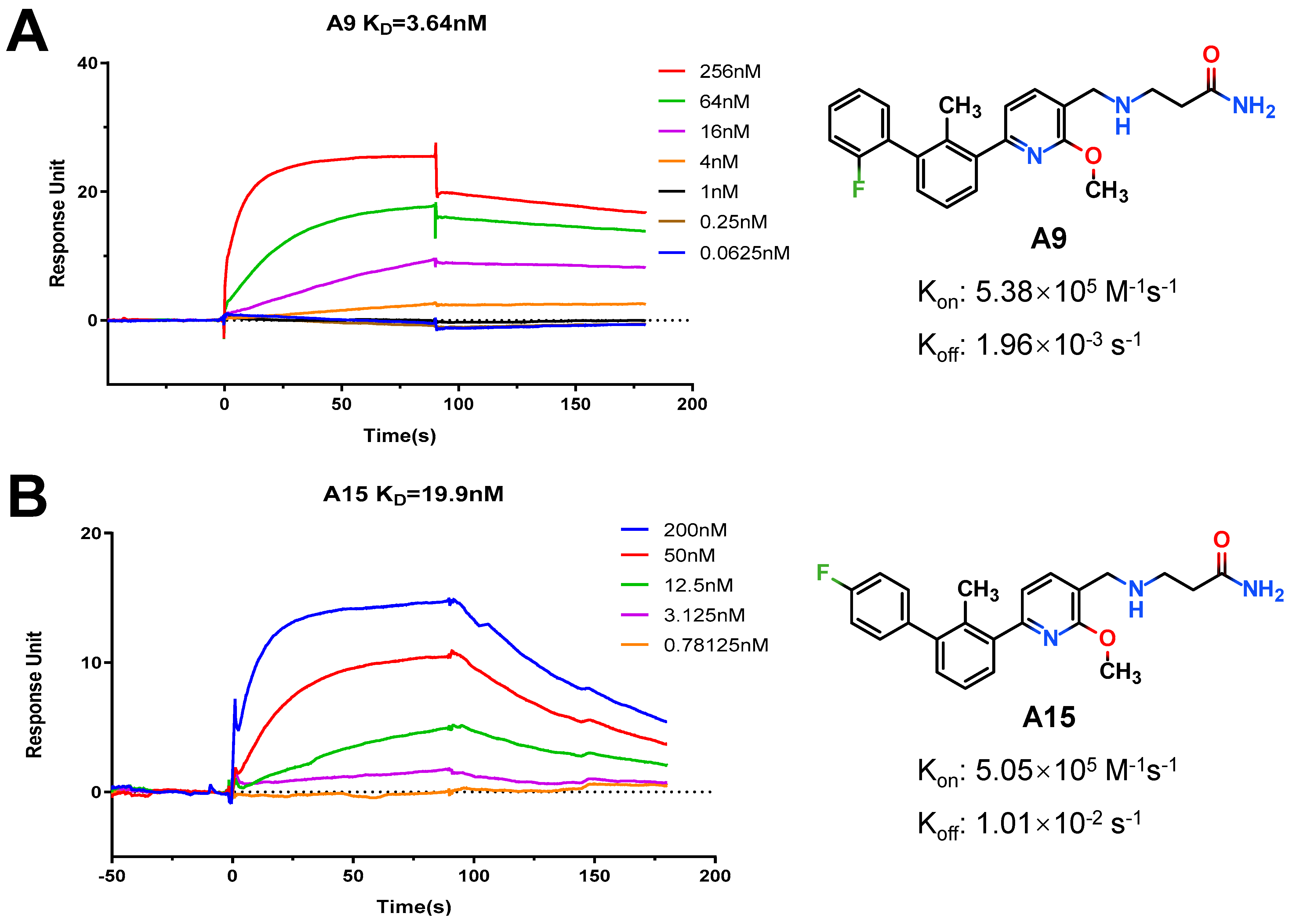
3-(((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A9)
White solid powder. Yield, 54.1%. m.p. 218.0–222.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 7.5 Hz, 1H, ArH), 7.52–7.45 (m, 2H, ArH), 7.45–7.33 (m, 5H, ArH, 1/2CONH2), 7.33–7.26 (m, 1H, ArH), 7.15 (d, J = 7.3 Hz, 1H, ArH), 6.82 (s, 1H, 1/2CONH2), 3.92 (s, 3H, OCH3), 3.72 (s, 2H, ArCH2), 2.77 (t, J = 6.8 Hz, 2H, NHCH2), 2.29 (t, J = 6.8 Hz, 2H, CH2CO), 2.14 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 172.08, 161.02, 158.13(F-C), 157.85(F-C), 141.56, 140.48, 136.87, 134.42, 132.12, 130.69,130.34, 130.24, 129.97, 129.01(d), 126.21, 125.11, 117.70, 115.85, 113.26, 54.12, 44.95, 43.43, 30.87, 18.16. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1929. LCMS: tR 8.19 min, purity, 96.7%.
3-(((6-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino) propenamide (A10).
