2.1. Structural Analysis
In this section, we report the crystal structure analysis for all the investigated systems accompanied by the computational QTAIMS analysis aimed at quantitatively describe the localized HBs and XBs. To this purpose, we have first investigated the actual existence of bond critical points (rank 3 and signature −1), hereafter simply termed as CPs, in correspondence to the pair interactions that emerged from the crystallographic structural analysis. Subsequently, for each CP, we have evaluated the topological and energetic features following the criterion by Espinosa et al. [
56,
57]. For the halogen bond interactions, the energy was estimated using a slightly different approach proposed by Tsirelson [
58]. The balance between the Lagrangian kinetic energy G(
r) and the potential energy density V(
r) at the CP were then utilized for quantifying the noncovalent character (−G/V > 1) of all the corresponding pair interactions.
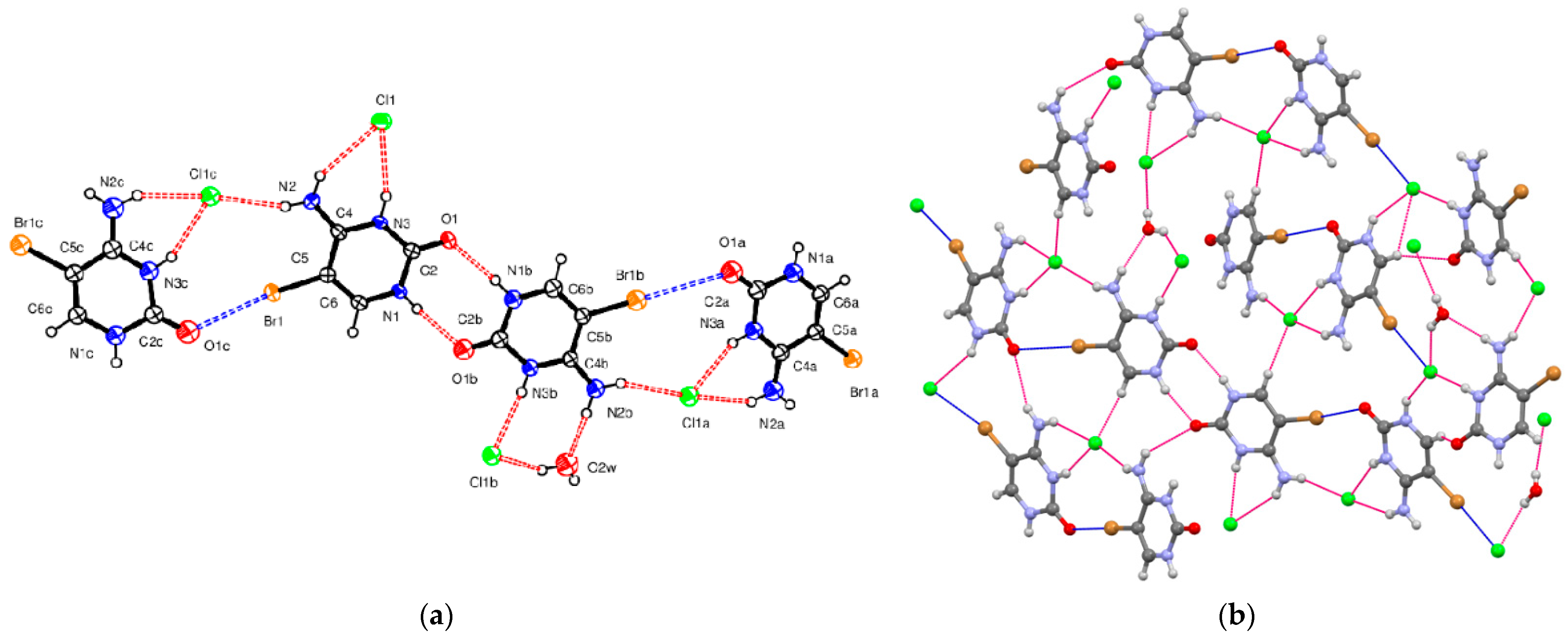
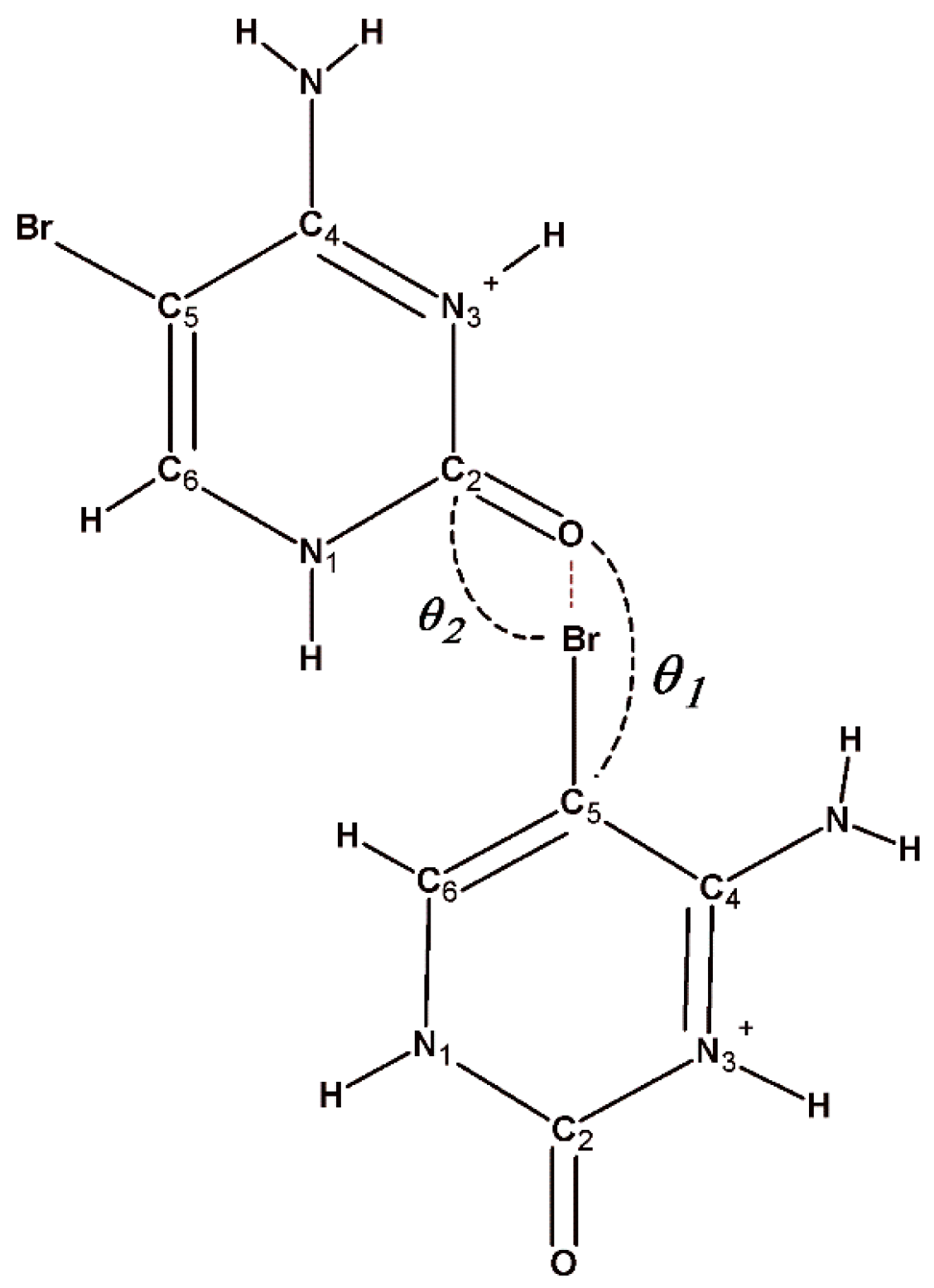
5-Bromocytosinium chloride, (
I), crystallizes in the orthorhombic space group Pbca in the cation/anion ratio 1:1, but the asymmetric unit consists of four 1
H,3
H-5-bromocytosinium cations protonated at the N3 atom, four chloride anions and one water molecule of crystallization joined together to form one-dimensional multicomponent planar aggregate (
Figure 1a). The occurrence of more than one molecule in the asymmetric unit of ionic compounds is rather unusual [
59,
60], but a similar situation for N
imino-protonated 5-bromocytosines have been encountered in the crystal structure of NUZKEY, which crystallizes with three 5-bromocytosinium cations, a hexachloro-platinum(IV) anion, one chloride anion and a water molecule in the asymmetric unit.
