
Enhancement of the Antioxidant and Antimicrobial Activities of Porphyran through Chemical Modification with Tyrosine Derivatives
 ,
,  , ,
, ,  , , and
, , and
Abstract
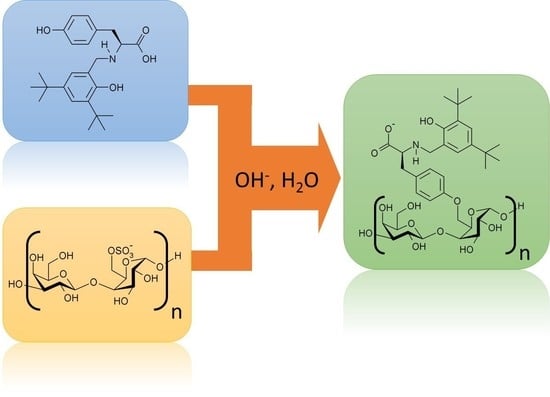
1. Introduction
2. Results and Discussion
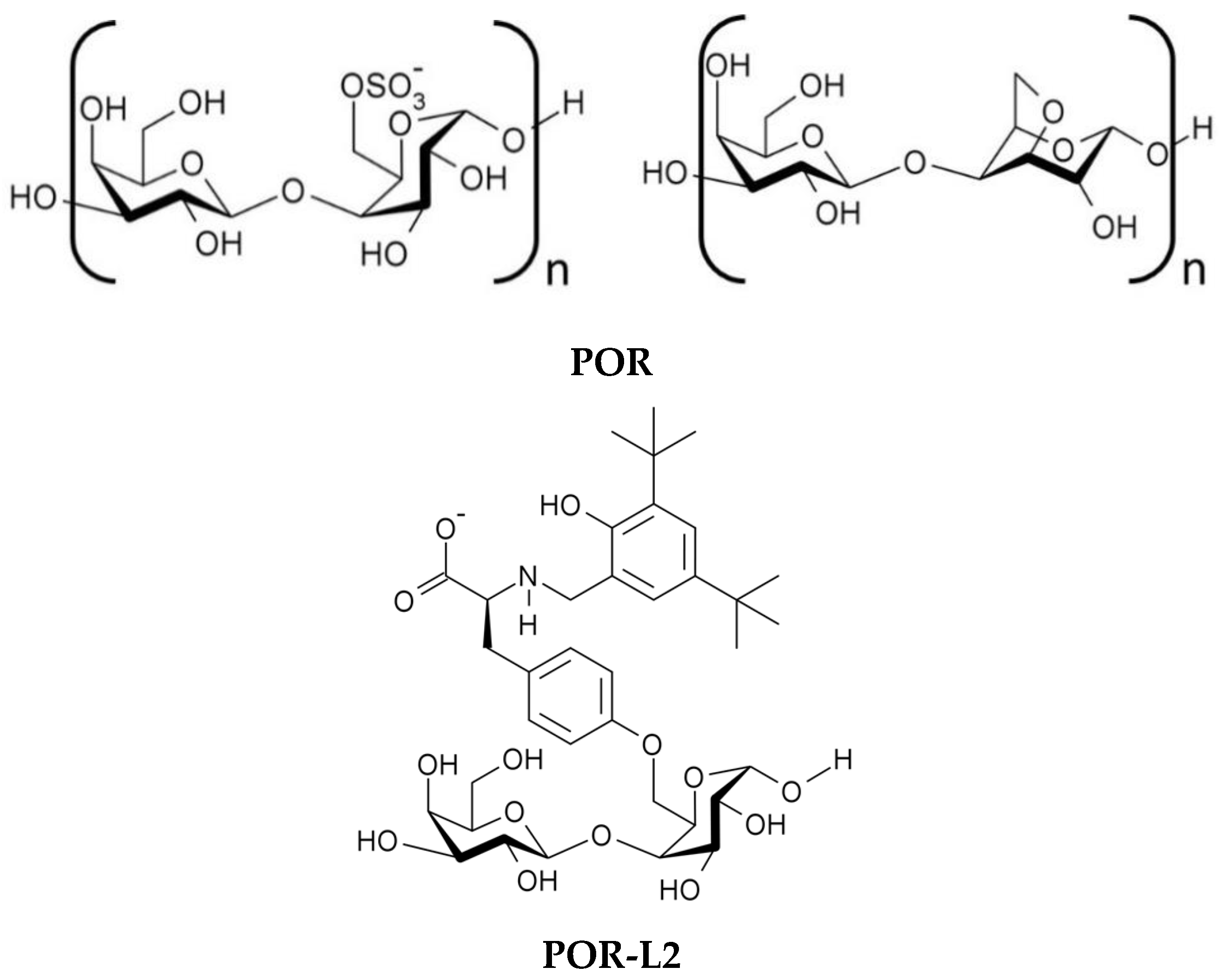
2.1. Extraction of Porphyran (POR) from Porphyra dioica
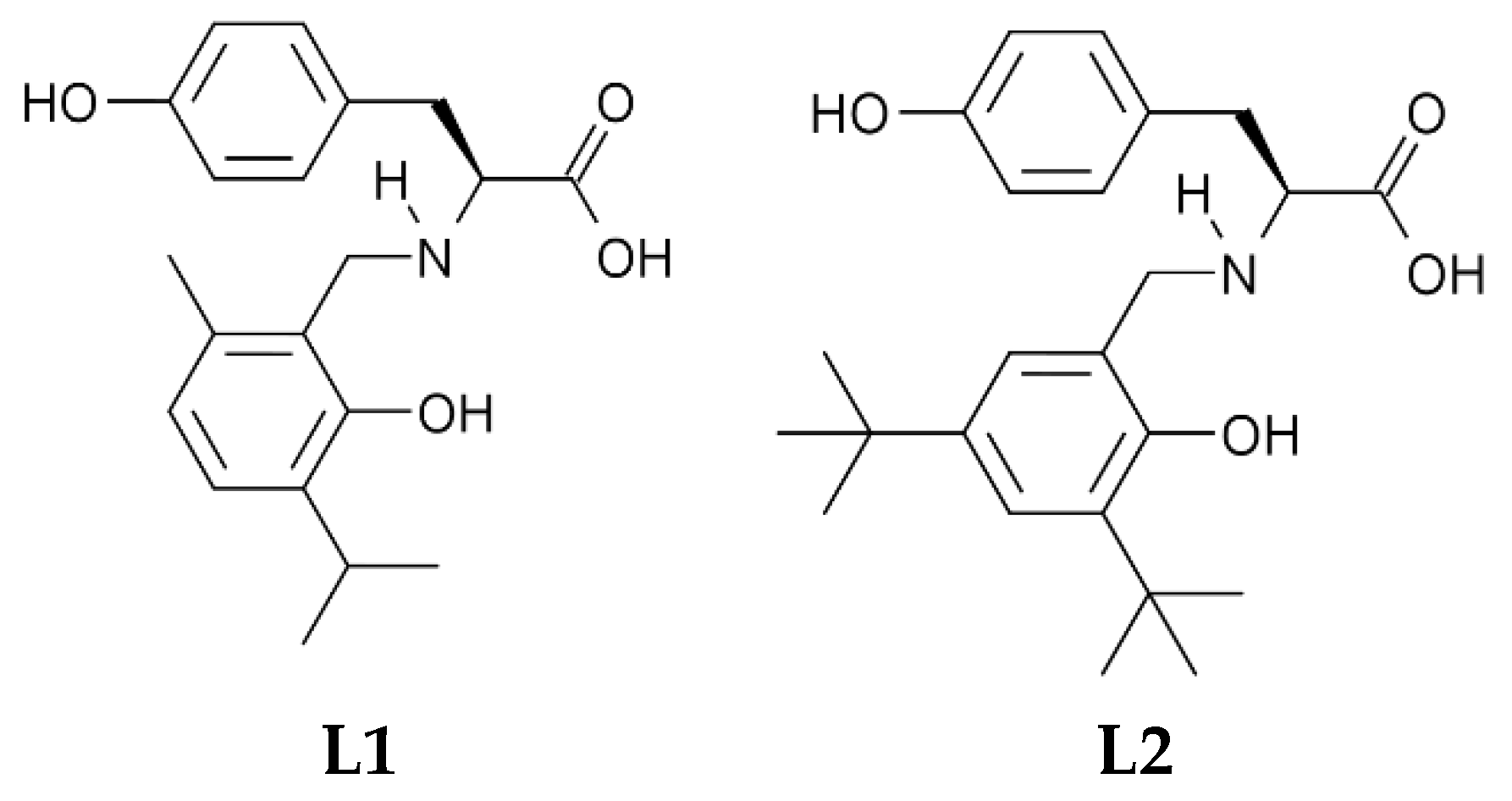
2.2. Synthesis of Modified Tyrosine
2.3. Treatment of Porphyran with the Modified Tyrosines
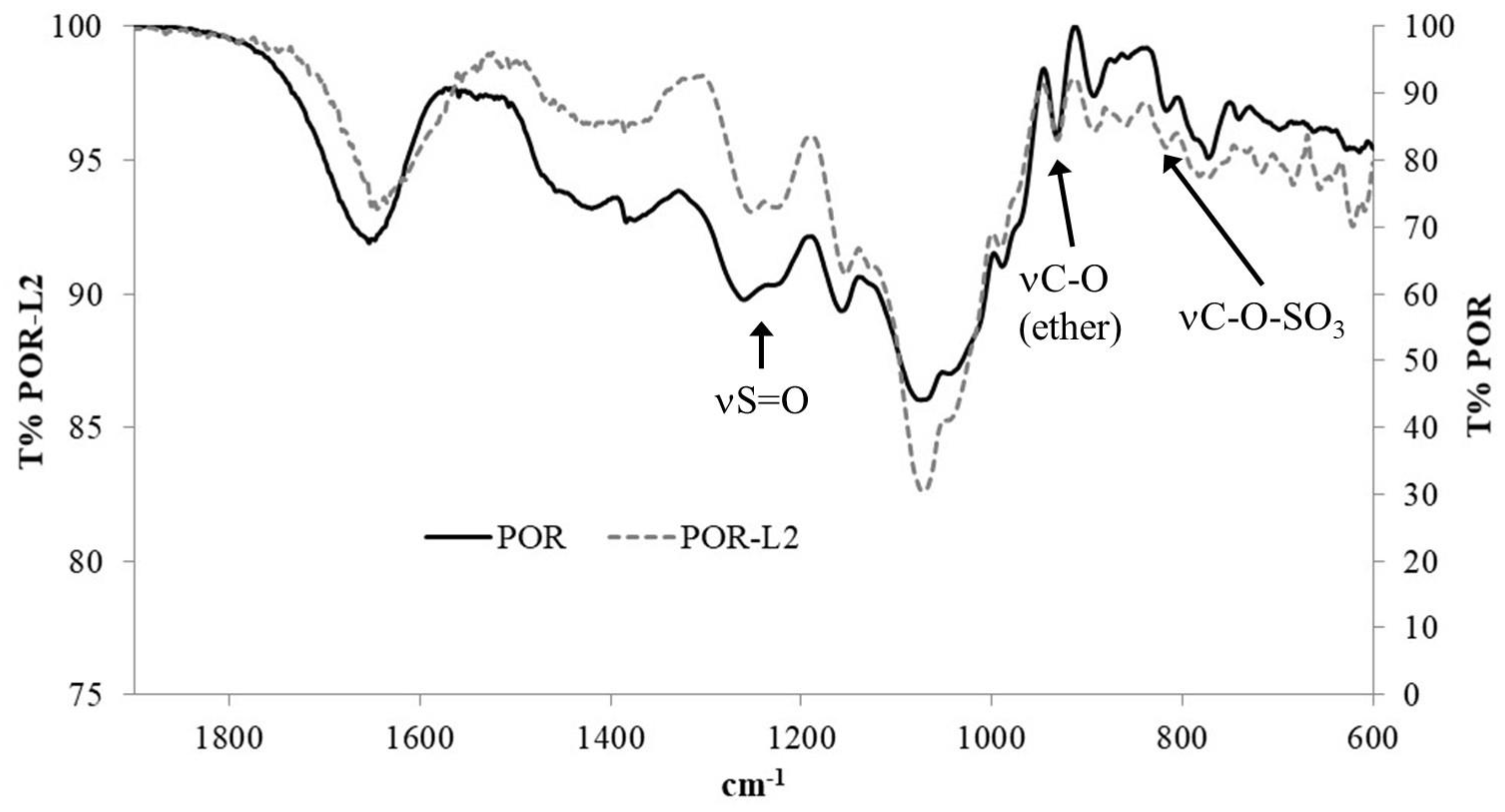
2.3.1. FTIR Analysis
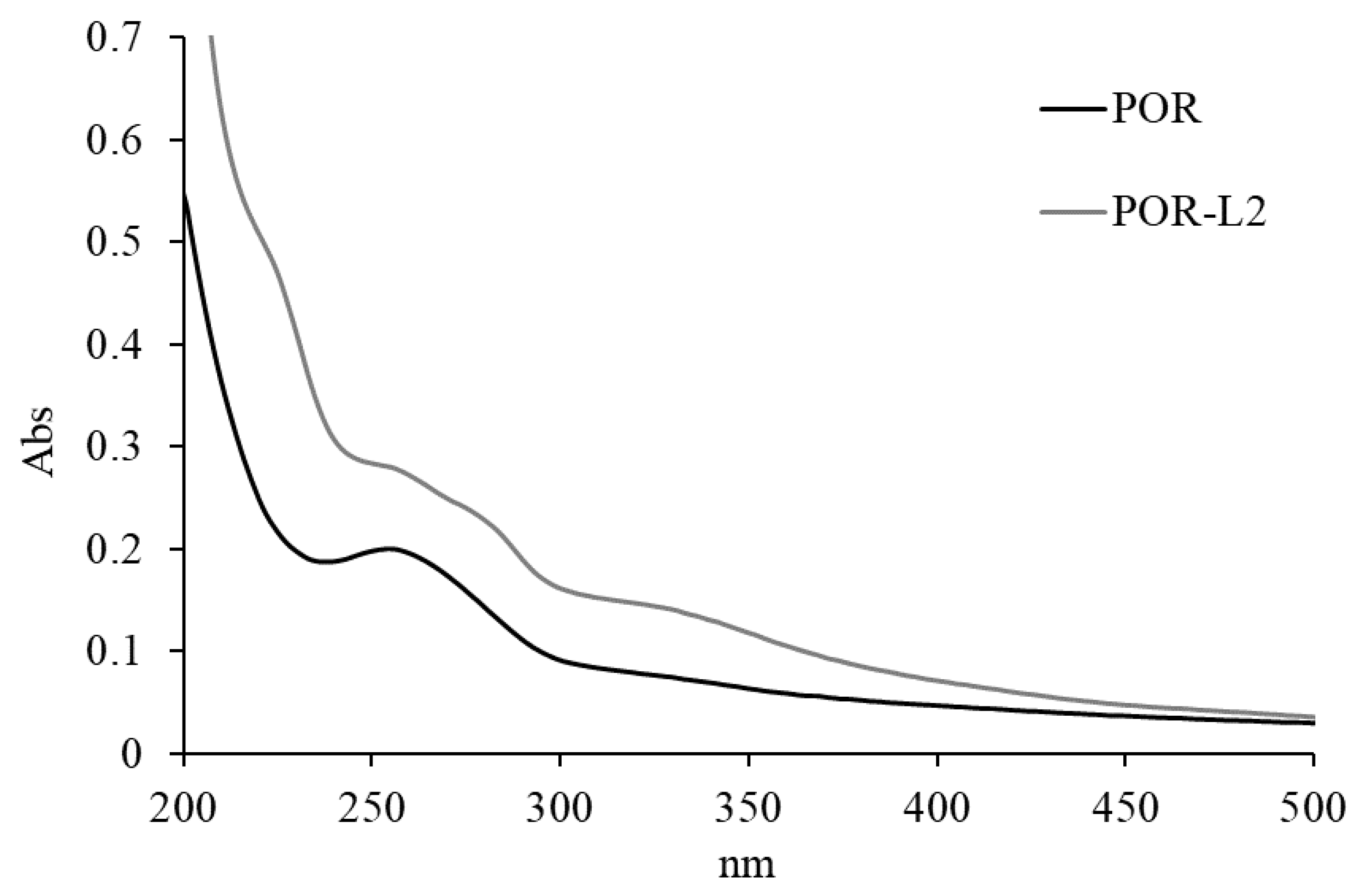
2.3.2. UV–Vis Analysis
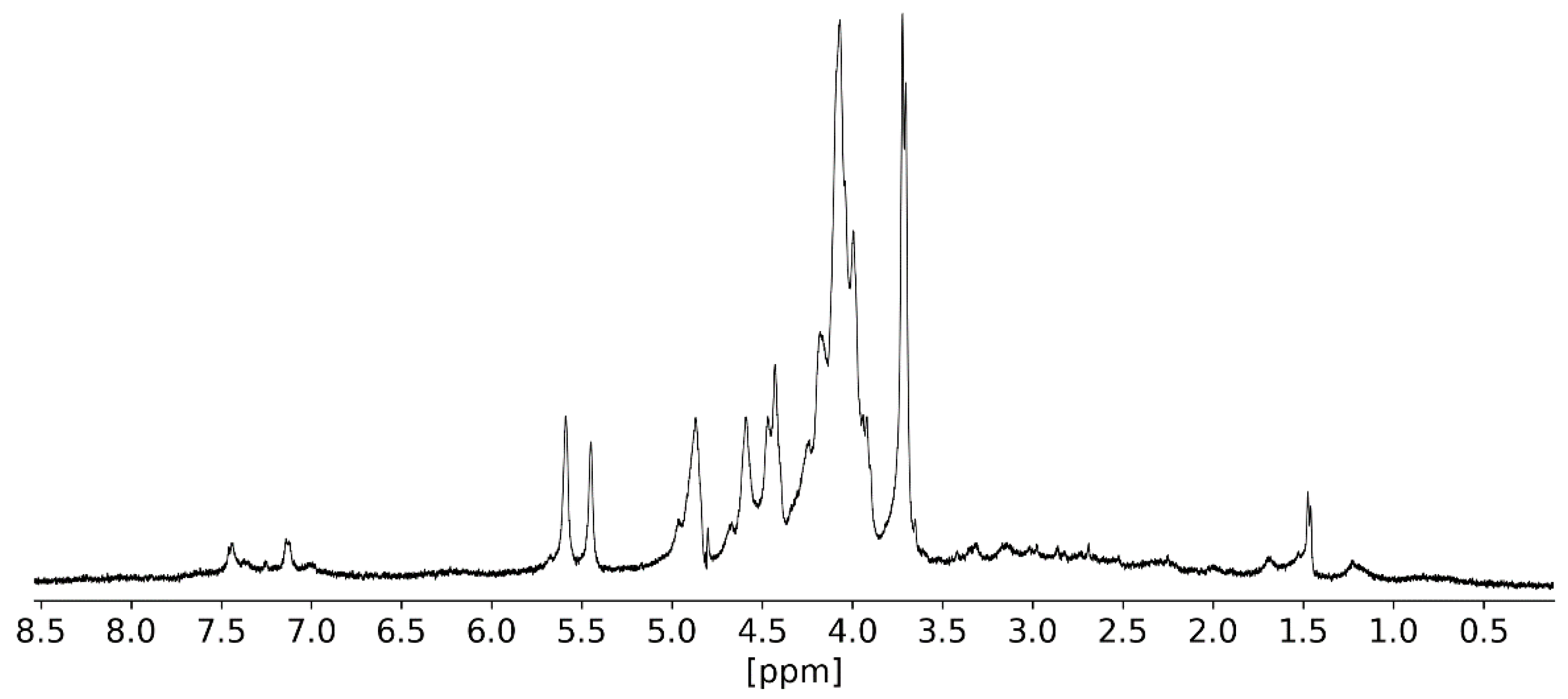
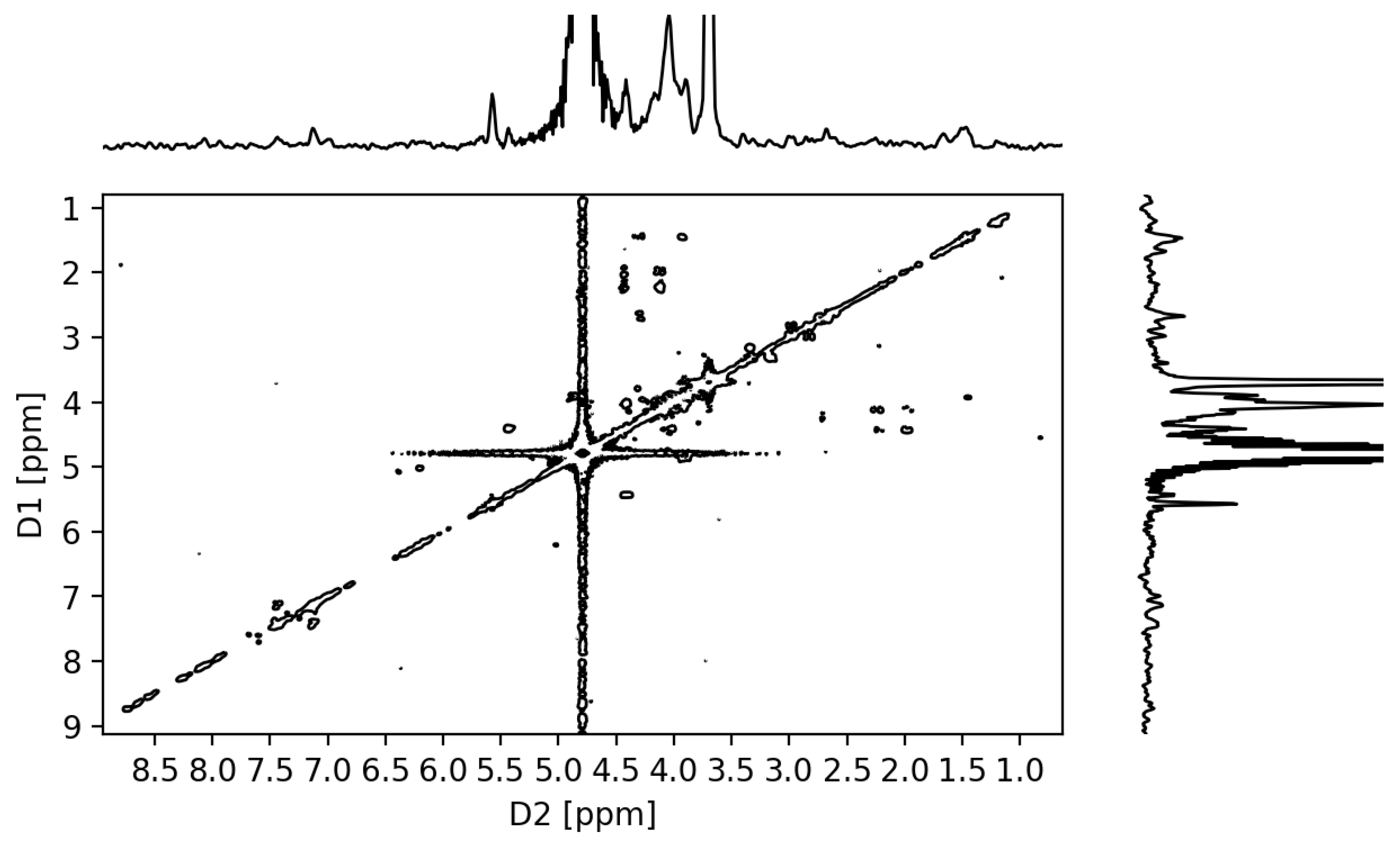
2.3.3. NMR Analysis
2.3.4. Elemental Analysis
2.4. Determination of Antioxidant Activity
2.4.1. Ferric Reducing Activity
2.4.2. DPPH and ABTS•+ Scavenging Activity
2.4.3. Suppression of the Na[FeEDTA]-Catalysed Oxidation of Methyl Red Dye with H2O2
2.4.4. Suppression of the Aerobic Oxidation of Methyl Red Dye with the Na[FeEDTA]/AcOH/Benzaldehyde Oxidant System
2.5. Microstructural Features of Produced Films
2.6. Antimicrobial Activity
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Vera, J.; Castro, J.; Gonzalez, A.; Moenne, A. Seaweed Polysaccharides and Derived Oligosaccharides Stimulate Defense Responses and Protection against Pathogens in Plants. Mar. Drugs 2011, 9, 2514–2525. [Google Scholar] [CrossRef]
- Suganya, A.M.; Sanjivkumar, M.; Chandran, M.N.; Palavesam, A.; Immanuel, G. Pharmacological Importance of Sulphated Polysaccharide Carrageenan from Red Seaweed Kappaphycus Alvarezii in Comparison with Commercial Carrageenan. Biomed. Pharmacother. 2016, 84, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.B.; Frota, A.F.; Silva, J.; Alves, C.; Neugebauer, A.Z.; Pinteus, S.; Rodrigues, J.A.G.; Cordeiro, E.M.S.; de Almeida, R.R.; Pedrosa, R.; et al. In Vitro Activities of Kappa-Carrageenan Isolated from Red Marine Alga Hypnea Musciformis: Antimicrobial, Anticancer and Neuroprotective Potential. Int. J. Biol. Macromol. 2018, 112, 1248–1256. [Google Scholar] [CrossRef]
- Madruga, L.Y.C.; Sabino, R.M.; Santos, E.C.G.; Popat, K.C.; Balaban, R.D.C.; Kipper, M.J. Carboxymethyl-Kappa-Carrageenan: A Study of Biocompatibility, Antioxidant and Antibacterial Activities. Int. J. Biol. Macromol. 2020, 152, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Rocha de Souza, M.C.; Marques, C.T.; Guerra Dore, C.M.; Ferreira da Silva, F.R.; Oliveira Rocha, H.A.; Leite, E.L. Antioxidant Activities of Sulfated Polysaccharides from Brown and Red Seaweeds. J. Appl. Phycol. 2007, 19, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S. Carrageenan for Encapsulation and Immobilization of Flavor, Fragrance, Probiotics, and Enzymes: A Review. J. Carbohydr. Chem. 2017, 36, 1–19. [Google Scholar] [CrossRef]
- Rostamnia, S.; Zeynizadeh, B.; Doustkhah, E.; Baghban, A.; Aghbash, K.O. The Use of κ-Carrageenan/Fe3O4 Nanocomposite as a Nanomagnetic Catalyst for Clean Synthesis of Rhodanines. Catal. Commun. 2015, 68, 77–83. [Google Scholar] [CrossRef]
- Yamazaki, S.; Mori, T.; Ogino, I.; Mukai, S.R. Flexible Film-Type Catalysts Encapsulating Urease within κ-Carrageenan Hydrogel Network. Chem. Eng. J. 2015, 278, 122–128. [Google Scholar] [CrossRef]