White solid powder. Yield, 55.5%. m.p. 70.0–73.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 7.5 Hz, 1H, ArH), 7.42 (dd, J = 7.6, 1.6 Hz, 2H, ArH), 7.36–7.25 (m, 4H, ArH, 1/2CONH2), 7.17–7.10 (m, 3H, ArH), 6.82 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.88 (s, 1H, NH), 3.70 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.05 (s, 3H, ArCH3), 2.00 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 174.03, 160.68, 155.56, 142.51, 141.86, 140.88, 138.20, 135.65, 133.61, 130.29, 129.58, 129.37, 129.18, 127.78, 126.25, 125.92, 120.84, 117.49, 53.58, 47.04, 45.50, 35.68, 20.10, 18.03. ESI-HRMS: m/z [M+H]+ calculated for C24H28N3O2, 390.2182; Found: 390.2183. LCMS: tR 9.04 min, purity, 95.1%.
3-(((6-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A11).
White solid powder. Yield, 52.9%. m.p. 49.0–51.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 7.5 Hz, 1H, ArH), 7.58–7.54 (m, 1H, ArH), 7.50 (s, 1H, 1/2CONH2), 7.46–7.40 (m, 3H, ArH), 7.40–7.31 (m, 2H, ArH), 7.24–7.02 (m, 2H, ArH), 6.89 (s, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.74 (s, 2H, ArCH2), 2.80 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.33 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.07 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 174.15, 160.70, 155.26, 140.83, 140.69, 140.38, 138.15, 134.20, 133.99, 132.82, 131.76, 129.87, 129.71, 129.59, 127.74, 125.94, 121.13, 117.52, 53.57, 47.11, 45.57, 35.80, 17.99. ESI-HRMS: m/z [M+H]+ calculated for C23H25ClN3O2, 410.1635; Found: 410.1633. LCMS: tR 9.02 min, purity, 98.7%.
3-(((2-Methoxy-6-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)pyridin-3-yl)methyl)amino)propenamide (A12).
White solid powder. Yield, 57.5%. m.p. 50.5–53.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.85 (t, J = 7.5 Hz, 2H, ArH), 7.74 (t, J = 7.2 Hz, 1H, ArH), 7.63 (t, J = 7.7 Hz, 1H, ArH), 7.50 (s, 1H, 1/2CONH2), 7.46 (d, J = 7.7Hz, 1H, ArH), 7.40 (d, J = 7.3 Hz, 1H, ArH), 7.33 (t, J = 7.7 Hz, 1H, ArH), 7.15 (dd, J = 7.6, 5.2 Hz, 2H, ArH), 6.90 (s, 1H, 1/2CONH2), 3.90 (s, 3H, OCH3), 3.82 (s, 2H, ArCH2), 2.85 (t, J = 6.9 Hz, 2H, NHCH2CH2), 2.36 (t, J = 6.8 Hz, 2H, NHCH2CH2), 1.98 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 173.58, 160.77, 155.86, 140.71, 140.43, 140.03, 138.98, 133.93, 132.84, 132.31, 129.91, 129.65, 128.58, 128.16(β-CF3), 127.77(β-CF3), 127.39(β-CF3), 127.00 (β-CF3), 126.47(CF3), 126.39, 125.26, 122.75(CF3), 120.61(CF3), 119.24, 117.59, 53.68, 46.60, 45.06, 34.75, 18.53. ESI-HRMS: m/z [M+H]+ calculated for C24H25F3N3O2, 444.1899; Found: 444.1894. LCMS: tR 6.68 min, purity, 95.9%.
3-(((2-Methoxy-6-(2-methyl-[1,1′-biphenyl]-3-yl)pyridin-3-yl)methyl)amino)propenamide (A13).
White solid powder. Yield, 50.3%. m.p. 127.0–128.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.78 (d, J = 7.4 Hz, 1H, ArH), 7.50–7.43 (m, 2H, ArH), 7.43–7.31 (m, 6H, ArH, 1/2CONH2), 7.24 (dd, J = 7.4, 1.7 Hz, 1H, ArH), 7.12 (d, J = 7.4 Hz, 1H, ArH), 6.81 (s, 1H, 1/2CONH2), 3.90 (s, 3H, OCH3), 3.71 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.18 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.15, 161.09, 156.99, 143.21, 142.21, 140.93, 138.21, 133.65, 129.94, 129.42(2C), 128.91, 128.08(2C), 126.84, 125.44, 119.35, 117.15, 53.52, 48.30, 44.85, 35.16, 18.64. ESI-HRMS: m/z [M+H]+ calculated for C23H26N3O2, 376.2025; Found: 376.2022. LCMS: tR 8.51 min, purity, 95.6%.
