
Emerging Molecular Receptors for the Specific-Target Delivery of Ruthenium and Gold Complexes into Cancer Cells




Abstract
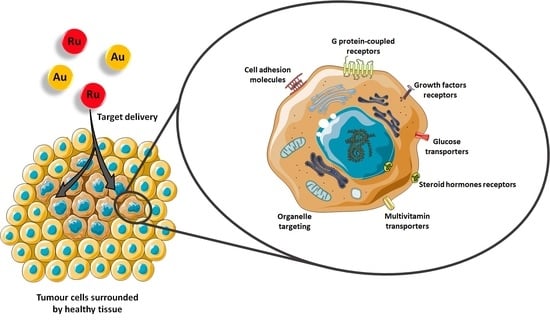
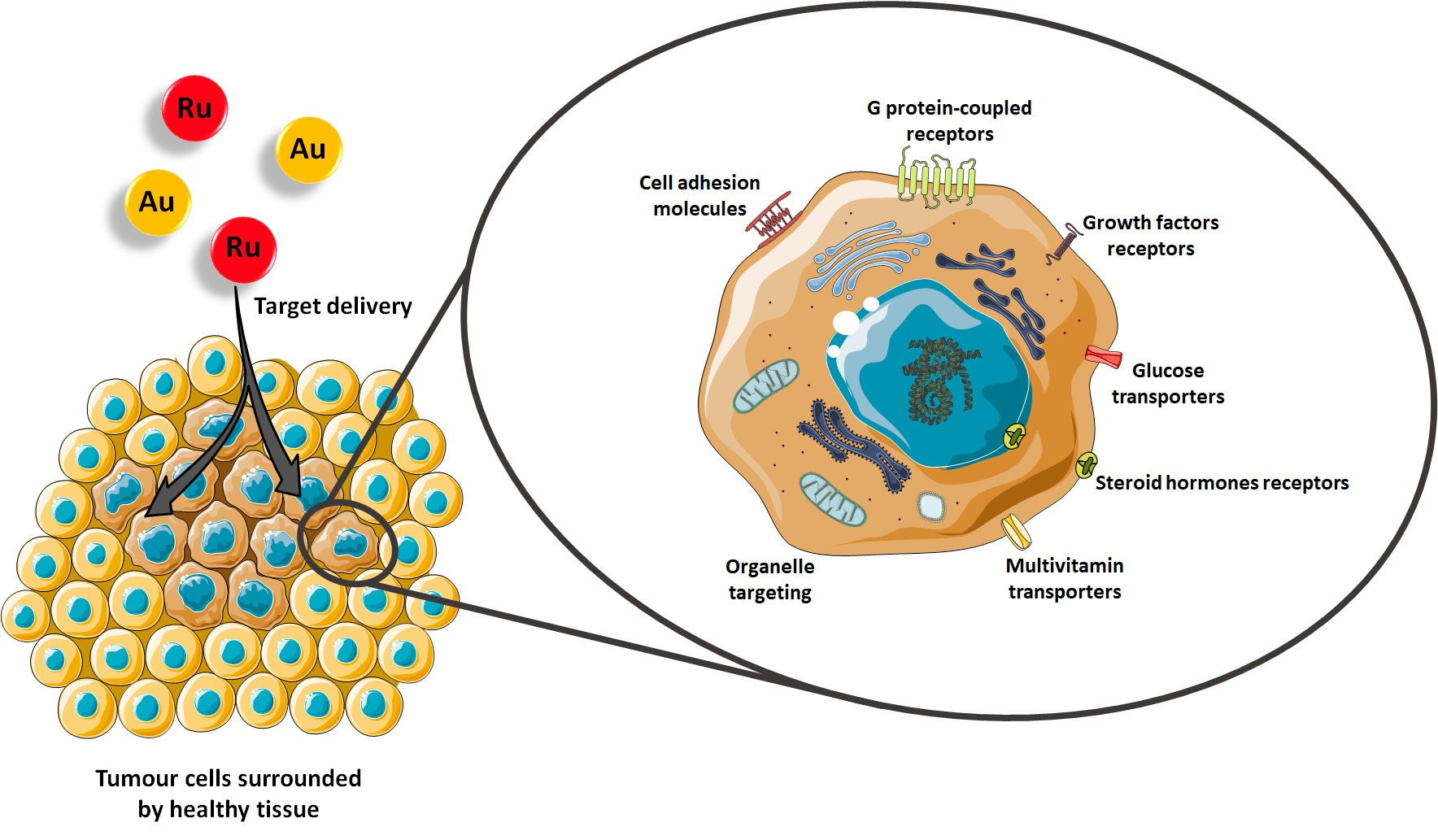
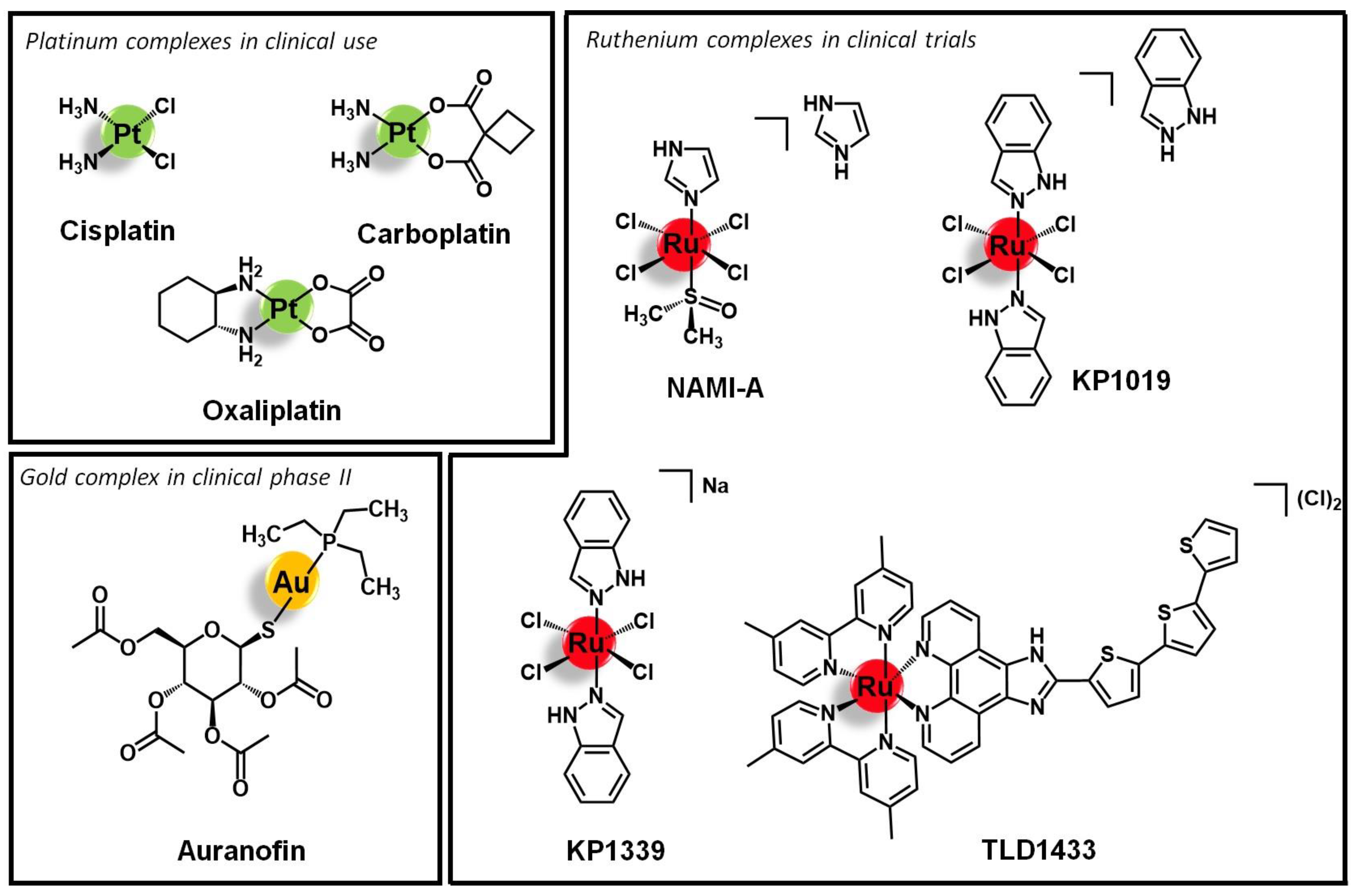
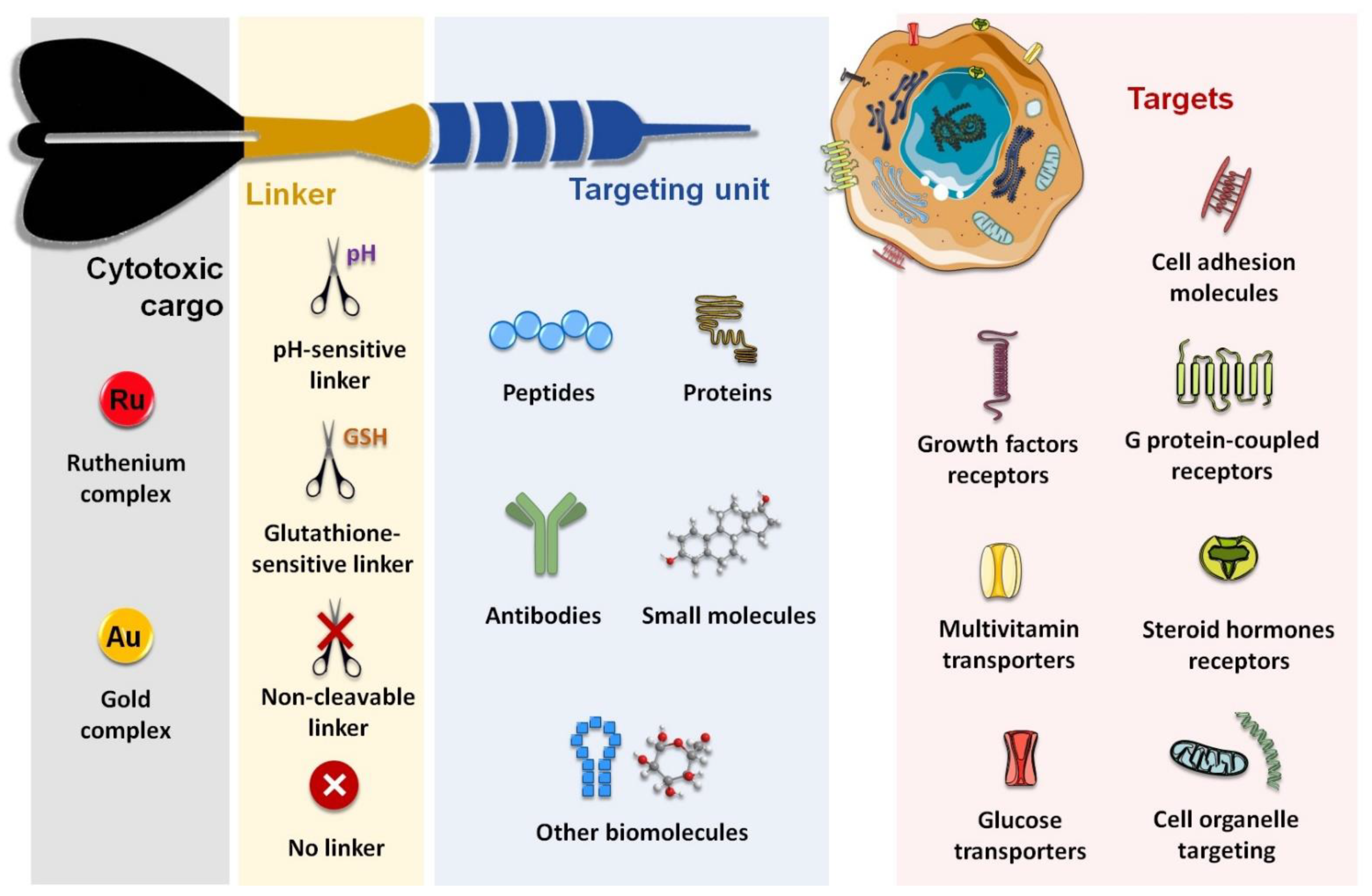
1. Introduction
2. Cell Adhesion Molecules (CAM)
2.1. Integrins
2.2. Cadherins
3. G Protein-Coupled Receptors (GPCR)
3.1. Somatostatin Receptors (SSTR)
3.2. Bombesin Receptors (BBR)
3.3. Opioid Receptors (OPR)
3.4. G Protein-Coupled Estrogen Receptors (GPER)
4. Growth Factors Receptors (GFR)
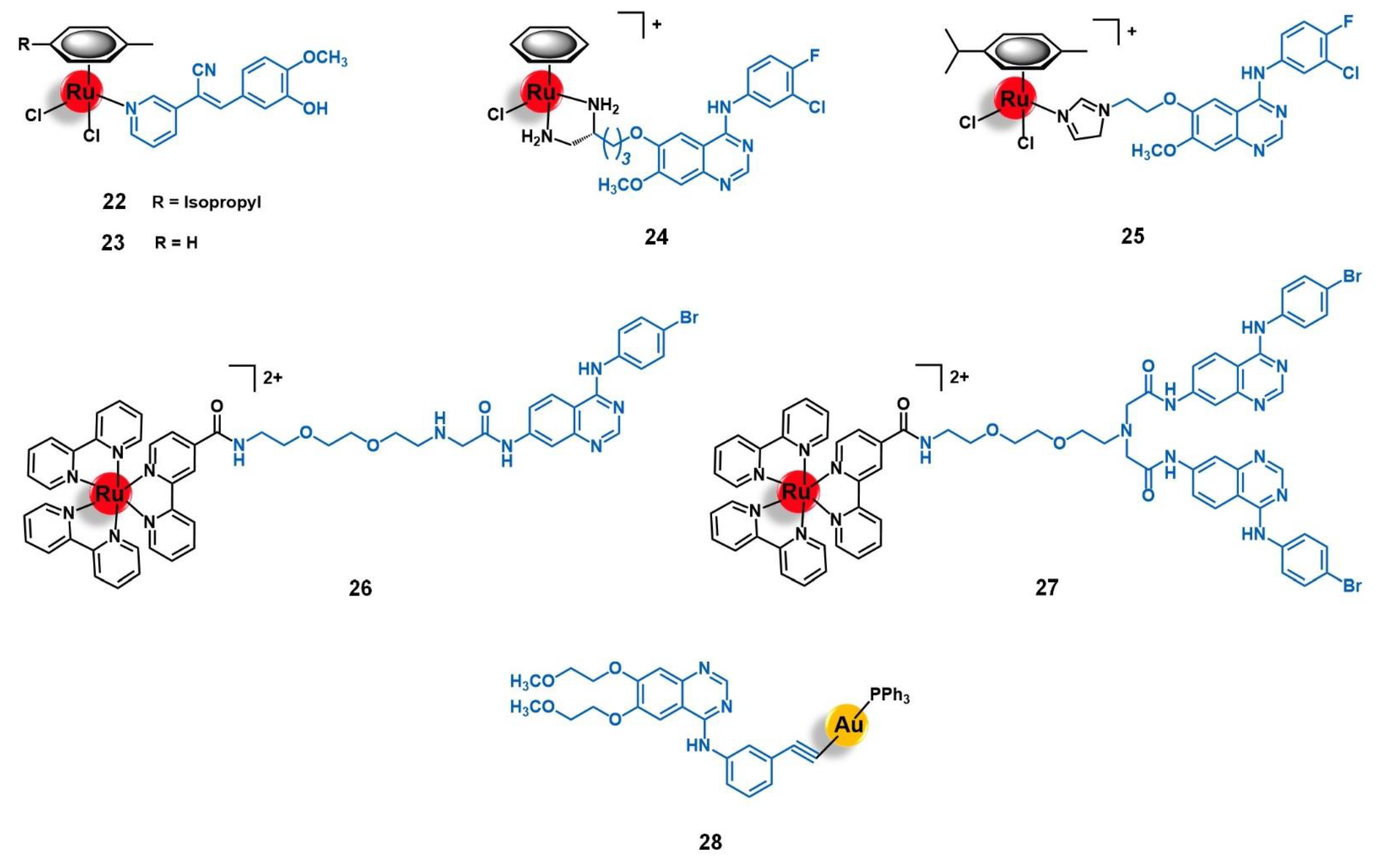
4.1. Epidermal Growth Factor Receptor (EGFR)
4.2. Human Epidermal Growth Factor Receptor 2 (HER2)
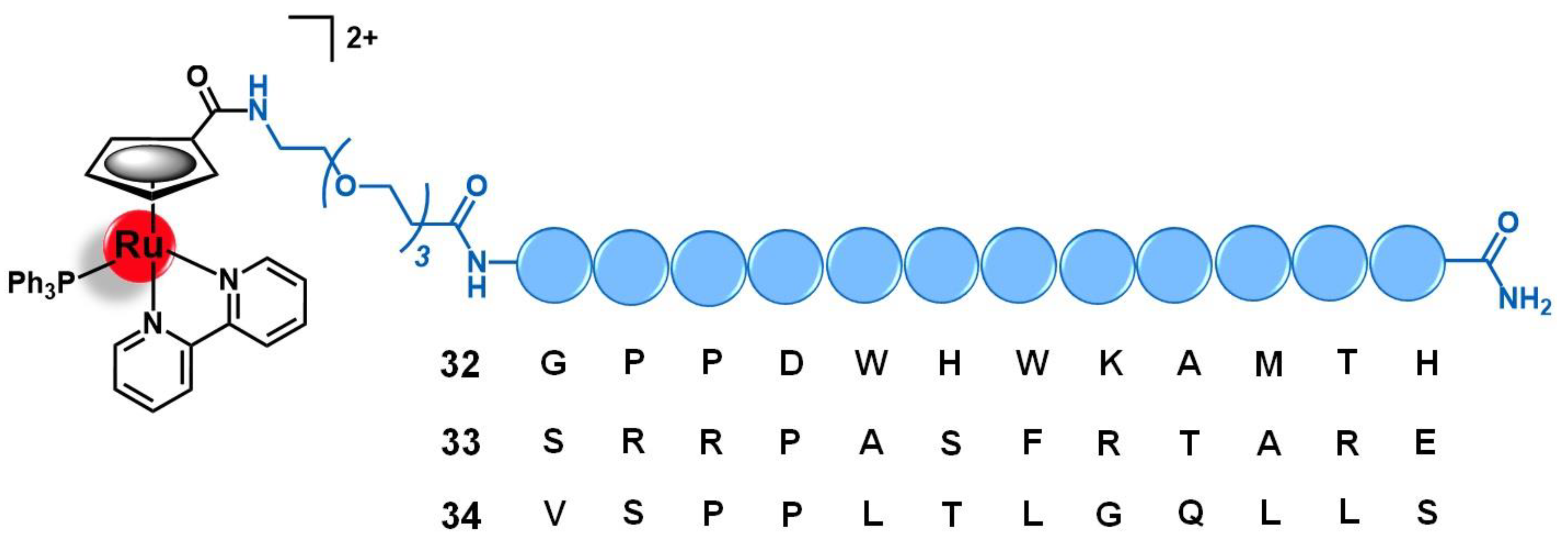
4.3. Fibroblast Growth Factor Receptor (FGFR)
5. Other Emerging Targets
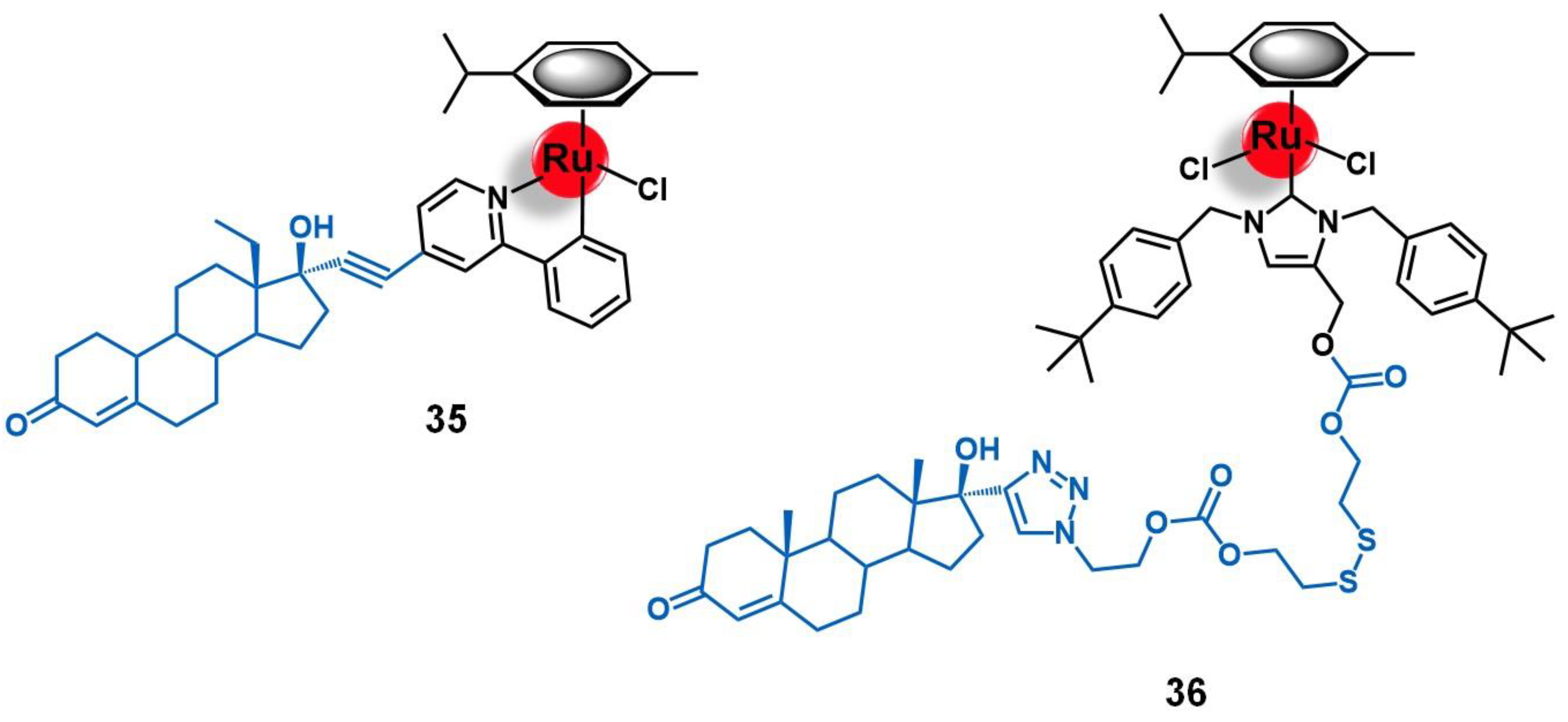
5.1. Progesterone Receptors (PR)
5.2. Targets Involved in Metabolic Pathways
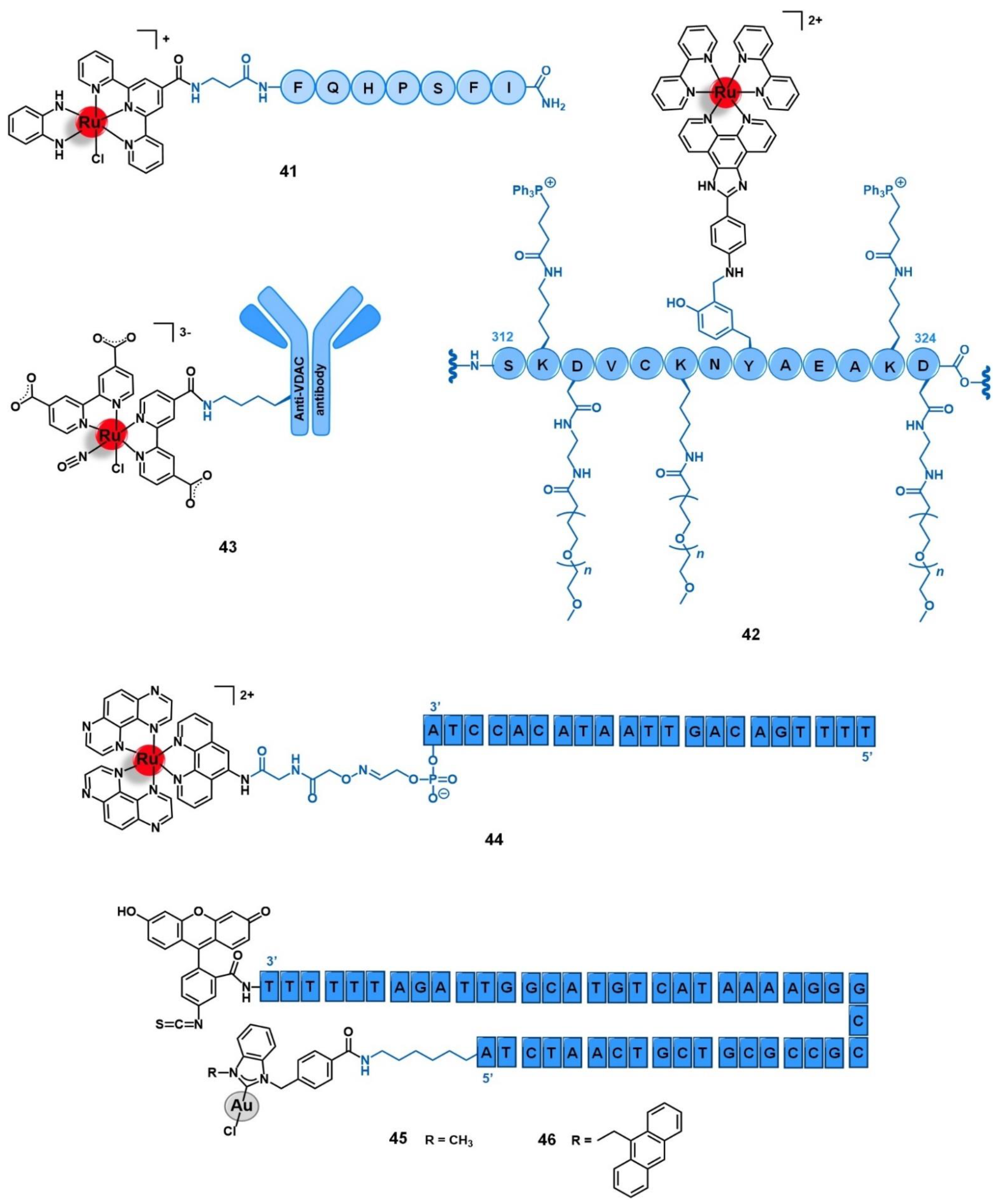
5.3. Cell Organelles and Targeted Gene Therapy
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Englinger, B.; Pirker, C.; Heffeter, P.; Terenzi, A.; Kowol, C.R.; Keppler, B.K.; Berger, W. Metal drugs and the anticancer immune response. Chem. Rev. 2019, 119, 1519–1624. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; Casini, A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalt. Trans. 2014, 43, 4209–4219. [Google Scholar] [CrossRef] [PubMed]
- Glišić, B.; Djuran, M.I. Gold complexes as antimicrobial agents: An overview of different biological activities in relation to the oxidation state of the gold ion and the ligand structure. J. Chem. Soc. Dalt. Trans. 2014, 43, 5950–5969. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.N.; Zhang, J.J.; Che, C.M. Chemical biology of anticancer gold(III) and gold(i) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef]
- Wester, H.J. Nuclear imaging probes: From bench to bedside. Clin. Cancer Res. 2007, 13, 3470–3481. [Google Scholar] [CrossRef]
- Schottelius, M.; Wester, H.J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef]
- Tweedle, M.F. Peptide-Targeted Diagnostics and Radiotherapeutics. Acc. Chem. Res. 2009, 42, 958–968. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef]
- Wang, J.; Wang, T.T.; Gao, P.F.; Huang, C.Z. Biomolecules-conjugated nanomaterials for targeted cancer therapy. J. Mater. Chem. B 2014, 2, 8452–8465. [Google Scholar] [CrossRef]
- Keenan, M.M.; Chi, J.T. Alternative fuels for cancer cells. J. Cancer 2015, 21, 49–55. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Guan, S.; Zhang, Q.; Bao, J.; Hu, R.; Czech, T.; Tang, J. Recognition Sites for Cancer-targeting Drug Delivery Systems. Curr. Drug Metab. 2019, 20, 815–834. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef]
- Krȩcisz, P.; Czarnecka, K.; Królicki, L.; Mikiciuk-Olasik, E.; Szymański, P. Radiolabeled Peptides and Antibodies in Medicine. Bioconjug. Chem. 2021, 32, 25–42. [Google Scholar] [CrossRef]
- Oliveira, M.C.; Correia, J.D.G. Biomedical applications of radioiodinated peptides. Eur. J. Med. Chem. 2019, 179, 56–77. [Google Scholar] [CrossRef]
- Correia, J.D.G.; Paulo, A.; Raposinho, P.D.; Santos, I. Radiometallated peptides for molecular imaging and targeted therapy. Dalt. Trans. 2011, 40, 6144–6167. [Google Scholar] [CrossRef] [PubMed]