- Makas, Y.G.; Kalkan, N.A.; Aksoy, S.; Altinok, H.; Hasirci, N. Immobilization of Laccase in κ-Carrageenan Based Semi-Interpenetrating Polymer Networks. J. Biotechnol. 2010, 148, 216–220. [Google Scholar] [CrossRef]
- Wahba, M.I.; Hassan, M.E. Agar-Carrageenan Hydrogel Blend as a Carrier for the Covalent Immobilization of β-D-Galactosidase. Macromol. Res. 2017, 25, 913–923. [Google Scholar] [CrossRef]
- Mak, C.A.; Ranjbar, S.; Riente, P.; Rodríguez-Escrich, C.; Pericàs, M.A. Hybrid Magnetic Materials (Fe3O4–κ-Carrageenan) as Catalysts for the Michael Addition of Aldehydes to Nitroalkenes. Tetrahedron 2014, 70, 6169–6173. [Google Scholar] [CrossRef]
- Manivasagan, P.; Oh, J. Marine Polysaccharide-Based Nanomaterials as a Novel Source of Nanobiotechnological Applications. Int. J. Biol. Macromol. 2016, 82, 315–327. [Google Scholar] [CrossRef]
- Anderson, N.S.; Rees, D.A. 1104. Porphyran: A Polysaccharide with a Masked Repeating Structure. J. Chem. Soc. 1965, 5880. [Google Scholar] [CrossRef]
- Morrice, L.M.; McLEAN, M.W.; Long, W.F.; Williamson, F.B. Porphyran Primary Structure. An Investigation Using Beta-Agarase I from Pseudomonas Atlantica and 13C-NMR Spectroscopy. Eur. J. Biochem. 1983, 133, 673–684. [Google Scholar] [CrossRef]
- Zhang, Q.; Qi, H.; Zhao, T.; Deslandes, E.; Ismaeli, N.M.; Molloy, F.; Critchley, A.T. Chemical Characteristics of a Polysaccharide from Porphyra Capensis (Rhodophyta). Carbohydr. Res. 2005, 340, 2447–2450. [Google Scholar] [CrossRef]
- Liu, J.; Zhan, X.; Wan, J.; Wang, Y.; Wang, C. Review for Carrageenan-Based Pharmaceutical Biomaterials: Favourable Physical Features versus Adverse Biological Effects. Carbohydr. Polym. 2015, 121, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Wang, J.; Song, H.; Zhang, H.; Niu, X. Regioselective Syntheses of Sulfated Porphyrans from Porphyra Haitanensis and Their Antioxidant and Anticoagulant Activities in Vitro. Carbohydr. Polym. 2010, 79, 1124–1129. [Google Scholar] [CrossRef]
- Isaka, S.; Cho, K.; Nakazono, S.; Abu, R.; Ueno, M.; Kim, D.; Oda, T. Antioxidant and Anti-Inflammatory Activities of Porphyran Isolated from Discolored Nori (Porphyra Yezoensis). Int. J. Biol. Macromol. 2015, 74, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Nogawa, M.; Siddiqui, R.; Watashi, K.; Wakita, T.; Kato, N.; Ikeda, M.; Okimura, T.; Isaka, S.; Oda, T.; et al. Acidic Polysaccharides Isolated from Marine Algae Inhibit the Early Step of Viral Infection. Int. J. Biol. Macromol. 2019, 124, 282–290. [Google Scholar] [CrossRef]
- Bhatia, S.; Sharma, K.; Nagpal, K.; Bera, T. Investigation of the Factors Influencing the Molecular Weight of Porphyran and Its Associated Antifungal Activity. Bioact. Carbohydr. Diet. Fibre 2015, 5, 153–168. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Wang, J.; Zhang, H.; Niu, X.; Li, P. Preparation of the Different Derivatives of the Low-Molecular-Weight Porphyran from Porphyra Haitanensis and Their Antioxidant Activities in Vitro. Int. J. Biol. Macromol. 2009, 45, 22–26. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Wang, J.; Song, H.; Zhang, H.; Niu, X. Chemical Modification and Influence of Function Groups on the in Vitro-Antioxidant Activities of Porphyran from Porphyra Haitanensis. Carbohydr. Polym. 2010, 79, 290–295. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Xiao, L.; Liu, C.; Qi, H.; Zhang, Z. In Vivo Antihyperlipidemic and Antioxidant Activity of Porphyran in Hyperlipidemic Mice. Carbohydr. Polym. 