
TiCl4 Dissolved in Ionic Liquid Mixtures from Аb Initio Molecular Dynamics Simulations


 and
and
Abstract
1. Introduction
2. Results
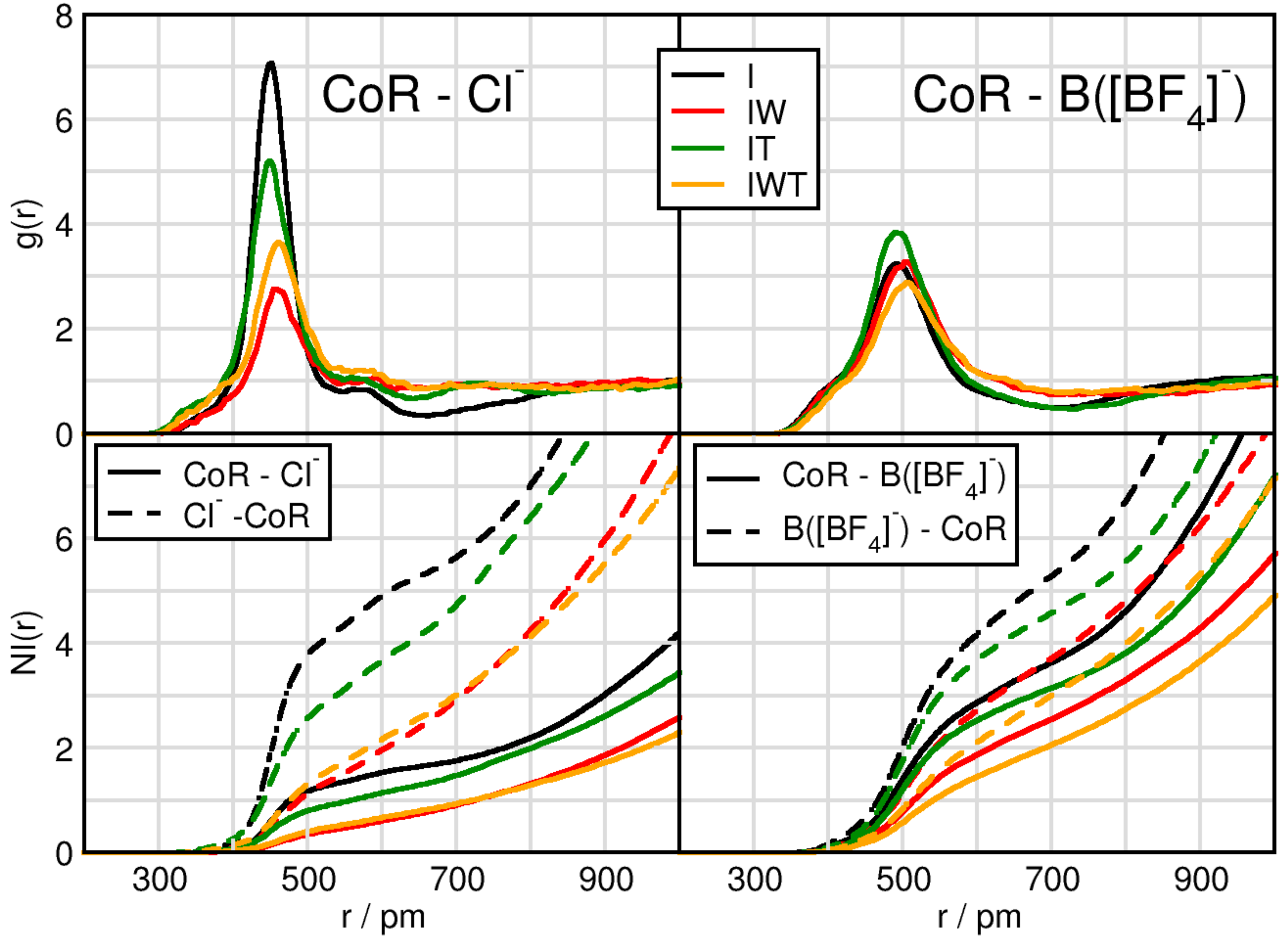
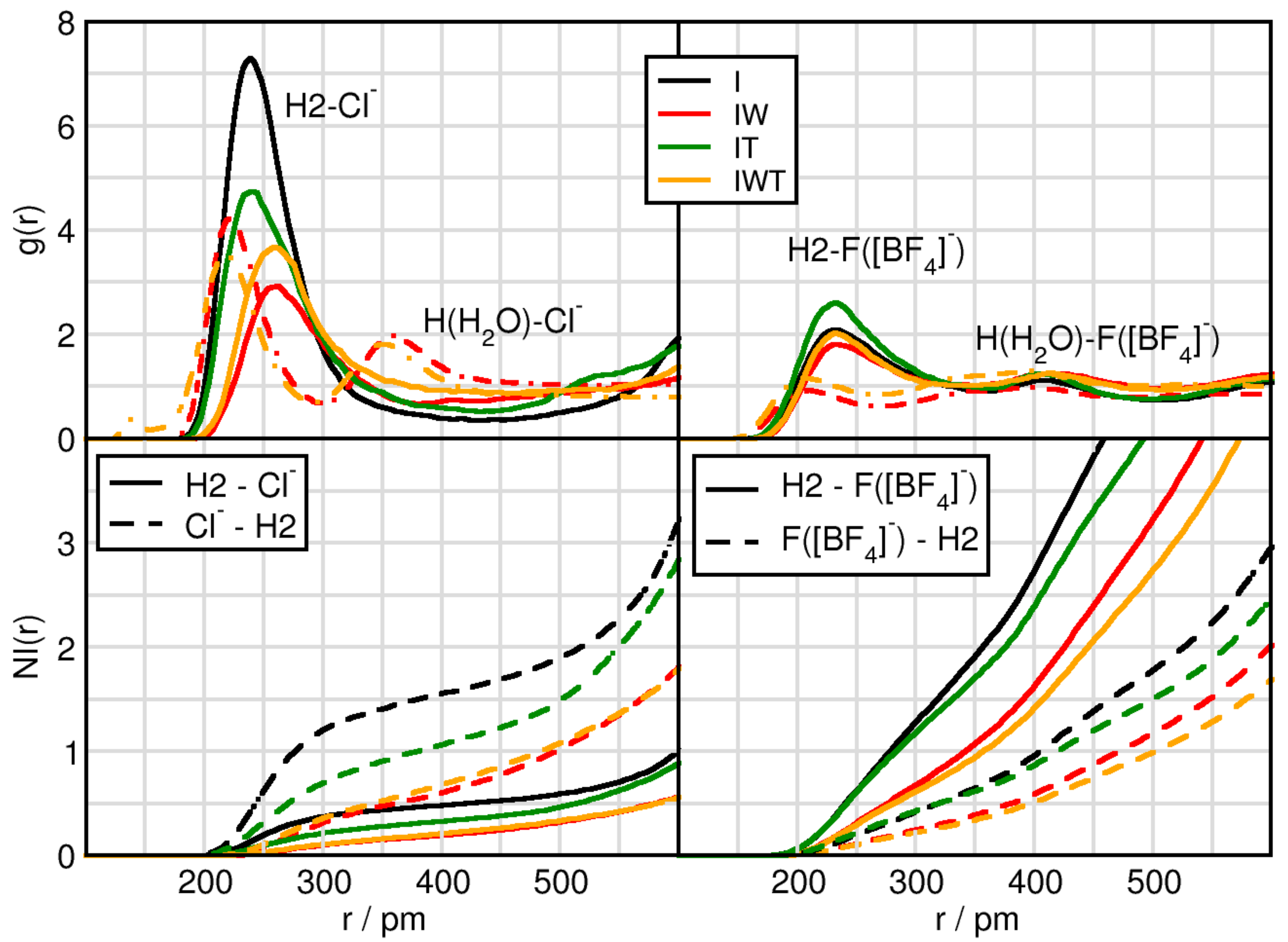
2.1. Cation-Anion Interplay
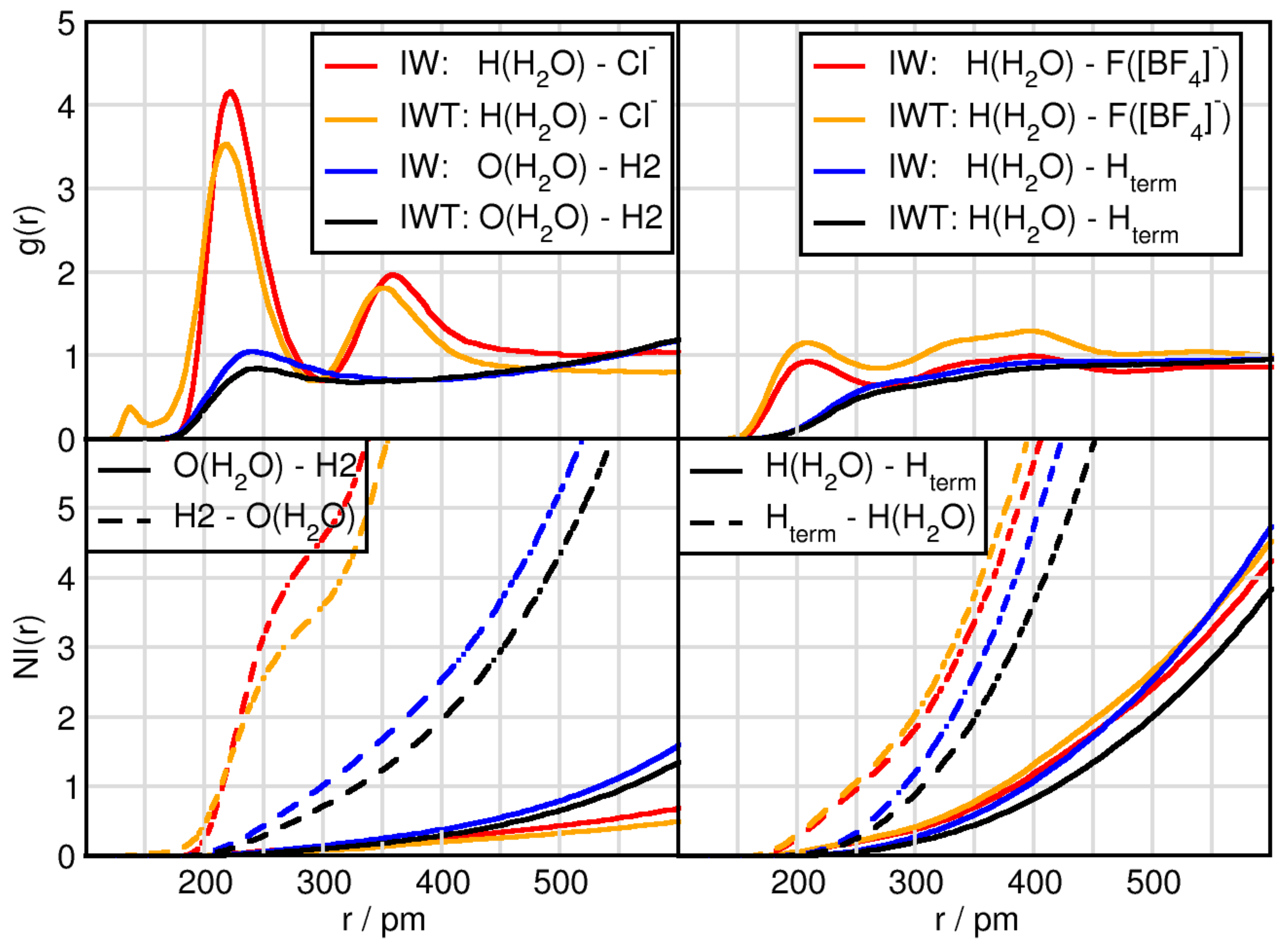
2.2. Solvation Structure of Water
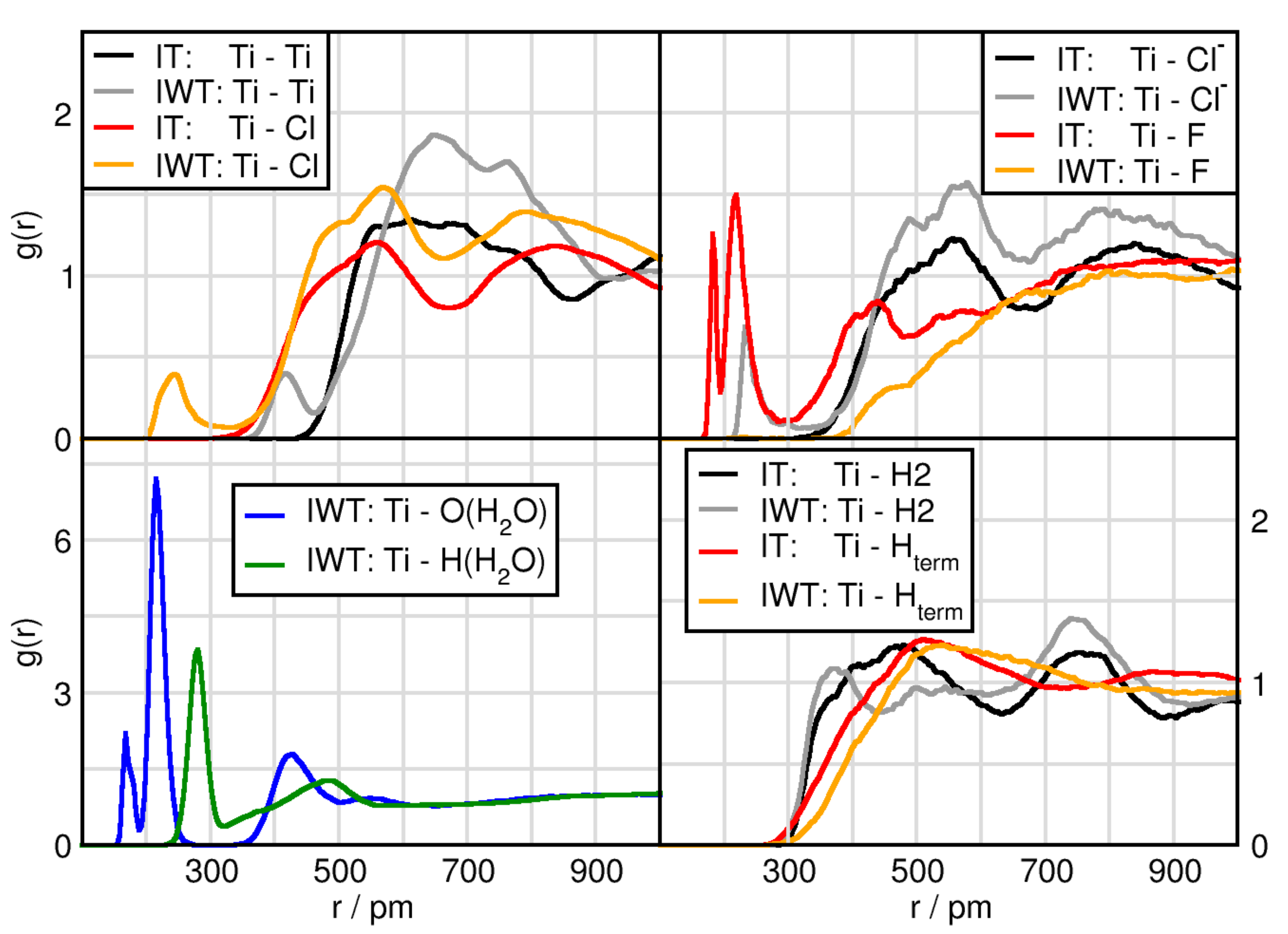
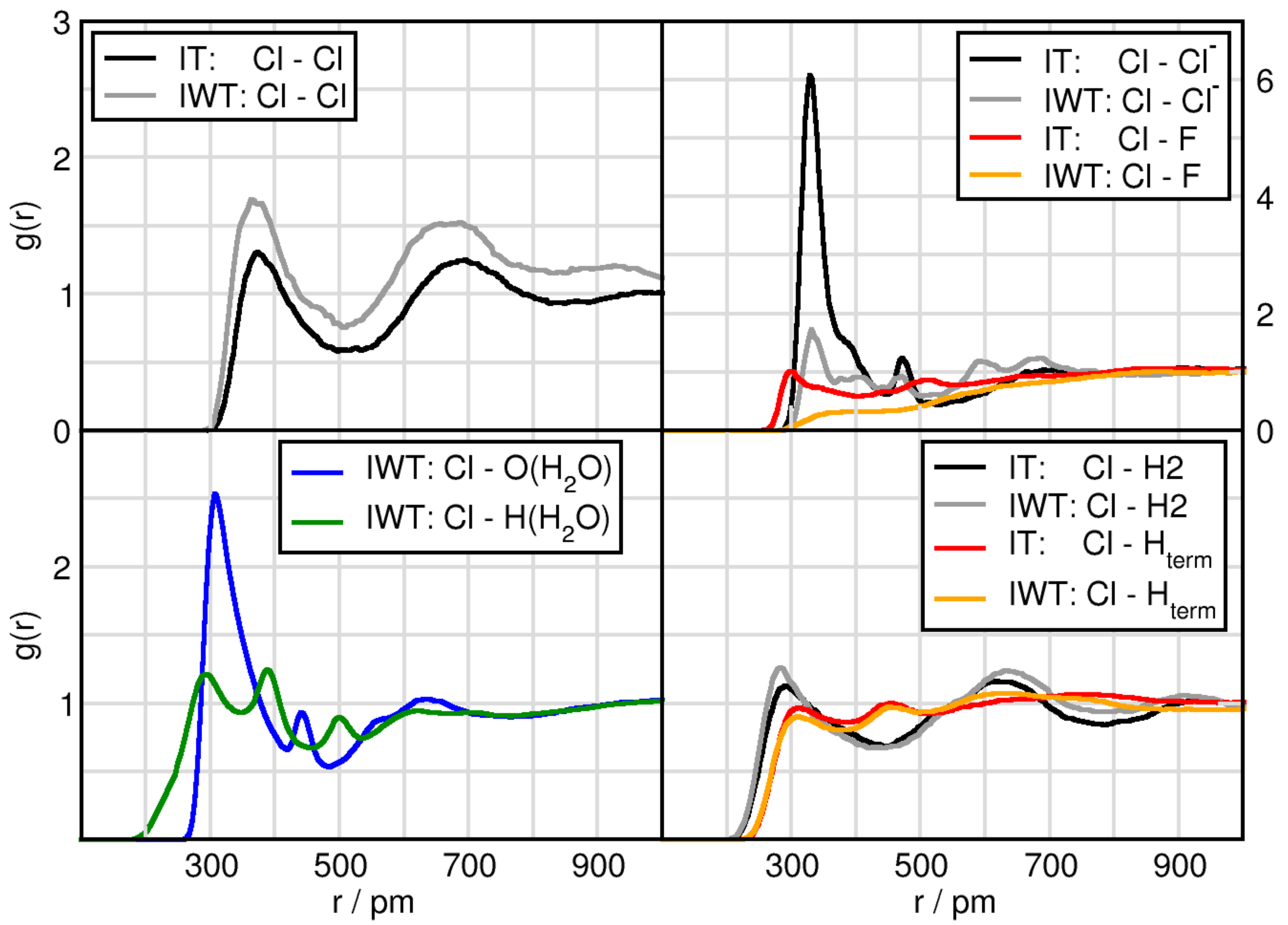
2.3. Solvation Structure of TiCl
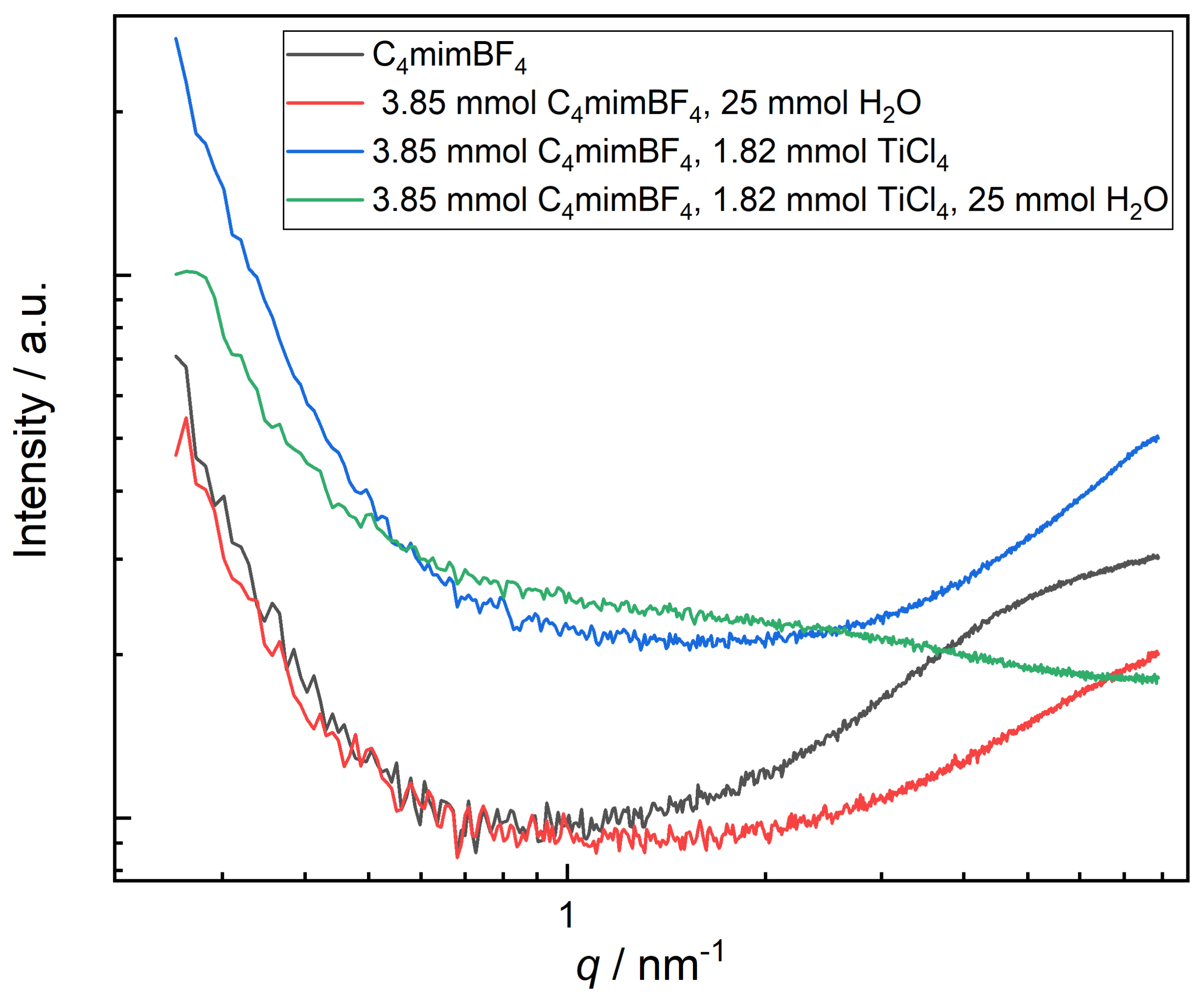
2.4. Small-Angle X-Ray Scattering (SAXS)
2.5. Domain and Voronoi Analysis
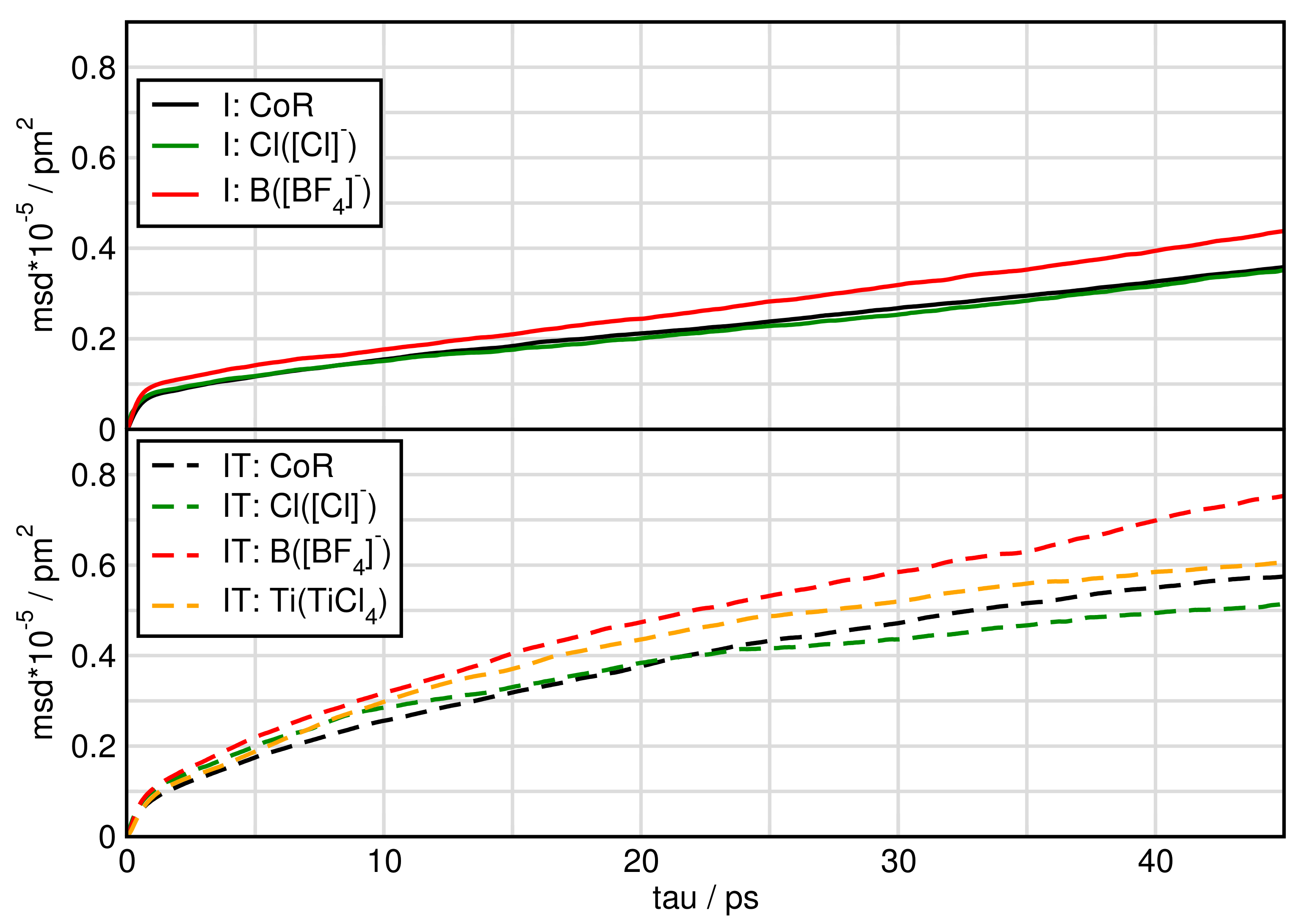
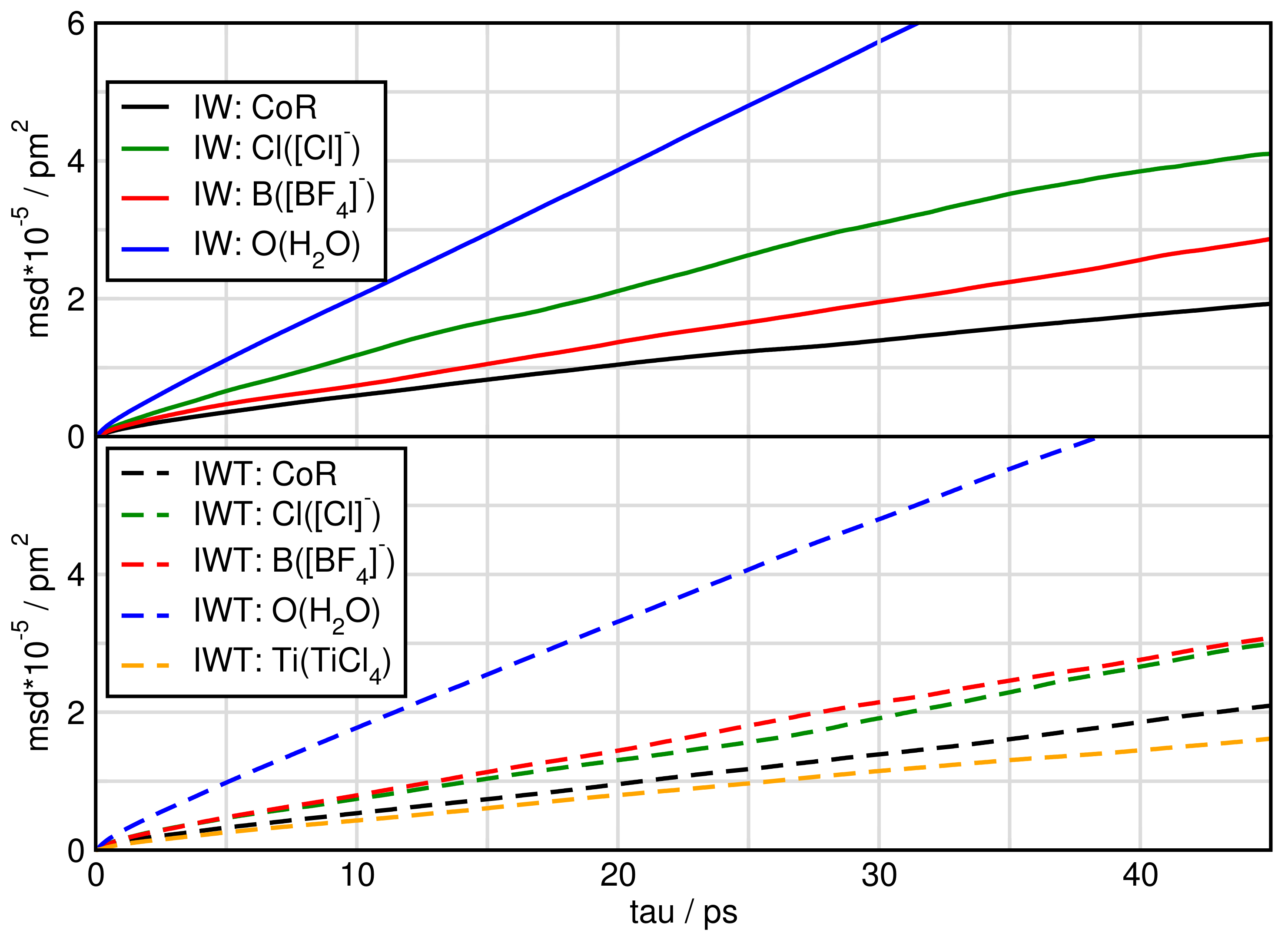
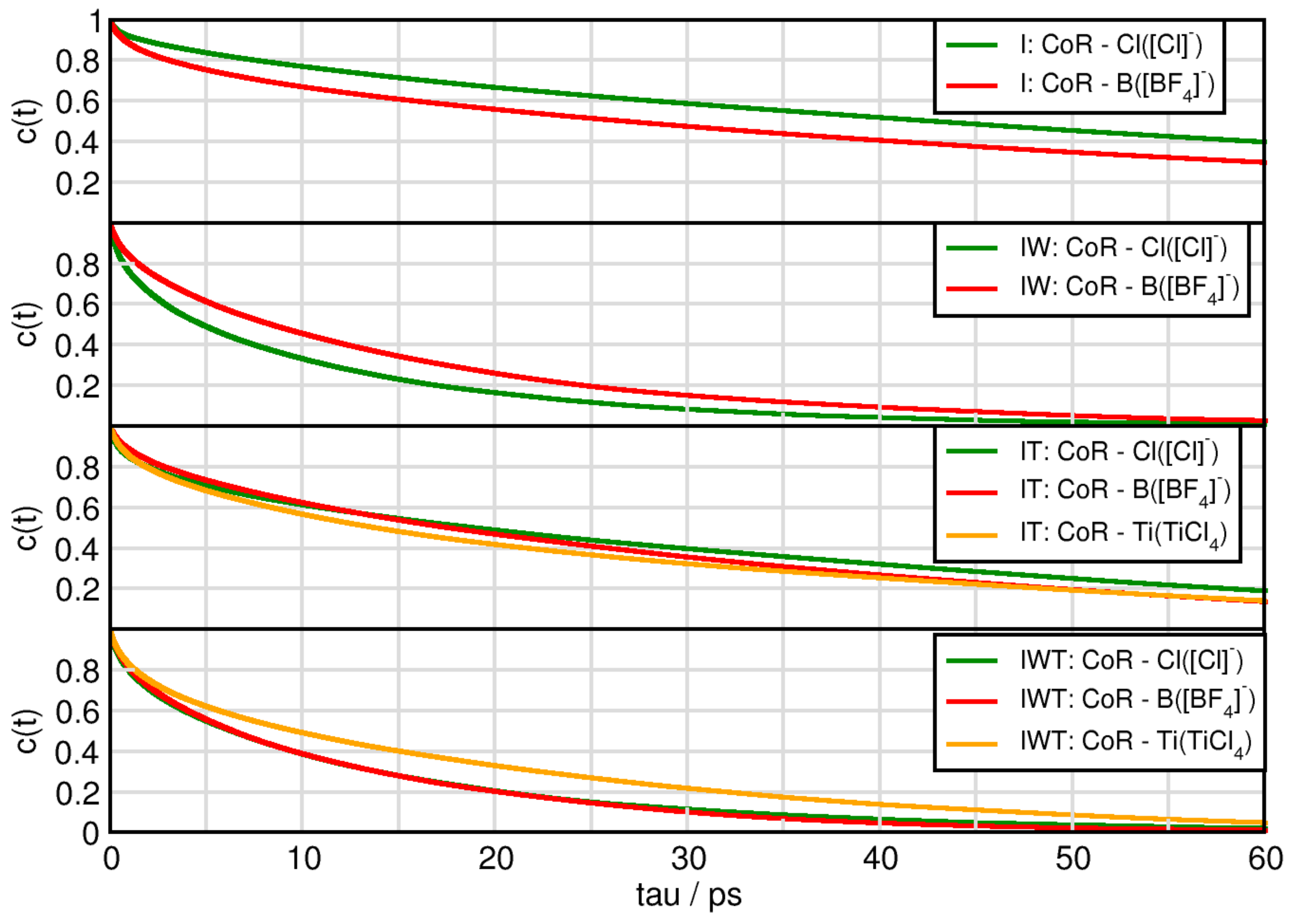
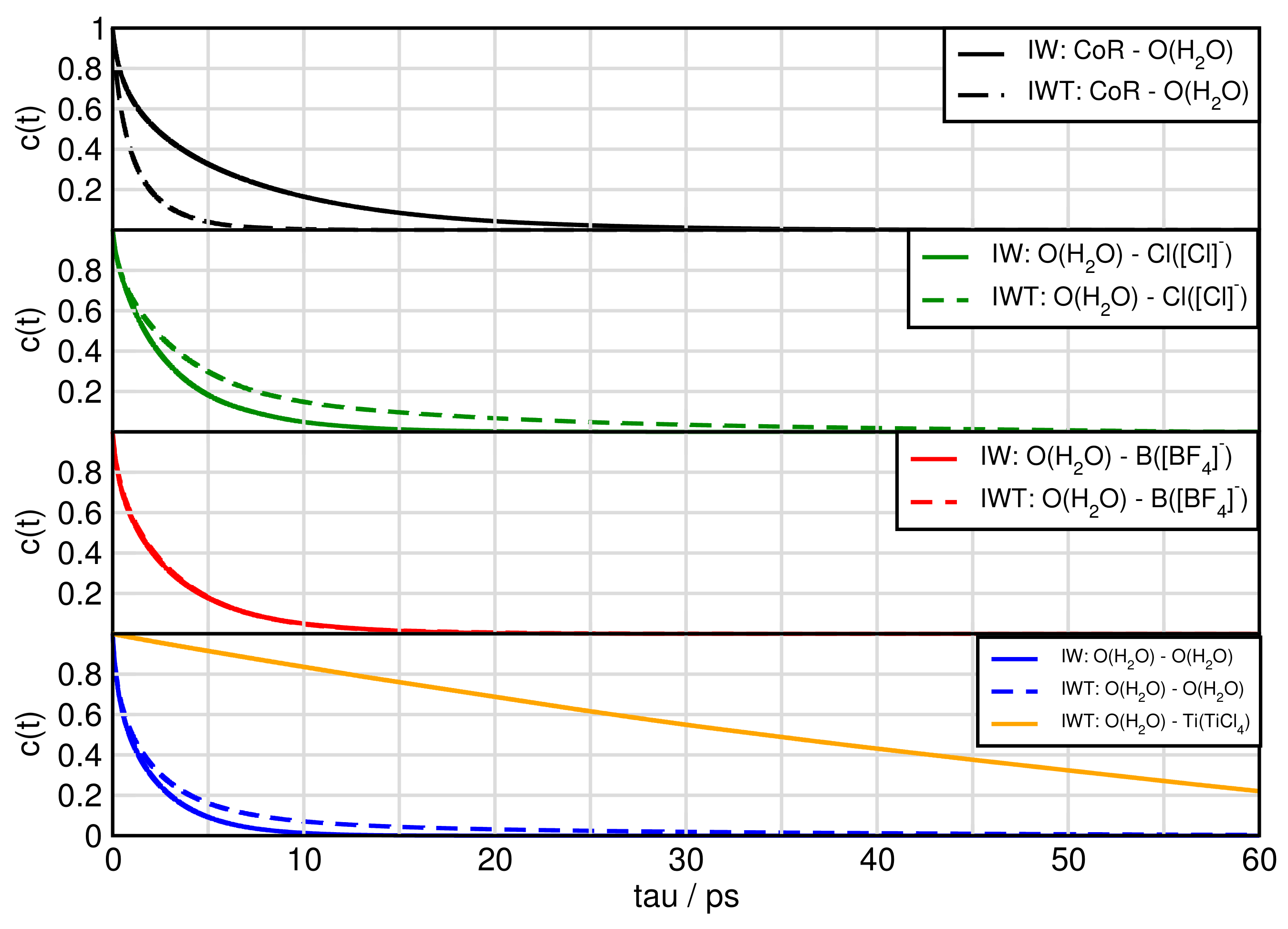
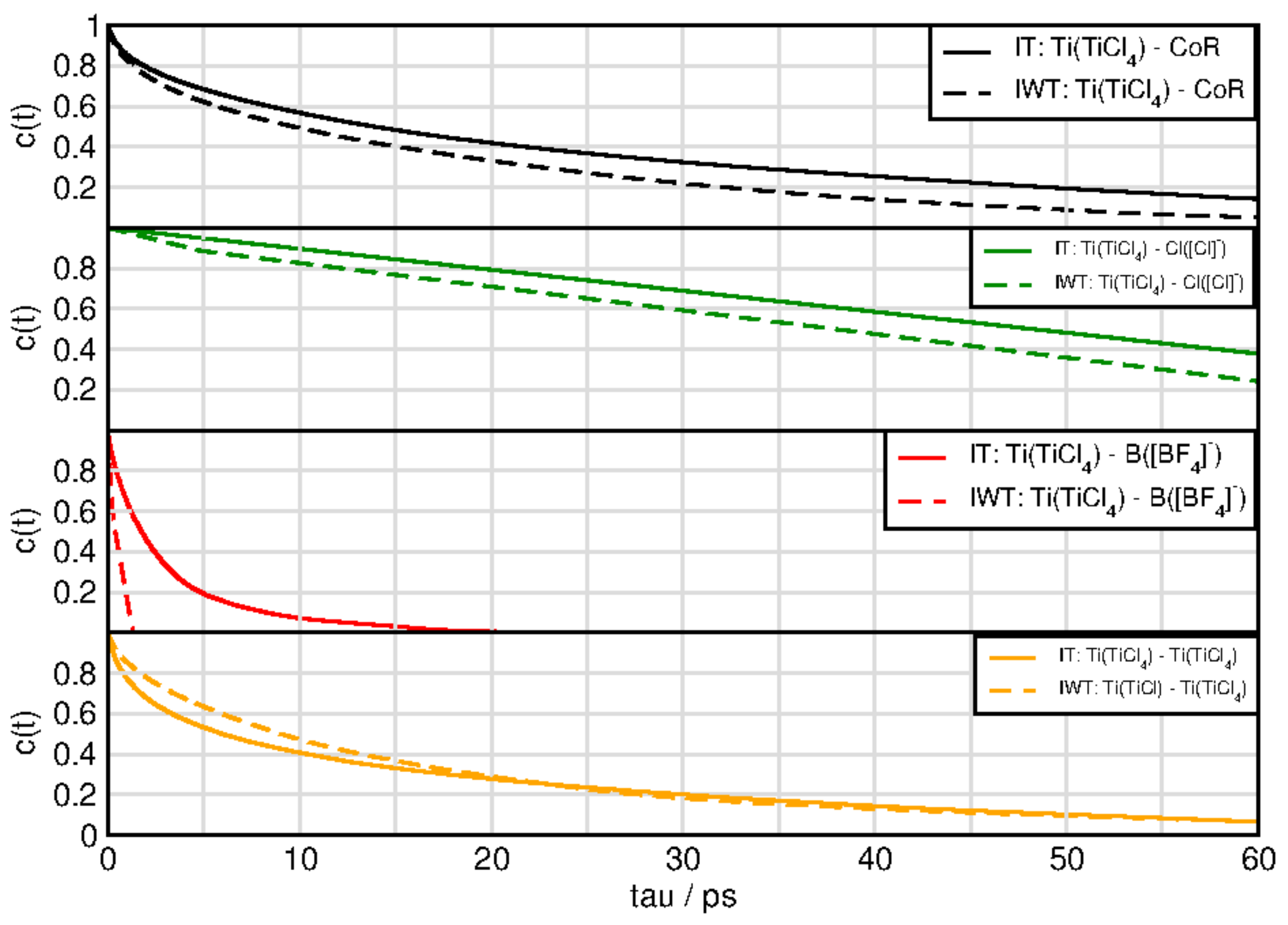
2.6. Dynamical Properties
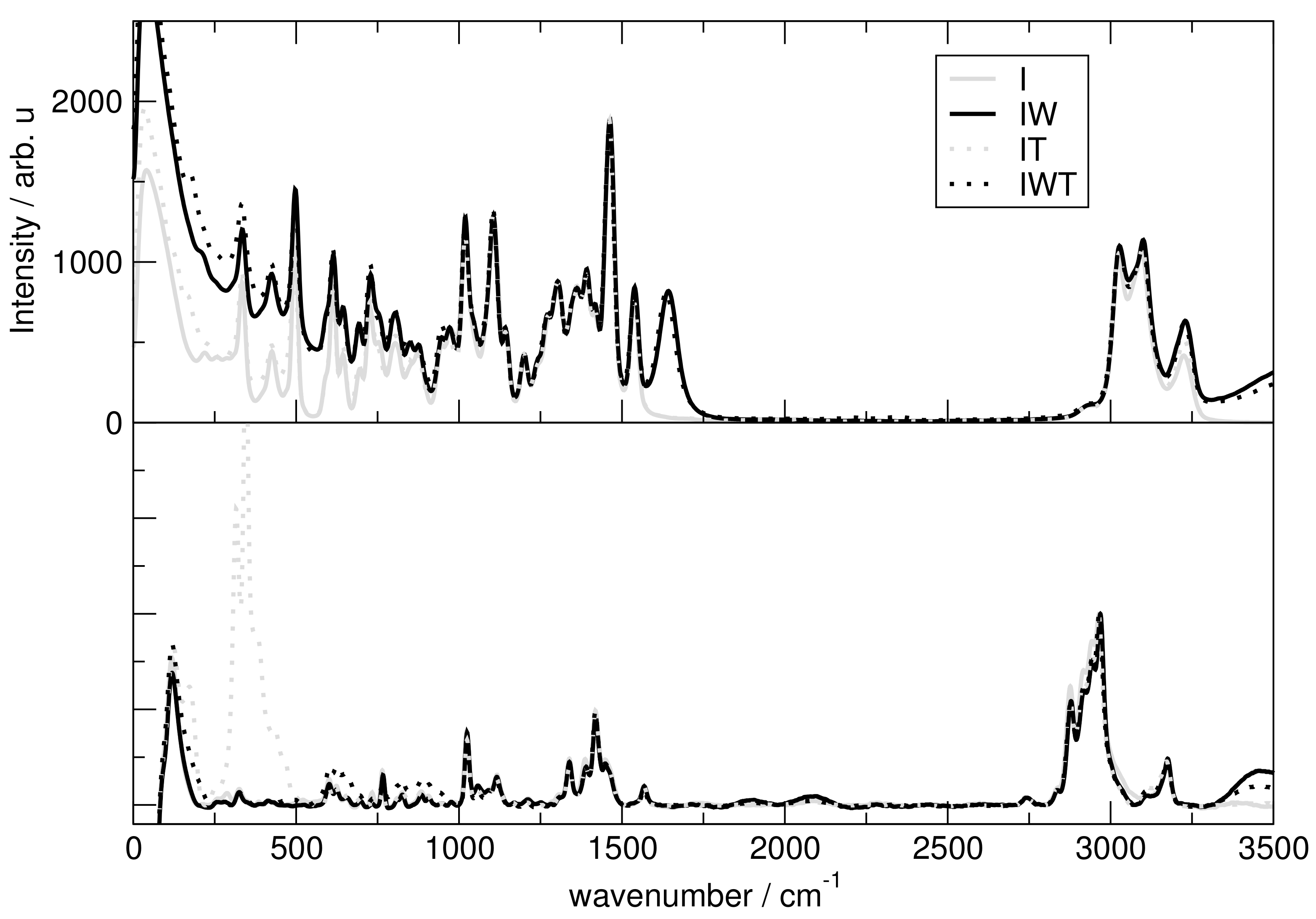
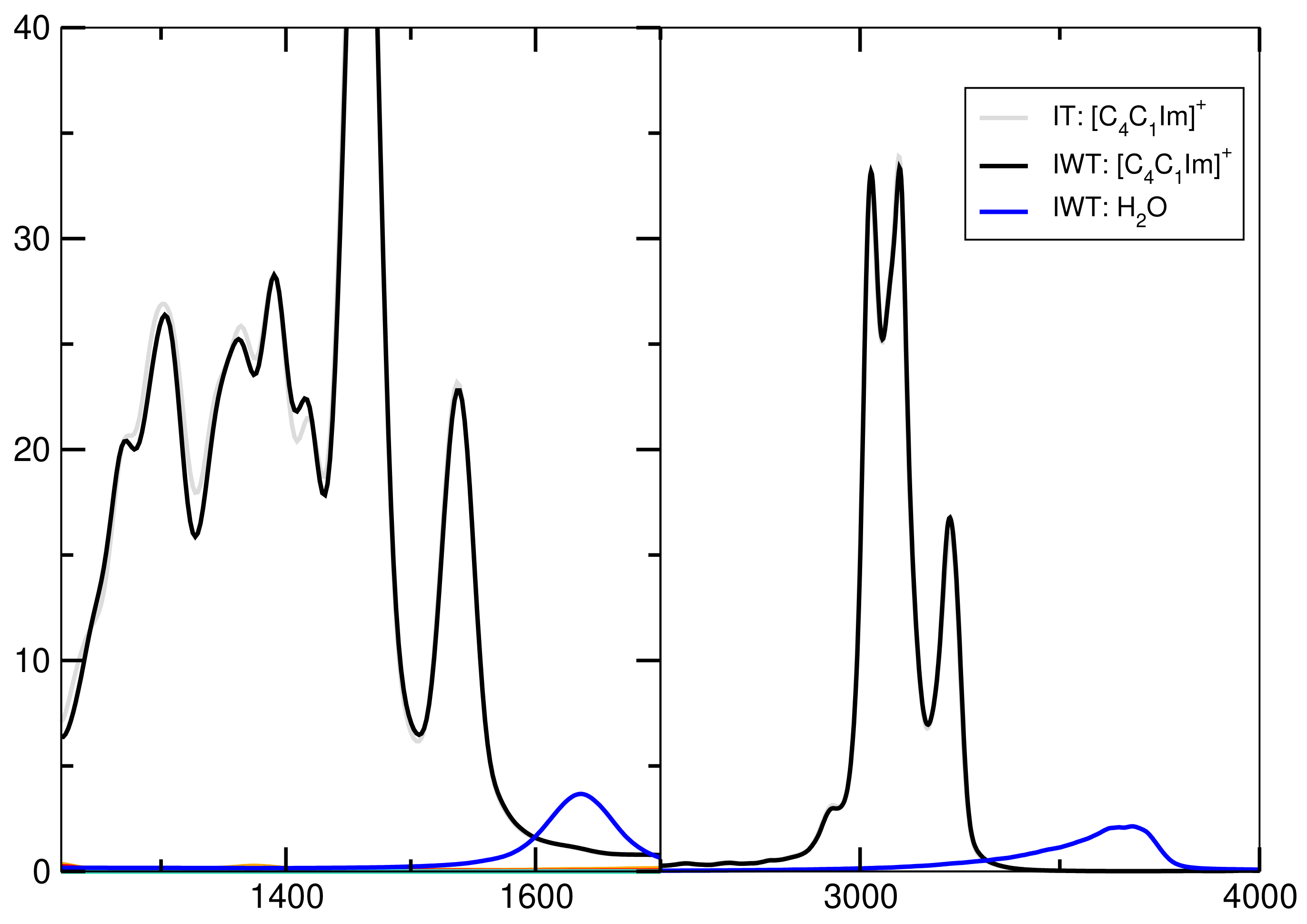
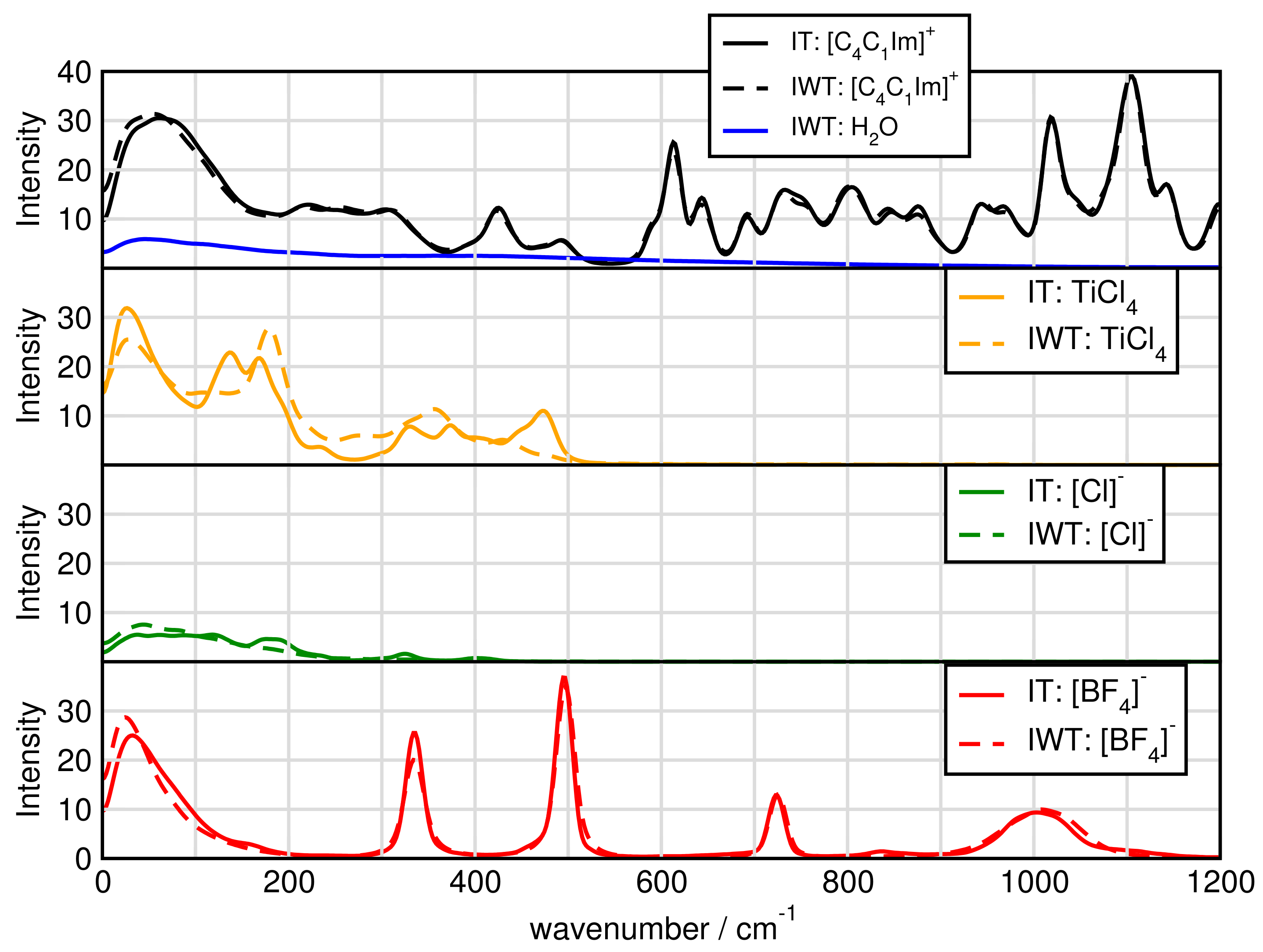
2.7. Spectroscopy
3. Discussion
4. Materials and Methods
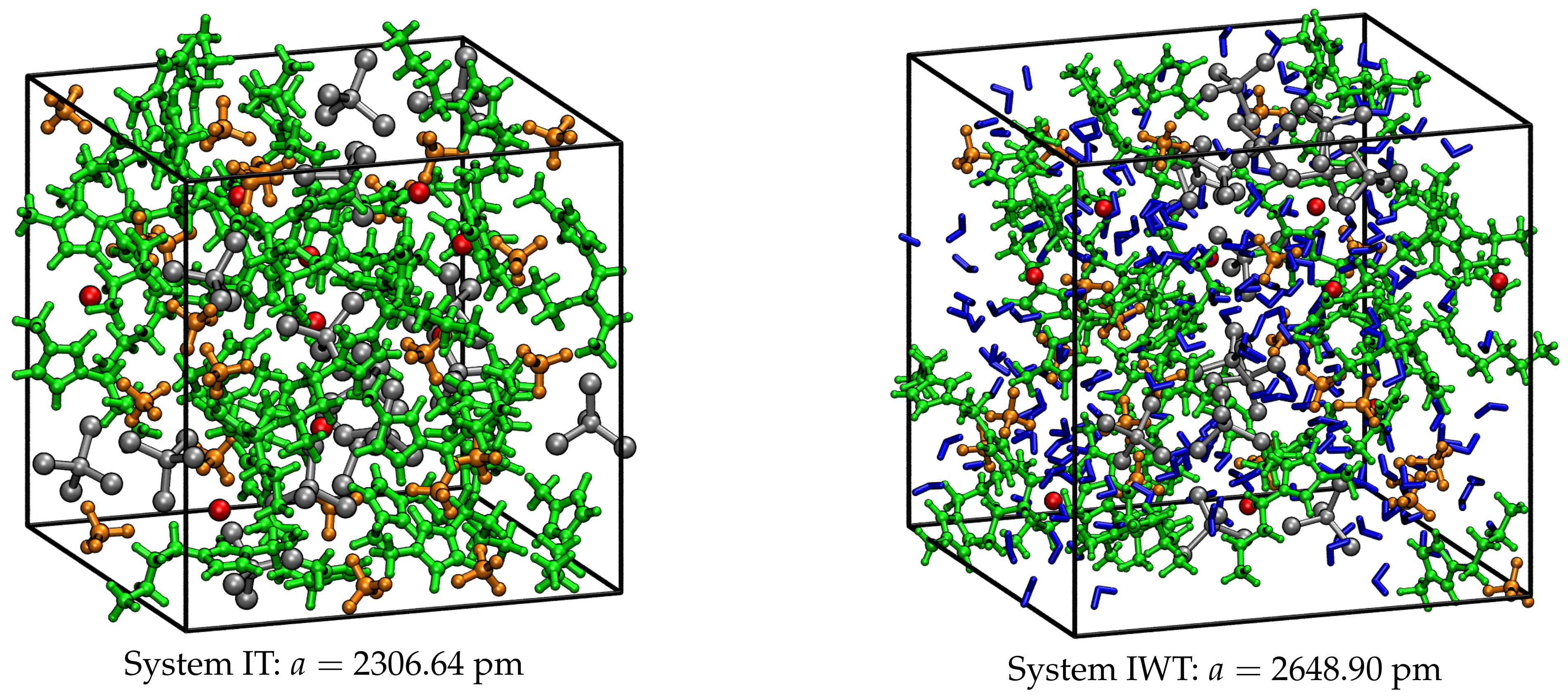
4.1. Composition and Preparation of Simulation Boxes
4.2. Pre-equilibration via Classical MD Simulations
4.3. AIMD Simulations
4.4. Small-Angle X-Ray Scattering (SAXS)
4.5. Raman Spectroscopy
4.6. Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IL | ionic liquid |
| AIMD | ab initio molecular dynamics |
| RDF | radial distribution function |
| NI | number integral |
| CoR | center of ring |
| IR | infrared |
| SAXS | small angle X-ray scattering |
References
- Urahata, S.M.; Ribeiro, M.C.C. Structure of ionic liquids of 1-alkyl-3-methylimidazolium cations: A systematic computer simulation study. J. Chem. Phys. 2004, 120, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Voth, G. Unique spatial heterogeneity in ionic liquids. J. Am. Chem. Soc. 2005, 127, 12192–12193. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.; Pádua, A.A. Nanostructural organization in ionic liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Elfgen, R.; Hollóczki, O.; Kirchner, B. A molecular level understanding of template effects in ionic liquids. Acc. Chem. Res. 2017, 50, 2949–2957. [Google Scholar] [CrossRef]
