
Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models

 ,
,  and
and
Abstract
1. Introduction
2. Results
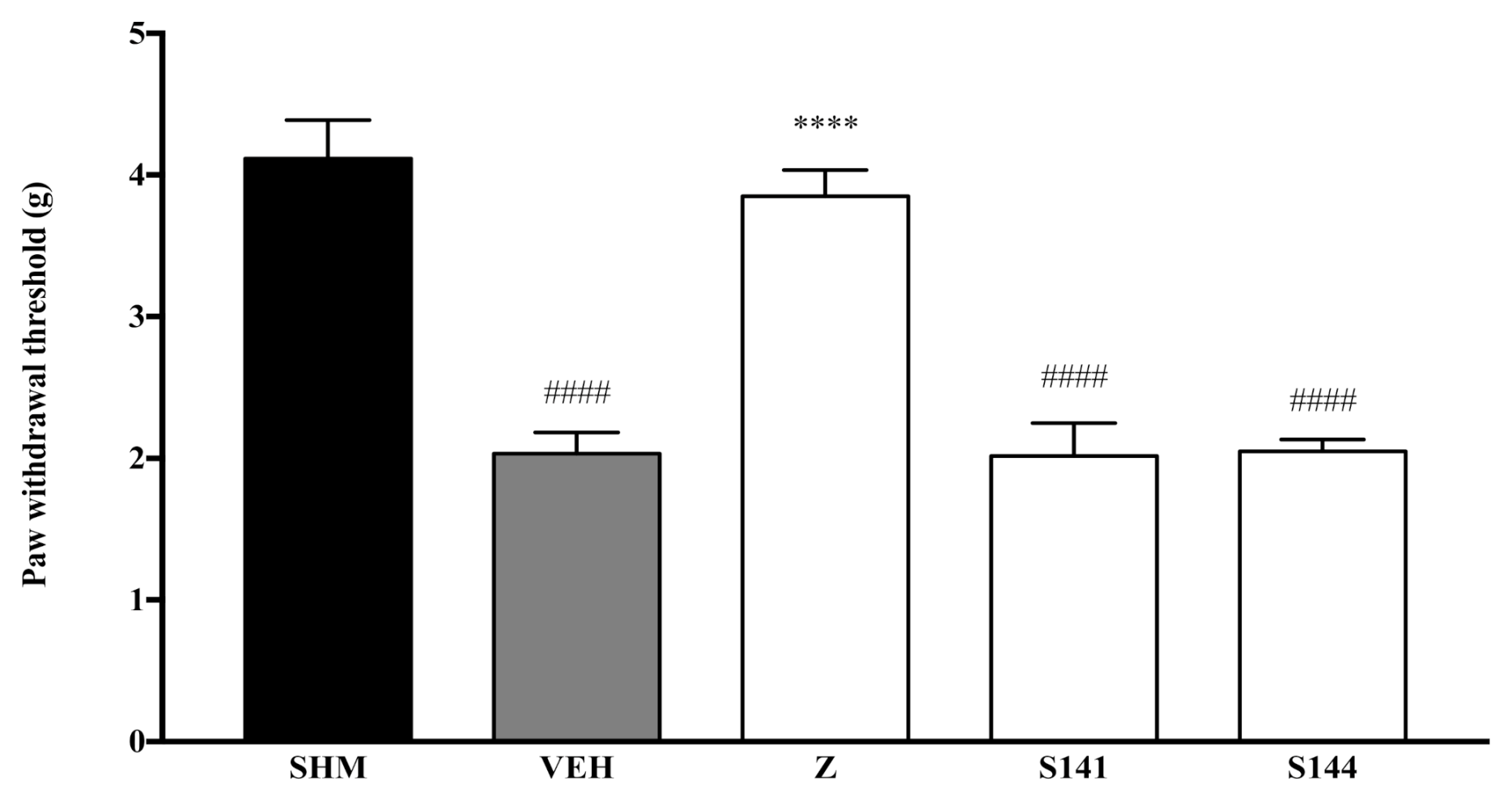
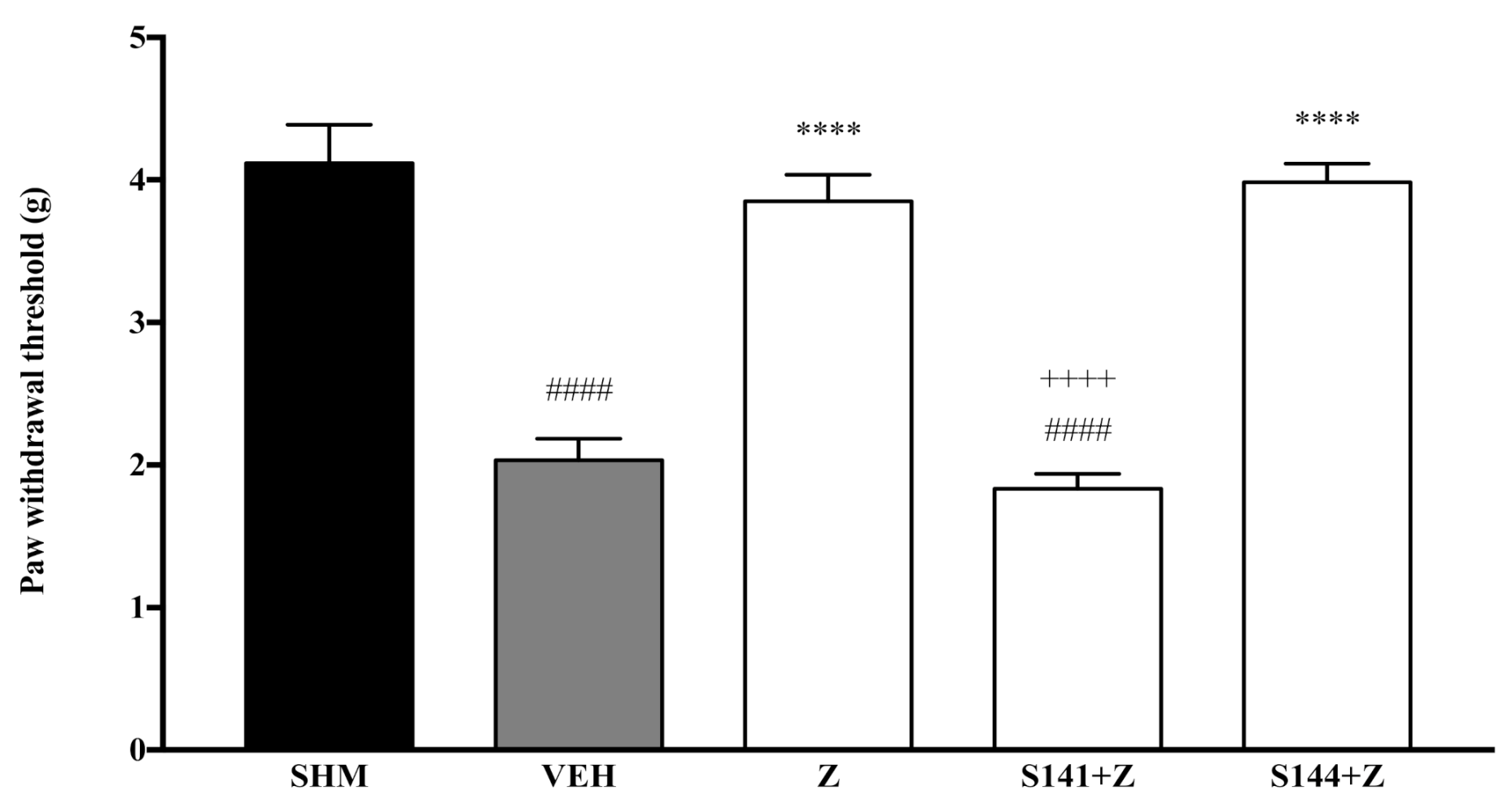
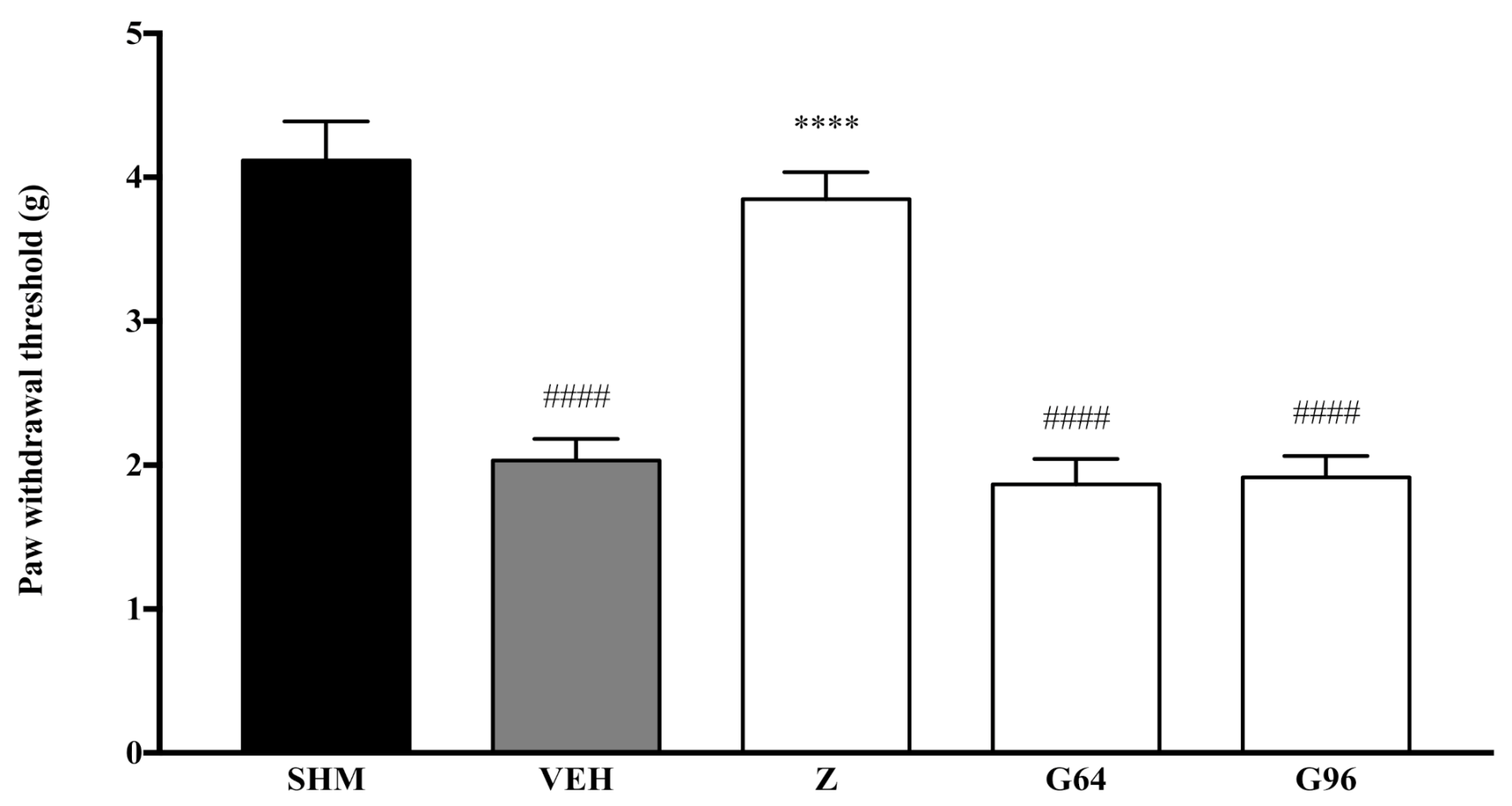
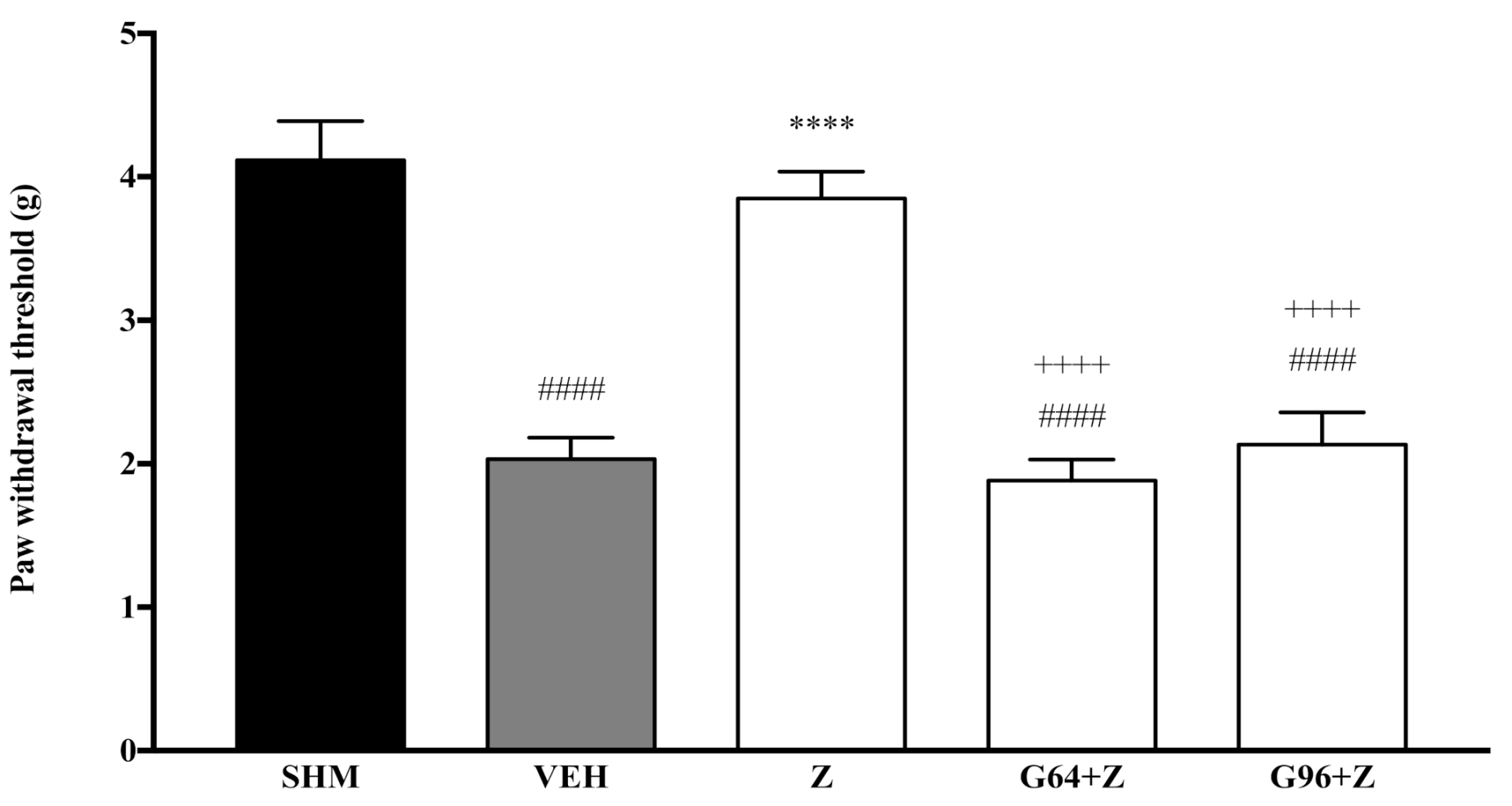
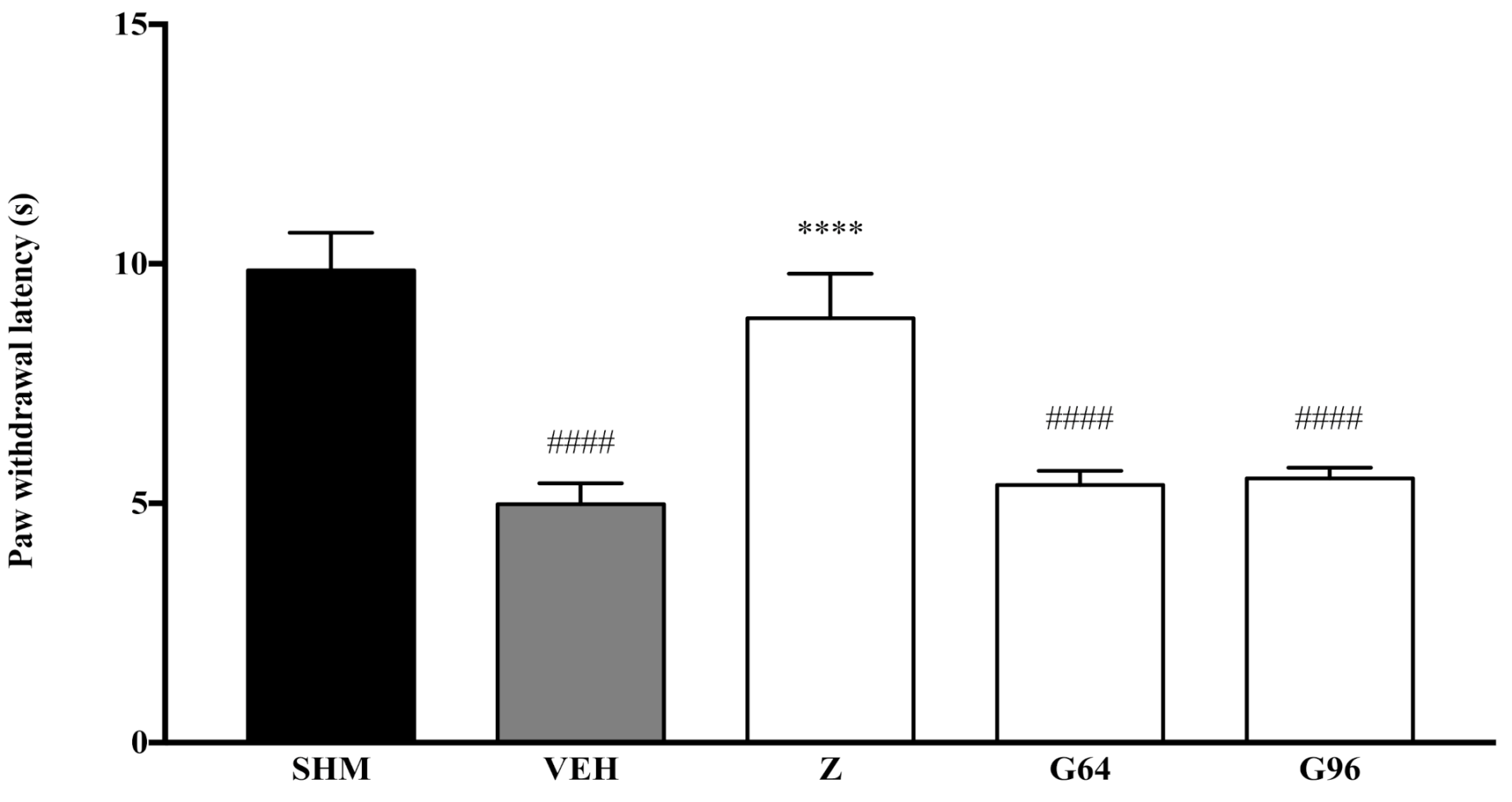
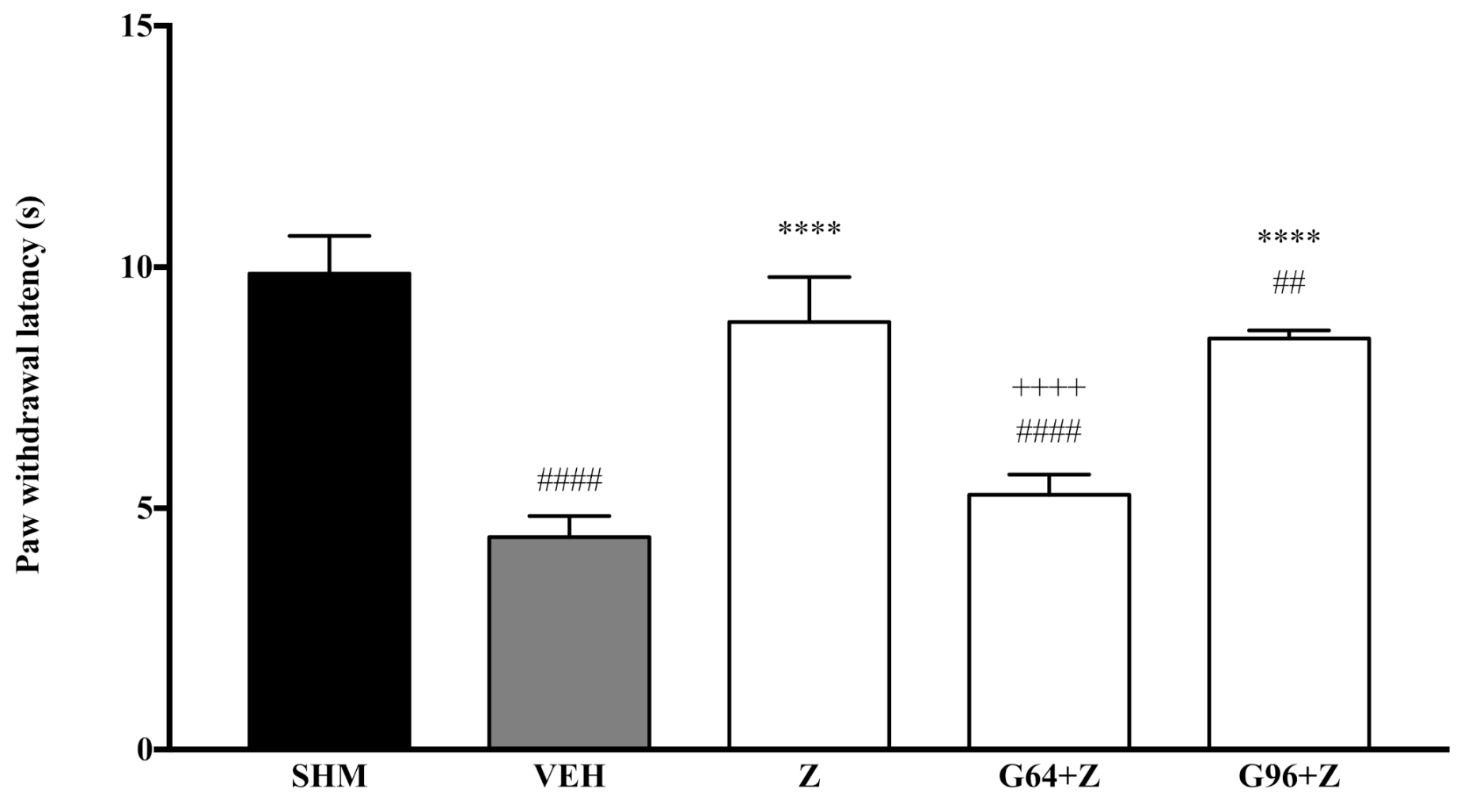
2.1. Involvement of Cannabinoid Receptors
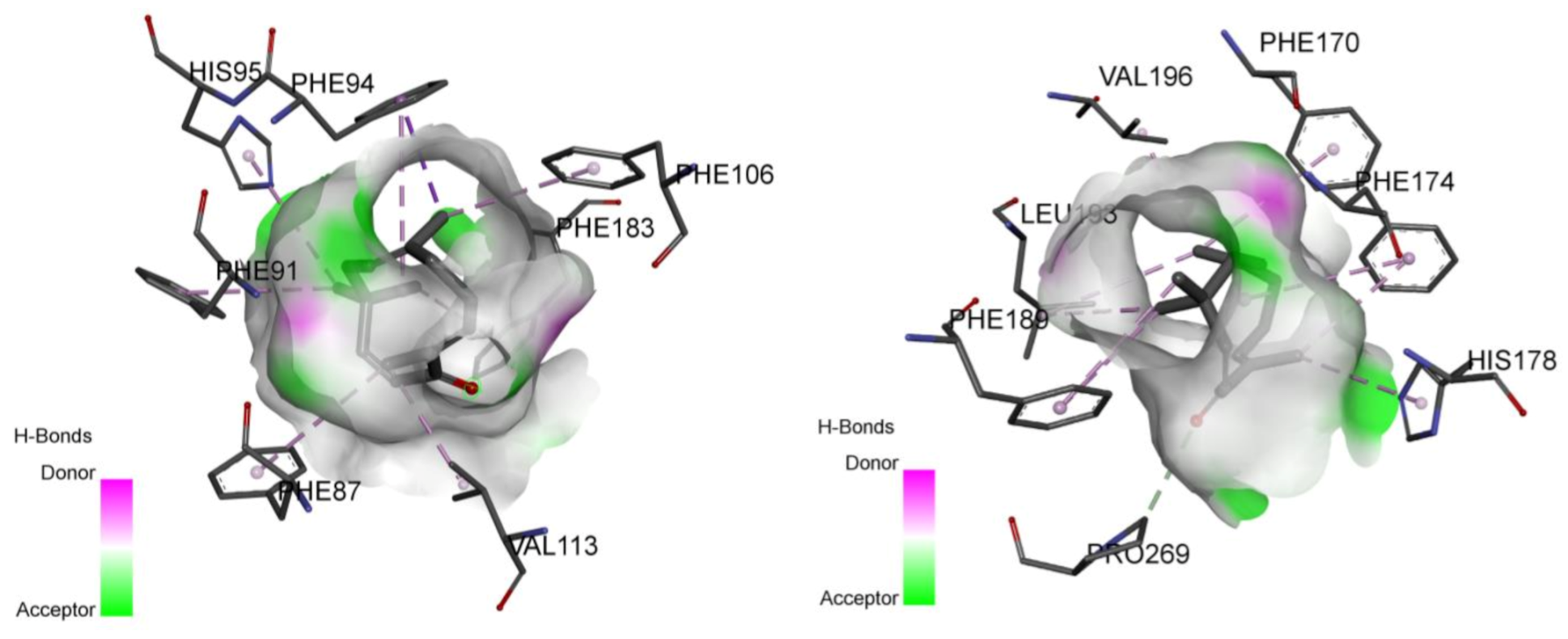
2.2. Involvement of Cannabinoid Receptors through Molecular Docking Studies
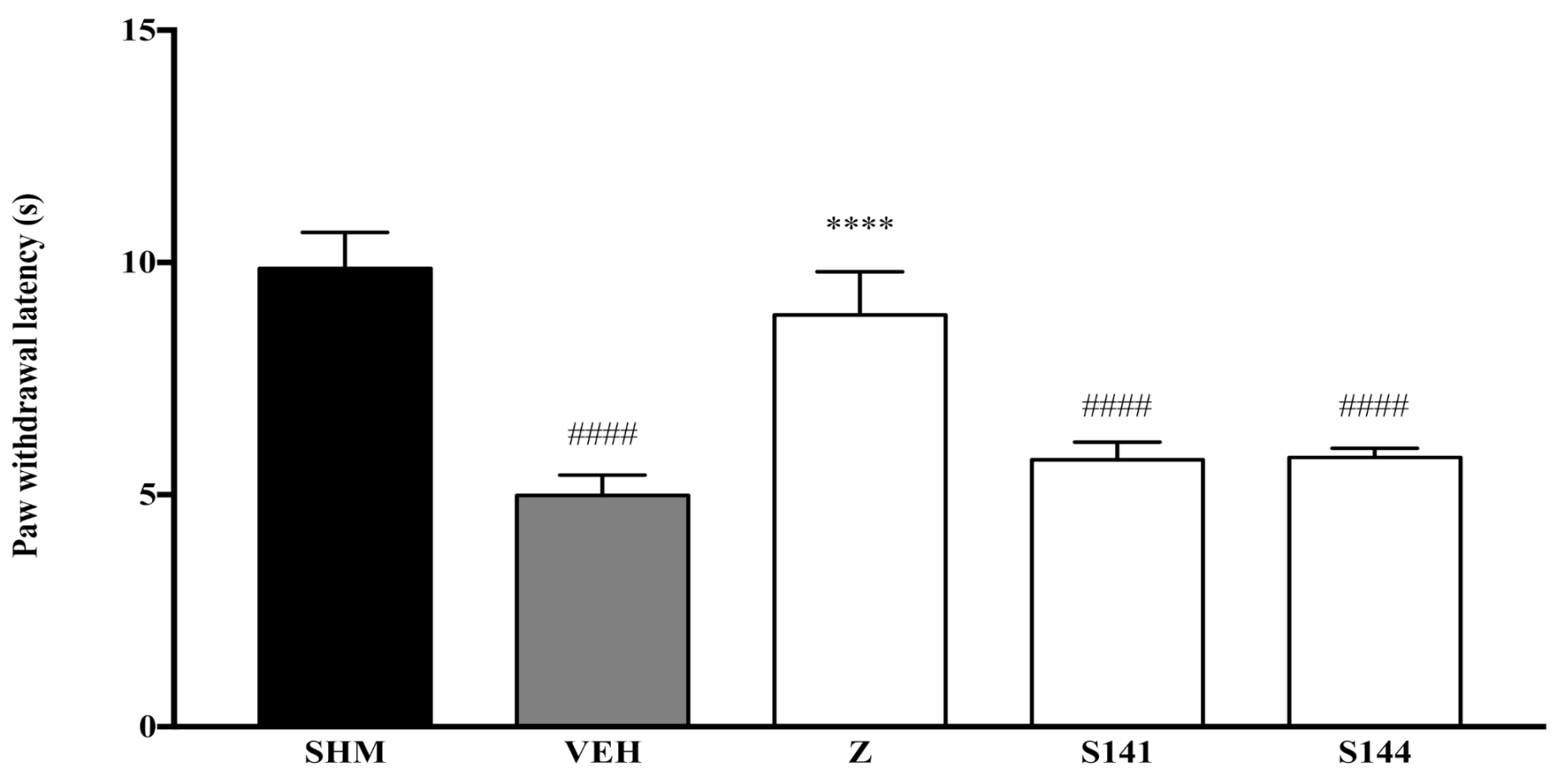
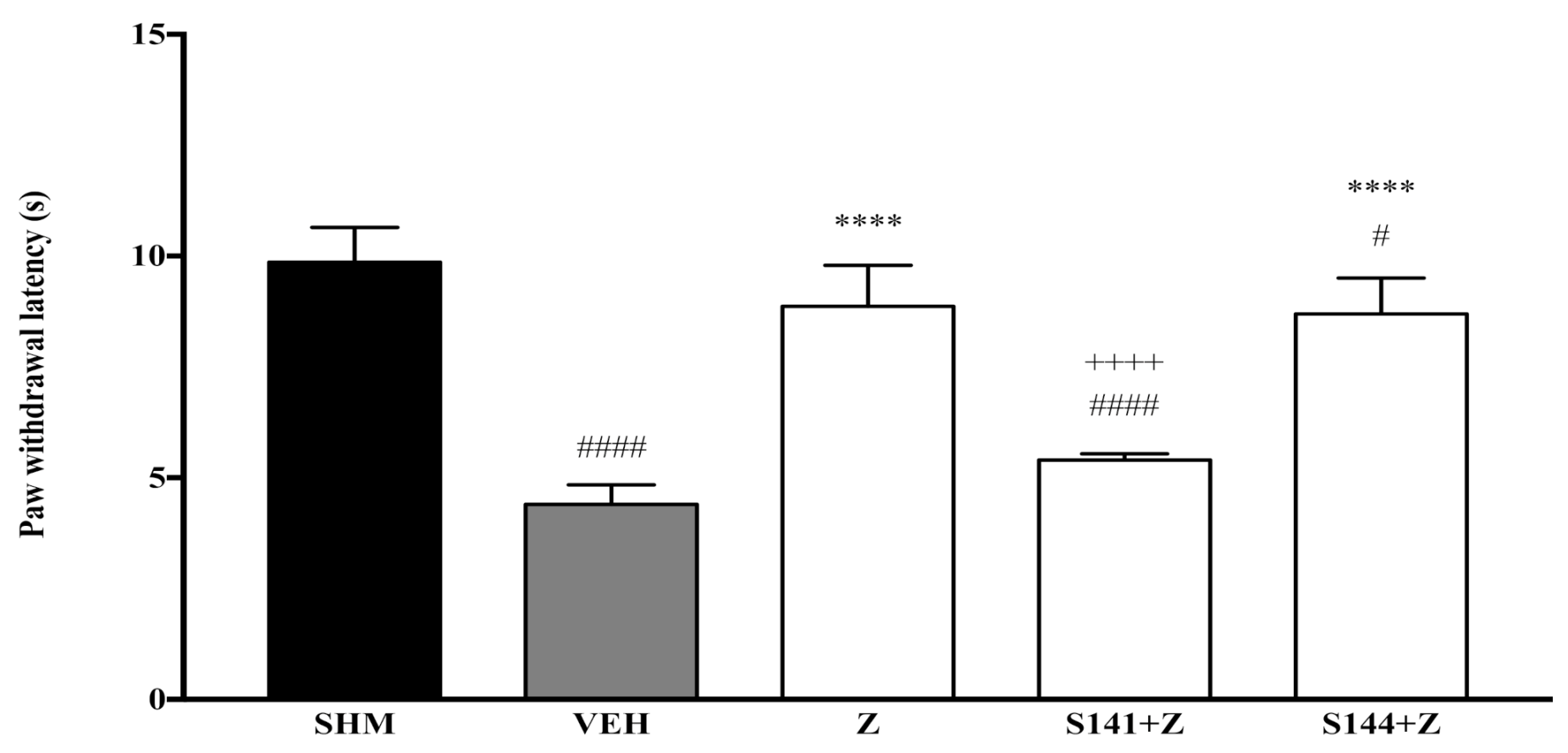
2.3. Involvement of Peroxisome Proliferator-Activated Receptors
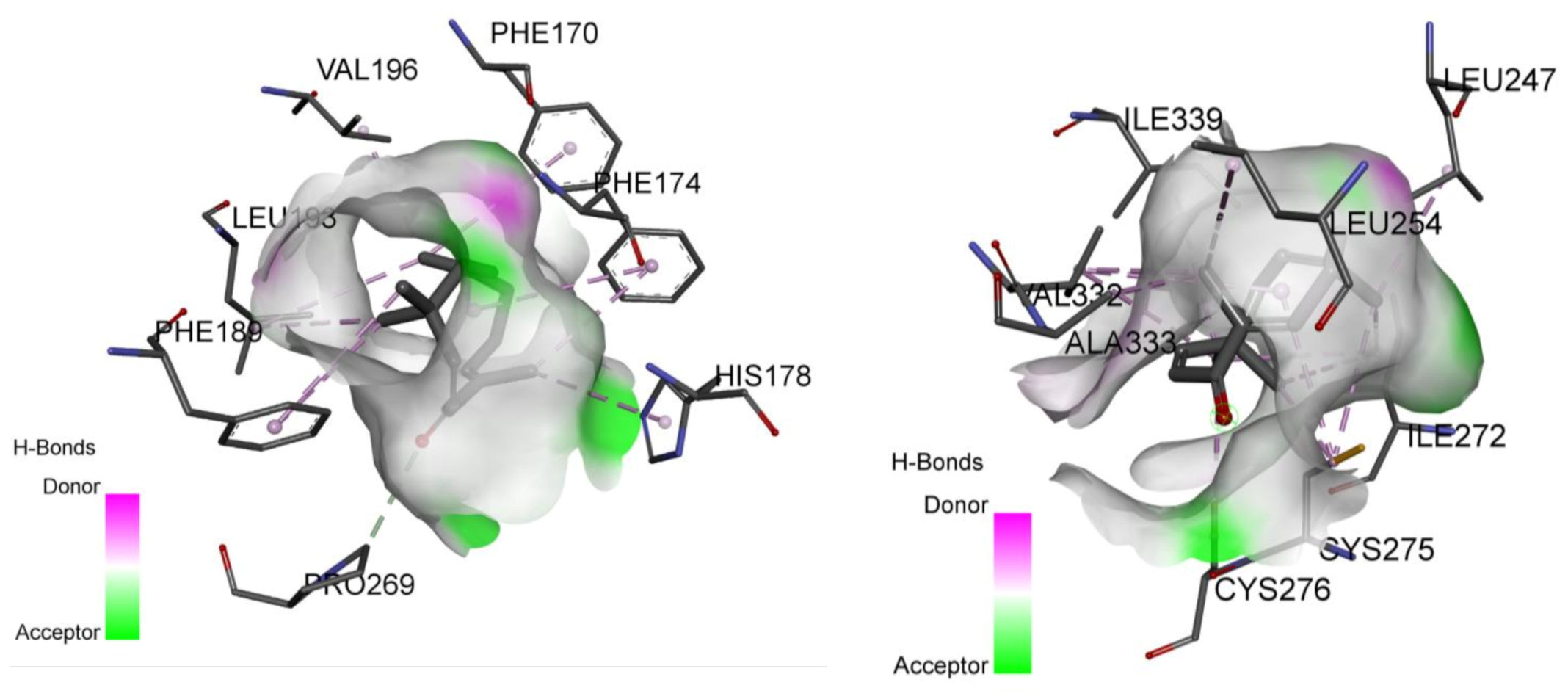
2.4. Involvement of Peroxisome Proliferator-Activated Receptors through Molecular Docking Studies
3. Discussion
4. Materials and Methods
4.1. Zerumbone
4.2. Animals
4.3. Neuropathic Pain Induction
4.4. Assessment of Mechanical Allodynia
4.5. Assessment of Thermal Hyperalgesia
4.6. Drugs and Chemicals
4.7. Experimental Design
4.8. Molecular Structure Preparation
4.9. Molecular Docking
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| AP-1 | Activator protein-1 |
| CB | Cannabinoid |
| CCI | Chronic constriction injury |
| COX-2 | Cyclooxygenase-2 |
| DMSO | Dimethylsulfoxide |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-κB |
| PDB | Protein data bank |
| PEA | Palmitoylethanolamide |
| PGE2 | Prostaglandin E2 |
| PPARs | Peroxisome proliferator-activated receptors |
| RXR | Retinoid X receptor |
| TNF-α | Tumour necrosis factor-α |
| TRPV1 | Vanilloid receptor 1 |
| WDR | Wide dynamic range neurons |
References
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Posso, I.d.P.; Palmeira, C.C.d.A.; Vieira, É.B.d.M. Epidemiology of neuropathic pain. Rev. Dor. 2016, 17, 11–14. [Google Scholar] [CrossRef]
- Yob, N.J.; Jofrry, S.M.; Affandi, M.M.; Teh, L.K.; Salleh, M.Z.; Zakaria, Z.A. Zingiber zerumbet (L.) Smith: A Review of Its Ethnomedicinal, Chemical, and Pharmacological Uses. Evid. Based Complement. Altern. Med. 2011, 2011, 543216. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.R.; Perimal, E.K.; Zakaria, Z.A.; Mokhtar, F.; Akhtar, M.N.; Lajis, N.H.; Israf, D.A. Preliminary analysis of the antinociceptive activity of zerumbone. Fitoterapia 2009, 80, 230–232. [Google Scholar] [CrossRef]