3-(((6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A14)
White solid powder. Yield, 48.6%. m.p. 48.0–51.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.58–7.48 (m, 1H, ArH), 7.48–7.41 (m, 3H, ArH, 1/2CONH2), 7.33–7.18 (m, 4H, ArH), 7.14 (d, J = 7.3 Hz, 1H, ArH), 6.81 (s, 1H, 1/2CONH2), 3.92 (s, 3H, OCH3), 3.71 (s, 2H, ArCH2), 2.76 (t, J = 6.9 Hz, 2H, NHCH2), 2.28 (t, J = 6.8 Hz, 2H, CH2CONH2), 2.21 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 172.31, 164.00, 161.02, 160.77, 158.03, 144.37, 141.78, 141.33, 140.85, 133.33, 130.78, 130.19, 129.67, 126.24, 117.71, 116.33, 114.26, 113.69, 54.11, 45.81, 43.61, 31.17, 18.77. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1926. LCMS: tR 4.95 min, purity, 96.1%.
3-(((6-(4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A15)
White solid powder. Yield, 49.3%. m.p. 120.0–122.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 7.5 Hz, 1H, ArH), 7.47–7.42 (m, 4H, ArH, 1/2CONH2), 7.40–7.25 (m, 4H, ArH), 7.15 (d, J = 7.5 Hz, 1H, ArH), 6.85 (s, 1H, 1/2CONH2), 3.93 (s, 3H, OCH3), 3.76 (s, 2H, ArCH2), 2.80 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.31 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.19 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.34, 163.17(F-C), 161.08(F-C), 160.17, 156.78, 142.14, 141.07, 138.18(2C), 133.72, 130.96(2C), 129.93, 129.07, 125.50, 119.67, 117.11, 115.09, 114.85, 53.50, 48.27, 44.93, 35.32, 18.61. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1931. LCMS: tR 8.47 min, purity, 95.6%.
3-(((6-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A16)
White solid powder. Yield, 43.9%. m.p. 113.0–115.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 7.4 Hz, 1H, ArH), 7.49–7.43 (m, 1H, ArH), 7.43–7.39 (m, 2H, ArH), 7.39–7.33 (m, 2H, ArH), 7.27–7.16 (m, 2H, ArH, 1/2CONH2), 7.12 (d, J = 7.3 Hz, 1H, ArH), 6.80 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.64 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2CO), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2CO), 2.10 (s, 3H, ArCH3). 13C NMR (100 MHz, DMSO-d6) δ 174.01, 160.71, 155.38, 141.04, 138.31, 135.85, 134.58, 133.03(d, 2C), 130.38, 130.10, 126.07, 125.59(d), 120.88, 117.46, 112.21(d), 104.38(d, 2C), 53.58, 47.00, 45.49, 35.60, 18.09. ESI-HRMS: m/z [M+H]+ calculated for C23H24F2N3O2, 412.1837; Found: 412.1831. LCMS: tR 8.92 min, purity, 98.7%.
3-(((6-(4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A17)
White solid powder. Yield, 46.9%. m.p. 122.0–124.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.56 (d, J = 9.4 Hz, 1H, ArH), 7.50–7.37 (m, 5H, ArH,1/2CONH2), 7.25 (d, J = 7.5 Hz, 1H, ArH), 7.13 (d, J = 7.3 Hz, 1H, ArH), 6.83 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.73 (s, 2H, ArCH2), 2.77 (t, J = 5.9 Hz, 2H, NHCH2CH2), 2.28 (t, J = 6.2 Hz, 2H, NHCH2CH2), 2.11 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 173.95, 161.05, 160.70, 157.77(F-C), 155.20(F-C), 141.08, 138.23, 135.61, 134.43, 133.69, 133.55, 133.37(d), 130.27, 128.33(d), 126.19, 125.35(d), 121.18, 117.51, 116.66(d), 53.63, 47.04, 45.55, 35.76, 18.15. ESI-HRMS: m/z [M+H]+ calculated for C23H24ClFN3O2, 428.1541; Found: 428.1537. LCMS: tR 9.62 min, purity, 95.2%.
3-(((6-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A18)
White solid powder. Yield, 50.9%. m.p. 137.0–139.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.87–7.76 (m, 2H, ArH), 7.71 (dd, J = 8.5, 1.8 Hz, 1H, ArH), 7.64 (t, J = 7.6 Hz, 1H, ArH), 7.50 (dd, J = 7.7, 1.6 Hz, 1H, ArH), 7.45–7.37 (m, 2H, ArH, 1/2CONH2), 7.30 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.13 (d, J = 7.3 Hz, 1H, ArH), 6.80 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.69(s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.12 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.29, 161.15, 160.63(F-C),158.16(F-C), 156.22, 140.95, 138.15, 135.48, 134.68, 133.49(d), 132.47(d), 131.51(dd), 130.23, 127.44(CF3), 125.69, 124.72(CF3), 122.04(CF3), 121.03, 119.95,119.33(CF3), 117.17, 113.13(d), 53.52, 48.33, 44.90, 35.35, 18.00. ESI-HRMS: m/z [M+H]+ calculated for C24H24F4N3O2, 462.1805; Found: 462.1799. LCMS: tR 9.72 min, purity, 96.8%.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