- Hollóczki, O.; Macchiagodena, M.; Weber, H.; Thomas, M.; Brehm, M.; Stark, A.; Russina, O.; Triolo, A.; Kirchner, B. Triphilic Ionic-Liquid Mixtures: Fluorinated and Non-fluorinated Aprotic Ionic-Liquid Mixtures. ChemPhysChem 2015, 16, 3325–3333. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Thomas, M.; Hollóczki, O.; Kirchner, B. Domain Analysis in Nanostructured Liquids: A Post-Molecular Dynamics Study at the Example of Ionic Liquids. ChemPhysChem 2015, 16, 3271–3277. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Maginn, E.J. Ionic liquids: Innovative fluids for chemical processing. AIChE J. 2001, 47, 2384–2389. [Google Scholar] [CrossRef]
- Kirchner, B.; Hollóczki, O.; Canongia Lopes, J.N.; Pádua, A.A.H. Multiresolution calculation of ionic liquids. WIREs Comp. Mol. Sci. 2014, 202–214. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef]
- Wishart, J.F. Energy applications of ionic liquids. Energy Environ. Sci. 2009, 2, 956–961. [Google Scholar] [CrossRef]
- Galiński, M.; Lewandowski, A.; Stępniak, I. Ionic liquids as electrolytes. Electrochim. Acta 2006, 51, 5567–5580. [Google Scholar] [CrossRef]
- Béguin, F.; Presser, V.; Balducci, A.; Frackowiak, E. Carbons and electrolytes for advanced supercapacitors. Adv. Mater. 2014, 26, 2219–2251. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Bredow, T.; Kirchner, B. Adsorption Behavior of the 1,3-Dimethylimidazolium Thiocyanate and Tetracyanoborate Ionic Liquids at Anatase (101) Surface. J. Phys. Chem. C 2015, 119, 15137–15149. [Google Scholar] [CrossRef]
- Weber, H.; Kirchner, B. Ionic liquid induced band shift of titanium dioxide. ChemSusChem 2016, 9, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Balducci, A.; Bardi, U.; Caporali, S.; Mastragostino, M.; Soavi, F. Ionic liquids for hybrid supercapacitors. Electrochem. Commun. 2004, 6, 566–570. [Google Scholar] [CrossRef]
- Balducci, A.; Henderson, W.A.; Mastragostino, M.; Passerini, S.; Simon, P.; Soavi, F. Cycling stability of a hybrid activated carbon//poly (3-methylthiophene) supercapacitor with N-butyl-N-methylpyrrolidinium bis (trifluoromethanesulfonyl) imide ionic liquid as electrolyte. Electrochim. Acta 2005, 50, 2233–2237. [Google Scholar] [CrossRef]
- Salanne, M.; Madden, P.A. Polarization effects in ionic solids and melts. Mol. Phys. 2011, 109, 2299–2315. [Google Scholar] [CrossRef]