- Morais, T.S.; Valente, A.; Tomaz, A.I.; Marques, F.; Garcia, M.H. Tracking antitumor metallodrugs: Promising agents with the Ru(II)- and Fe(II)-cyclopentadienyl scaffolds. Future Med. Chem. 2016, 8, 527–544. [Google Scholar] [CrossRef]
- Jia, P.; Ouyang, R.; Cao, P.; Tong, X.; Zhou, X.; Lei, T.; Zhao, Y.; Guo, N.; Chang, H.; Miao, Y.; et al. Review: Recent advances and future development of metal complexes as anticancer agents. J. Coord. Chem. 2017, 70, 2175–2201. [Google Scholar] [CrossRef]
- Chen, D.; Milacic, V.; Frezza, M.; Dou, Q. Metal Complexes, their Cellular Targets and Potential for Cancer Therapy. Curr. Pharm. Des. 2009, 15, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Ping Dou, Q. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy—An update from drug design perspective. Drug Des. Devel. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Sudhindra, P.; Ajay Sharma, S.; Roy, N.; Moharana, P.; Paira, P. Recent advances in cytotoxicity, cellular uptake and mechanism of action of ruthenium metallodrugs: A review. Polyhedron 2020, 192, 114827. [Google Scholar] [CrossRef]
- Cardo, L.; Hannon, M.J. Non-covalent metallo-drugs: Using shape to target DNA and RNA junctions and other nucleic acid structures. In Metallo-Drugs: Development and Action of Anticancer Agents; Walter de Gruyter GmbH: Wustermark, Germany, 2018; Volume 18, pp. 303–321. ISBN 9783110470734. [Google Scholar]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase i study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Dominelli, B.; Correia, J.D.G.; Kühn, F.E. Medicinal Applications of Gold(I/III)-Based Complexes Bearing N-Heterocyclic Carbene and Phosphine Ligands. J. Organomet. Chem. 2018, 866, 153–164. [Google Scholar] [CrossRef]
- Home—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 3 May 2021).
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Dunehoo, A.L.; Anderson, M.; Majumdar, S.; Kobayashi, N.; Berkland, C.; Siahaan, T.J. Cell Adhesion Molecules for Targeted Drug Delivery. J. Pharm. Sci. 2006, 95, 1856–1872. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Millard, M.; Odde, S.; Neamati, N. Integrin Targeted Therapeutics. Theranostics 2012, 1, 154–188. [Google Scholar] [CrossRef]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef]
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Updat. 2005, 8, 381–402. [Google Scholar] [CrossRef]
- Katsamakas, S.; Chatzisideri, T.; Thysiadis, S.; Sarli, V. RGD-mediated delivery of small-molecule drugs. Future Med. Chem. 2017, 9, 579–604. [Google Scholar] [CrossRef]
- Barragán, F.; López-Senín, P.; Salassa, L.; Betanzos-Lara, S.; Habtemariam, A.; Moreno, V.; Sadler, P.J.; Marchán, V. Photocontrolled DNA binding of a receptor-targeted organometallic ruthenium(II) complex. J. Am. Chem. Soc. 2011, 133, 14098–14108. [Google Scholar] [CrossRef][Green Version]
- Hahn, E.M.; Estrada-Ortiz, N.; Han, J.; Ferreira, V.F.C.; Kapp, T.G.; Correia, J.D.G.; Casini, A.; Kühn, F.E. Functionalization of Ruthenium(II) Terpyridine Complexes with Cyclic RGD Peptides To Target Integrin Receptors in Cancer Cells. Eur. J. Inorg. Chem. 2017, 2017, 1667–1672. [Google Scholar] [CrossRef]
- Hu, J.J.; Lei, Q.; Zhang, X.Z. Recent advances in photonanomedicines for enhanced cancer photodynamic therapy. Prog. Mater. Sci. 2020, 114, 100685. [Google Scholar] [CrossRef]