2017, 174, 417–420. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Su, H.; Pan, Y.; Han, J.; Zhang, T.; Mao, G. Effect of Sulfated Galactan from Porphyra Haitanensis on H2O2-Induced Premature Senescence in WI-38 Cells. Int. J. Biol. Macromol. 2018, 106, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Sedayu, B.B.; Cran, M.J.; Bigger, S.W. A Review of Property Enhancement Techniques for Carrageenan-Based Films and Coatings. Carbohydr. Polym. 2019, 216, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, H.; Wang, P.; Guo, M.; Jiang, S.; Li, X.; Jiang, S. Films Based on κ-Carrageenan Incorporated with Curcumin for Freshness Monitoring. Food Hydrocoll. 2018, 83, 134–142. [Google Scholar] [CrossRef]
- Roy, S.; Rhim, J.-W. Agar-Based Antioxidant Composite Films Incorporated with Melanin Nanoparticles. Food Hydrocoll. 2019, 94, 391–398. [Google Scholar] [CrossRef]
- Roy, S.; Rhim, J.-W.; Jaiswal, L. Bioactive Agar-Based Functional Composite Film Incorporated with Copper Sulfide Nanoparticles. Food Hydrocoll. 2019, 93, 156–166. [Google Scholar] [CrossRef]
- Arfat, Y.A.; Ahmed, J.; Jacob, H. Preparation and Characterization of Agar-Based Nanocomposite Films Reinforced with Bimetallic (Ag-Cu) Alloy Nanoparticles. Carbohydr. Polym. 2017, 155, 382–390. [Google Scholar] [CrossRef]
- Davidović, S.; Lazić, V.; Miljković, M.; Gordić, M.; Sekulić, M.; Marinović-Cincović, M.; Ratnayake, I.S.; Ahrenkiel, S.P.; Nedeljković, J.M. Antibacterial Ability of Immobilized Silver Nanoparticles in Agar-Agar Films Co-Doped with Magnesium Ions. Carbohydr. Polym. 2019, 224, 115187. [Google Scholar] [CrossRef]
- Giménez, B.; López de Lacey, A.; Pérez-Santín, E.; López-Caballero, M.E.; Montero, P. Release of Active Compounds from Agar and Agar–Gelatin Films with Green Tea Extract. Food Hydrocoll. 2013, 30, 264–271. [Google Scholar] [CrossRef]
- Shojaee-Aliabadi, S.; Hosseini, H.; Mohammadifar, M.A.; Mohammadi, A.; Ghasemlou, M.; Ojagh, S.M.; Hosseini, S.M.; Khaksar, R. Characterization of Antioxidant-Antimicrobial κ-Carrageenan Films Containing Satureja Hortensis Essential Oil. Int. J. Biol. Macromol. 2013, 52, 116–124. [Google Scholar] [CrossRef]
- Malagurski, I.; Levic, S.; Nesic, A.; Mitric, M.; Pavlovic, V.; Dimitrijevic-Brankovic, S. Mineralized Agar-Based Nanocomposite Films: Potential Food Packaging Materials with Antimicrobial Properties. Carbohydr. Polym. 2017, 175, 55–62. [Google Scholar] [CrossRef]
- Wolfenden, R.; Yuan, Y. Monoalkyl Sulfates as Alkylating Agents in Water, Alkylsulfatase Rate Enhancements, and the “Energy-Rich” Nature of Sulfate Half-Esters. Proc. Natl. Acad. Sci. USA 2007, 104, 83–86. [Google Scholar] [CrossRef]
- Varsha, K.K.; Devendra, L.; Shilpa, G.; Priya, S.; Pandey, A.; Nampoothiri, K.M. 2,4-Di-tert-butyl Phenol as the Antifungal, Antioxidant Bioactive Purified from a Newly Isolated Lactococcus Sp. Int. J. Food Microbiol. 2015, 211, 44–50. [Google Scholar] [CrossRef]