- Merlet, C.; Rotenberg, B.; Madden, P.A.; Taberna, P.L.; Simon, P.; Gogotsi, Y.; Salanne, M. On the molecular origin of supercapacitance in nanoporous carbon electrodes. Nat. Mat. 2012, 11, 306. [Google Scholar] [CrossRef]
- Salanne, M.; Siqueira, L.J.A.; Seitsonen, A.P.; Madden, P.A.; Kirchner, B. From molten salts to room temperature ionic liquids: Simulation studies on chloroaluminate systems. Faraday Discuss. 2012, 154, 171–188. [Google Scholar] [CrossRef]
- Wu, B.; Kuroda, K.; Takahashi, K.; Castner, E.W., Jr. Structural analysis of zwitterionic liquids vs. homologous ionic liquids. J. Chem. Phys. 2018, 148, 193807. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Anthony, J.L.; Shah, J.K.; Morrow, T.I.; Brennecke, J.F.; Maginn, E.J. Why is CO2 so soluble in imidazolium-based ionic liquids? J. Am. Chem. Soc. 2004, 126, 5300–5308. [Google Scholar] [CrossRef] [PubMed]
- Elfgen, R.; Hollóczki, O.; Ray, P.; Groh, M.F.; Ruck, M.; Kirchner, B. Theoretical Investigation of the Te4Br2 Molecule in Ionic Liquids. Z. Anorg. Allg. Chem. 2017, 643, 41–52. [Google Scholar] [CrossRef]
- Voepel, P.; Seitz, C.; Waack, J.M.; Zahn, S.; Leichtweiß, T.; Zaichenko, A.; Mollenhauer, D.; Amenitsch, H.; Voggenreiter, M.; Polarz, S. Peering into the mechanism of low-temperature synthesis of bronze-type TiO2 in ionic liquids. Cryst. Growth Des. 2017, 17, 5586–5601. [Google Scholar] [CrossRef]
- Cooper, E.R.; Andrews, C.D.; Wheatley, P.S.; Webb, P.B.; Wormald, P.; Morris, R.E. Ionic liquids and eutectic mixtures as solvent and template in synthesis of zeolite analogues. Nature 2004, 430, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.V.; Peterson, D.C.; Zaworotko, M.J. Ternary hydrogen halide/base/benzene mixtures: A new generation of liquid clathrates. J. Incl. Phenom. 1988, 6, 425–428. [Google Scholar] [CrossRef]
- Blanchard, L.A.; Brennecke, J.F. Recovery of organic products from ionic liquids using supercritical carbon dioxide. Ind. Eng. Chem. Res. 2001, 40, 287–292. [Google Scholar] [CrossRef]
- Holbrey, J.D.; Reichert, W.M.; Nieuwenhuyzen, M.; Sheppard, O.; Hardacre, C.; Rogers, R.D. Liquid clathrate formation in ionic liquid–aromatic mixtures. Chem. Commun. 2003, 476–477. [Google Scholar] [CrossRef]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Möck, A.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef]