In the crystal packing of (
I), shown in
Figure 1b, in the planar aggregates, a pair of 5-bromocytosinium cations are linked together by N–H···O hydrogen bonds from the CONH pyrimidinium groups forming a cyclic hydrogen-bonded ring motif represented by graph-set notation R
22(8) [
61]. These ion pairs are further connected into a zigzag chain via N–H
… Cl
− hydrogen bonds and two almost linear C–Br
…O contacts (3.03 Å, 165°, and 3.26 Å, 175°) involving the carbonyl oxygen atom of adjacent peripheral 5-bromocytosinium cations, to give 1D chains. The XB ratios
RXB vary between 0.90 and 0.96, indicating weak interaction strength. In this respect, the XB interaction energies (between 1.5 and 2.0 kcal/mol) and −G(
r)/V(
r) ratio (between 1.2 and 1.3), evaluated based on topological analysis on the corresponding CPs, clearly confirm the low, but not negligible strength of these interactions, as well as their genuine noncovalent character. These chains are then united by N–H
…Cl
−, N
+–H
…Cl
−, N–H
…O
w and O
w–H
…Cl
− hydrogen bonds and two C–Br
…Cl
− contacts involving Cl
− ions (3.07 Å, 171°,
RXB = 0.86 and 3.22 Å, 165°,
RXB = 0.90, respectively) into sheets. In correspondence with the latter interactions, CPs have been localized by the topological analysis with an estimated interaction energy of 1.2 kcal/mol. No relevant intermolecular HBs involving the Br atom were observed (
Table 1). From the topological analysis, all the HBs turn out to be characterized by −G(
r)/V(
r) close to unity with interaction energies varying from 3.0 to 10.0 kcal/mol. In particular, rather strong interactions are found for all the N
+–H
…Cl
−, and although to a lesser extent, even N–H
…Cl
− HBs. Rather surprisingly, our calculations have revealed significantly E
int (Internal Energy) values also for the HBs involving uncharged N1–H1
…O1B and N1B–H1B
…O1. The latter result can be partially rationalized based on the relatively high value of the Laplacian of the density observed for the corresponding pair interactions.
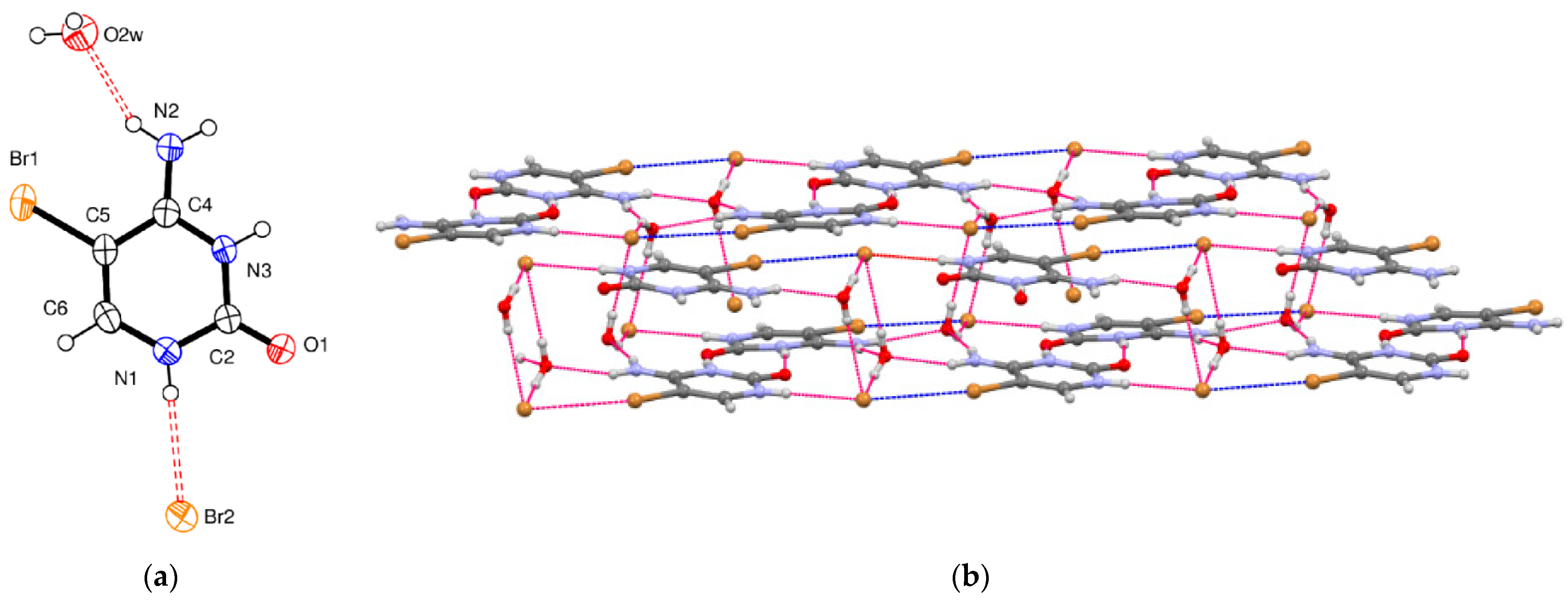
5-Bromocytosinium bromide, (
II), crystallizes in the monoclinic space group
C2/
c, and the asymmetric unit consists of one 1
H,3
H-5-bromocytosinium cation protonated at the N3 atom, one bromide anion, and one water molecule of crystallization (
Figure 2a).