- Viszwapriya, D.; Prithika, U.; Deebika, S.; Balamurugan, K.; Pandian, S.K. In Vitro and in Vivo Antibiofilm Potential of 2,4-Di- Tert -Butylphenol from Seaweed Surface Associated Bacterium Bacillus Subtilis against Group A Streptococcus. Microbiol. Res. 2016, 191, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.; Giacomini, D. Antibacterial and Antioxidant Activities for Natural and Synthetic Dual-Active Compounds. Eur. J. Med. Chem. 2018, 158, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Zabka, M.; Pavela, R. Antifungal Efficacy of Some Natural Phenolic Compounds against Significant Pathogenic and Toxinogenic Filamentous Fungi. Chemosphere 2013, 93, 1051–1056. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, P.; Lucardi, R.D.; Su, Z.; Li, S. Natural Sources and Bioactivities of 2,4-Di-Tert-Butylphenol and Its Analogs. Toxins 2020, 12, 35. [Google Scholar] [CrossRef]
- Marchese, A.; Orhan, I.E.; Daglia, M.; Barbieri, R.; Di Lorenzo, A.; Nabavi, S.F.; Gortzi, O.; Izadi, M.; Nabavi, S.M. Antibacterial and Antifungal Activities of Thymol: A Brief Review of the Literature. Food Chem. 2016, 210, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Jitsukawa, K.; Masuda, H.; Einaga, H. Thermodynamic Stabilities and Formation Mechanisms of Iron(III) Complexes with Nα-Carboxymethyl-Nα-Salicyl-l-Phenylalanine and Nα-Salicyl-l-Phenylalanine: Enhanced Thermodynamic Stability by Additional Methylcarboxylate Group. Polyhedron 1994, 13, 2343–2351. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Calhorda, M.J.; Cavaco, I.; Costa, P.J.; Correia, I.; Costa, D.; Vilas-Boas, L.F.; Félix, V.; Gillard, R.D.; Henriques, R.T.; et al. N-Salicylideneamino Acidato Complexes of Oxovanadium(IV). The Cysteine and Penicillamine Complexes. Dalton Trans. 2004, 2855. [Google Scholar] [CrossRef] [PubMed]
- Safaei, E.; Sheykhi, H.; Wojtczak, A.; Jagličić, Z.; Kozakiewicz, A. Synthesis and Characterization of an Iron(III) Complex of Glycine Derivative of Bis(Phenol)Amine Ligand in Relevance to Catechol Dioxygenase Active Site. Polyhedron 2011, 30, 1219–1224. [Google Scholar] [CrossRef]
- Núñez-Montenegro, A.; Pino-Cuevas, A.; Carballo, R.; Vázquez-López, E.M. Structural Evidence of an Intramolecular Proton Transfer Leading to Keto-Amine Tautomer in the Crystals of Schiff Bases Derived from Tyrosine and Histidine Esters. J. Mol. Struct. 2014, 1062, 110–115. [Google Scholar] [CrossRef]
- Muche, S.; Hołyńska, M. New Insights into the Coordination Chemistry of Schiff Bases Derived from Amino Acids: Planar [Ni 4 ] Complexes with Tyrosine Side-Chains. J. Mol. Struct. 2017, 1142, 168–174. [Google Scholar] [CrossRef]
- Magoński, J.; Pawlak, Z.; Jasiński, T. Dissociation Constants of Substituted Phenols and Homoconjugation Constants of the Corresponding Phenol–Phenolate Systems in Acetonitrile. J. Chem. Soc. Faraday Trans. 1993, 89, 119–122. [Google Scholar] [CrossRef]
- Hanai, T. Quantitative Structure–Retention Relationships of Phenolic Compounds without Hammett’s Equations. J. Chromatogr. A 2003, 985, 343–349. [Google Scholar] [CrossRef]
- Roy, K.; Popelier, P.L.A. Predictive QSPR Modeling of the Acidic Dissociation Constant (PKa) of Phenols in Different Solvents. J. Phys. Org. Chem. 2009, 22, 186–196. [Google Scholar] [CrossRef]
- Penhoat, M. Scope and Limitations of a 1H NMR Method for the Prediction of Substituted Phenols PKa Values in Water, CH3CN, DMF, DMSO and i-PrOH. Tetrahedron Lett. 2013, 54, 2571–2574. [Google Scholar] [CrossRef]
- Wischang, D.; Hartung, J. Bromination of Phenols in Bromoperoxidase-Catalyzed Oxidations. Tetrahedron 2012, 68, 9456–9463. [Google Scholar] [CrossRef]