- Nyce, G.W.; Lamboy, J.A.; Connor, E.F.; Waymouth, R.M.; Hedrick, J.L. Expanding the catalytic activity of nucleophilic N-heterocyclic carbenes for transesterification reactions. Org. Lett. 2002, 4, 3587–3590. [Google Scholar] [CrossRef]
- Reichert, W.M.; Holbrey, J.D.; Vigour, K.B.; Morgan, T.D.; Broker, G.A.; Rogers, R.D. Approaches to crystallization from ionic liquids: Complex solvents-complex results, or, a strategy for controlled formation of new supramolecular architectures? Chem. Commun. 2006, 4767–4779. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E. Ionothermal synthesis—Ionic liquids as functional solvents in the preparation of crystalline materials. Chem. Commun. 2009, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yu, J.; Dai, S. Preparation of Inorganic Materials Using Ionic Liquids. Adv. Mater. 2010, 22, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Mudring, A.V.; Tang, S. Ionic Liquids for Lanthanide and Actinide Chemistry. Eur. J. Inorg. Chem. 2010, 2010, 2569–2581. [Google Scholar] [CrossRef]
- Taubert, A. Heavy Elements in Ionic Liquids. In Ionic Liquids; Kirchner, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 127–159. [Google Scholar]
- Ahmed, E.; Ruck, M. Homo- and heteroatomic polycations of groups 15 and 16. Recent advances in synthesis and isolation using room temperature ionic liquids. Coord. Chem. Rev. 2011, 255, 2892–2903. [Google Scholar] [CrossRef]
- Vollmer, C.; Janiak, C. Naked metal nanoparticles from metal carbonyls in ionic liquids: Easy synthesis and stabilization. Coord. Chem. Rev. 2011, 255, 2039–2057. [Google Scholar] [CrossRef]
- Freudenmann, D.; Wolf, S.; Wolff, M.; Feldmann, C. Ionic Liquids: New Perspectives for Inorganic Synthesis? Angew. Chem. Int. Ed. 2011, 50, 11050–11060. [Google Scholar] [CrossRef]
- Ahmed, E.; Breternitz, J.; Groh, M.F.; Ruck, M. Ionic liquids as crystallisation media for inorganic materials. CrystEngComm 2012, 14, 4874–4885. [Google Scholar] [CrossRef]
- Groh, M.F.; Wolff, A.; Grasser, M.A.; Ruck, M. Controlled Synthesis of Polyions of Heavy Main-Group Elements in Ionic Liquids. Int. J. Mol. Sci. 2016, 17, 1452. [Google Scholar] [CrossRef]
