
New Disulfiram Derivatives as MAGL-Selective Inhibitors


Abstract
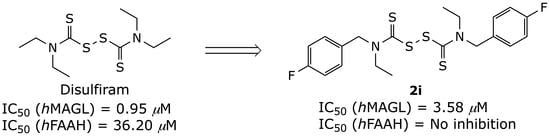
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Compounds 2g and 2l
3.2. MAGL Enzyme Inhibition Assay
3.3. FAAH Enzyme Inhibition Assay
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vandevoorde, S.; Saha, B.; Mahadevan, A.; Razdan, R.K.; Pertwee, R.G.; Martin, B.R.; Fowler, C.J. Influence of the degree of unsaturation of the acyl side chain upon the interaction of analogues of 1-arachidonoylglycerol with monoacylglycerol lipase and fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 2005, 337, 104–109. [Google Scholar] [CrossRef]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nat. Cell Biol. 1998, 394, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.; Valverde, O.; Berrendero, F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006, 29, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; García-Merino, A. Neuroprotective agents: Cannabinoids. Clin. Immunol. 2012, 142, 57–67. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nat. Cell Biol. 2001, 410, 822–825. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.-P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef]
- Berdan, C.A.; Erion, K.A.; Burritt, N.E.; Corkey, B.E.; Deeney, J.T. Inhibition of Monoacylglycerol Lipase Activity Decreases Glucose-Stimulated Insulin Secretion in INS-1 (832/13) Cells and Rat Islets. PLoS ONE 2016, 11, e0149008. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol Lipase Regulates a Fatty Acid Network that Promotes Cancer Pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, B.; Seviour, E.; Tao, K.-X.; Liu, X.-H.; Ling, Y.; Chen, J.-Y.; Wang, G.-B. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011, 307, 6–17. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef]
- Sticht, M.; Long, J.Z.; Rock, E.M.; Limebeer, C.L.; Mechoulam, R.; Cravatt, B.F.; Parker, L. Inhibition of monoacylglycerol lipase attenuates vomiting in Suncus murinus and 2-arachidonoyl glycerol attenuates nausea in rats. Br. J. Pharmacol. 2012, 165, 2425–2435. [Google Scholar] [CrossRef]
- LaBar, G.; Bauvois, C.; Muccioli, G.G.; Wouters, J.; Lambert, D.M. Disulfiram is an Inhibitor of Human Purified Monoacylglycerol Lipase, the Enzyme Regulating 2-Arachidonoylglycerol Signaling. ChemBioChem 2007, 8, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Kapanda, C.N.; Muccioli, G.G.; LaBar, G.; Poupaert, J.H.; Lambert, D.M. Bis(dialkylaminethiocarbonyl)disulfides as Potent and Selective Monoglyceride Lipase Inhibitors. J. Med. Chem. 2009, 52, 7310–7314. [Google Scholar] [CrossRef]
- Saario, S.M.; Salo-Ahen, O.M.H.; Nevalainen, T.; Poso, A.; Laitinen, J.T.; Järvinen, T.; Niemi, R. Characterization of the Sulfhydryl-Sensitive Site in the Enzyme Responsible for Hydrolysis of 2-Arachidonoyl-Glycerol in Rat Cerebellar Membranes. Chem. Biol. 2005, 12, 649–656. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nat. Cell Biol. 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef]
- Muccioli, G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; Lopez-Rodriguez, M.L.; Bajjalieh, S.; Stella, N. Identification of a Novel Endocannabinoid-Hydrolyzing Enzyme Expressed by Microglial Cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Omran, Z. Development of new disulfiram analogues as ALDH1a1-selective inhibitors. Bioorganic Med. Chem. Lett. 2021, 40, 127958. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}

| Compound. | R1 | R2 | IC50 (µM) ± SE (hMAGL) | IC50 (µM) ± SE (hFAAH) |
|---|---|---|---|---|
| 2a (Disulfiram) | Et | Et | 0.95 ± 0.26 | 36.20 ± 10.99 |
| 2b | -CH(CH3)2 | -CH(CH3)2 | 1.89 ± 0.37 | NI |
| 2c | -CH2CH2CH2CH3 | -CH2CH2CH2CH3 | 7.14 ± 0.21 | NI |
| 2d | -CHCH2(CH3)2 | -CHCH2(CH3)2 | 13.42 ± 4.79 | NI |
| 2e | Et | -CH2CH2OH | 0.72 ± 0.20 | 25.35 ± 3.26 |
| 2f | Et | -CH2CH2COOH | 0.87 ± 0.21 | 12.76 ± 1.57 |
| 2g | Et | -CH2CH2COOEt | 3.53 ± 1.32 | 10.05 ± 0.68 |
| 2h | Et |  | 5.53 ± 2.62 | NI |
| 2i | Et |  | 3.58 ± 1.54 | NI |
| 2j | Et |  | 3.71 ± 1.28 | 38.26 ± 7.96 |
| 2k | Et |  | 22.63 ± 16.83 | NI |
| 2l | Et |  | 5.03 ± 2.35 | NI |
| 2m |  |  | 3.58 ± 0.95 | NI |
| JZL-184 | – | 0.02 ± 0.00 | – | |
| JZL-195 | – | 0.04± 0.02 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omran, Z. New Disulfiram Derivatives as MAGL-Selective Inhibitors. Molecules 2021, 26, 3296. https://doi.org/10.3390/molecules26113296
Omran Z. New Disulfiram Derivatives as MAGL-Selective Inhibitors. Molecules. 2021; 26(11):3296. https://doi.org/10.3390/molecules26113296
Chicago/Turabian StyleOmran, Ziad. 2021. "New Disulfiram Derivatives as MAGL-Selective Inhibitors" Molecules 26, no. 11: 3296. https://doi.org/10.3390/molecules26113296
APA StyleOmran, Z. (2021). New Disulfiram Derivatives as MAGL-Selective Inhibitors. Molecules, 26(11), 3296. https://doi.org/10.3390/molecules26113296