- Guo, G.; Lin, F. The Bromination Kinetics of Phenolic Compounds in Aqueous Solution. J. Hazard. Mater. 2009, 170, 645–651. [Google Scholar] [CrossRef]
- Gallard, H.; Pellizzari, F.; Croué, J.P.; Legube, B. Rate Constants of Reactions of Bromine with Phenols in Aqueous Solution. Water Res. 2003, 37, 2883–2892. [Google Scholar] [CrossRef]
- Díaz-Oviedo, C.; Quevedo, R. Role of Hydrogen Bonding in the Selectivity of Aromatic Mannich Reaction of Tyramines: Macrocyclization vs. Linear Condensation. J. Mol. Struct. 2020, 1202, 127283. [Google Scholar] [CrossRef]
- Norton, R.S.; Bradbury, J.H. Carbon-13 Nuclear Magnetic Resonance Study of Tyrosine Titrations. J. Chem. Soc. Chem. Commun. 1974, 870b. [Google Scholar] [CrossRef]
- International Union of Pure and Applied Chemistry; Serjeant, E.P.; Dempsey, B. (Eds.) Ionisation Constants of Organic Acids in Aqueous Solution; Pergamon Press: Oxford, UK; New York, NY, USA, 1979. [Google Scholar]
- Conner, A.H. Predicting the reactivity of phenolic compounds with formaldehyde under basic conditions: An ab initio study. J. Appl. Polym. Sci. 2000, 78, 355–363. [Google Scholar] [CrossRef]
- Mitsunaga, T.; Conner, A.H.; Hill, C.G., Jr. Predicting the reactivity of phenolic compounds with formaldehyde. II. Continuation of an ab initio study. J. Appl. Polym. Sci. 2002, 86, 135–140. [Google Scholar] [CrossRef]
- Noseda, M.D.; Viana, A.G.; Duarte, M.E.R.; Cerezo, A.S. Alkali modification of carrageenans. Part IV. Porphyrans as model compounds. Carbohydr. Polym. 2000, 42, 301–305. [Google Scholar] [CrossRef]
- Viana, A.; Noseda, M.; Duarte, M.; Cerezo, A. Alkali Modification of Carrageenans. Part V. The Iota–Nu Hybrid Carrageenan from and Its Cyclization to Iota-Carrageenan. Carbohydr. Polym. 2004, 58, 455–460. [Google Scholar] [CrossRef]
- Tranquilan-Aranilla, C.; Nagasawa, N.; Bayquen, A.; Dela Rosa, A. Synthesis and Characterization of Carboxymethyl Derivatives of Kappa-Carrageenan. Carbohydr. Polym. 2012, 87, 1810–1816. [Google Scholar] [CrossRef]
- Pereira, L.; Amado, A.M.; Critchley, A.T.; van de Velde, F.; Ribeiro-Claro, P.J.A. Identification of Selected Seaweed Polysaccharides (Phycocolloids) by Vibrational Spectroscopy (FTIR-ATR and FT-Raman). Food Hydrocoll. 2009, 23, 1903–1909. [Google Scholar] [CrossRef]
- He, D.; Wu, S.; Yan, L.; Zuo, J.; Cheng, Y.; Wang, H.; Liu, J.; Zhang, X.; Wu, M.; Choi, J.; et al. Antitumor Bioactivity of Porphyran Extracted from Pyropia Yezoensis Chonsoo2 on Human Cancer Cell Lines. J. Sci. Food Agric. 2019, 99, 6722–6730. [Google Scholar] [CrossRef]
- Gómez-Ordóñez, E.; Rupérez, P. FTIR-ATR Spectroscopy as a Tool for Polysaccharide Identification in Edible Brown and Red Seaweeds. Food Hydrocoll. 2011, 25, 1514–1520. [Google Scholar] [CrossRef]
- Van de Velde, F.; Pereira, L.; Rollema, H.S. The Revised NMR Chemical Shift Data of Carrageenans. Carbohydr. Res. 2004, 339, 2309–2313. [Google Scholar] [CrossRef] [PubMed]
- Tojo, E.; Prado, J. Chemical Composition of Carrageenan Blends Determined by IR Spectroscopy Combined with a PLS Multivariate Calibration Method. Carbohydr. Res. 2003, 338, 1309–1312. [Google Scholar] [CrossRef]
- Correc, G.; Hehemann, J.-H.; Czjzek, M.; Helbert, W. Structural Analysis of the Degradation Products of Porphyran Digested by Zobellia Galactanivorans β-Porphyranase A. Carbohydr. Polym. 2011, 83, 277–283. [Google Scholar] [CrossRef]