- Santner, S.; Heine, J.; Dehnen, S. Synthesis of Crystalline Chalcogenides in Ionic Liquids. Angew. Chem. Int. Ed. 2016, 55, 876–893. [Google Scholar] [CrossRef]
- Parnham, E.R.; Morris, R.E. Ionothermal synthesis of zeolites, metal–organic frameworks, and inorganic–organic hybrids. Accounts Chem. Res. 2007, 40, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Feldmann, C. Structural Variety in the Pseudo-ternary System M/Ge/I: α-[NMe(nBu)3](GeI4)I, β-[NMe(nBu)3](GeI4)I and [ImMe(nBu)][N(nBu)4](GeI4)3I2. Z. Anorg. Allg. Chem. 2009, 635, 1179–1186. [Google Scholar] [CrossRef]
- Okrut, A.; Feldmann, C. Ein neues cis-[Bi3I12]3‒-Anion in Tri(n-butyl)methylammonium Dodecaiodotribismutat. Z. Anorg. Allg. Chem. 2006, 632, 409–412. [Google Scholar] [CrossRef]
- Rabenau, A.; Rau, H.; Rosenstein, G. Tellurium Halide Systems: Te2Br and Te3Cl2. Angew. Chem. Int. Ed. 1970, 9, 802–803. [Google Scholar] [CrossRef]
- Groh, M.F.; Mueller, U.; Ahmed, E.; Rothenberger, A.; Ruck, M. Substitution of conventional high-temperature syntheses of inorganic compounds by near-room-temperature syntheses in ionic liquids. Z. Naturforsch. B 2013, 68, 1108–1122. [Google Scholar] [CrossRef]
- Ahmed, E.; Beck, J.; Daniels, J.; Doert, T.; Eck, S.J.; Heerwig, A.; Isaeva, A.; Lidin, S.; Ruck, M.; Schnelle, W.; et al. Halbleiter oder eindimensionales Metall und Supraleiter durch Tellur-p-Stapelung. Angew. Chem. 2012, 124, 8230–8233. [Google Scholar] [CrossRef]
- Ahmed, E.; Beck, J.; Daniels, J.; Doert, T.; Eck, S.J.; Heerwig, A.; Isaeva, A.; Lidin, S.; Ruck, M.; Schnelle, W.; et al. A Semiconductor or A One-Dimensional Metal and Superconductor through Tellurium p Stacking. Angew. Chem. Int. Ed. 2012, 51, 8106–8109. [Google Scholar] [CrossRef]
- Ahmed, E.; Ahrens, E.; Heise, M.; Ruck, M. A Facile Route for the Synthesis of Polycationic Tellurium Cluster Compounds: Synthesis in Ionic Liquid Media and Characterization by Single-Crystal X-ray Crystallography and Magnetic Susceptibility. Z. Anorg. Allg. Chem. 2010, 636, 2602–2606. [Google Scholar] [CrossRef]
- Schulz, C.; Daniels, J.; Bredow, T.; Beck, J. Die elektrochemische Synthese polykationischer Cluster. Angew. Chem. 2016, 128, 1188–1192. [Google Scholar] [CrossRef]