- Zhao, Z.; Qiu, K.; Liu, J.; Hao, X.; Wang, J. Two-photon photodynamic ablation of tumour cells using an RGD peptide-conjugated ruthenium(ii) photosensitiser. Chem. Commun. 2020, 56, 12542–12545. [Google Scholar] [CrossRef]
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, X.; Li, C.; Chen, T. Designing luminescent ruthenium prodrug for precise cancer therapy and rapid clinical diagnosis. Biomaterials 2019, 192, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, J.; Song, S.; Beljaars, L.; Groothuis, G.M.M.; Permentier, H.; Bischoff, R.; Halmos, G.B.; Verhoeven, C.J.; Amstalden van Hove, E.R.; et al. Targeted imaging of integrins in cancer tissues using photocleavable Ru(ii) polypyridine complexes as mass-tags. Chem. Commun. 2020, 56, 5941–5944. [Google Scholar] [CrossRef] [PubMed]
- Śmiłowicz, D.; Slootweg, J.C.; Metzler-Nolte, N. Bioconjugation of Cyclometalated Gold(III) Lipoic Acid Fragments to Linear and Cyclic Breast Cancer Targeting Peptides. Mol. Pharm. 2019, 16, 4572–4581. [Google Scholar] [CrossRef] [PubMed]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Blaschuk, O.W. N-cadherin antagonists as oncology therapeutics. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- On, N.H.; Kiptoo, P.; Siahaan, T.J.; Miller, D.W. Modulation of blood-brain barrier permeability in mice using synthetic E-cadherin peptide. Mol. Pharm. 2014, 11, 974–981. [Google Scholar] [CrossRef]
- Bihari, Z.; Vultos, F.; Fernandes, C.; Gano, L.; Santos, I.; Correia, J.D.G.; Buglyó, P. Synthesis, characterization and biological evaluation of a 67Ga-labeled (η6-Tyr)Ru(η5-Cp) peptide complex with the HAV motif. J. Inorg. Biochem. 2016, 160, 189–197. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal. Transduct. Target. Ther. 2021, 6, 1–27. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Franco Machado, J.; Silva, R.; Melo, R.G.; Correia, J. Less Exploited GPCRs in Precision Medicine: Targets for Molecular Imaging and Theranostics. Molecules 2018, 24, 49. [Google Scholar] [CrossRef]
- Moody, T.W.; Ramos-Alvarez, I.; Jensen, R.T. Neuropeptide G protein-coupled receptors as oncotargets. Front. Endocrinol. (Lausanne). 2018, 9, 345. [Google Scholar] [CrossRef]
- Sun, L.-C.; Coy, D.H. Somatostatin Receptor-Targeted Anti-Cancer Therapy. Curr. Drug Deliv. 2011, 8, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zabarska, N.; Wu, Y.; Lamla, M.; Fischer, S.; Monczak, K.; Ng, D.Y.W.; Rau, S.; Weil, T. Receptor selective ruthenium-somatostatin photosensitizer for cancer targeted photodynamic applications. Chem. Commun. 2015, 51, 12552–12555. [Google Scholar] [CrossRef] [PubMed]
- Vegi, N.M.; Chakrabortty, S.; Zegota, M.M.; Kuan, S.L.; Stumper, A.; Rawat, V.P.S.; Sieste, S.; Buske, C.; Rau, S.; Weil, T.; et al. Somatostatin receptor mediated targeting of acute myeloid leukemia by photodynamic metal complexes for light induced apoptosis. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Barragán, F.; Carrion-Salip, D.; Gómez-Pinto, I.; González-Cantó, A.; Sadler, P.J.; De Llorens, R.; Moreno, V.; González, C.; Massaguer, A.; Marchán, V. Somatostatin subtype-2 receptor-targeted metal-based anticancer complexes. Bioconjug. Chem. 2012, 23, 1838–1855. [Google Scholar] [CrossRef]
- Moreno, P.; Ramos-Álvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef]
- Joshi, T.; Pierroz, V.; Ferrari, S.; Gasser, G. Bis(dipyridophenazine)(2-(2′-pyridyl)pyrimidine-4-carboxylic acid)ruthenium(II) Hexafluorophosphate: A Lesson in Stubbornness. ChemMedChem 2014, 9, 1419–1427. [Google Scholar] [CrossRef]