- Maciel, J.; Chaves, L.; Souza, B.; Teixeira, D.; Freitas, A.; Feitosa, J.; Depaula, R. Structural Characterization of Cold Extracted Fraction of Soluble Sulfated Polysaccharide from Red Seaweed Gracilaria Birdiae. Carbohydr. Polym. 2008, 71, 559–565. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (OH/O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Billany, M.R.; Khatib, K.; Gordon, M.; Sugden, J.K. Alcohols and Ethanolamines as Hydroxyl Radical Scavengers. Int. J. Pharm. 1996, 137, 143–147. [Google Scholar] [CrossRef]
- Nie, M.; Wang, Q.; Qiu, G. Enhancement of Ultrasonically Initiated Emulsion Polymerization Rate Using Aliphatic Alcohols as Hydroxyl Radical Scavengers. Ultrason. Sonochemistry 2008, 15, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Mollah, M.Y.A.; Susan, M.A.B.H.; Islam, M.M. Role of in Situ Electrogenerated Reactive Oxygen Species towards Degradation of Organic Dye in Aqueous Solution. Electrochim. Acta 2020, 344, 136146. [Google Scholar] [CrossRef]
- Kalinowski, M.K.; Klimkiewicz, J. Solvation Effects in the Electrochemistry of Diphenylpicrylhydrazyl. Monatsh. Chem. 1983, 114, 1035–1043. [Google Scholar] [CrossRef]
- Scott, S.L.; Chen, W.J.; Bakac, A.; Espenson, J.H. Spectroscopic Parameters, Electrode Potentials, Acid Ionization Constants, and Electron Exchange Rates of the 2,2′-Azinobis(3-Ethylbenzothiazoline-6-Sulfonate) Radicals and Ions. J. Phys. Chem. 1993, 97, 6710–6714. [Google Scholar] [CrossRef]
- Bourbonnais, R.; Leech, D.; Paice, M.G. Electrochemical Analysis of the Interactions of Laccase Mediators with Lignin Model Compounds. Biochim. Biophys. Acta Gen. Subj. 1998, 1379, 381–390. [Google Scholar] [CrossRef]
- Ley, C.; Zengin Çekiç, S.; Kochius, S.; Mangold, K.-M.; Schwaneberg, U.; Schrader, J.; Holtmann, D. An Electrochemical Microtiter Plate for Parallel Spectroelectrochemical Measurements. Electrochim. Acta 2013, 89, 98–105. [Google Scholar] [CrossRef]
- Gramss, G. Reappraising a Controversy: Formation and Role of the Azodication (ABTS2+) in the Laccase-ABTS Catalyzed Breakdown of Lignin. Fermentation 2017, 3, 27. [Google Scholar] [CrossRef]
- Nam, S.; Renganathan, V.; Tratnyek, P.G. Substituent Effects on Azo Dye Oxidation by the Fe(III)–EDTA–H2O2 System. Chemosphere 2001, 45, 59–65. [Google Scholar] [CrossRef]
- Mas-Ballesté, R.; Que, L. Iron-Catalyzed Olefin Epoxidation in the Presence of Acetic Acid: Insights into the Nature of the Metal-Based Oxidant. J. Am. Chem. Soc. 2007, 129, 15964–15972. [Google Scholar] [CrossRef]
- Duban, E.A.; Bryliakov, K.P.; Talsi, E.P. The Active Intermediates of Non-Heme-Iron-Based Systems for Catalytic Alkene Epoxidation with H2O2/CH3COOH. Eur. J. Inorg. Chem. 2007, 2007, 852–857. [Google Scholar] [CrossRef]
- Cussó, O.; Ribas, X.; Costas, M. Biologically Inspired Non-Heme Iron-Catalysts for Asymmetric Epoxidation; Design Principles and Perspectives. Chem. Commun. 2015, 51, 14285–14298. [Google Scholar] [CrossRef]
- Hu, Y.; Li, Y.; He, J.; Liu, T.; Zhang, K.; Huang, X.; Kong, L.; Liu, J. EDTA-Fe(III) Fenton-like Oxidation for the Degradation of Malachite Green. J. Environ. Manag. 2018, 226, 256–263. [Google Scholar] [CrossRef]
- Sankar, M.; Nowicka, E.; Carter, E.; Murphy, D.M.; Knight, D.W.; Bethell, D.; Hutchings, G.J. The Benzaldehyde Oxidation Paradox Explained by the Interception of Peroxy Radical by Benzyl Alcohol. Nat. Commun. 2014, 5, 3332. [Google Scholar] [CrossRef]