- Schulz, C.; Daniels, J.; Bredow, T.; Beck, J. The Electrochemical Synthesis of Polycationic Clusters. Angew. Chem. Int. Ed. 2016, 55, 1173–1177. [Google Scholar] [CrossRef]
- D’Angelo, P.; Serva, A.; Aquilanti, G.; Pascarelli, S.; Migliorati, V. Structural properties and aggregation behavior of 1-hexyl-3-methylimidazolium iodide in aqueous solutions. J. Phys. Chem. B 2015, 119, 14515–14526. [Google Scholar] [CrossRef] [PubMed]
- Serva, A.; Migliorati, V.; Lapi, A.; Aquilanti, G.; Arcovito, A.; D’Angelo, P. Structural properties of geminal dicationic ionic liquid/water mixtures: A theoretical and experimental insight. Phys. Chem. Chem. Phys. 2016, 18, 16544–16554. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, B.; Seitsonen, A.P. Ionic liquids from Car-Parrinello simulations. 2. Structural diffusion leading to large anions in chloraluminate ionic liquids. Inorg. Chem. 2007, 46, 2751–2754. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, B.; Seitsonen, A.P. Green Chemistry from Supercomputers: Car–Parrinello Simulations of Emim-Chloroaluminates Ionic Liquids. In High Performance Computing in Science and Engineering 07; Springer: Berlin/Heidelberg, Germany, 2008; pp. 157–171. [Google Scholar]
- Hollóczki, O. Toward Anionic Structural Diffusion and Highly Conducting Ionic Liquid Electrolytes. ACS Sustain. Chem. Eng. 2018, 7, 2626–2633. [Google Scholar] [CrossRef]
- Hollóczki, O.; Wolff, A.; Pallmann, J.; Whiteside, R.E.; Hartley, J.; Grasser, M.A.; Nockemann, P.; Brunner, E.; Doert, T.; Ruck, M. Spontaneous Substitutions at Phosphorus Trihalides in Imidazolium Halide Ionic Liquids: Grotthuss Diffusion of Anions? Chem. Eur. J. 2018, 24, 16323–16331. [Google Scholar] [CrossRef]
- Thorsmølle, V.K.; Rothenberger, G.; Topgaard, D.; Brauer, J.C.; Kuang, D.B.; Zakeeruddin, S.M.; Lindman, B.; Grätzel, M.; Moser, J.E. Extraordinarily Efficient Conduction in a Redox-Active Ionic Liquid. ChemPhysChem 2011, 12, 145–149. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Yethiraj, A. Grotthuss Transport of Iodide in EMIM/I3 Ionic Crystal. J. Phys. Chem. B 2018, 122, 250–257. [Google Scholar] [CrossRef]
- Abe, H.; Aono, M.; Kiyotani, T.; Tsuzuki, S. Polyiodides in room-temperature ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 32337–32344. [Google Scholar] [CrossRef]