The packing of (
II) is shown in
Figure 2b. Pairs of 5-bromocytosinium cations are linked together by equivalent N
+–H···O hydrogen bonds, which are significantly shorter than normal N–H
…O intermolecular interactions (
Table 2). These pairs then assemble in layers held together by N–H
…O
w of R
24(8) graph motif involving the amino group of one protonated nucleobase and a nearby oxygen atom of a water molecule, and by N–H
…Br
− and O
w–H
…Br
− hydrogen bonds to reconstruct the 3D network. All the HBs found in the crystal are confirmed by the topological analyses, which also indicates interaction energies in the range 3.0–9.0 kcal/mol. C–Br
…O interactions do not emerge from the crystal structure analysis nor the topological analysis. On the other hand, weak (
RXB = 0.96) and almost linear C–Br
…Br
− contacts (3.52 Å, 160°) involving Br
− counterions are present in the crystal and are also localized and characterized by topological analysis as relatively weak (E
int = 1.5 kcal/mol) and markedly noncovalent interactions (−G(r)/V(r) > 1). Moreover, in this case, as expected, the strongest interactions turn out to be in correspondence with the HBs involving the N
+–H moiety.
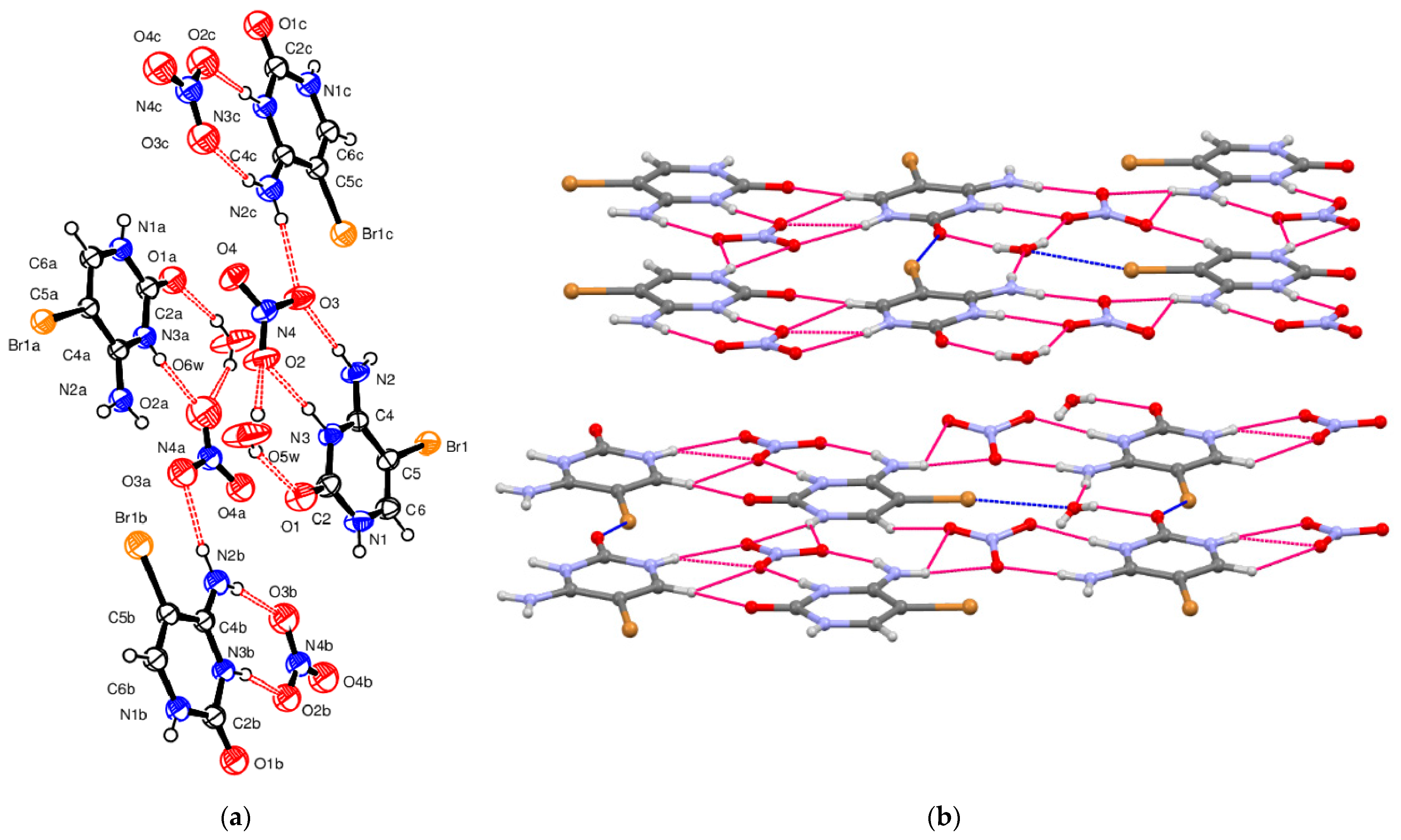
5-Bromocytosinium nitrate, (
III), crystallizes in the triclinic space group
P-1 in the cation/anion ratio 1:1. As already mentioned above, for compound (
I) and NUZKEY, multiple molecules are present in the asymmetric unit. Four 1
H,3
H-5-bromocytosinium cations protonated at the N3 atom, four nitrate anions, and two water molecules of crystallization are joined together to form a multicomponent aggregate (
Figure 3a).
The packing of compound (
III) is shown in
Figure 3b. The ions assemble in layers through N–H
…O
− and N
+–HO
− hydrogen bonds (