- Serra, A.C.; Rocha Gonsalves, A.M.d.A. Mild Oxygen Activation with Isobutyraldehyde Promoted by Simple Salts. Tetrahedron Lett. 2011, 52, 3489–3491. [Google Scholar] [CrossRef]
- Basiak, E.; Lenart, A.; Debeaufort, F. How Glycerol and Water Contents Affect the Structural and Functional Properties of Starch-Based Edible Films. Polymers 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Boulogne, F.; Giorgiutti-Dauphiné, F.; Pauchard, L. How to Reduce the Crack Density in Drying Colloidal Material? Oil Gas Sci. Technol. Rev. IFP Energ. Nouv. 2014, 69, 397–404. [Google Scholar] [CrossRef]
- Aissaoui, N.; Mahjoubi, M.; Nas, F.; Mghirbi, O.; Arab, M.; Souissi, Y.; Hoceini, A.; Masmoudi, A.S.; Mosbah, A.; Cherif, A.; et al. Antibacterial Potential of 2,4-Di-Tert-Butylphenol and Calixarene-Based Prodrugs from Thermophilic Bacillus Licheniformis Isolated in Algerian Hot Spring. Geomicrobiol. J. 2019, 36, 53–62. [Google Scholar] [CrossRef]
- Mishra, R.; Kushveer, J.S.; Khan, M.I.K.; Pagal, S.; Meena, C.K.; Murali, A.; Dhayalan, A.; Venkateswara Sarma, V. 2,4-Di-Tert-Butylphenol Isolated From an Endophytic Fungus, Daldinia Eschscholtzii, Reduces Virulence and Quorum Sensing in Pseudomonas Aeruginosa. Front. Microbiol. 2020, 11, 1668. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, A.R.; Periyasamy, M.; Pandian, S.K. Assessment of 2,4-Di-Tert-Butylphenol Induced Modifications in Extracellular Polymeric Substances of Serratia Marcescens. Bioresour. Technol. 2015, 188, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Govender, T.; Govinden, U.; Mocktar, C.; Kruger, H.G.; Veljkovic, J.; Cindro, N.; Bobinac, D.; Zabcic, I.; MlinariC-Majerski, K.; Basaric, N. In Vitro Investigation of the Antimicrobial Activity of a Series of Lipophilic Phenols and Naphthols. S. Afr. J. Chem. 2016, 69, 44–50. [Google Scholar] [CrossRef][Green Version]
- Padmavathi, A.R.; Bakkiyaraj, D.; Thajuddin, N.; Pandian, S.K. Effect of 2, 4-Di-Tert-Butylphenol on Growth and Biofilm Formation by an Opportunistic Fungus Candida Albicans. Biofouling 2015, 31, 565–574. [Google Scholar] [CrossRef]
- Kroll, T.M.; Bommiasamy, H.; Boissy, R.E.; Hernandez, C.; Nickoloff, B.J.; Mestril, R.; Poole, I.C.L. 4-Tertiary Butyl Phenol Exposure Sensitizes Human Melanocytes to Dendritic Cell-Mediated Killing: Relevance to Vitiligo. J. Investig. Dermatol. 2005, 124, 798–806. [Google Scholar] [CrossRef]
- Kadoma, Y.; Ito, S.; Atsumi, T.; Fujisawa, S. Mechanisms of Cytotoxicity of 2- or 2,6-Di-Tert-Butylphenols and 2-Methoxyphenols in Terms of Inhibition Rate Constant and a Theoretical Parameter. Chemosphere 2009, 74, 626–632. [Google Scholar] [CrossRef]
- Steiner, A.A.; van Winden, H. Recipe for Ferric Salts of Ethylenediaminetetraacetic Acid. Plant. Physiol. 1970, 46, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Van Meerten, S.G.J.; Franssen, W.M.J.; Kentgens, A.P.M. SsNake: A Cross-Platform Open-Source NMR Data Processing and Fitting Application. J. Magn. Reson. 2019, 301, 56–66. [Google Scholar] [CrossRef]
- Lewis, I.A.; Schommer, S.C.; Markley, J.L. RNMR: Open Source Software for Identifying and Quantifying Metabolites in NMR Spectra: RNMR: Metabolomics Software. Magn. Reson. Chem. 2009, 47, S123–S126. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 30 November 2020).