- Zulazmi, N.A.; Gopalsamy, B.; Farouk, A.A.O.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Antiallodynic and antihyperalgesic effects of zerumbone on a mouse model of chronic constriction injury-induced neuropathic pain. Fitoterapia 2015, 105, 215–221. [Google Scholar] [CrossRef]
- Ibrahim, M.Y.; Abdul, A.; Ibrahim, T.A.T.; Abdelwahab, S.I.; Elhassan, M.M.; Syam, M. Evaluation of acute toxicity and the effect of single injected doses of zerumbone on the kidney and liver functions in Sprague Dawley rats. Afr. J. Biotechnol. 2010, 9, 4442–4450. [Google Scholar]
- Rahman, H.S.; Rasedee, A.; Othman, H.H.; Chartrand, M.S.; Namvar, F.; Yeap, S.K.; Abdul Samad, N.; Andas, R.J.; Muhammad Nadzri, N.; Anasamy, T.; et al. Acute Toxicity Study of Zerumbone-Loaded Nanostructured Lipid Carrier on BALB/c Mice Model. BioMed Res. Int. 2014, 2014, 15. [Google Scholar] [CrossRef]
- Sulaiman, M.R.; Perimal, E.K.; Akhtar, M.N.; Mohamad, A.S.; Khalid, M.H.; Tasrip, N.A.; Mokhtar, F.; Zakaria, Z.A.; Lajis, N.H.; Israf, D.A. Anti-inflammatory effect of zerumbone on acute and chronic inflammation models in mice. Fitoterapia 2010, 81, 855–858. [Google Scholar] [CrossRef]
- Abel, E.L. Marihuana: The First Twelve Thousand Years; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Zlas, J.; Stark, H.; Seligman, J.; Levy, R.; Werker, E.; Breuer, A.; Mechoulam, R. Early medical use of cannabis. Nature 1993, 363, 215. [Google Scholar] [CrossRef]
- Zuardi, A.W. History of cannabis as a medicine: A review. Rev. Bras. Psiquiatr. 2006, 28, 153–157. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Rana, V.S.; Verdeguer, M.; Blazquez, M.A. Chemical composition of the essential oil of Zingiber zerumbet var. darcyi. Nat. Prod. Commun. 2012, 7, 1369–1370. [Google Scholar] [CrossRef]
- Koga, A.Y.; Beltrame, F.L.; Pereira, A.V. Several aspects of Zingiber zerumbet: A review. Rev. Bras. Farmacogn. 2016, 26, 385–391. [Google Scholar] [CrossRef]
- Hendriks, H.; Malingré, T.M.; Batterman, S.; Bos, R. Mono-and sesqui-terpene hydrocarbons of the essential oil of Cannabis sativa. Phytochemistry 1975, 14, 814–815. [Google Scholar] [CrossRef]
- Escher, P.; Wahli, W. Peroxisome proliferator-activated receptors: Insight into multiple cellular functions. Mutat. Res. 2000, 448, 121–138. [Google Scholar] [CrossRef]