- Kirchner, B.; Intemann, B. Molecular transport: Catch the carbon dioxide. Nat. Chem. 2016, 8, 401–402. [Google Scholar] [CrossRef]
- Corradini, D.; Coudert, F.X.; Vuilleumier, R. Carbon dioxide transport in molten calcium carbonate occurs through an oxo-Grotthuss mechanism via a pyrocarbonate anion. Nat. Chem. 2016, 8, 454. [Google Scholar] [CrossRef]
- Tokuda, H.; Tsuzuki, S.; Susan, M.A.B.H.; Hayamizu, K.; Watanabe, M. How ionic are room-temperature ionic liquids? An indicator of the physicochemical properties. J. Phys. Chem. B 2006, 110, 19593–19600. [Google Scholar] [CrossRef] [PubMed]
- Pensado, A.S.; Brehm, M.; Thar, J.; Seitsonen, A.P.; Kirchner, B. Effect of Dispersion on the Structure and Dynamics of the Ionic Liquid 1-Ethyl-3-methylimidazolium Thiocyanate. ChemPhysChem 2012, 13, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.B.C.; Roatsch, M.; Schoppke, M.; Kirchner, B. On the physical origin of the cation-anion intermediate bond in ionic liquids Part I. Placing a (weak) hydrogen bond between two charges. Phys. Chem. Chem. Phys. 2010, 12, 7473–7486. [Google Scholar] [CrossRef] [PubMed]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Design 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.M.; Martinez, L. Packing optimization for automated generation of complex system’s initial configurations for molecular dynamics and docking. J. Comp. Chem. 2003, 24, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comp. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A. Modeling ionic liquids using a systematic all-atom force field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A. Molecular force field for ionic liquids composed of triflate or bistriflylimide anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Lorentz, H. Nachtrag zu der Abhandlung: Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Der Phys. 1881, 248, 660–661. [Google Scholar] [CrossRef]
- Berthelot, D. Sur le mélange des gaz. Compt. Rendus 1898, 126, 1703–1706. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- CP2k, A General Program to Perform Molecular Dynamics Simulations. CP2k Developers Group Under the Terms of the GNU General Public License. Available online: http://www.cp2k.org (accessed on 1 November 2020).