Table 3). For all of these HBs, the topological analysis has allowed us to locate as many CPs. The corresponding values of −G(r)/V(r) and E
int, falling in the range 0.9–1.2 and 3.0–8.0 kcal/mol, respectively, describe these pair interactions as genuine noncovalent and rather stable HBs. As expected, the highest E
int values are found in correspondence with the N
+–H
…O interactions. There are two kinds of C–Br
…O interactions (see also
Table S6 in the Supplementary Information). The first one involves two carbonyl oxygen atoms (3.05 Å, 157°,
RXB = 0.90 and 3.16 Å, 150°,
RXB = 0.94, respectively), and according to QTAIMS analysis, is characterized by a rather high –G(
r)/V(
r) values (1.20 and 1.22) and by an appreciable interaction energy (3.6 and 2.4 kcal/mol). The second kind of C–Br
…O interactions involves two oxygen atoms of water molecules (2.97 Å, 164°,
RXB = 0.76 and 3.03 Å, 161°,
RXB = 0.78, respectively), and similarly to the previous case, has revealed an appreciable interaction energy of 2.9 and 2.1 kcal/mol, respectively. No relevant intermolecular HBs involving the Br atom were observed. On the other hand, topological analysis has also located (see
Table S7 in the Supplementary Information) quantitatively relevant noncovalent interlayers interactions (−G(
r)/V(
r) close to 1.0) involving N4, O2, and O3 atoms of nitrate anions with a relatively high E
int (between 7.0 and 8.0 kcal/mol).
5-Bromocytosinium sulfate, (
IV), crystallizes in the monoclinic space group
P2
1/
c, and the asymmetric unit consists of one 1
H,3
H-5-bromocytosinium cation protonated at the N3 atom, one sulfate anion, and one protonated water molecule of crystallization (
Figure 4a). Short S–O bonds (1.458–1.483 (1) Å) in the slightly distorted tetrahedral sulfate anion indicate the absence of H atoms and delocalization of negative charge between them.