- Kota, B.P.; Huang, T.H.; Roufogalis, B.D. An overview on biological mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef]
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Chia, J.S.; Omar Farouk, A.A.; Mohamad, A.S.; Sulaiman, M.R.; Perimal, E.K. Zerumbone alleviates chronic constriction injury-induced allodynia and hyperalgesia through serotonin 5-HT receptors. Biomed. Pharmacother. 2016, 83, 1303–1310. [Google Scholar] [CrossRef]
- Chia, J.S.M.; Izham, N.A.M.; Farouk, A.A.O.; Sulaiman, M.R.; Mustafa, S.; Hutchinson, M.R.; Perimal, E.K. Zerumbone Modulates alpha2A-Adrenergic, TRPV1, and NMDA NR2B Receptors Plasticity in CCI-Induced Neuropathic Pain In Vivo and LPS-Induced SH-SY5Y Neuroblastoma In Vitro Models. Front. Pharmacol. 2020, 11, 92. [Google Scholar] [CrossRef]
- Gopalsamy, B.; Chia, J.S.M.; Farouk, A.A.O.; Sulaiman, M.R.; Perimal, E.K. Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain. Molecules 2020, 25, 3880. [Google Scholar] [CrossRef]
- Seyrek, M.; Kahraman, S.; Deveci, M.S.; Yesilyurt, O.; Dogrul, A. Systemic cannabinoids produce CB(1)-mediated antinociception by activation of descending serotonergic pathways that act upon spinal 5-HT(7) and 5-HT(2A) receptors. Eur. J. Pharmacol. 2010, 649, 183–194. [Google Scholar] [CrossRef]
- Gutierrez, T.; Nackley, A.G.; Neely, M.H.; Freeman, K.G.; Edwards, G.L.; Hohmann, A.G. Effects of neurotoxic destruction of descending noradrenergic pathways on cannabinoid antinociception in models of acute and tonic nociception. Brain Res. 2003, 987, 176–185. [Google Scholar] [CrossRef]
- Bambico, F.R.; Katz, N.; Debonnel, G.; Gobbi, G. Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J. Neurosci. 2007, 27, 11700–11711. [Google Scholar] [CrossRef]
- Muntoni, A.L.; Pillolla, G.; Melis, M.; Perra, S.; Gessa, G.L.; Pistis, M. Cannabinoids modulate spontaneous neuronal activity and evoked inhibition of locus coeruleus noradrenergic neurons. Eur. J. Neurosci. 2006, 23, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; LoVerme, J.; La Rana, G.; D’Agostino, G.; Sasso, O.; Calignano, A.; Piomelli, D. Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR-alpha receptor agonist GW7647. Eur. J. Pharmacol. 2007, 566, 117–119. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sorgard, M.; Di Marzo, V.; Julius, D.; Hogestatt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Perimal, E.K.; Akhtar, M.N.; Mohamad, A.S.; Khalid, M.H.; Ming, O.H.; Khalid, S.; Tatt, L.M.; Kamaldin, M.N.; Zakaria, Z.A.; Israf, D.A.; et al. Zerumbone-induced antinociception: Involvement of the L-arginine-nitric oxide-cGMP -PKC-K+ ATP channel pathways. Basic Clin. Pharmacol. Toxicol. 2011, 108, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zulazmi, N.A.; Gopalsamy, B.; Min, J.C.; Farouk, A.A.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Zerumbone Alleviates Neuropathic Pain through the Involvement of l-Arginine-Nitric Oxide-cGMP-K(+) ATP Channel Pathways in Chronic Constriction Injury in Mice Model. Molecules 2017, 22, 555. [Google Scholar] [CrossRef]
- Gopalsamy, B.; Farouk, A.A.O.; Tengku, T.A.S.; Sulaiman, M.R.; Perimal, E.K. Antiallodynic and antihyperalgesic activities of zerumbone via the suppression of IL-1β, IL-6, and TNF-α in a mouse model of neuropathic pain. J. Pain Res. 2017, 10, 2605–2619. [Google Scholar] [CrossRef]
- Howlett, A.C.; Mukhopadhyay, S. Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem. Phys. Lipids 2000, 108, 53–70. [Google Scholar] [CrossRef]
- Guindon, J.; Hohmann, A. Cannabinoid CB2 receptors: A therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef]
- Croxford, J.L.; Yamamura, T. Cannabinoids and the immune system: Potential for the treatment of inflammatory diseases? J. Neuroimmunol. 2005, 166, 3–18. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martin, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef] [PubMed]
- Fine, P.G.; Rosenfeld, M.J. Cannabinoids for neuropathic pain. Curr. Pain Headache Rep. 2014, 18, 451. [Google Scholar] [CrossRef]
- Glass, M.; Faull, R.; Dragunow, M. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Bodor, Á.L.; Katona, I.; Nyíri, G.; Mackie, K.; Ledent, C.; Hájos, N.; Freund, T.F. Endocannabinoid signaling in rat somatosensory cortex: Laminar differences and involvement of specific interneuron types. J. Neurosci. 2005, 25, 6845–6856. [Google Scholar] [CrossRef]
- Ishac, E.J.; Jiang, L.; Lake, K.D.; Varga, K.; Abood, M.E.; Kunos, G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB receptors on peripheral sympathetic nerves. Br. J. Pharmacol. 1996, 118, 2023–2028. [Google Scholar] [CrossRef]