- Doulain, P.E.; Decréau, R.; Racoeur, C.; Goncalves, V.; Dubrez, L.; Bettaieb, A.; Le Gendre, P.; Denat, F.; Paul, C.; Goze, C.; et al. Towards the elaboration of new gold-based optical theranostics. Dalt. Trans. 2015, 44, 4874–4883. [Google Scholar] [CrossRef]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid Receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Fichna, J.; Zielińska, M. Opioids in Cancer Development, Progression and Metastasis: Focus on Colorectal Cancer. Curr. Treat. Options Oncol. 2020, 21, 6. [Google Scholar] [CrossRef]
- Meier, S.M.; Novak, M.; Kandioller, W.; Jakupec, M.A.; Arion, V.B.; Metzler-Nolte, N.; Keppler, B.K.; Hartinger, C.G. Identification of the Structural Determinants for Anticancer Activity of a Ruthenium Arene Peptide Conjugate. Chem. Eur. J. 2013, 19, 9297–9307. [Google Scholar] [CrossRef]
- Luengo, A.; Marzo, I.; Reback, M.; Daubit, I.M.; Fernández-Moreira, V.; Metzler-Nolte, N.; Gimeno, M.C. Luminescent Bimetallic Ir III /Au I Peptide Bioconjugates as Potential Theranostic Agents. Chem. Eur. J. 2020, 26, 12158–12167. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Hsu, L.-H.; Chu, N.-M.; Lin, Y.-F.; Kao, S.-H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; O’Connell, M.A.; Waller, Z.A.E.; Bochmann, M. A Gold(III) Pincer Ligand Scaffold for the Synthesis of Binuclear and Bioconjugated Complexes: Synthesis and Anticancer Potential. Chem. Eur. J. 2018, 24, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, M.; Sun, W.; Fan, J.; Du, J.; Peng, X. An estrogen receptor targeted ruthenium complex as a two-photon photodynamic therapy agent for breast cancer cells. Chem. Commun. 2018, 54, 7038–7041. [Google Scholar] [CrossRef] [PubMed]
- Golbaghi, G.; Haghdoost, M.M.; Yancu, D.; López De Los Santos, Y.; Doucet, N.; Patten, S.A.; Sanderson, J.T.; Castonguay, A. Organoruthenium(II) Complexes Bearing an Aromatase Inhibitor: Synthesis, Characterization, in Vitro Biological Activity and in Vivo Toxicity in Zebrafish Embryos. Organometallics 2019, 38, 702–711. [Google Scholar] [CrossRef]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for Growth Factors in Cancer Progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef]
- Krause, D.S.; Van Etten, R.A. Tyrosine Kinases as Targets for Cancer Therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochem. 2020, 85, 967–993. [Google Scholar] [CrossRef]
- Biersack, B.; Zoldakova, M.; Effenberger, K.; Schobert, R. (Arene)Ru(II) complexes of epidermal growth factor receptor inhibiting tyrphostins with enhanced selectivity and cytotoxicity in cancer cells. Eur. J. Med. Chem. 2010, 45, 1972–1975. [Google Scholar] [CrossRef]
- Zheng, W.; Luo, Q.; Lin, Y.; Zhao, Y.; Wang, X.; Du, Z.; Hao, X.; Yu, Y.; Lü, S.; Ji, L.; et al. Complexation with organometallic ruthenium pharmacophores enhances the ability of 4-anilinoquinazolines inducing apoptosis. Chem. Commun. 2013, 49, 10224–10226. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, W.; Luo, Q.; Zhao, Y.; Zhang, E.; Liu, S.; Wang, F. Dual-targeting organometallic ruthenium(ii) anticancer complexes bearing EGFR-inhibiting 4-anilinoquinazoline ligands. Dalt. Trans. 2015, 44, 13100–13111. [Google Scholar] [CrossRef]