In the crystal structure of compound (
IV), the ionic components are joined together essentially by two kinds of short-contact interactions. The first one corresponds to N–H
…O
− hydrogen bonds involving the oxygen atoms of the sulfate anion. These interactions were also localized by QTAIMS analysis and characterized by interaction energy between 5.0 and 7.0 kcal/mol. The second type of interaction corresponds to an almost linear (2.96 Å, 175°) and rather weak (
RXB = 0.88) C–Br
…O halogen bond involving bromine and carbonyl oxygen atoms of two adjacent 5-bromocytosinium cations, to give 1D zigzag chains (
Figure 4b). The latter interaction has been indeed described by the topological analysis, and results in an interaction energy of 2.1 kcal/mol and a G(r)/V(r) value slightly larger than 1.20. These chains are then connected by N–H
…O
− and N
+–H
…O
− hydrogen bonds with 1D chains formed by alternating sulfate and hydronium ions linked by two very strong O
w–H
…O
− interactions (
Table 4) to form 2D layers. A short contact, C6–H6
…O
5 [3.169 (2) Å] has been confirmed by topological analysis showing a relatively low E
int (1.7 kcal/mol). The 2D layers, thus, formed are themselves linked into a three-dimensional network by the remaining very strong O
w–H
…O
− hydrogen bond. More precisely, the analysis of this part of the structure reveals a peculiar interaction characterized by an H
3O
+ moiety coordinating three sulfate anions, as also described in
Table S10 in the Supporting Information, with strong HBs contacts whose interaction energies parallel the −G(
r)/V(
r) values of 0.90, 0.59 and 0.69. No relevant intermolecular HBs involving the Br atom were observed.
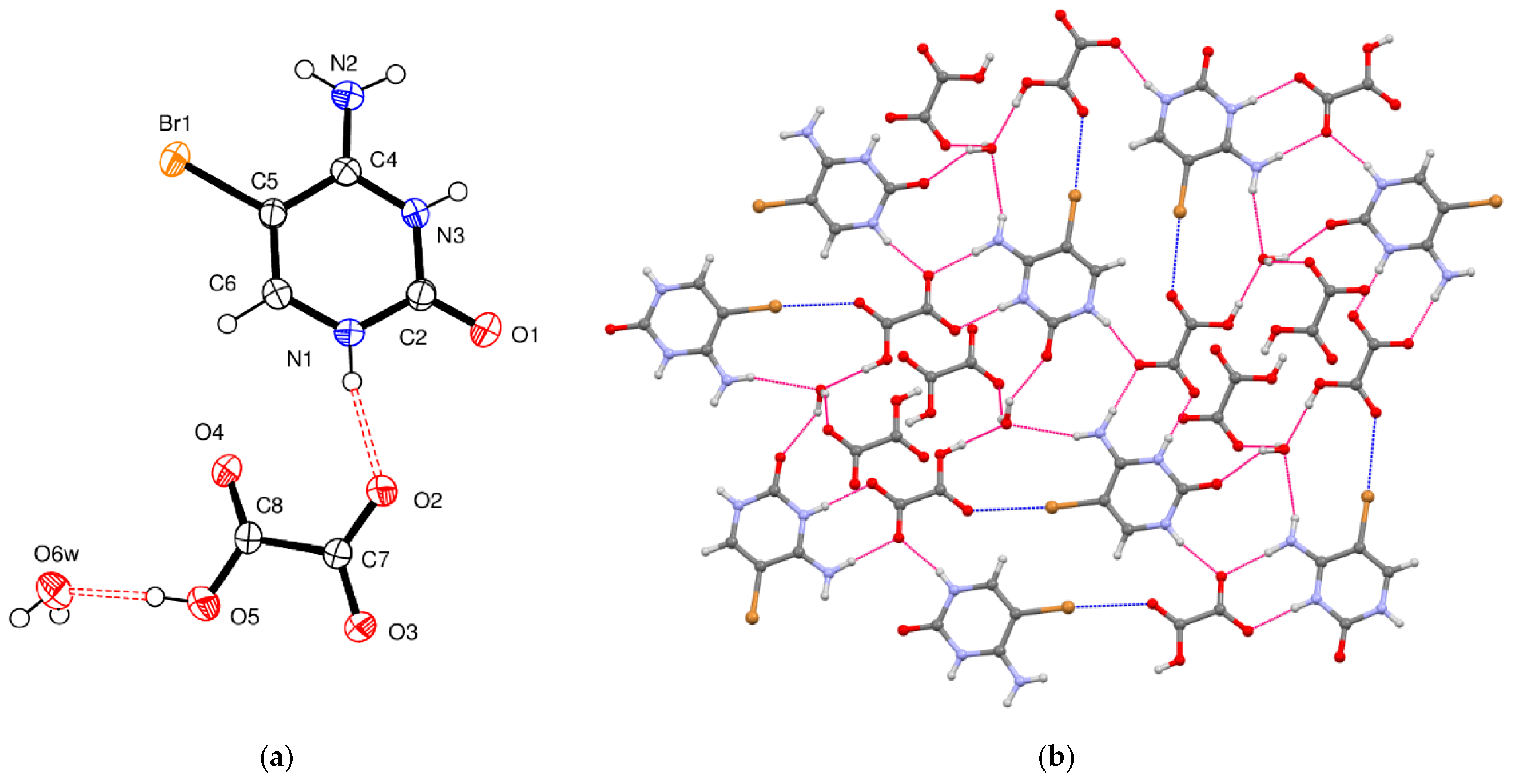
5-Bromocytosinium dihydrogen monophosphate, (
V), crystallizes in the monoclinic space group
P2
1/
c, and the asymmetric unit contains one 1
H,3
H-5-bromocytosinium cation protonated at the N3 atom and one dihydrogen monophosphate anion (
Figure 5a).