- Galiegue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carriere, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Yu, X.H.; Cao, C.Q.; Martino, G.; Puma, C.; Morinville, A.; St-Onge, S.; Lessard, É.; Perkins, M.N.; Laird, J.M. A peripherally restricted cannabinoid receptor agonist produces robust anti-nociceptive effects in rodent models of inflammatory and neuropathic pain. Pain 2010, 151, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Martin, W.J.; Tsou, K.; Walker, J.M. Cannabinoid receptor-mediated inhibition of the rat tail-flick reflex after microinjection into the rostral ventromedial medulla. Neurosci. Lett. 1998, 242, 33–36. [Google Scholar] [CrossRef]
- Monhemius, R.; Azami, J.; Green, D.; Roberts, M. CB1 receptor mediated analgesia from the Nucleus Reticularis Gigantocellularis pars alpha is activated in an animal model of neuropathic pain. Brain Res. 2001, 908, 67–74. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- Sain, N.M.; Liang, A.; Kane, S.A.; Urban, M.O. Antinociceptive effects of the non-selective cannabinoid receptor agonist CP 55,940 are absent in CB1(-/-) and not CB2(-/-) mice in models of acute and persistent pain. Neuropharmacology 2009, 57, 235–241. [Google Scholar] [CrossRef]
- Doğrul, A.; Gül, H.; Yıldız, O.; Bilgin, F.; Güzeldemir, M.E. Cannabinoids blocks tactile allodynia in diabetic mice without attenuation of its antinociceptive effect. Neurosci. Lett. 2004, 368, 82–86. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Singh, S.P.; Singh, N.I.; Nongalleima, K.; Doley, P.; Singh, C.B.; Sahoo, D. Molecular docking, MD simulation, DFT and ADME-toxicity study on analogs of zerumbone against IKK-β enzyme as anti-cancer agents. Netw. Model. Anal. Health Inform. Bioinform. 2018, 7, 1–8. [Google Scholar] [CrossRef]
- Maeda, T.; Kishioka, S. PPAR and Pain. Int. Rev. Neurobiol. 2009, 85, 165–177. [Google Scholar]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Lambert, M.H.; Kliewer, S.A. Peroxisome proliferator–activated receptor γ and metabolic disease. Annu. Rev. Biochem. 2001, 70, 341–367. [Google Scholar] [CrossRef] [PubMed]
- Berghe, W.V.; Vermeulen, L.; Delerive, P.; De Bosscher, K.; Staels, B.; Haegeman, G. A paradigm for gene regulation: Inflammation, NF-κB and PPAR. In Peroxisomal Disorders and Regulation of Genes; Springer: Boston, MA, USA, 2003; pp. 181–196. [Google Scholar]