- Du, J.; Zhang, E.; Zhao, Y.; Zheng, W.; Zhang, Y.; Lin, Y.; Wang, Z.; Luo, Q.; Wu, K.; Wang, F. Discovery of a dual-targeting organometallic ruthenium complex with high activity inducing early stage apoptosis of cancer cells. Metallomics 2015, 7, 1573–1583. [Google Scholar] [CrossRef]
- Ilmi, R.; Tseriotou, E.; Stylianou, P.; Christou, Y.A.; Ttofi, I.; Dietis, N.; Pitris, C.; Odysseos, A.D.; Georgiades, S.N. A Novel Conjugate of Bis[((4-bromophenyl)amino)quinazoline], a EGFR-TK Ligand, with a Fluorescent Ru(II)-Bipyridine Complex Exhibits Specific Subcellular Localization in Mitochondria. Mol. Pharm. 2019, 16, 4260–4273. [Google Scholar] [CrossRef]
- Ortega, E.; Zamora, A.; Basu, U.; Lippmann, P.; Rodríguez, V.; Janiak, C.; Ott, I.; Ruiz, J. An Erlotinib gold(I) conjugate for combating triple-negative breast cancer. J. Inorg. Biochem. 2020, 203, 110910. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Curado, N.; Dewaele-Le Roi, G.; Poty, S.; Lewis, J.S.; Contel, M. Trastuzumab gold-conjugates: Synthetic approach and: In vitro evaluation of anticancer activities in breast cancer cell lines. Chem. Commun. 2019, 55, 1394–1397. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, J.; Muñoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Franco Machado, J.; Machuqueiro, M.; Marques, F.; Robalo, M.P.; Piedade, M.F.M.; Garcia, M.H.; Correia, J.D.G.; Morais, T.S. Novel “ruthenium cyclopentadienyl”—peptide conjugate complexes against human FGFR(+) breast cancer. Dalt. Trans. 2020, 49, 5974–5987. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Rodríguez, V.; Cutillas, N.; Espinosa, A.; Hannon, M.J. A potent ruthenium(II) antitumor complex bearing a lipophilic levonorgestrel group. Inorg. Chem. 2011, 50, 9164–9171. [Google Scholar] [CrossRef]
- Lv, G.; Qiu, L.; Li, K.; Liu, Q.; Li, X.; Peng, Y.; Wang, S.; Lin, J. Enhancement of therapeutic effect in breast cancer with a steroid-conjugated ruthenium complex. New, J. Chem. 2019, 43, 3419–3427. [Google Scholar] [CrossRef]
- Poynton, F.E.; Bright, S.A.; Blasco, S.; Williams, D.C.; Kelly, J.M.; Gunnlaugsson, T. The development of ruthenium(II) polypyridyl complexes and conjugates for: In vitro cellular and in vivo applications. Chem. Soc. Rev. 2017, 46, 7706–7756. [Google Scholar] [CrossRef]
- Flamme, M.; Clarke, E.; Gasser, G.; Hollenstein, M. Applications of Ruthenium Complexes Covalently Linked to Nucleic Acid Derivatives. Molecules 2018, 23, 1515. [Google Scholar] [CrossRef]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and cancer-targeting design of ruthenium complexes for precise cancer therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef]
- Yeo, C.; Ooi, K.; Tiekink, E. Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy? Molecules 2018, 23, 1410. [Google Scholar] [CrossRef]
- Andrejević, T.P.; Glišić, B.Đ.; Djuran, M.I. Amino Acids and Peptides as Versatile Ligands in the Synthesis of Antiproliferative Gold Complexes. Chemistry 2020, 2, 13. [Google Scholar] [CrossRef]
- Grimm, S.L.; Hartig, S.M.; Edwards, D.P. Progesterone Receptor Signaling Mechanisms. J. Mol. Biol. 2016, 428, 3831–3849. [Google Scholar] [CrossRef]
- Daniel, A.R.; Hagan, C.R.; Lange, C.A. Progesterone receptor action: Defining a role in breast cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 359–369. [Google Scholar] [CrossRef]
- Valadez-Cosmes, P.; Vázquez-Martínez, E.R.; Cerbón, M.; Camacho-Arroyo, I. Membrane progesterone receptors in reproduction and cancer. Mol. Cell. Endocrinol. 2016, 434, 166–175. [Google Scholar] [CrossRef]