The single deprotonation of the phosphoric acid is reflected in the bond distances involving the P1–O bonds in the slightly distorted tetrahedral phosphate group. There are two short P1–O bonds of 1.493 (2) and 1.503 (2) Å to terminal atoms O2 and O3, respectively, and two longer bonds of 1.564 (2) and 1.569 (2) Å to atoms O4 and O5, bearing H atoms. These values correlate well with those reported for dihydrogen monophosphate anion in the crystal structure of 6-methylisocytosinium dihydrogen monophosphate adduct [
51]. The similarity of P1–O2 and P1–O3 bond distances suggests delocalization of negative charge between the two bonds.
The supramolecular structure of compound (
V) is based on a vast three-dimensional hydrogen-bonded network, which includes a combination of rather strong O–H
…O and N–H
…O hydrogen bonds (
Figure 5b), as confirmed by the topological analysis. The hydrogen-bonding scheme involves all available donor/acceptor sites, but carbonyl O1, which remains partially unsaturated (
Table 5). Within the cation chains, adjacent 5-bromocytosinium ions are linked head-to-tail through N–H
…O hydrogen bonds, so forming infinite chains. Each dihydrogen monophosphate ion participates in five N–H
…O and O–H
…O hydrogen bonds with neighboring cations and anions as acceptor and in two O–H
…O hydrogen bonds with neighboring anions as a donor. Through these interactions, dihydrogen monophosphates form pairs, all showing a rather high E
int, of infinite chains cross-linked to cation chains. An approximately linear (3.04 Å, 171°) and rather weak (
RXB = 0.90) C–Br
…O halogen bond involving the O5 oxygen atom connects pairs of 5-bromocytosinium and dihydrogen monophosphate ions. Similar XB interactions (3.00–3.01 Å, 172–173°) have been observed in the crystal structures of U2AF
65-RNA complexes (U2AF = U2 auxiliary protein factor), where 5-bromouracil units are implicated in intramolecular XBs between Br atoms and O atoms belonging to nearby phosphate anions [
62,
63]. Topological analysis carried out on the latter contact has revealed interaction energy of 2.2 kcal/mol and a genuinely noncovalent character (−G(r)/V(r) > 1.2).