- Sousa, A.M.M.; Borges, J.; Silva, F.; Ramos, A.M.; Cabrita, E.J.; Gonçalves, M.P. Shaping the Molecular Assemblies of Native and Alkali-Modified Agars in Dilute and Concentrated Aqueous Media via Microwave-Assisted Extraction. Soft Matter 2013, 9, 3131. [Google Scholar] [CrossRef]
- Bax, A.; Freeman, R. Investigation of Complex Networks of Spin-Spin Coupling by Two-Dimensional NMR. J. Magn. Reson. (1969) 1981, 44, 542–561. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-Based Two-Dimensional Homonuclear Magnetization Transfer Spectroscopy. J. Magn. Reson. (1969) 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Cian, R.E.; Salgado, P.R.; Drago, S.R.; Mauri, A.N. Effect of Glycerol and Ca+2 Addition on Physicochemical Properties of Edible Carrageenan/Porphyran-Based Films Obtained from the Red Alga, Pyropia Columbina. J. Appl. Phycol. 2015, 27, 1699–1708. [Google Scholar] [CrossRef]
- Berker, K.I.; Güçlü, K.; Tor, İ.; Apak, R. Comparative Evaluation of Fe(III) Reducing Power-Based Antioxidant Capacity Assays in the Presence of Phenanthroline, Batho-Phenanthroline, Tripyridyltriazine (FRAP), and Ferricyanide Reagents. Talanta 2007, 72, 1157–1165. [Google Scholar] [CrossRef]
- Miceli, M.; Roma, E.; Rosa, P.; Feroci, M.; Loreto, M.; Tofani, D.; Gasperi, T. Synthesis of Benzofuran-2-One Derivatives and Evaluation of Their Antioxidant Capacity by Comparing DPPH Assay and Cyclic Voltammetry. Molecules 2018, 23, 710. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Applying an Improved ABTS Radical Cation Decolorization Assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, Y.; Li, T.; Dai, F.; Luo, G.; Xiao, G.; Tang, C. Phenolic Profiles and Antioxidant Capacities of Mulberry (Morus Atropurpurea Roxb.) Juices from Different Cultivars. Int. J. Food Prop. 2019, 22, 1340–1352. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | υ(O-H) | υ(C-H) | υ(S=O) | υ(C-O, ether) | υ(C-O-SO3) |
|---|---|---|---|---|---|
| POR | 3444.7 (b) | 2929.3 (w) | 1259.8 (m) 1230.4 (sh) | 931.9 (w) | 815.5 (w) |
| POR-L2 | 3418.2 (b) | 2932.2 (w) | 1252.1 (m) 1225.5 (sh) | 931.4 (w) | 817.2 (w) |
| Sample a | t (min) | Abs 510 nm b |
|---|---|---|
| Control c | 0 | 0.042 ± 0.003 |
| 30 | 0.045 ± 0.002 | |
| POR d | 30 | 0.108 ± 0.002 |
| POR-L2d | 30 | 0.161 ± 0.002 |
| Sample a | Δ Abs 524 nm b |
|---|---|
| Control | 0.0886 ± 0.0043 |
| POR | 0.0663 ± 0.0018 |
| POR-L2 | 0.0464 ± 0.0031 |
| Sample a | Δ Abs 500 nm b |
|---|---|
| Control c | 0.0351 ± 0.0034 |
| POR | 0.0065 ± 0.0031 |
| POR-L2 | 0.0012 ± 0.0010 |
| POR | POR/gly | POR-L2 | POR-L2/gly | |
|---|---|---|---|---|
| C (at%) | 59.2 ± 1.1 | 45.8 ± 1.2 | 57.8 ± 2.1 | 46.9 ± 5.8 |
| O (at%) | 38.4 ± 1.6 | 46.5 ± 3.3 | 37.8 ± 2.8 | 39.4 ± 4.9 |
| S (at%) | 1.4 ± 0.3 | 3.3 ± 0.2 | 1.0 ± 0.1 | 4.3 ± 0.7 |
| Mg (at%) | 0.2 ± 0.1 | 0.5 ± 0.1 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| Na (at%) | 0.4 ± 0.1 | 0.7 ± 0.2 | 0.4 ± 0.1 | 1.4 ± 0.1 |
| K (at%) | nd | 0.9 ± 0.3 | 2.9 ± 0.7 | 7.6 ± 2.2 |
| Ca (at%) | nd | 0.6 ± 0.2 | nd | 0.3 ± 0.1 |
| Solution Concentration (mg/mL) | % Growth Inhibition | |
|---|---|---|
| S. aureus | E. coli | |
| POR | ||
| 0.2 | nd | nd |
| 0.4 | nd | nd |
| 0.8 | 4.9 ± 0.7 | nd |
| 1.6 | 6.6 ± 0.4 | nd |
| POR-L2 | ||
| 0.2 | nd | nd |
| 0.4 | nd | nd |
| 0.8 | 27.3 ± 5.8 | 3.1 ±2.5 |
| 1.6 | 56.3 ± 3.0 | 4.5 ± 2.62 |
| POR-L2 | ||
| 0.2 | nd | nd. |
| 0.4 | 18.9 ± 2.1 | nd |
| 0.8 | 43.3 ± 4.7 | nd |
| 1.6 | 65.7 ± 2.1 | nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adão, P.; Reboleira, J.; Teles, M.; Santos, B.; Ribeiro, N.; Teixeira, C.M.; Guedes, M.; Pessoa, J.C.; Bernardino, S. Enhancement of the Antioxidant and Antimicrobial Activities of Porphyran through Chemical Modification with Tyrosine Derivatives. Molecules 2021, 26, 2916. https://doi.org/10.3390/molecules26102916
Adão P, Reboleira J, Teles M, Santos B, Ribeiro N, Teixeira CM, Guedes M, Pessoa JC, Bernardino S. Enhancement of the Antioxidant and Antimicrobial Activities of Porphyran through Chemical Modification with Tyrosine Derivatives. Molecules. 2021; 26(10):2916. https://doi.org/10.3390/molecules26102916
Chicago/Turabian StyleAdão, Pedro, João Reboleira, Marco Teles, Beatriz Santos, Nádia Ribeiro, Carlos M. Teixeira, Mafalda Guedes, João Costa Pessoa, and Susana Bernardino. 2021. "Enhancement of the Antioxidant and Antimicrobial Activities of Porphyran through Chemical Modification with Tyrosine Derivatives" Molecules 26, no. 10: 2916. https://doi.org/10.3390/molecules26102916
APA StyleAdão, P., Reboleira, J., Teles, M., Santos, B., Ribeiro, N., Teixeira, C. M., Guedes, M., Pessoa, J. C., & Bernardino, S. (2021). Enhancement of the Antioxidant and Antimicrobial Activities of Porphyran through Chemical Modification with Tyrosine Derivatives. Molecules, 26(10), 2916. https://doi.org/10.3390/molecules26102916