- Bregman, H.; Meggers, E. Ruthenium half-sandwich complexes as protein kinase inhibitors: An N-succinimidyl ester for rapid derivatizations of the cyclopentadienyl moiety. Org. Lett. 2006, 8, 5465–5468. [Google Scholar] [CrossRef]
- Almeida, M.A.P.; Do Nascimento, F.B.; Graminha, A.E.; Ferreira, A.G.; Ellena, J.; Mello, F.M.D.S.; De Lima, A.P.; Silveira-Lacerda, E.D.P.; Batista, A.A. Structural features and cytotoxic activities of [Ru(AA-H)(dppb)(bipy)] PF6 complexes. Polyhedron 2014, 81, 735–742. [Google Scholar] [CrossRef]
- Lima, A.P.; Pereira, F.C.; Almeida, M.A.P.; Mello, F.M.S.; Pires, W.C.; Pinto, T.M.; Delella, F.K.; Felisbino, S.L.; Moreno, V.; Batista, A.A.; et al. Cytoxicity and Apoptotic Mechanism of Ruthenium(II) Amino Acid Complexes in Sarcoma-180 Tumor Cells. PLoS ONE 2014, 9, e105865. [Google Scholar] [CrossRef]
- de Sousa, I.H.; Campos, V.N.S.; Vale, A.A.M.; Maciel-Silva, V.L.; Leite, C.M.; Lopes, A.J.O.; Mourão, P.S.; das Chagas Alves Lima, F.; Batista, A.A.; de Azevedo dos Santos, A.P.S.; et al. Ruthenium (II) complexes with N, O-chelating proline and threonine ligands cause selective cytotoxicity by the induction of genomic instability, cell cycle arrest and apoptosis in breast and prostate tumor cells. Toxicol. Vitr. 2020, 62, 104679. [Google Scholar] [CrossRef]
- Nardon, C.; Schmitt, S.M.; Yang, H.; Zuo, J.; Fregona, D.; Dou, Q.P. Gold(III)-Dithiocarbamato Peptidomimetics in the Forefront of the Targeted Anticancer Therapy: Preclinical Studies against Human Breast Neoplasia. PLoS ONE 2014, 9, e84248. [Google Scholar] [CrossRef]
- Boscutti, G.; Nardon, C.; Marchiò, L.; Crisma, M.; Biondi, B.; Dalzoppo, D.; Via, L.D.; Formaggio, F.; Casini, A.; Fregona, D. Anticancer Gold(III) Peptidomimetics: From Synthesis to in vitro and ex vivo Biological Evaluations. ChemMedChem 2018, 13, 1131–1145. [Google Scholar] [CrossRef]
- Pettenuzzo, N.; Brustolin, L.; Coltri, E.; Gambalunga, A.; Chiara, F.; Trevisan, A.; Biondi, B.; Nardon, C.; Fregona, D. Cu II and Au III Complexes with Glycoconjugated Dithiocarbamato Ligands for Potential Applications in Targeted Chemotherapy. ChemMedChem 2019, 14, 1162–1172. [Google Scholar] [CrossRef]
- Liu, J.; Liao, X.; Xiong, K.; Kuang, S.; Jin, C.; Ji, L.; Chao, H. Boosting two-photon photodynamic therapy with mitochondria-targeting ruthenium-glucose conjugates. Chem. Commun. 2020, 56, 5839–5842. [Google Scholar] [CrossRef]
- Li, J.; Zeng, L.; Xiong, K.; Rees, T.W.; Jin, C.; Wu, W.; Chen, Y.; Ji, L.; Chao, H. A biotinylated ruthenium(ii) photosensitizer for tumor-targeted two-photon photodynamic therapy. Chem. Commun. 2019, 55, 10972–10975. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Karas, B.; Gírio, P.; Moreno, A.; Avecilla, F.; Marques, F.; Buckley, B.T.; Cooper, K.R.; Doherty, C.; Falson, P.; et al. Unprecedented inhibition of P-gp activity by a novel ruthenium-cyclopentadienyl compound bearing a bipyridine-biotin ligand. Eur. J. Med. Chem. 2019, 163, 853–863. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Karas, B.; Brás, A.R.; Pilon, A.; Avecilla, F.; Marques, F.; Preto, A.; Buckley, B.T.; Cooper, K.R.; Doherty, C.; et al. Ruthenium-Cyclopentadienyl Bipyridine-Biotin Based Compounds: Synthesis and Biological Effect. Inorg. Chem. 2019, 58, 9135–9149. [Google Scholar] [CrossRef]