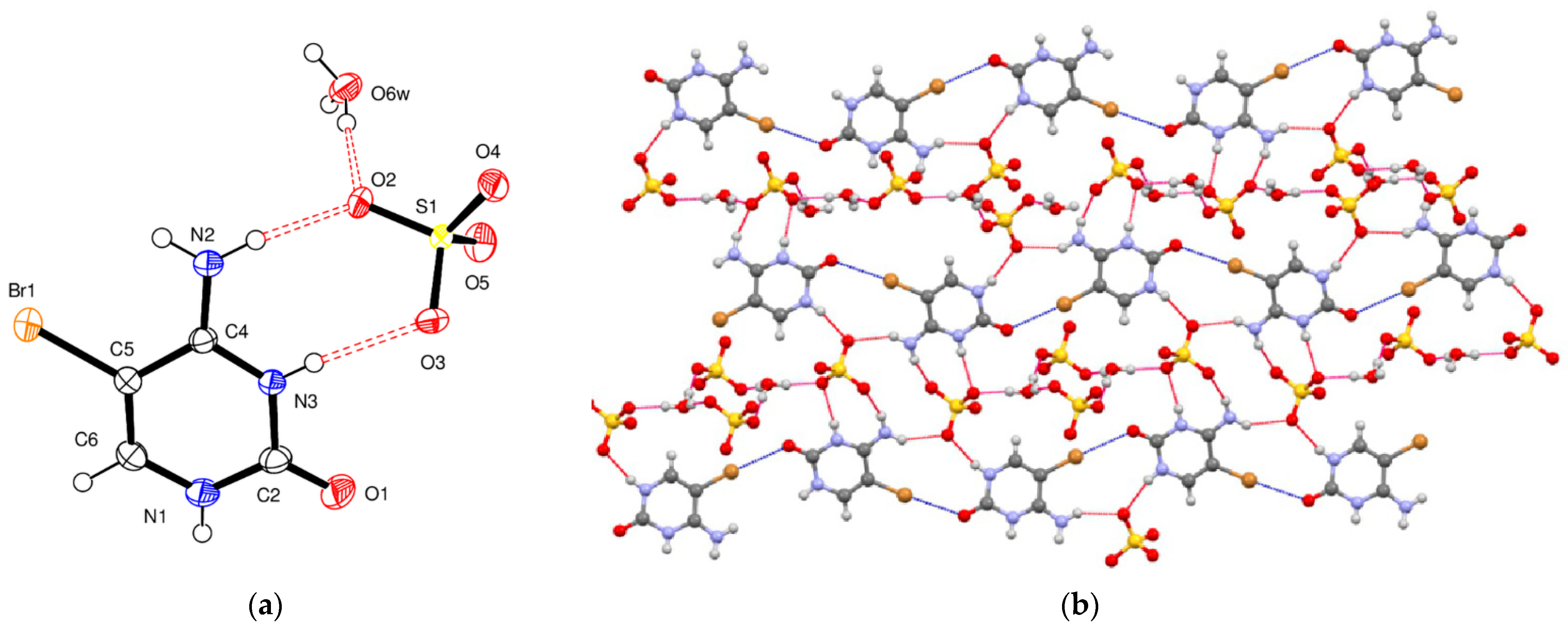
5-Bromocytosinium hydrogen oxalate, (
VI), crystallizes in the monoclinic space group
P2
1/
c, and the asymmetric unit consists of one 1
H,3
H-5-bromocytosinium cation protonated at the N3 atom, one hydrogen oxalate anion, and one water molecule of crystallization (
Figure 6a). The semi-oxalate anion is almost planar, as the dihedral angle formed by the planes defined by the CO
2H and CO
2− non-H atoms is 5.2 (4)°. Bond distances around atom C7 [1.248–1.250 (3) Å] are typical of a carboxylate group, and the differences between them of 0.002 (3) Å indicates delocalization of the negative charge between the two O atoms. Bond distances around atom C8 (1.209(4) and 1.300 (4) Å) are consistent with the presence of a carboxylic acid group.
The crystal structure of compound (
VI) is dominated by different structurally significant hydrogen bonds, formed by neutral and charged N–H
…O and O–H
…O interactions (
Figure 6b). Two semi-oxalate anions are engaged in hydrogen bonds with two 5-bromocytosinium cations and two water molecules, forming an R
66(22) hydrogen-bonded ring. This subunit is then linked into a two-dimensional network by multiple hydrogen bonds, whose E
int is relatively strong by QTAIMS analysis, and a weak C–Br
…O halogen bond (2.97 Å, 166°,
RXB = 0.88) between 5-bromocytosinium cations and hydrogen oxalate anions. This latter interaction is characterized by a relatively high −G(r)/V(r) value (1.20) and a value on E
int of 2.1 kcal/mol. As previously mentioned, water molecules play an important role in the cohesion of the crystal structure, as they maximize HB interactions with all potential donors and acceptors utilized. They form one N–H
…O
w and one O–H
…O
w hydrogen bonds connecting one 5-bromocytosinium cation and one semi-oxalate anion as donors, and two O
w–H
…O hydrogen bonds connecting one 5-bromocytosinium cation and one semi-oxalate anion as acceptors (
Table 6). No relevant intermolecular HBs involving the Br atom were observed.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}