- Jackson, S.M.; Parhami, F.; Xi, X.-P.; Berliner, J.A.; Hsueh, W.A.; Law, R.E.; Demer, L.L. Peroxisome proliferator—activated receptor activators target human endothelial cells to inhibit leukocyte—endothelial cell interaction. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2094–2104. [Google Scholar] [CrossRef]
- Taylor, B.K.; Dadia, N.; Yang, C.B.; Krishnan, S.; Badr, M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation 2002, 26, 121–127. [Google Scholar] [CrossRef]
- LoVerme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Taylor, B.; Kriedt, C.; Nagalingam, S.; Dadia, N.; Badr, M. Central administration of perfluorooctanoic acid inhibits cutaneous inflammation. Inflamm. Res. 2005, 54, 235–242. [Google Scholar] [CrossRef]
- LoVerme, J.; Russo, R.; La Rana, G.; Fu, J.; Farthing, J.; Mattace-Raso, G.; Meli, R.; Hohmann, A.; Calignano, A.; Piomelli, D. Rapid Broad-Spectrum Analgesia through Activation of Peroxisome Proliferator-Activated Receptor-α. J. Pharmacol. Exp. Ther. 2006, 319, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Faber, E.L.; Sah, P. Calcium-activated potassium channels: Multiple contributions to neuronal function. Neuroscientist 2003, 9, 181–194. [Google Scholar] [CrossRef]
- Costa, B.; Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: Involvement of CBTRPV1 and PPARgamma receptors and neurotrophic factors. Pain 2008, 139, 541–550. [Google Scholar] [CrossRef]
- Morgenweck, J.; Abdel-Aleem, O.; McNamara, K.; Donahue, R.; Badr, M.; Taylor, B. Activation of peroxisome proliferator-activated receptor γ in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology 2010, 58, 337–345. [Google Scholar] [CrossRef][Green Version]
- Maeda, T.; Kiguchi, N.; Kobayashi, Y.; Ozaki, M.; Kishioka, S. Pioglitazone attenuates tactile allodynia and thermal hyperalgesia in mice subjected to peripheral nerve injury. J. Pharmacol. Sci. 2008, 108, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Churi, S.B.; Abdel-Aleem, O.S.; Tumber, K.K.; Scuderi-Porter, H.; Taylor, B.K. Intrathecal Rosiglitazone Acts at Peroxisome Proliferator–Activated Receptor-γ to Rapidly Inhibit Neuropathic Pain in Rats. J. Pain 2008, 9, 639–649. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Martucci, C.; Trovato, A.E.; Costa, B.; Borsani, E.; Franchi, S.; Magnaghi, V.; Panerai, A.E.; Rodella, L.F.; Valsecchi, A.E.; Sacerdote, P.; et al. The purinergic antagonist PPADS reduces pain related behaviours and interleukin-1 beta, interleukin-6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain 2008, 137, 81–95. [Google Scholar] [CrossRef]
- Sambasevam, Y.; Omar Farouk, A.A.; Tengku Mohamad, T.A.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Cardamonin attenuates hyperalgesia and allodynia in a mouse model of chronic constriction injury-induced neuropathic pain: Possible involvement of the opioid system. Eur. J. Pharmacol. 2017, 796, 32–38. [Google Scholar] [CrossRef]
- Ming-Tatt, L.; Khalivulla, S.I.; Akhtar, M.N.; Lajis, N.; Perimal, E.K.; Akira, A.; Ali, D.I.; Sulaiman, M.R. Anti-hyperalgesic effect of a benzilidine-cyclohexanone analogue on a mouse model of chronic constriction injury-induced neuropathic pain: Participation of the kappa-opioid receptor and KATP. Pharmacol. Biochem. Behav. 2013, 114–115, 58–63. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Voon, F.L.; Sulaiman, M.R.; Akhtar, M.N.; Idris, M.F.; Akira, A.; Perimal, E.K.; Israf, D.A.; Ming-Tatt, L. Cardamonin (2′,4′-dihydroxy-6′-methoxychalcone) isolated from Boesenbergia rotunda (L.) Mansf. inhibits CFA-induced rheumatoid arthritis in rats. Eur. J. Pharmacol. 2017, 794, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Wright, C.E.; Angus, J.A. Evidence that CB-1 and CB-2 cannabinoid receptors mediate antinociception in neuropathic pain in the rat. Pain 2004, 109, 124–131. [Google Scholar] [CrossRef]
- Donvito, G.; Bagdas, D.; Toma, W.; Rahimpour, E.; Jackson, A.; Meade, J.A.; AlSharari, S.; Kulkarni, A.R.; Carroll, F.I.; Lichtman, A.H. The interaction between alpha 7 nicotinic acetylcholine receptor and nuclear peroxisome proliferator-activated receptor-α represents a new antinociceptive signaling pathway in mice. Exp. Neurol. 2017, 295, 194–201. [Google Scholar] [CrossRef]
- Mansouri, M.T.; Naghizadeh, B.; Ghorbanzadeh, B.; Rajabi, H.; Pashmforoush, M. Pharmacological evidence for systemic and peripheral antinociceptive activities of pioglitazone in the rat formalin test: Role of PPARγ and nitric oxide. Eur. J. Pharmacol. 2017, 805, 84–92. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Best Binding Energy (kcal/mol) for | Ligands | |||