- Paul, S.; Kundu, P.; Bhattacharyya, U.; Garai, A.; Maji, R.C.; Kondaiah, P.; Chakravarty, A.R. Ruthenium(II) Conjugates of Boron-Dipyrromethene and Biotin for Targeted Photodynamic Therapy in Red Light. Inorg. Chem. 2020, 59, 913–924. [Google Scholar] [CrossRef]
- Zhao, Z.; Gao, P.; You, Y.; Chen, T. Cancer-Targeting Functionalization of Selenium-Containing Ruthenium Conjugate with Tumor Microenvironment-Responsive Property to Enhance Theranostic Effects. Chem. Eur. J. 2018, 24, 3289–3298. [Google Scholar] [CrossRef]
- Ren, W.X.; Han, J.; Uhm, S.; Jang, Y.J.; Kang, C.; Kim, J.H.; Kim, J.S. Recent development of biotin conjugation in biological imaging, sensing, and target delivery. Chem. Commun. 2015, 51, 10403–10418. [Google Scholar] [CrossRef]
- Dutt Vadlapudi, A.; Krishna Vadlapatla, R.; Mitra, A.K. Sodium Dependent Multivitamin Transporter (SMVT): A Potential Target for Drug Delivery. Curr. Drug Targets 2012, 13, 994–1003. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Kardani, K.; Milani, A.H.; Shabani, S.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef]
- Neugebauer, U.; Pellegrin, Y.; Devocelle, M.; Forster, R.J.; Signac, W.; Moran, N.; Keyes, T.E. Ruthenium polypyridyl peptide conjugates: Membrane permeable probes for cellular imaging. Chem. Commun. 2008, 5307–5309. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Dolan, C.; Moriarty, R.D.; Martin, A.; Neugebauer, U.; Forster, R.J.; Davies, A.; Volkov, Y.; Keyes, T.E. Osmium(II) polypyridyl polyarginine conjugate as a probe for live cell imaging; A comparison of uptake, localization and cytotoxicity with its ruthenium(II) analogue. Dalt. Trans. 2015, 44, 14323–14332. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Burke, C.S.; Keyes, T.E. Precision targeted ruthenium(II) luminophores; Highly effective probes for cell imaging by stimulated emission depletion (STED) microscopy. Chem. Sci. 2016, 7, 6551–6562. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.S.; Byrne, A.; Keyes, T.E. Targeting Photoinduced DNA Destruction by Ru(II) Tetraazaphenanthrene in Live Cells by Signal Peptide. J. Am. Chem. Soc. 2018, 140, 6945–6955. [Google Scholar] [CrossRef]
- Burke, C.S.; Byrne, A.; Keyes, T.E. Highly Selective Mitochondrial Targeting by a Ruthenium(II) Peptide Conjugate: Imaging and Photoinduced Damage of Mitochondrial DNA. Angew. Chemie Int. Ed. 2018, 57, 12420–12424. [Google Scholar] [CrossRef]
- Cullinane, D.; Gkika, K.S.; Byrne, A.; Keyes, T.E. Photostable NIR emitting ruthenium(II) conjugates; uptake and biological activity in live cells. J. Inorg. Biochem. 2020, 207, 111032. [Google Scholar] [CrossRef]
- Burke, C.S.; Keyes, T.E. An efficient route to asymmetrically diconjugated tris(heteroleptic) complexes of Ru(ii). RSC Adv. 2016, 6, 40869–40877. [Google Scholar] [CrossRef]
- Köster, S.D.; Alborzinia, H.; Can, S.; Kitanovic, I.; Wölfl, S.; Rubbiani, R.; Ott, I.; Riesterer, P.; Prokop, A.; Merz, K.; et al. A spontaneous gold(i)-azide alkyne cycloaddition reaction yields gold-peptide bioconjugates which overcome cisplatin resistance in a p53-mutant cancer cell line. Chem. Sci. 2012, 3, 2062–2072. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, W.; Shi, H.; Chen, H.; Chai, X.; Liu, J.; Zhang, P.; Li, Z.; Zhang, Q. A HCBP1 peptide conjugated ruthenium complex for targeted therapy of hepatoma. Dalt. Trans. 2020, 49, 972–976. [Google Scholar] [CrossRef]