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr Optimized for Gradient-Corrected Exchange-Correlation Functionals. Theor. Chem. Accounts 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Kirchner, B. TRAVIS—A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | [CCIm] | [BF] | [Cl] | TiCl | HO |
|---|---|---|---|---|---|
 |  |  |  |  | |
| I | 32 | 22 | 10 | − | − |
| IW | 32 | 22 | 10 | − | 211 |
| IT | 32 | 22 | 10 | 15 | − |
| IWT | 32 | 22 | 10 | 15 | 211 |
| r(CoR-Cl) | NI | r(H2-Cl) | NI | |||
|---|---|---|---|---|---|---|
| r/pm | r/pm | |||||
| max min | max min | |||||
| I | 452 700 | 1.8 | 5.6 | 239 455 | 0.5 | 1.7 |
| IW | 458 700 | 0.9 | 2.9 | 259 402 | 0.1 | 0.4 |
| IT | 449 700 | 1.5 | 4.8 | 240 455 | 0.4 | 1.3 |
| IWT | 462 700 | 0.9 | 3.0 | 257 402 | 0.2 | 0.7 |
| (I-IW) | −6 0 | 0.9 | 2.7 | −20 53 | 0.4 | 1.3 |
| (IT-IWT) | −13 0 | 0.6 | 1.8 | −17 53 | 0.2 | 0.6 |
| (I-IT) | 3 0 | 0.3 | 0.8 | −1 0 | 0.1 | 0.4 |
| (IW-IWT) | −4 0 | 0.0 | −0.1 | 2 0 | −0.1 | −0.3 |
| r(CoR−B([BF])) | NI | r(H2- F([BF])) | NI | |||
| r/pm | r/pm | |||||
| max min | max min | |||||
| I | 492 700 | 3.6 | 5.3 | 233 355 | 2.0 | 0.7 |
| IW | 502 700 | 2.5 | 3.7 | 232 330 | 0.9 | 0.3 |
| IT | 490 700 | 3.1 | 4.6 | 233 355 | 1.8 | 0.6 |
| IWT | 505 700 | 2.0 | 3.0 | 232 330 | 0.8 | 0.3 |
| (I-IW) | −10 0 | 1.1 | 1.6 | 1 25 | 1.1 | 0.4 |
| (IT-IWT) | −15 0 | 1.1 | 1.6 | 1 25 | 1.0 | 0.3 |
| (I-IT) | 2 0 | 0.5 | 0.7 | 0 0 | 0.2 | 0.1 |
| (IW-IWT) | −3 0 | 0.5 | 0.7 | 0 0 | 0.1 | 0.0 |
| r(O(HO)-H2) | O(HO)-H(HO) | r(O(HO)-O(HO) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| max | min | NI | max | min | NI | max | min | NI | ||||
| IW | 240 | 400 | 0.4 | 2.6 | 186 | 254 | − | − | 286 | 400 | 4.6 | 4.6 |
| IWT | 245 | 400 | 0.3 | 2.0 | 184 | 254 | − | − | 284 | 400 | 4.0 | 4.0 |
| (IW-IWT) | 5 | 0 | −0.1 | −0.6 | 2 | 0 | − | − | 2.0 | 0.0 | 0.6 | 0.6 |
| r(H(HO)-H) | r(H(HO)-Cl) | r(H(HO)-F([BF]) | ||||||||||
| max | min | NI | max | min | NI | max | min | NI | ||||
| IW | − | − | − | − | 222 | 300 | 0.1 | 4.6 | 210 | 270 | 0.3 | 1.3 |
| IWT | − | − | − | − | 219 | 300 | 0.1 | 3.6 | 206 | 270 | 0.3 | 1.4 |
| (IW-IWT) | − | − | − | − | −3 | 0 | 0 | 1.0 | 4 | 0 | 0 | −0.1 |
| P | NP | TiCl | HO | |
|---|---|---|---|---|
| I | 1.0 | 1.0 | − | − |
| IW | 1.2 | 5.3 | − | 1.2 |
| IT | 1.0 | 1.5 | 1.4 | − |
| IWT | 1.1 | 5.4 | 2.3 | 1.1 |
| ring | chain | [BF] | [Cl] | TiCl | HO | |
|---|---|---|---|---|---|---|
| IT | 1.1 | 1.5 | 9.1 | 8.9 | 1.4 | − |
| IWT | 2.0 | 5.4 | 13.8 | 9.3 | 2.3 | 1.1 |
| Ring | CH | [BF] | [Cl] | TiCl | HO | |
|---|---|---|---|---|---|---|
| system I | ||||||
| ring | 27.8 | 49.8 | 65.3 | 81.0 | − | − |
| CH | 35.7 | 28.9 | 31.3 | 16.8 | − | − |
| [BF] | 28.4 | 19.0 | 3.0 | 1.8 | − | − |
| [Cl] | 8.1 | 2.3 | 0.4 | 0.4 | − | − |
| system IW | ||||||
| ring | 12.3 | 36.5 | 39.0 | 25.5 | − | 20.0 |
| CH | 25.9 | 15.9 | 17.4 | 10.7 | − | 12.0 |
| [BF] | 16.8 | 10.5 | 2.2 | 0.6 | − | 8.4 |
| [Cl] | 2.5 | 1.5 | 0.1 | 0.0 | − | 2.9 |
| HO | 42.5 | 35.6 | 41.3 | 63.2 | − | 56.7 |
| system IT | ||||||
| ring | 19.7 | 41.0 | 58.1 | 61.9 | 41.3 | − |
| CH | 29.5 | 24.0 | 25.8 | 13.6 | 26.8 | − |
| [BF] | 25.1 | 15.5 | 3.0 | 2.4 | 11.4 | − |
| [Cl] | 6.0 | 1.8 | 1.0 | 0.8 | 4.4 | − |
| TiCl | 19.7 | 17.7 | 12.6 | 21.3 | 16.2 | − |
| system IWT | ||||||
| ring | 9.1 | 34.5 | 32.2 | 30.6 | 27.1 | 17.1 |
| CH | 24.6 | 13.6 | 15.9 | 10.6 | 17.0 | 9.9 |
| [BF] | 13.8 | 9.5 | 1.5 | 1.3 | 2.3 | 9.5 |
| [Cl] | 3.0 | 1.5 | 0.3 | 0.3 | 1.0 | 2.4 |
| TiCl | 12.7 | 11.2 | 2.5 | 4.9 | 12.2 | 8.8 |
| HO | 36.8 | 29.7 | 47.6 | 52.3 | 40.5 | 52.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esser, L.; Macchieraldo, R.; Elfgen, R.; Sieland, M.; Smarsly, B.M.; Kirchner, B. TiCl4 Dissolved in Ionic Liquid Mixtures from Аb Initio Molecular Dynamics Simulations. Molecules 2021, 26, 79. https://doi.org/10.3390/molecules26010079
Esser L, Macchieraldo R, Elfgen R, Sieland M, Smarsly BM, Kirchner B. TiCl4 Dissolved in Ionic Liquid Mixtures from Аb Initio Molecular Dynamics Simulations. Molecules. 2021; 26(1):79. https://doi.org/10.3390/molecules26010079
Chicago/Turabian StyleEsser, Lars, Roberto Macchieraldo, Roman Elfgen, Melanie Sieland, Bernd Michael Smarsly, and Barbara Kirchner. 2021. "TiCl4 Dissolved in Ionic Liquid Mixtures from Аb Initio Molecular Dynamics Simulations" Molecules 26, no. 1: 79. https://doi.org/10.3390/molecules26010079
APA StyleEsser, L., Macchieraldo, R., Elfgen, R., Sieland, M., Smarsly, B. M., & Kirchner, B. (2021). TiCl4 Dissolved in Ionic Liquid Mixtures from Аb Initio Molecular Dynamics Simulations. Molecules, 26(1), 79. https://doi.org/10.3390/molecules26010079