|---|---|---|---|---|
| Zerumbone | Amino Acid Residue | Agonists CB1 (AM11542), CB2 (AM841) | Amino Acid Residue | |
| CB1 receptor | −7.8 | Val196, Phe170, Phe174, Leu193, His178, Pro269, Phe189 | −12.7 | Phe177, Phe174, Phe170, Phe379, Ser383, Cys386, Leu359, Phe200, Val196, Leu193, Phe268, Pro269, Phe189, Ile271, Leu276, Tyr275 |
| CB2 receptor | −9.4 | His95, Phe94 Phe91, Phe87 Val113, Phe 183 Phe106 | −10.9 | Phe94, Ile110, Phe87, Val113, Phe183, Phe117, Cys288, Phe281, Val261, Met265, Trp258 |
| Best Binding Energy (kcal/mol) for | Ligands | |||
|---|---|---|---|---|
| Zerumbone | Amino Acid Residue | Agonists PPARα (APHM13), PPARγ (JKPL53) | Amino Acid Residue | |
| PPARα receptor | −5.1 | Leu254, Ala333, Val332, Cys275, Ile339, Cys276, Ile272, Leu247 | −12.2 | His 440, Tyr 464, Tyr 314, Ser280, Cys276, Phe273, Ile272, Cys275, Cys276, Leu321, Val255, Val332, Ala333, Ile339 |
| PPARγ receptors | −6.1 | Leu333, Val339, Met364, Cys285 Arg288, Ile341, Leu330, Ile326 | −10.0 | Tyr473, Gly284, Phe282, Arg280, Arg288, Cya285, Tyr327, Ile341, Leu333, Leu330 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chia, J.S.M.; Farouk, A.A.O.; Mohamad, T.A.S.T.; Sulaiman, M.R.; Zakaria, H.; Hassan, N.I.; Perimal, E.K. Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models. Molecules 2021, 26, 3849. https://doi.org/10.3390/molecules26133849
Chia JSM, Farouk AAO, Mohamad TAST, Sulaiman MR, Zakaria H, Hassan NI, Perimal EK. Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models. Molecules. 2021; 26(13):3849. https://doi.org/10.3390/molecules26133849
Chicago/Turabian StyleChia, Jasmine Siew Min, Ahmad Akira Omar Farouk, Tengku Azam Shah Tengku Mohamad, Mohd Roslan Sulaiman, Hanis Zakaria, Nurul Izzaty Hassan, and Enoch Kumar Perimal. 2021. "Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models" Molecules 26, no. 13: 3849. https://doi.org/10.3390/molecules26133849
APA StyleChia, J. S. M., Farouk, A. A. O., Mohamad, T. A. S. T., Sulaiman, M. R., Zakaria, H., Hassan, N. I., & Perimal, E. K. (2021). Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models. Molecules, 26(13), 3849. https://doi.org/10.3390/molecules26133849