- Chakrabortty, S.; Agrawalla, B.K.; Stumper, A.; Vegi, N.M.; Fischer, S.; Reichardt, C.; Kögler, M.; Dietzek, B.; Feuring-Buske, M.; Buske, C.; et al. Mitochondria Targeted Protein-Ruthenium Photosensitizer for Efficient Photodynamic Applications. J. Am. Chem. Soc. 2017, 139, 2512–2519. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
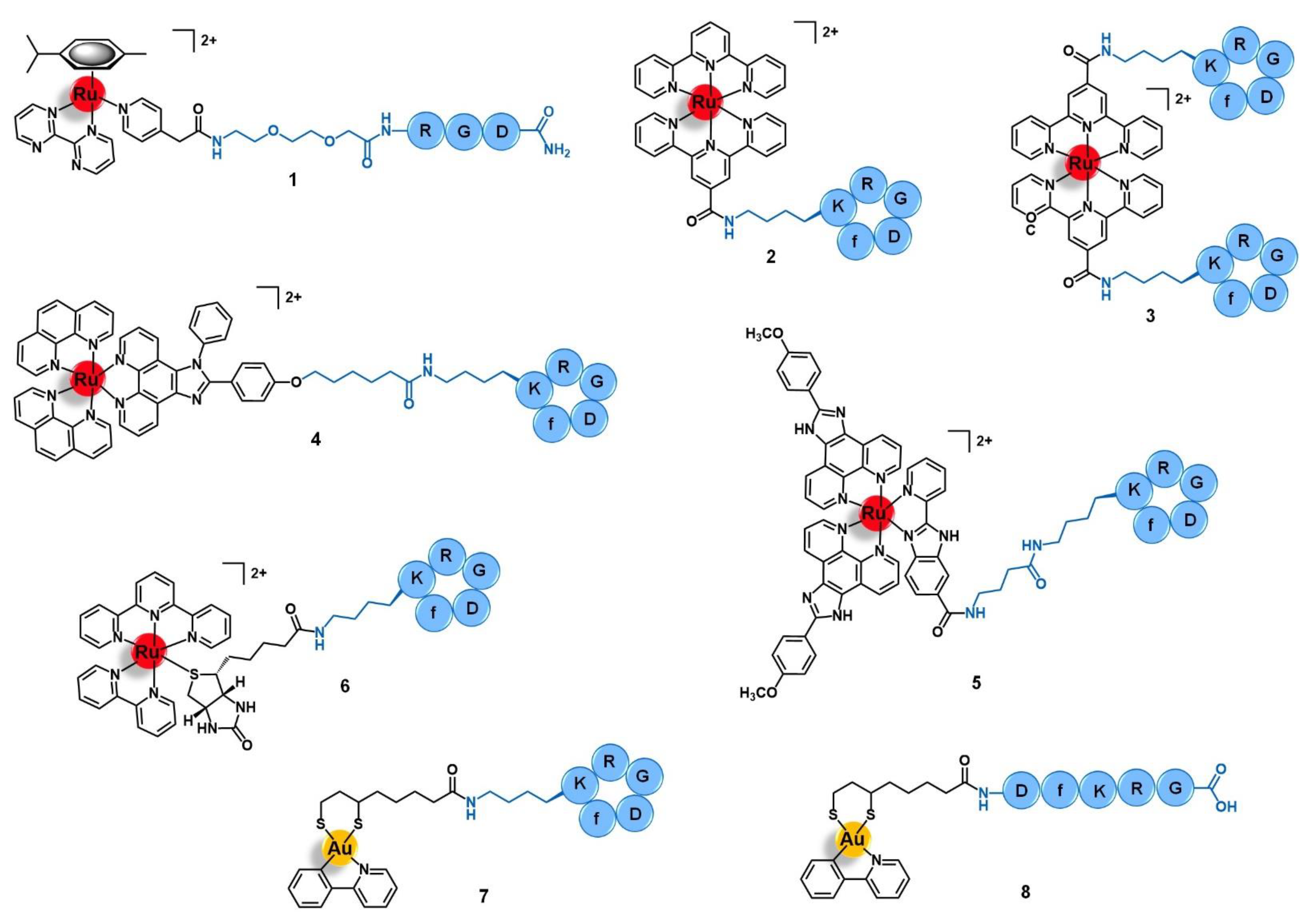{kind=link}
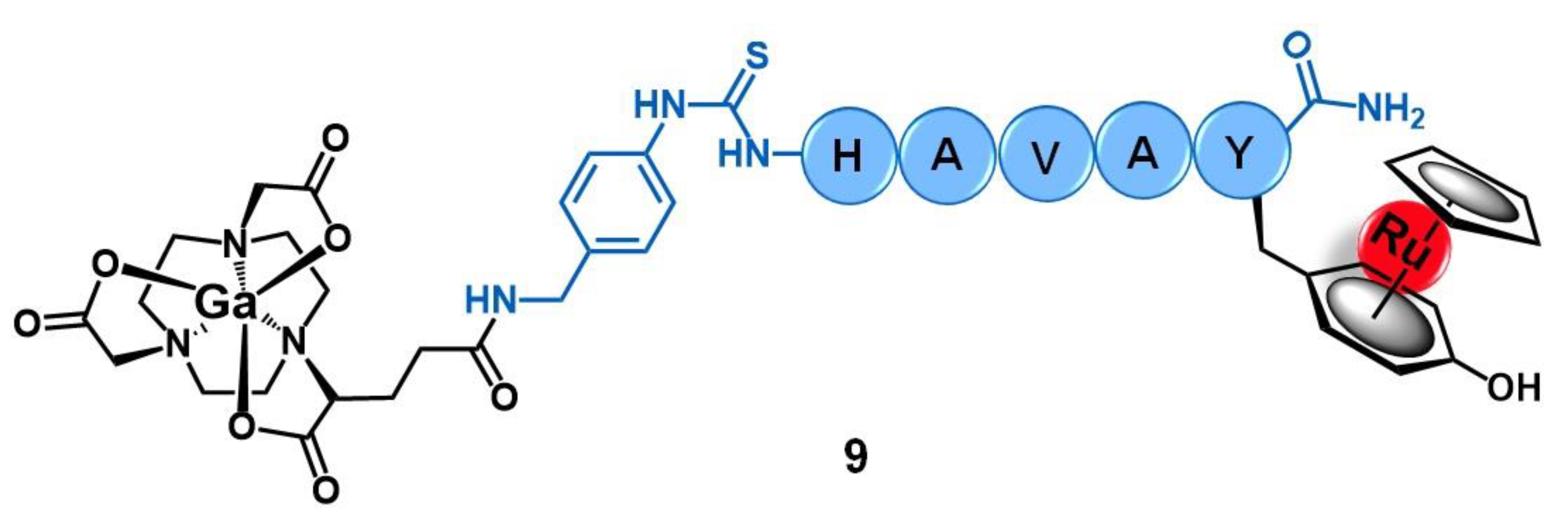{kind=link}
{kind=link}
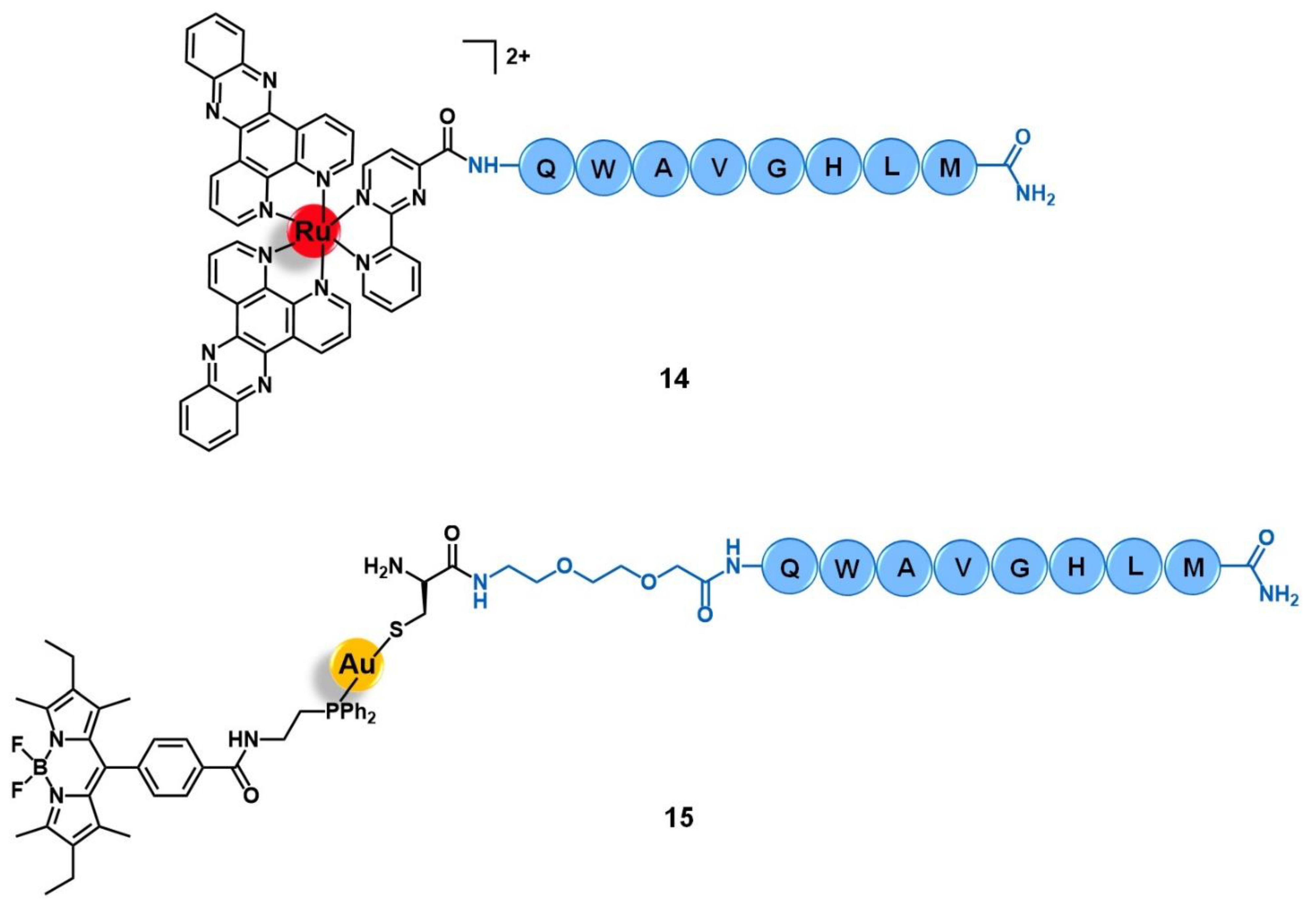{kind=link}
{kind=link}
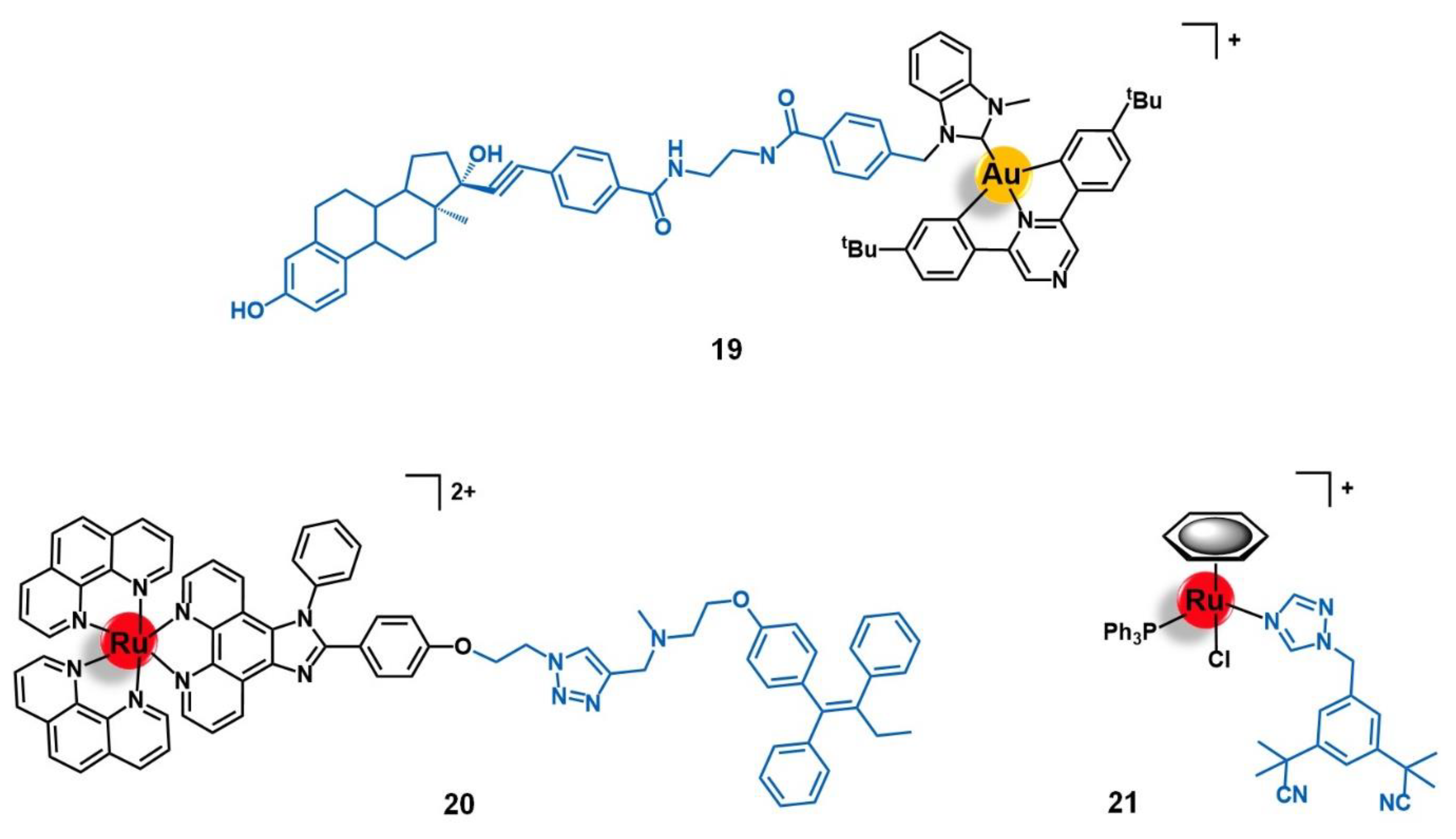{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Vector | Metal Complex | Ref. | |
|---|---|---|---|---|
| Cell Adhesion Molecules | Integrins | RGD | [Ru(h6-p-cym)(bpm)(pyac)]2+ | [45] |
| cyclo-RGDfK peptide | [Ru(terpy)(terpyCOOH)]2+ [Ru(terpyCOOH)2]2+ [Ru(phen)2(phenimi)]2+ [Ru(POP)2(pbiz)]2+ [Ru(terpy)(bipy)(D-biotin)]2+ [Au(ppy)(Lpa)] | [46,47,48,49,51,52] | ||
| DfKRG peptide | [Au(ppy)(Lpa)] | [52] | ||
| Cadherins | HAV peptide | 67Ga-NODAGA- [(η6-Tyr-RuCp)] | [57] | |
| G Protein-Coupled Receptors | Somatostatin receptors | AGCKNFFWKTFTSC peptide | [Ru(bipy)3]2+ | [64] |
| Cyclic-fCFwKTCT peptide | [Ru(η6-p-cym)(bpm)(pyac)]2+ [Ru(η6-p-cym)(dap)Cl]+ [Ru(η6-p-cym)(PPh3)(imbez)Cl]+ | [45,66] | ||
| Bombesin receptors | QWAVGHLM peptide | [Ru(dppz)2(CppH)]2+ [Au(DPPEB-BODIPY)Cl] | [69] | |
| Opioid receptors | leu-enkephalin peptide (YGGFL) | [Ru(η6-p-cym)(azapyr)Cl] | [72] | |
| met-enkephalin peptide (YGGFM) | [AuN3PPh3]+ [Ir(ppy)2(phenCOONa)] | [73] | ||
| G protein-coupled estrogen receptors | 17α-ethinylestradiol | [Au(bbfpz)(acbim)]+ | [77] | |
| Tamoxifen | Ru(phen)2(phenimi)]2+ | [78] | ||
| Anastrozole | [Ru(η6-C6H6)(PPh3)(η1-ATZ)Cl]+ | [79] | ||
| Growth Factors Receptors | Epidermal growth factor receptor | Tyrphostin (TYR) peptide | [Ru(η6-p-cym)(TYR)Cl2] [Ru(η6-toluene)(TYR)Cl2] | [84] |
| 4-anilinoquinazoline derivatives: AQZ, AcetAQZ | [Ru(η6-benzene)(enAQZ)Cl]+ [Ru(η6-p-cym)(enAQZ)Cl]+ [Ru(bipy)2(bipyCOOH)]2+ | [85,86,87,88] | ||
| erlotinib | [Au(erlotinib)(PPh3)] | [89] | ||
| Human epidermal growth factor receptor 2 | Trastuzumab antibody | [Au(PPh3)(DPTP)] [Au(PPh3)(MBANHS)] | [91] | |
| LTVSPWY peptide | [Au(ppy)(Lpa)] | [52] | ||
| Fibroblast growth factor receptor | Peptides: GPPDWHWKAMTH SRRPASFRTARE VSPPLTLGQLLS | [Ru(CpCOOH)(bipy)(PPh3)]2+ | 96 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, J.F.; Correia, J.D.G.; Morais, T.S. Emerging Molecular Receptors for the Specific-Target Delivery of Ruthenium and Gold Complexes into Cancer Cells. Molecules 2021, 26, 3153. https://doi.org/10.3390/molecules26113153
Machado JF, Correia JDG, Morais TS. Emerging Molecular Receptors for the Specific-Target Delivery of Ruthenium and Gold Complexes into Cancer Cells. Molecules. 2021; 26(11):3153. https://doi.org/10.3390/molecules26113153
Chicago/Turabian StyleMachado, João Franco, João D. G. Correia, and Tânia S. Morais. 2021. "Emerging Molecular Receptors for the Specific-Target Delivery of Ruthenium and Gold Complexes into Cancer Cells" Molecules 26, no. 11: 3153. https://doi.org/10.3390/molecules26113153
APA StyleMachado, J. F., Correia, J. D. G., & Morais, T. S. (2021). Emerging Molecular Receptors for the Specific-Target Delivery of Ruthenium and Gold Complexes into Cancer Cells. Molecules, 26(11), 3153. https://doi.org/10.3390/molecules26113153